

Summary
Natural products are the most historically significant source of compounds for drug development. However, unacceptably high rates of compound rediscovery associated with large-scale screening of common microbial producers have resulted in the abandonment of many natural product drug discovery efforts, despite the increasing prevalence of clinically-problematic antibiotic resistance. Screening of underexplored taxa represents one strategy to avoid rediscovery. Herein we report the discovery, isolation, and structural elucidation of streptomonomicin (STM), an antibiotic lasso peptide from Streptomonospora alba, and report the genome for its producing organism. STM-resistant clones of Bacillus anthracis harbor mutations to walR, the gene encoding a response regulator for the only known widely-distributed and essential two-component signal transduction system in Firmicutes. Streptomonospora had been hitherto biosynthetically and genetically uncharacterized, with STM being the first reported compound from the genus. Our results demonstrate that understudied microbes remain fruitful reservoirs for the rapid discovery of novel, bioactive natural products.
Introduction
Microbial biosynthesis represents the most important historical source of chemical matter for the understanding of biology and the development of medicines (Newman and Cragg, 2012). However, despite the value of natural products as medicinal leads, the frequent rediscovery of known compounds renders traditional screening methods (i.e. bioassay-guided isolation) increasingly unappealing and economically disadvantageous (Baltz, 2006). In response, a number of modern natural product discovery strategies have been developed, including genome-guided discovery (Doroghazi et al., 2014), antibiotic resistance-mediated isolation (Thaker et al., 2014), reactivity-based screening (Cox et al., 2014), PCR-based strain prioritization (Hindra et al., 2014), mass spectrometry-based network analysis (Nguyen et al., 2013), heterologous expression (Feng et al., 2010), and metagenomics (Kang and Brady, 2013) with the general aim of reducing the burden of rediscovery and thereby accelerating the drug discovery process.
In widely-studied organisms, it is frequently the case that the abundant natural products are already known; undiscovered compounds are often believed to be silent or at least below a detection threshold. This can complicate identification, purification, structural elucidation, and mechanism-of-action determination efforts. Moreover, broad metabolic and bioinformatic analysis shows that natural product biosynthetic capability tends to parallel bacterial speciation (Doroghazi et al., 2014); hence, rather than to solely concentrate on screening many strains of one particular known producing species, it may also be useful to seek new species in underexplored taxa. Understudied organisms—those that are lab-cultivable yet unsequenced or metabolically uncharacterized—thus present the prospect of discovery of novel, abundant bioactive metabolites with lower rates of rediscovery (Pidot et al., 2014). Among these organisms are the Streptomonospora, a genus within the family Nocardiopsaceae (Cui et al., 2001). Members of the genus are slow-growing halophiles, typically requiring cultivation for a month or more in high-salt media (10–25% w/v NaCl). At least nine distinct Streptomonospora species have been reported; however, no corresponding genome sequences are available. These characteristics have rendered Streptomonospora sp. markedly unattractive both from the standpoint of traditional high-throughput natural product screening campaigns and more modern genome-driven discovery efforts. Perhaps for this reason, the biosynthetic capacity of the Streptomonospora has gone entirely neglected in the 13 years since their first description. As actinomycetes in general have been shown to be unusually talented biosynthetic chemists, we reasoned that investigating an understudied, yet tractable, genus would shed light on the natural product repertoire of these organisms and guide future genome-mining programs.
Taking into account the above, we cultured Streptomonospora alba YIM 90003 (Li et al., 2003) and found an abundant natural product, streptomonomicin (STM), which was isolated and subjected to structural and biological characterization. We also performed whole-genome sequencing of S. alba, revealing STM’s biosynthetic origin and shedding light into the biosynthetic capability of the genus.
Results and Discussion
Isolation of streptomonomicin
The MALDI-TOF mass spectra of intact S. alba YIM 90003 cells and a non-lytic methanol extract were dominated by the presence of an intense peak (m/z 2256, [M+Na]+), indicating an abundant, exported natural product (Figure 1A). Extracts from the organism were fractionated using a C18 Sep-Pak, and the resulting fractions containing m/z 2256 displayed antibacterial activity against Bacillus subtilis in a preliminary bioassay. Accordingly, we named the compound “streptomonomicin”, and by subjecting MeOH extracts of scaled-up cultures of S. alba to purification via C18 Sep-Pak and high-performance liquid chromatography (HPLC), isolated additional material for structural elucidation and further refinement of bioactivity (Figure S1). Yields obtained on solid media ranged from 10–14 mg/l.
Figure 1. Mass spectrometry analysis.
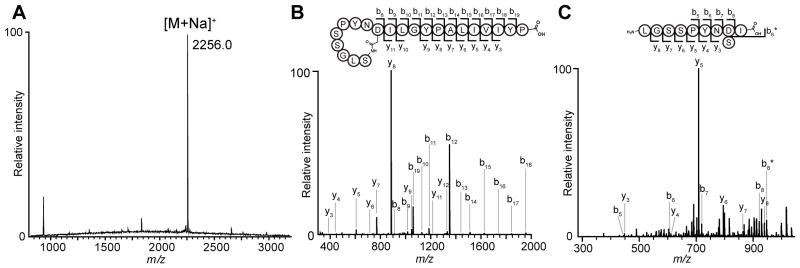
(A) MALDI-TOF mass spectrum of an extract of Streptomonospora alba YIM 90003 showing the compound as the dominant peak (m/z 2256, [M+Na]+). (B) An m/z scan of purified STM that was directly infused into an 11T FTMS resulted in a 2+ charge ion consistent with the calculated m/z for STM (Figure S2A). The ion was fragmented and produced the resulting spectra. A diagram of STM is shown with the observed b- and y-ions labeled on the structure and in the spectra. (C) An m/z scan of purified, carboxypeptidase Y- and thermolysin-digested STM that was directly infused into an 11T FTMS resulted in a 1+ charge ion consistent with the 10 N-terminal residues of STM (Figure S2B). The ion was fragmented and produced the resulting spectra. A diagram of carboxypeptidase Y- and thermolysin-digested STM is shown with the observed b- and y-ions labeled on the structure and in the spectra. See also Figures S1 and S2.
Structure elucidation of streptomonomicin
The structure of STM was determined by the synergistic use of mass spectrometry (MS), nuclear magnetic resonance (NMR) spectroscopy, amino acid analysis, and genome sequencing. The compound was submitted to a full suite of NMR experiments (1H; 1H-1H DQF-COSY, TOCSY, NOESY; 1H-13C HSQC and HMBC; Figure S3) as well as high-resolution Fourier transform (FT) MS/MS analysis (Figures 1 and S2). The FT-MS/MS data (Figure 2 and S2) and 1H-1H TOCSY (Figure S3I) revealed STM to be a peptide, consistent with its UV absorbance spectrum (Figure S1C). Analysis of the TOCSY spectrum revealed the presence of 21 amino acids, which were predominantly hydrophobic: 3 each Leu/Ile/Tyr/Ser/Pro, 2 Gly, 2 Asx (1 Asn, 1 Asp, as later determined, vide infra), 1 Ala, 1 Val (Figure S3I and Table S1). This amino acid composition was consistent with subsequent quantitative amino acid (QAA) analysis (Table S2). Mild hydrolysis and chiral analysis of STM indicated that all residues were the L stereoisomer (Table S2). Resonances of the spin systems were assigned primarily by analysis of TOCSY, HSQC, and HMBC experiments; intra-residue connectivity was established by TOCSY and DQF-COSY.
Figure 2. NMR analysis.

(A) Integrations of amide protons from 1H NMR spectrum in CD3OH indicate a single NH proton is present for each residue. Residue assignments are shown above the spectrum. Peaks (ppm) are shown above residue assignments. Integrations are shown below the spectrum. (B) Expansion of 1H-1H NOESY spectrum in CD3OD showing amide/side chain through-space correlations. The isopeptide Ser1-Asp9 linkage is indicated by the strong NOE crosspeak between the Ser1 NH and the β protons of Asp9, as highlighted in red. (C) Overlaid 1H NMR spectra taken in CD3OD shortly after sample dissolution (black) and 1 d later (red). Residue assignments are shown above the spectrum. Significant resistance to deuterium exchange is evident for many peaks. See also Figure S3 and Table S1.
Importantly, initial MS/MS analysis provided a partial sequence of the C-terminal 11 residues of the peptide (residues 10–21): Ile/Leu-Ile/Leu-Gly-Tyr-Pro-Ala-Ile/Leu-Ile/Leu-Val-Ile/Leu-Tyr-Pro (Figures 1B and S2). This sequence was supported by the HMBC/NOESY data (Figure S3). Interestingly, additional MS/MS fragmentation was not seen N-terminal to this sequence. Moreover, the FT-MS data supported a chemical formula of C107H160N22O30 with an error of <1 ppm, corresponding to the above 21 amino acids with the loss of a single H2O. Integration of the 1H NMR spectrum taken in CD3OH revealed that all the backbone amide resonances corresponded to a single proton each, with the absence of a free N-terminal amine (Figure 2A). Taken together with the lack of MS/MS fragmentation, this indicated that the 9 N-terminal residues were cyclized. Analysis of NOESY spectra revealed one of the earlier-identified Asx residues to be Asn; the other residue displayed a strong NOE correlation to one of the Ser NH resonances, and was thus assigned as a Ser-Asp isopeptide linkage, forming a 9-residue ring (Figure 2B).
To establish the amino acid sequence of the 9-residue ring, we first treated STM with carboxypeptidase Y. Although this did not result in complete C-terminal tail truncation, the enzymatic digestion yielded a mixture primarily containing three products wherein either residues 11–21, 18–21, or 19–21 were removed to give N-terminal macrocycle-containing fragments of 10, 17, or 18 residues, respectively. The 10-residue peptide was further treated with thermolysin, providing a linearized fragment of STM. The MS/MS fragmentation pattern of digested STM allowed partial sequence assignment of the N-terminus and supported the presence of the isopeptide Ser-Asp linkage (Figures 1C and S2).
With the constituents of the macrocycle established, we sought to fully assign the sequence. Using HMBC and NOESY correlations, each of the earlier-identified NMR spin systems was unambiguously assigned, clarifying the identity of the triplicate residues (Tyr, Ser, Pro) and MS-ambiguous Ile/Leu residues and confirming the assigned sequence within the ring (Figure S3 and Table S1). In conjunction, we also sequenced the full genome of S. alba YIM 90003 (vide infra). Using our spectrometrically-obtained primary sequence as a query in a TBLASTN search, we identified a peptide encoded in the S. alba genome whose C-terminal 21 residues corresponded to our structure, thereby corroborating our sequence assignment (Figures 3A and 4).
Figure 3. Structure of streptomonomicin.

(A) Schematic representation of the structure of STM with the isopeptide Ser1-Asp9 linkage indicated. Residues 1–9 (gray) comprise the ring and residues 10–21 (white) are the tail. (B) Ensemble of the 15 lowest energy conformers of NMR solution structure; backbone atoms are depicted. (C) Stereo view of one conformer of STM with side chains shown. Backbone atoms are depicted in red for the ring and blue for the tail. Side chains are depicted in orange for residues 14–16, which thread the ring. Images in panels B and C were generating using PyMOL. See also Figures S4 and S5 and Tables S2, S3, and S4.
Figure 4. Biosynthetic gene cluster for streptomonomicin.

Streptomonomicin’s gene cluster architecture closely resembles that of lariatin. The stm operon containing the biosynthetic gene cluster for STM (stmABCDE) is shown above the characterized larABCDE gene cluster responsible for lariatin production in Rhodococcus jostii. Several genes, stmF-M, are present in the operon but have not been predicted to be required for biosynthesis. Predicted function/homology is indicated in Table 2 and Figure S10. See also Figure S6 and Table S5.
Three-dimensional solution structure of streptomonomicin
Two aspects of the NMR data pointed to a highly-ordered three-dimensional structure. First, many amide protons in the ring and the N-terminal half of the tail showed resistance to deuterium exchange (Figure 2C). Second, a large number of long-range NOE correlations were apparent, particularly from the methyl group of Ala15 to a number of the ring residues (e.g. Ser1, Ser5, Tyr7, Asn8, Asp9) (Figures S3D and S3J). Additionally, the ring/tail topology is characteristic of lasso peptides, a class of conformationally-constrained peptidic natural products wherein a 7- to 9-membered N-terminal ring is nearly always threaded by a C-terminal tail; this conformational lock protects them to a degree from degradation and has resulted in recent interest in these scaffolds (Arnison et al., 2013; Blond et al., 2002; Maksimov et al., 2012). However, lasso peptide conformation cannot be assumed from sequence alone, as one recently-described lasso peptide is reported to be unthreaded (Gavrish et al., 2014). As such, we sought to determine the three-dimensional solution structure of STM.
Using NOE distance constraints derived from the NOESY spectra and dihedral angle restraints extracted from the DQF-COSY, an ensemble solution structure was calculated using XPLOR-NIH (Figures 3B, 3C, S4, and Table S3) (Protein Data Bank [PDB] code 2MW3). The resulting STM structure contains the characteristic right-handed lasso; the ring encircles the tail between residues 14–16 (Figure 3B). The constrained ring and loop of STM are well-ordered (0.26 Å rmsd against lowest conformer for the backbone for residues 1–17), and occlude solvent from inward-oriented amide protons, rationalizing the observed slow deuterium exchange of many residues of the ring and the N-terminal half of the tail (Figures 2C and 3C). The C-terminal portion of the tail (residues 18–21) is more disordered, consistent with the observed rapid deuterium exchange (Figure 2C). In lasso peptides without disulfide bonds, bulky residues on the tail immediately flanking the ring serve as steric locks (Maksimov and Link, 2014) (Table S4). Inspection of the van der Waals surface of the ring (residues 1–9) and the tail residues that thread the ring suggests that Leu16 serves as this steric lock, preventing the ring from unthreading (Figures 3C and S5A). Ala15 is in close contact with the ring but is unlikely to be bulky enough to prevent unthreading. Pro14 may also play a role in biasing the conformation of the tail, although mutational studies would be necessary to confirm this. Threaded lasso peptides are typically resistant to carboxypeptidase Y digestion (Hegemann et al., 2013; Zimmermann et al., 2013); in contrast, STM is unusually susceptible (vide supra). However, the complete tail digestion expected for unthreaded lasso peptides was not observed, and a mixture of partially truncated peptides was instead obtained. We hypothesize that the comparatively small steric lock of STM (Leu/Ala) combined with its large, 9-member macrocycle render it more prone to proteolysis.
Unlike other lasso peptides, STM is unusually hydrophobic, consisting of 5% hydrophilic and 52% hydrophobic residues. Known class II lasso peptides (those without disulfide bonds, like STM) are 10–43% hydrophilic and almost always contain a charged residue (Table S4). Interestingly, STM’s amino acid composition is closer to that common for class I and III lasso peptides (those conformationally restrained by two or one ring-tail disulfide bonds, respectively). RP-71955, the siamycins, and BI-32169, like STM, contain 5% hydrophilic and 43–53% hydrophobic residues, being rich in Leu/Ile/Val/Ala (Table S4). Additionally, despite little primary sequence identity, the structure of STM aligns closely (backbone rmsd 1.03 Å for residues 1–17 against lowest energy conformer of STM) to the crystal structure of BI-32169 (PDB code 3NJW), a lasso peptide of streptomycete origin (Knappe et al., 2010; Nar et al., 2010) (Figure S5B).
Markedly, although a Ser-Glu isopeptide linkage has been recently reported in the caulonodins, no lasso peptide with a Ser-Asp isopeptide linkage was formerly known (Zimmermann et al., 2014). Before the caulonodins and STM, it was thought that the first residue of lasso peptides was invariantly Gly or Cys, as amino acid substitutions at this site had not been found in nature; even conservative substitutions at this site were not tolerated by the enzymatic machinery for microcin J25, capistruin, or astexin-1, for instance (Knappe et al., 2009; Pavlova et al., 2008; Zimmermann et al., 2013).
Biosynthetic gene cluster for streptomonomicin
Lasso peptides belong to a class of ribosomally synthesized and post-translationally modified peptides (RiPPs) whose biosynthetic enzymes and precursor peptides are readily identified through bioinformatic analysis (Arnison et al., 2013). The biosynthesis of highly-ordered lasso topologies like that of STM is a compelling topic of study because of the general inaccessibility of such structures by chemical peptide synthesis. In the interest of characterizing STM’s biosynthetic origin and shedding light on the biosynthetic potential of the genus Streptomonospora, we performed whole-genome sequencing of S. alba YIM 90003. This revealed a gene cluster (stm) containing biosynthetic genes (stmABCDE) homologous to those known to be involved in lasso peptide biosynthesis (Figures 4 and S6). Of the characterized lasso peptide gene clusters, stmABCDE most resemble larABCDE, the genes in Rhodococcus jostii K01-B0171 responsible for production of lariatin (Inokoshi et al., 2012); however, we have annotated the STM gene cluster in agreement with the community consensus (Arnison et al., 2013). The gene stmA encodes the 41-residue precursor peptide (StmA) comprised of a 20-amino acid N-terminal leader sequence and the 21-member C-terminal core peptide that is subsequently modified to give the mature natural product. StmB is a transglutaminase homolog; its homologs in lasso peptide genes clusters are believed to function as ATP-dependent cysteine proteases implicated in leader peptidolysis. StmC is homologous to asparagine synthase B and putatively catalyzes macrolactam formation via adenylation of the Asp9 side chain, activating it towards nucleophilic attack by the N-terminus of the core peptide. StmD is an ATP-binding cassette (ABC) transporter likely involved in exporting the mature STM product. Last, StmE is homologous to a protein of unknown, but essential, function that is present in the lariatin biosynthetic cluster but not in the clusters for capistruin or microcin J25 (Inokoshi et al., 2012; Maksimov and Link, 2014). In addition to the requisite biosynthetic machinery, the stm cluster includes a number of other genes stmF-M (Figures 4 and S6). Although these genes have not previously been reported, they are conserved across a series of additional predicted lasso peptide gene clusters (Figure S6).
Analysis of biosynthetic gene clusters within the S. alba genome
Streptomonospora resides in the Streptosporangiales, a suborder of the Actinobacteria; these organisms, based on a survey of 14 known genomes, harbor on average about 15 natural product biosynthetic gene clusters, although some appear to contain zero (Doroghazi et al., 2014). Using the bioinformatics-based prediction algorithm antiSMASH (Blin et al., 2013; Medema et al., 2011), we analyzed the genome of S. alba to evaluate its biosynthetic repertoire. This revealed the presence of at least eleven other putative biosynthetic gene clusters, including those containing terpenoid, type I and III polyketide synthase (PKS), non-ribosomal peptide synthase (NRPS), lanthipeptide, and thiopeptide machinery (Table S5). As the biosynthetic capability of an organism has been closely linked to phylogeny (Doroghazi et al., 2014), this result suggests by extension that the genus Streptomonospora harbors the ability to produce a diverse array of secondary metabolites.
Streptomonomicin bioactivity and resistance
STM’s antibiotic activity was tested against a panel of Gram-positives, Gram-negatives, and fungal species via a microbroth dilution assay (Table 1). Although STM was inactive against fungi and Gram-negatives, it was active against several Gram-positives. STM most potently inhibited the growth of Bacillus anthracis, the causative agent of anthrax, with minimum inhibitory concentrations (MICs) of 2–4 μM. MICs against other members of the Bacillus genus ranged from 4–7 μM with the exception of B. subtilis (29 μM). Weak activity was seen against other Firmicutes. A time-course growth curve of B. anthracis str. Sterne challenged mid-exponential phase with STM showed an immediate cessation of growth upon addition of the compound (Figure 5A). Additionally, the minimum bactericidal concentration (MBC) was determined to be 24 μM.
Table 1. Spectrum of antimicrobial activity for streptomonomicin.
The top 10 strains are bacteria from the Firmicutes phylum, which is followed by a Mycobacterium. The next two species are from the Proteobacteria phylum. The lowest 4 organisms are Ascomycota fungi.
| Strain | MIC (μg/ml) | MIC (μM) |
|---|---|---|
| Bacillus anthracis str. Sterne ΔLF | 4 | 2 |
| Bacillus anthracis str. Sterne | 8 | 4 |
| Bacillus halodurans | 8 | 4 |
| Bacillus cereus ATCC 4342 | 8 | 4 |
| Bacillus cereus ATCC 13472 | 16 | 7 |
| Bacillus sp. Al Hakam | 16 | 7 |
| Bacillus subtilis | 64 | 29 |
| Listeria monocytogenes | 32 | 14 |
| Enterococcus faecalis | 64 | 29 |
| Staphylococcus aureus | 128 | 57 |
| Mycobacterium smegmatis | >128 | >57 |
| Escherichia coli | >128 | >57 |
| Pseudomonas putida | >128 | >57 |
| Saccharomyces cerevisiae | >128 | >57 |
| Talaromyces stipitatus | >128 | >57 |
| Aspergillus niger | >128 | >57 |
| Trichoderma longibrachiatum | >128 | >57 |
Figure 5. Bioactivity of streptomonomicin.

(A) Growth curve (595 nm absorbance) depicting effect of addition of STM to wild-type B. anthracis str. Sterne ΔLF. Cultures were grown at 37 °C to mid-exponential phase (OD600 = 0.4) before the addition of 1×, 2×, or 4×MIC of STM. (B) Wild-type B. anthracis forms chains of usually fewer than 8 cells under standard culturing conditions, as evidenced by DIC microscopy. (C) A representative DIC image of one STM-resistant mutant (WalR-H215P) shows the resistant strains display an exaggerated chaining phenotype. Scale bars, 10 μm. See also Figure S7 and Table S5.
To gain preliminary insight into the mechanism of action by means of investigating bacterial resistance to STM, we selected STM-resistant mutants in B. anthracis. Whole-genome sequencing revealed that all resistant strains harbored mutations to walR, including five instances in the coding region and one in the region upstream of walR (Figure S7A). WalR, a response regulator, forms a two-component regulatory system (TCS) with the histidine kinase WalK that in characterized cases (e.g. B. subtilis) controls genes related to cell wall metabolism and cell division (Dhiman et al., 2014; Dubrac and Msadek, 2008). In the WalK/WalR TCS, formerly known as YycG/YycF (Dubrac and Msadek, 2004), the membrane-associated WalK is involved in extracellular signal sensing and subsequent His autophosphorylation. This phosphoryl group is then transferred to a conserved aspartate (Asp54) in WalR, a cytosolic response regulator composed of an N-terminal receiver domain (containing Asp54) and a C-terminal DNA-binding ‘effector’ domain. Transcription is activated by the dimeric binding of the activated response regulator to specific repeated DNA sequences and to RNA polymerase (Dubrac and Msadek, 2008). The WalK/WalR TCS is highly conserved in Firmicutes and been shown to be the only widely-distributed TCS essential for cell viability (Dubrac and Msadek, 2008; Fabret and Hoch, 1998). Although no formal demonstration of essentiality has been reported for B. anthracis, it is generally thought that WalK/WalR is essential in Firmicutes (Dubrac and Msadek, 2008).
Three of the substitutions (V176P, H215P, P216S) in our STM-resistant strains are in the DNA-binding domain of WalR, whereas two substitutions (D84Y, D88N) are in the receiver domain (Figure S7C). Homology modeling with MtrA (PDB 2GWR; 50% identity to WalR) allowed visualization of the point mutation locations (Arnold et al., 2006; Biasini et al., 2014). In the receiver domain, Asp84 and Asp88 are located on the same surface in spatial proximity to the active site, Asp54, but distant enough that the mutants would be expected to remain somewhat functional (ca. 14 and 10 Å, respectively) (Figure S7D). In the effector domain, which adopts a winged helix-turn-helix DNA-binding motif, two of the mutated residues (His215, Pro216) are found in the α3-β5 loop immediately following the α2/α3 helix-turn-helix motif (Figure S7D); this loop (especially Ser214-Pro216) has been previously implicated in DNA binding (Doi et al., 2010). The other mutated residue (Val176) is found immediately preceding the helix-turn-helix (Supplemental Figure S7D).
WalR substitutions have previously been shown to correlate to growth defects or perturbation in protein function. WalR bearing a H215A or H215P substitution exhibits significantly attenuated DNA-binding, and B. subtilis with WalR-H215P are thermosensitive (Doi et al., 2010; Fabret and Hoch, 1998; Watanabe et al., 2003). Amino acid changes in the helix-loop-helix region of WalR often display phenotypes consistent with the alteration of cell wall metabolism, including intermediate vancomycin resistance and cell wall thickening (Hafer et al., 2012; Howden et al., 2011). STM-resistant B. anthracis mutants displayed obvious phenotypic defects, including visible clumping of cells in liquid culture and recalcitrance to pelleting by centrifugation. Differential interference contrast (DIC) microscopy revealed that STM-resistant strains displayed extremely long chain lengths, indicative of a septal cleavage defect (Figures 5B, 5C, and S7B). This chaining phenotype in Bacillus sp. is well known from deletion studies of particular autolysins (peptidoglycan hydrolases), such as LytE (formerly CwlF), LytF, CwlS, and BslO (Anderson et al., 2011; Fukushima et al., 2006; Hashimoto et al., 2012; Ishikawa et al., 1998). In addition to cell separation, LytE, in collaboration with the CwlO autolysin, has been implicated in cell elongation (Meisner et al., 2013).
Intriguingly, the WalK/WalR TCS in B. subtilis is known to control the expression of the autolysin lytE (Dubrac and Msadek, 2008; Fukuchi et al., 2000; Salzberg et al., 2013) and in B. anthracis the predicted regulon includes also the cell division ABC transporter ftsE (Dhiman et al., 2014); this suggests a likely mechanism for the chaining phenotype displayed by the STM-resistant strains. To confirm if the WalK/WalR TCS in B. anthracis controls the expression of these genes, we compared the expression levels between wild-type and STM-resistant B. anthracis strains by qRT-PCR. These experiments confirmed that, relative to wild-type, the STM-resistant strains downregulated both ftsE and lytE (Table S6). A similar analysis of STM-treated wild type B. anthracis showed an upregulation of liaI and liaH, genes that are induced as part of the cell wall stress regulon (Table S6) (Dominguez-Escobar et al., 2014; Wolf et al., 2010). These combined data are consistent with STM directly or indirectly inducing cell wall stress and with resistance arising by a strategic, but costly, perturbation in cell wall metabolism. Excitingly, this potential mechanism stands in contrast to the antimicrobial activities of the lasso peptides for which the mechanism of action has been characterized, which target RNA polymerase (capistruin and microcin J25) or the ATP-dependent ClpC1 protease (lassomycin) (Bellomio et al., 2007; Gavrish et al., 2014; Kuznedelov et al., 2011). However, as our data primarily relates to a resistant mechanism rather than a direct target, further studies will be required to ascribe a precise mechanism of action to STM.
Significance
The discovery of streptomonomicin (STM) illustrates that understudied lab-cultivable bacteria (here, Streptomonospora alba) remain sources of readily-discoverable natural products. The long cultivation times required for the Streptomonospora, compared to most commonly-screened antibiotic producers, has limited their exploration. Here, we report the first genome sequence of any Streptomonospora revealing a diversity of biosynthetic gene clusters and setting the stage for genome-guided discovery within this genus. STM itself is significant in several regards. First, it represents the first reported natural product from Streptomonospora. Second, STM adopts a threaded lasso peptide structure with an unprecedented isopeptide linkage between Ser-Asp. Third, STM is among the most hydrophobic of the lasso peptides and more hydrophobic than any previously reported class II lasso peptide; hydrophobicity is linked to in vivo bioavailability and cell permeability, and STM thus represents a potentially useful expansion of lasso peptide chemical space. Fourth, STM exhibits antibiotic activity against several Gram-positive organisms, most notably Bacillus anthracis, the causative agent of anthrax. Last, spontaneous resistant mutants to STM exhibit visible growth defects by mutations to walR, a gene encoding a response regulator in a broadly-distributed two-component signal transduction system involved in cell wall metabolism which has been shown to be essential in several Gram-positive bacteria. Our data are consistent with STM inducing cell envelope stress, suggesting a biological activity distinct from the activities reported for all other lasso peptides.
Experimental procedures
Bacterial strains and growth conditions
Streptomonospora alba YIM 90003 was obtained from the USDA Agricultural Research Service (ARS) Culture Collection and grown on ISP medium no. 5 (1 l contains 1 g L-asparagine, 1 g K2HPO4, 20 g agar, 10 g glycerol, 1 mg FeSO4·7H2O, 1 mg ZnSO4·7H2O, 1 mg MnCl2·7H2O) with 10% (w/v) NaCl at 30 °C. Bacillus anthracis strain Sterne ΔLF, Bacillus anthracis strain Sterne, Bacillus halodurans, Bacillus cereus ATCC 4342, Bacillus cereus ATCC 13472, Bacillus sp. Al Hakam, Bacillus subtilis strain 168, Escherichia coli DH5α, Pseudomonas putida KT2440 were grown in Luria-Bertani (LB) medium (1 l contains 10 g NaCl, 5 g yeast extract, 10 g tryptone) at 37 °C. Listeria monocytogenes strain 4b F2365, Enterococcus faecalis U503 (vancomycin-resistant), Staphylococcus aureus USA300 (methicillin-resistant) were grown in brain-heart infusion (BHI) medium at 37 °C. Mycobacterium smegmatis was grown in ATCC medium no. 172 (1 l contains 10 g glucose, 5 g yeast extract, 5 g N-Z Amine Type A, 20 g soluble starch, 1 g CaCO3) at 30 °C. Saccharomyces cerevisiae, Talaromyces stipitatus, Aspergillus niger and Trichoderma longibrachiatum were grown in yeast extract-peptone-dextrose (YPD) medium (1 l contains 10 g yeast extract, 20 g peptone, 20 g glucose) at 30 °C.
Evaluation of bioactivity
The broth microdilution assay for determination of minimum inhibitory concentration (MIC) was used for evaluation of STM activity. Bacterial strains were grown in 10 ml of their corresponding media at 37 °C to stationary phase. The cultures were adjusted to an OD600 of 0.01 in fresh media. STM was serially diluted 2-fold in a 96-well microplate and an equal volume of prepared bacterial culture was added to each well (final concentration of STM in wells ranged from 0.5–128 μg/ml [0.2–57 μM]; final volume, 100 μl/well). Plates were covered and incubated at 37 °C with shaking. The MIC reported is the concentration of STM that resulted in no visible growth after 18 h.
Fungal strains S. cerevisiae, T. stipitatus, A. niger, and T. longibrachiatum were grown in YPD media for 48 h at 30 °C. Cultures were diluted 1:100 into fresh media. STM was serially diluted 2-fold in 96-well microplates, and an equal volume of fungal culture was added to each well (final concentration of STM in wells ranged from 0.5–128 μg/ml, 0.2 – 57 μM). Plates were covered and incubated at 30 °C with shaking. The MIC reported is the concentration of STM that resulted in no visible growth after 36 h.
Mass spectrometry
MALDI-TOF MS analysis
Mass spectra were obtained using a Bruker Daltonics UltrafleXtreme MALDI TOF/TOF mass spectrometer in positive reflector mode. The instrument was calibrated using a peptide calibration kit (AnaSpec – Peptide Mass Standard Kit). Samples (0.5 μl) were spotted on a steel plate with 3 μl of α-cyano-4-hydroxycinnamic acid (CHCA) matrix solution (sat., 1:1 MeCN/H2O containing 0.1% trifluoroacetic acid [TFA]) and air-dried at rt. Data were analyzed using flexAnalysis 3.3 (Bruker).
FT-MS/MS analysis
STM was dissolved in 80% aq. MeCN containing 1% (v/v) formic acid (FA). Using an Advion Nanomate 100, STM was directly infused into a Thermo Scientific LTQ-FT hybrid linear ion trap operating at 11T (calibrated weekly). The FT-MS was operated using the following parameters: minimum target signal counts, 5,000; resolution, 100,000; m/z range detected, dependent on target m/z; isolation width (MS/MS), 5 m/z; normalized collision energy (MS/MS), 35; activation q value (MS/MS), 0.4; activation time (MS/MS), 30 ms. Data were analyzed using the Qualbrowser application of Xcalibur (Thermo Scientific).
Screening of cell extract
S. alba was grown on ISP medium no. 5 containing 10% (w/v) NaCl for 21 days at 30 °C. Exported metabolites were extracted from cells using 70% aq. MeOH at rt for 1 h. The intact cells were removed by centrifugation (4000 × g, 10 min), and the extract was analyzed by MALDI-TOF MS.
Purification of streptomonomicin
S. alba was grown on agar plates of ISP medium no. 5 containing 10% NaCl for 30 days at 30 °C (90 plates × 15 cm diameter; 5 l in total, performed in 5 portions). Plates were air-dried for 2 days, and exported metabolites were extracted with MeOH (2 l) for 12 h. The extract was centrifuged (4000 × g, 10 min), filtered, and dried by rotary evaporation. The dried material was dissolved in 25% aq. MeOH (500 ml), filtered, loaded onto a Waters Sep-Pak C18 cartridge (125 Å pore size; 2 g sorbent; 55–106 μm particle size), and washed with 20% aq. MeCN (ca. 100 ml) containing 0.1% (v/v) TFA. The STM-containing fraction was eluted with 60% aq. MeCN containing 0.1% (v/v) TFA (20 ml). Subsequent purification employed an Agilent 1200 HPLC system outfitted with a Thermo Scientific Betasil C18 column (100 Å; 250 × 10 mm; 5 μm particle size) operating at 4.0 ml/min. The column was equilibrated with 5% aq. MeCN containing 0.1% (v/v) FA and purified via a linear gradient of 5–60% aq. MeCN containing 0.1% (v/v) FA over 40 min. Absorbance was monitored at 220 nm. The STM-containing fraction was dried using a vacuum concentrator to afford a slightly yellow solid (yields ranged from 10–14 mg/l of media, 0.6–0.8 mg/plate).
Protease treatment
100 μg of purified STM was treated with 0.5 U carboxypeptidase Y (from baker’s yeast; Sigma-Aldrich) in a buffer containing 50 mM MES and 1 mM CaCl2 at pH 6.7 for 6 h at 25 °C. The reaction mixture was then purified with an Agilent 1200 HPLC system outfitted with an Agilent G1956B single quadrupole mass analyzer and a Thermo Scientific Biobasic C18 column (300 Å, 250 × 4.6 mm, 5 μM particle size) operating at a flow rate of 1.0 ml/min and separated with a linear gradient of 5–60% aq. MeCN containing 0.1% (v/v) FA over 40 min. Absorbance was monitored at 220 nm. Fractions were collected manually and analyzed by MALDI-TOF MS. The fraction containing a mass ion of m/z 1057.1 ([M+Na]+) was dried, dissolved in 100 μl of buffer solution (50 mM Tris-HCl pH 8, 100 mM NaCl), and digested by thermolysin (from Bacillus thermoproteolyticus rokko; Sigma-Aldrich) (1 U) for 1 h at 37 °C. The products of the reaction were separated by HPLC using a Biobasic C18 column (300 Å, 250 × 4.6 mm, 5 μM particle size) (Thermo Scientific) operating at a flow rate of 1.0 ml/min with a linear gradient of 5–60% aq. MeCN containing 0.1% (v/v) FA over 40 min. Fractions were collected manually, screened by MALDI-TOF MS, concentrated, and analyzed by FT-MS/MS.
NMR spectroscopy
Samples were prepared by dissolving ca. 4 mg of STM (HPLC-purified and lyophilized) in 500 μl of either methanol-d4 (CD3OD; 99.96 atom % D; Sigma-Aldrich) or methanol-d3 (CD3OH; 99.8 atom % D; Sigma-Aldrich). NMR spectra were recorded on an Agilent VNMRS 750 MHz narrow bore magnet spectrometer equipped with a 5mm triple resonance (1H-13C-15N) triaxial gradient probe and pulse-shaping capabilities. Samples were held at 25 °C during acquisition. Standard Varian pulse sequences were used for each of the following experiments: 1H, 1H-1H DQF-COSY, 1H-1H TOCSY (80 ms mixing time), 1H-13C HSQCAD, 1H-13C HMBCAD, and 1H-1H NOESY (400 ms mixing time). Solvent suppression by presaturation (PRESAT) was employed when CD3OH was used as the solvent. Spectra were recorded with VNMRJ 3.2A software and data was processed using MestReNova 8.1.1, nmrPipe (Delaglio et al., 1995), and Sparky (Goddard and Kneller). Resonances were referenced internally to the solvent peak (3.30 ppm, methanol).
Structure calculations
STM’s solution structure was calculated by simulated annealing using distance and angle restraints within XPLOR-NIH v 2.36 (Schwieters et al., 2006; Schwieters et al., 2003). Standard XPLOR-NIH potentials for bond angles, improper angles, van der Waals, and favored/allowed Ramachandran regions were used. The Ser1-Asp9 isopeptide linkage was generated using a manual patch of the “protein-1.0.top” XPLOR-NIH file (Supplemental Experimental Procedures). Distance restraints were derived from peak area integrations in the NOESY data sets, which were binned into specific distance categories (2.5, 3.5, 5.0, and 6.0 Å). nmrPipe was used for processing of raw NOESY data and conversion from Varian to UCSF format (Sparky), and Sparky was used for peak picking, volume integration, and creation of the XPLOR distance restraint table. Peptide backbone dihedral restraints were derived from coupling constants determined from DQF-COSY. Three hundred structures were calculated, of which the 15 structures of lowest energy were chosen for structural analysis. The quality of the NMR structures was evaluated using PROCHECK-NMR (Laskowski et al., 1993; Laskowski et al., 1996). A list of the number and type of restraints used (Table S3) and the PROCHECK quality report (Figure S4) are given in the Supplemental Information.
Generation of resistant mutants
Six independent cultures (10 ml) of B. anthracis str. Sterne ΔLF were grown overnight in LB media at 37 °C. A portion (100 μl) of each culture was spread onto LB agar plates containing 4, 7, or 14 μM STM. The plates containing 4 μM STM yielded resistant mutants after 24 h of incubation at 37 °C. No bacterial growth was detected at higher concentrations of STM after 48 h. The MIC of all six STM-resistant strains was 7 μM and was obtained as described above.
Whole-genome sequencing
Genomic DNA from S. alba YIM 90003 as well as wild-type and four STM-resistant mutants of B. anthracis str. Sterne ΔLF was isolated using an UltraClean Microbial DNA Isolation Kit (MO BIO). The shotgun genomic libraries were then prepared with an Illumina-Compatible KAPA DNA Library Preparation Kit (Kapa Biosystems). Paired-end sequencing with read length 100 nt was conducted by the Roy J. Carver Biotechnology Center (Univ. Illinois, Urbana, IL, United States) using an Illumina HiSeq2500. The genome of S. alba YIM 90003 was assembled and mapped with CLC Assembly Cell software (CLC bio) and deposited in GenBank. Breseq software was used for finding mutations in the genomes of wild-type and STM-resistant mutants, relative to a reference genome (AE017225 for chromosomal DNA and NC_007322 for pXO1 plasmid) (Deatherage and Barrick, 2014). Nucleotide substitutions which were present in STM-resistant mutants but not in the wild-type strain, in comparison to the reference genome, were identified and verified with PCR amplification and Sanger sequencing. Nucleotide substitutions in two STM-resistant mutants whose genomes were not sequenced were identified by Sanger sequencing alone.
Microscopy
Stationary-phase cultures of B. anthracis str. Sterne ΔLF (wild-type and STM-resistant strains) were used to inoculate LB media (200 μl into 5 ml). Cultures were grown to an OD600 of 0.5 before an aliquot (1 ml) was removed, harvested by centrifugation, and resuspended in 250 μl phosphate-buffered saline (PBS). Due to the abnormal growth of the STM-resistant mutants, the wild-type culture was used to determine cell density. 10 μl aliquots of each resuspended culture were combined with 10 μl liquefied low gelling temperature agarose (10 μl, 2%) on a microscope slide. Cell morphology was analyzed by differential interference contrast (DIC) microscopy. DIC microscopy analyses were conducted using a Zeiss LSM 700 Confocal Microscope outfitted with a 405 nm laser. Linear contrast was applied when deconvoluting images in ZEN 2012 software (Carl Zeiss).
Supplementary Material
Table 2. List of Stm proteins.
Proteins are depicted along with identified biosynthetic roles based on homology to characterized enzymes in the lariatin biosynthetic cluster. For proteins without LarABCDE homologs, proposed functions are given based on homologous gene families; Pfam domains are given in parentheses.
| CDS | Size (aa) | Lar Homolog (identity, %/aa length coverage) | Proposed Function (Pfam) |
|---|---|---|---|
| StmA | 41 | LarA (34/35) | Precursor peptide |
| StmC | 628 | LarB (25/538) | Lasso peptide cyclase (PF00733) |
| StmE | 87 | LarC (28/83) | Unknown (PF05402) |
| StmB | 143 | LarD (35/107) | Lasso peptide protease (PF13471) |
| StmD | 628 | LarE (34/590) | ABC transporter (export; PF00005, PF00664) |
| StmF | 248 | - | ABC-type multidrug transport system (PF00005) |
| StmG | 224 | - | ABC-2 family transporter (PF12730) |
| StmH | 250 | - | ABC-2 family transporter (PF12730) |
| StmI | 190 | - | Hypothetical, no identifiable homologs |
| StmJ | 253 | - | Hypothetical, no identifiable homologs |
| StmK | 204 | - | Hypothetical, no identifiable homologs |
| StmL | 209 | - | Response regulator (PF00072, PF00196) |
| StmM | 418 | - | Signal transduction histidine kinase (PF07730, PF02518) |
Highlights.
Streptomonomicin (STM) is a lasso peptide with a unique Ser1-Asp9 linkage
STM exhibits activity against Gram-positive bacteria
Substitutions in the essential two-component regulator WalR endow STM resistance
Genome sequencing shows biosynthetic potential in the genus Streptomonospora
Acknowledgments
This work was supported in part by a NIH Director’s New Innovator Award Program (DP2 OD008463 to D.A.M.), the David and Lucile Packard Fellowship for Science and Engineering (to D.A.M.), the Robert C. and Carolyn J. Springborn Endowment (to J.I.T.), and the NIH (R01 NS031609 for I.L.). M.M. was supported by a Ministry of Education and Science of Russian Federation project (14.B25.31.0004 to K.S.). The Bruker UltrafleXtreme MALDI TOF/TOF mass spectrometer was purchased in part with a grant from the National Center for Research Resources, National Institutes of Health (S10 RR027109 A).
Footnotes
Author contributions
M.M. performed and analyzed genomic and microbiological experiments and compound isolation; J.I.T. performed and analyzed NMR experiments with critical input from L.Z.; J.O.M. performed and analyzed MS experiments; P.M.B. performed microscopy and microbiological experiments; I.L. performed chirality analysis; D.A.M. conceived of and managed the project; J.O.M., P.M.B., L.Z., I.L., and K.S. assisted in revisions during the writing process; J.I.T. wrote the paper with significant editorial assistance from M.M. and D.A.M.
Accession numbers
The PDB accession number for the streptomonomicin solution structure reported in this article is 2MW3. The genome of S. alba is desposited in GenBank under the accession number JROO00000000.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson VJ, Kern JW, McCool JW, Schneewind O, Missiakas D. The SLH-domain protein BslO is a determinant of Bacillus anthracis chain length. Mol Microbiol. 2011;81:192–205. doi: 10.1111/j.1365-2958.2011.07688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Baltz RH. Marcel Faber Roundtable: is our antibiotic pipeline unproductive because of starvation, constipation or lack of inspiration? J Ind Microbiol Biotechnol. 2006;33:507–513. doi: 10.1007/s10295-005-0077-9. [DOI] [PubMed] [Google Scholar]
- Bellomio A, Vincent PA, de Arcuri BF, Farias RN, Morero RD. Microcin J25 has dual and independent mechanisms of action in Escherichia coli: RNA polymerase inhibition and increased superoxide production. J Bacteriol. 2007;189:4180–4186. doi: 10.1128/JB.00206-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T. antiSMASH 2.0-a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41:W204–W212. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blond A, Cheminant M, Destoumieux-Garzon D, Segalas-Milazzo I, Peduzzi J, Goulard C, Rebuffat S. Thermolysin-linearized microcin J25 retains the structured core of the native macrocyclic peptide and displays antimicrobial activity. Eur J Biochem. 2002;269:6212–6222. doi: 10.1046/j.1432-1033.2002.03340.x. [DOI] [PubMed] [Google Scholar]
- Cox CL, Tietz JI, Sokolowski K, Melby JO, Doroghazi JR, Mitchell DA. Nucleophilic 1,4-additions for natural product discovery. ACS Chem Biol. 2014;9:2014–2022. doi: 10.1021/cb500324n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui XL, Mao PH, Zeng M, Li WJ, Zhang LP, Xu LH, Jiang CL. Streptimonospora salina gen. nov., sp nov., a new member of the family Nocardiopsaceae. Int J Syst Evol Microbiol. 2001;51:357–363. doi: 10.1099/00207713-51-2-357. [DOI] [PubMed] [Google Scholar]
- Deatherage DE, Barrick JE. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol. 2014;1151:165–188. doi: 10.1007/978-1-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Dhiman A, Bhatnagar S, Kulshreshtha P, Bhatnagar R. Functional characterization of WalRK: A two-component signal transduction system from Bacillus anthracis. FEBS Open Bio. 2014;4:65–76. doi: 10.1016/j.fob.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi A, Okajima T, Gotoh Y, Tanizawa K, Utsumi R. X-ray crystal structure of the DNA-binding domain of response regulator WalR essential to the cell viability of Staphylococcus aureus and interaction with target DNA. Biosci Biotechnol Biochem. 2010;74:1901–1907. doi: 10.1271/bbb.100307. [DOI] [PubMed] [Google Scholar]
- Dominguez-Escobar J, Wolf D, Fritz G, Hofler C, Wedlich-Soldner R, Mascher T. Subcellular localization, interactions and dynamics of the phage-shock protein-like Lia response in Bacillus subtilis. Mol Microbiol. 2014;92:716–732. doi: 10.1111/mmi.12586. [DOI] [PubMed] [Google Scholar]
- Doroghazi JR, Albright JC, Goering AW, Ju KS, Haines RR, Tchalukov KA, Labeda DP, Kelleher NL, Metcalf WW. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat Chem Biol. 2014;10:963–968. doi: 10.1038/nchembio.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrac S, Msadek T. Identification of genes controlled by the essential YycG/YycF two-component system of Staphylococcus aureus. J Bacteriol. 2004;186:1175–1181. doi: 10.1128/JB.186.4.1175-1181.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrac S, Msadek T. Tearing down the wall: peptidoglycan metabolism and the WalK/WalR (YycG/YycF) essential two-component system. Adv Exp Med Biol. 2008;631:214–228. doi: 10.1007/978-0-387-78885-2_15. [DOI] [PubMed] [Google Scholar]
- Fabret C, Hoch JA. A two-component signal transduction system essential for growth of Bacillus subtilis: Implications for anti-infective therapy. J Bacteriol. 1998;180:6375–6383. doi: 10.1128/jb.180.23.6375-6383.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng ZY, Kim JH, Brady SF. Fluostatins produced by the heterologous expression of a TAR reassembled environmental DNA derived type II PKS gene cluster. J Am Chem Soc. 2010;132:11902–11903. doi: 10.1021/ja104550p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi K, Kasahara Y, Asai K, Kobayashi K, Moriya S, Ogasawara N. The essential two-component regulatory system encoded by yycF and yycG modulates expression of the ftsAZ operon in Bacillus subtilis. Microbiology. 2000;146:1573–1583. doi: 10.1099/00221287-146-7-1573. [DOI] [PubMed] [Google Scholar]
- Fukushima T, Afkham A, Kurosawa S, Tanabe T, Yamamoto H, Sekiguchi J. A new D,L-Endopeptidase gene product, YojL (renamed CwlS), plays a role in cell separation with LytE and LytF in Bacillus subtilis. J Bacteriol. 2006;188:5541–5550. doi: 10.1128/JB.00188-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrish E, Sit CS, Cao S, Kandror O, Spoering A, Peoples A, Ling L, Fetterman A, Hughes D, Bissell A, et al. Lassomycin, a ribosomally synthesized cyclic peptide, kills Mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem Biol. 2014;21:509–518. doi: 10.1016/j.chembiol.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: [Google Scholar]
- Hafer C, Lin Y, Kornblum J, Lowy FD, Uhlemann AC. Contribution of selected gene mutations to resistance in clinical isolates of vancomycin-intermediate Staphylococcus aureus. Antimicrob Agents Chemother. 2012;56:5845–5851. doi: 10.1128/AAC.01139-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Ooiwa S, Sekiguchi J. Synthetic lethality of the lytE cwlO genotype in Bacillus subtilis is caused by lack of D,L-endopeptidase activity at the lateral cell wall. J Bacteriol. 2012;194:796–803. doi: 10.1128/JB.05569-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegemann JD, Zimmermann M, Xie XL, Marahiel MA. Caulosegnins I–III: A highly diverse group of lasso peptides derived from a single biosynthetic gene cluster. J Am Chem Soc. 2013;135:210–222. doi: 10.1021/ja308173b. [DOI] [PubMed] [Google Scholar]
- Hindra Huang T, Yang D, Rudolf JD, Xie P, Xie G, Teng Q, Lohman JR, Zhu X, Huang Y, et al. Strain prioritization for natural product discovery by a high-throughput real-time PCR method. J Nat Prod. 2014;77:2296–2303. doi: 10.1021/np5006168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden BP, McEvoy CR, Allen DL, Chua K, Gao W, Harrison PF, Bell J, Coombs G, Bennett-Wood V, Porter JL, et al. Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator WalKR. PLoS Pathog. 2011;7:e1002359. doi: 10.1371/journal.ppat.1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inokoshi J, Matsuhama M, Miyake M, Ikeda H, Tomoda H. Molecular cloning of the gene cluster for lariatin biosynthesis of Rhodococcus jostii K01-B0171. Appl Microbiol Biotechnol. 2012;95:451–460. doi: 10.1007/s00253-012-3973-8. [DOI] [PubMed] [Google Scholar]
- Ishikawa S, Hara Y, Ohnishi R, Sekiguchi J. Regulation of a new cell wall hydrolase gene, cwlF, which affects cell separation in Bacillus subtilis. J Bacteriol. 1998;180:2549–2555. doi: 10.1128/jb.180.9.2549-2555.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HS, Brady SF. Arimetamycin A: Improving clinically relevant families of natural products through sequence-guided screening of soil metagenomes. Angew Chem, Int Ed. 2013;52:11063–11067. doi: 10.1002/anie.201305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knappe TA, Linne U, Robbel L, Marahiel MA. Insights into the biosynthesis and stability of the lasso peptide capistruin. Chem Biol. 2009;16:1290–1298. doi: 10.1016/j.chembiol.2009.11.009. [DOI] [PubMed] [Google Scholar]
- Knappe TA, Linne U, Xie X, Marahiel MA. The glucagon receptor antagonist BI-32169 constitutes a new class of lasso peptides. FEBS Lett. 2010;584:785–789. doi: 10.1016/j.febslet.2009.12.046. [DOI] [PubMed] [Google Scholar]
- Kuznedelov K, Semenova E, Knappe TA, Mukhamedyarov D, Srivastava A, Chatterjee S, Ebright RH, Marahiel MA, Severinov K. The antibacterial threaded-lasso peptide capistruin inhibits bacterial RNA polymerase. J Mol Biol. 2011;412:842–848. doi: 10.1016/j.jmb.2011.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Li WJ, Xu P, Zhang LP, Tang SK, Cui XL, Mao PH, Xu LH, Schumann P, Stackebrandt E, Jiang CL. Streptomonospora alba sp nov., a novel halophilic actinomycete, and emended description of the genus Streptomonospora Cui et al 2001. Int J Syst Evol Microbiol. 2003;53:1421–1425. doi: 10.1099/ijs.0.02543-0. [DOI] [PubMed] [Google Scholar]
- Maksimov MO, Link AJ. Prospecting genomes for lasso peptides. J Ind Microbiol Biotechnol. 2014;41:333–344. doi: 10.1007/s10295-013-1357-4. [DOI] [PubMed] [Google Scholar]
- Maksimov MO, Pan SJ, Link AJ. Lasso peptides: structure, function, biosynthesis, and engineering. Nat Prod Rep. 2012;29:996–1006. doi: 10.1039/c2np20070h. [DOI] [PubMed] [Google Scholar]
- Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39:W339–W346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisner J, Llopis PM, Sham LT, Garner E, Bernhardt TG, Rudner DZ. FtsEX is required for CwlO peptidoglycan hydrolase activity during cell wall elongation in Bacillus subtilis. Mol Microbiol. 2013;89:1069–1083. doi: 10.1111/mmi.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nar H, Schmid A, Puder C, Potterat O. High-resolution crystal structure of a lasso peptide. ChemMedChem. 2010;5:1689–1692. doi: 10.1002/cmdc.201000264. [DOI] [PubMed] [Google Scholar]
- Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DD, Wu CH, Moree WJ, Lamsa A, Medema MH, Zhao XL, Gavilan RG, Aparicio M, Atencio L, Jackson C, et al. MS/MS networking guided analysis of molecule and gene cluster families. Proc Natl Acad Sci USA. 2013;110:E2611–E2620. doi: 10.1073/pnas.1303471110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova O, Mukhopadhyay J, Sineva E, Ebright RH, Severinov K. Systematic structure-activity analysis of microcin J25. J Biol Chem. 2008;283:25589–25595. doi: 10.1074/jbc.M803995200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidot SJ, Coyne S, Kloss F, Hertweck C. Antibiotics from neglected bacterial sources. Int J Med Microbiol. 2014;304:14–22. doi: 10.1016/j.ijmm.2013.08.011. [DOI] [PubMed] [Google Scholar]
- Salzberg LI, Powell L, Hokamp K, Botella E, Noone D, Devine KM. The WalRK (YycFG) and sigma(I) RsgI regulators cooperate to control CwlO and LytE expression in exponentially growing and stressed Bacillus subtilis cells. Mol Microbiol. 2013;87:180–195. doi: 10.1111/mmi.12092. [DOI] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Clore GM. Using Xplor-NIH for NMR molecular structure determination. Prog Nucl Magn Reson Spectrosc. 2006;48:47–62. [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- Thaker MN, Waglechner N, Wright GD. Antibiotic resistance-mediated isolation of scaffold-specific natural product producers. Nat Protoc. 2014;9:1469–1479. doi: 10.1038/nprot.2014.093. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Hashimoto Y, Umemoto Y, Tatebe D, Furuta E, Fukamizo T, Yamamoto K, Utsumi R. Molecular characterization of the essential response regulator protein YycF in Bacillus subtilis. J Mol Microbiol Biotechnol. 2003;6:155–163. doi: 10.1159/000077246. [DOI] [PubMed] [Google Scholar]
- Wolf D, Kalamorz F, Wecke T, Juszczak A, Mader U, Homuth G, Jordan S, Kirstein J, Hoppert M, Voigt B, et al. In-depth profiling of the LiaR response of Bacillus subtilis. J Bacteriol. 2010;192:4680–4693. doi: 10.1128/JB.00543-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Hegemann JD, Xie X, Marahiel MA. Characterization of caulonodin lasso peptides revealed unprecedented N-terminal residues and a precursor motif essential for peptide maturation. Chem Sci. 2014;5:4032–4043. [Google Scholar]
- Zimmermann M, Hegemann JD, Xie XL, Marahiel MA. The astexin-1 lasso peptides: biosynthesis, stability, and structural studies. Chem Biol. 2013;20:558–569. doi: 10.1016/j.chembiol.2013.03.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.