

Abstract
We report a novel cyclic-AMP (cAMP) response element (CRE) in the human BCRP promoter that is functional in human cancer cell lines of multiple lineages. 8Br-cAMP increased the activity of a BCRP promoter reporter construct and BCRP mRNA in human carcinoma cells. Activation of the epidermal growth factor receptor (EGFR) pathway also led to an increase in BCRP promoter reporter activity and to phosphorylation of the c-AMP response element binding protein (CREB) via two major downstream EGFR signaling pathways: the phosphotidylinositol-3-kinase (PI3K)/AKT pathway and the mitogen-activated protein kinase (MAPK) pathway. EGF treatment increased the phosphorylation of EGFR, AKT, ERK and CREB, while simultaneously enhancing BCRP mRNA and functional protein expression. EGF-stimulated CREB phosphorylation and BCRP induction were diminished by inhibition of EGFR, PI3K/AKT or RAS/MAPK signaling. CREB silencing using RNA interference reduced basal levels of BCRP mRNA and diminished the induction of BCRP by EGF. Chromatin immunoprecipitation assays confirmed that a putative CRE site on the BCRP promoter bound phospho-CREB; point mutation of the CRE site abolished EGF-induced stimulation of BCRP promoter reporter activity. Furthermore, the CREB co-activator, cAMP-regulated transcriptional co-activator (CRTC2), is also involved in CREB-mediated BCRP transcription: androgen depletion of LNCaP human prostate cancer cells increased both CREB phosphorylation and CRTC2 nuclear translocation, and enhanced BCRP expression. Silencing CREB or CRTC2 reduced basal BCRP expression and BCRP induction under androgen-depletion conditions. This novel CRE site plays a central role in mediating BCRP gene expression in multiple human cancer cell lines following activation of a variety of signaling pathways.
Introduction
Breast cancer resistance protein (BCRP) is a member of the G subfamily of the ATP-binding cassette (ABC) superfamily of membrane transporters, and is formally designated ABCG2. BCRP functions primarily as a xenobiotic transporter; as such, BCRP may play a role in the disposition of many drugs. When BCRP is overexpressed in cancer cells, it can cause or contribute to the resistance of these cells to antineoplastic drugs.
Several transcription factors and their respective cis-regulatory elements have been identified and characterized in the BCRP promoter (reviewed in [1, 2]). These include a hypoxia response element, an estrogen response element, progesterone response element, an aryl hydrocarbon response element, and an anti-oxidant response element. The BCRP/Bcrp1 promoter is complex in both humans and mice. In mice alternative promoter usage is clearly observed; alternative promoter usage is likely to occur in humans as well. The human E1b/c BCRP promoter corresponds to the mouse Bcrp1 E1B alternative promoter; these alternative promoters were previously found to control BCRP/Bcrp1 expression in human and mouse intestine, respectively [3]. In this same work, we established that the major alternative promoter controlling Bcrp1 expression in mouse intestine – E1B – contains a functional cyclic AMP (cAMP) response element (CRE) that binds to phospho-cAMP response element binding protein (p-CREB), resulting in enhanced Bcrp1 transcription [3].
The basic leucine zipper transcription factor p-CREB binds to CRE sequences in promoters, which leads to an increase or decrease in the transcription of the target genes. Initially, p-CREB was recognized as a cAMP-driven transcription factor generated by the cAMP-dependent protein kinase A (PKA) pathway. However, there are other mechanisms which augment nuclear levels of p-CREB independent of the cAMP/PKA pathway. CREB phosphorylation can also be driven by growth factors such as epidermal growth factor (EGF) and fibroblast growth factor (FGF) as a result of their activation of multiple downstream signaling pathways such as the phosphotidylinositol-3-kinase (PI3K) pathway and the mitogen activated protein kinase (MAPK) pathways, which phosphorylate CREB [4, 5].
EGF enhancement of BCRP expression via either the MAPK pathway or the PI3K/AKT-dependent pathway was reported previously [6, 7]. The latter study found that AKT-dependent phosphorylation of membrane EGFR caused EGFR to translocate to the nucleus where it interacted with the BCRP promoter to enhance transcription of BCRP in gefitinib-resistant cells [7]. However, at present it is not known whether EGF-mediated PI3K/AKT activity or MAPK activity can regulate BCRP expression via CREB in human cells.
In addition to transcriptional activation via p-CREB binding to CRE-site, two co-activators of p-CREB cAMP-regulated transcriptional co-activator (CRTC2 – also known as transducer of regulated CREB activity 2 [TORC2]) and P300/CBP – also enhance CREB target gene expression. CRTC2 enhances CREB target gene expression via nuclear translocation following its activation by de-phosphorylation [8]. Under basal conditions, CRTC2 is sequestered in the cytoplasm, maintained in an inactive phosphorylated state by AMP-dependent protein kinase (AMPK) [9]. Inactivation of AMPK results in de-phosphorylation of CRTC2, which causes it to translocate to the nucleus, where it binds to p-CREB and enhances CREB transcriptional activity. CRTC2 nuclear recruitment does not appear to modulate CREB DNA binding activity, but rather enhances CREB activity in the absence of a cAMP stimulus [10]. CRTC2 nuclear translocation is sufficient to activate CRE-dependent transcription; hence CRTC2 also plays an important role in the regulation of CREB activity [11].
Although the mouse BCRP promoter harbors a functional CRE, the structural organization of the mouse promoter differs significantly from the human promoter, and it is not known whether the latter can be regulated by CRE/CREB related pathways in cells of human origin, including human cancer cells. In this study, we sought to determine whether the human BCRP promoter contains a functional CRE that activates BCRP transcription upon p-CREB binding. We also examined in multiple human cancer cell lines whether important cancer-related signaling pathways that lead to either CREB phosphorylation or CRTC2 nuclear translocation can regulate BCRP transcription via this CRE.
Materials and Methods
Materials
8Br-cAMP, PD98059 and LY294002 were purchased from Sigma-Aldrich, Inc. (St. Louis, MO). EGF was purchased from Gemini Bioproducts (West Sacramento, CA). ZD1839 (gefitinib) was purchased from AstraZeneca. Anti-BCRP (BXP-21) and anti-EGFR antibodies were obtained from Millipore, Cambridge, MA; anti-p-CREB (Ser-133) and anti-CRTC2 antibodies were purchased from Santa Cruz Bio Technology (Santa Cruz, CA). The anti-p-AKT, p-ERK, and GAPDH antibodies and the horseradish peroxidase labeled secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA). Fumitremorgin C (FTC) was kindly provided by Dr. Susan Bates of the Medicine Branch, National Cancer Institute.
Cell Culture
IGROV1 cells were cultured in RPMI1640 medium (Biosource, Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Biosource), 50 units/mL penicillin, and 50 μg/mL streptomycin. MDAMB468 cells were grown in DMEM/F12 (1:1) media (Biosource) supplemented with 10% FBS (Biosource), 50 units/mL penicillin and 50 μg/mL streptomycin. LNCaP cells were cultured in either RPMI1640 medium (Biosource) supplemented with 10% FBS (Biosource), 50 units/mL penicillin, and 50 μg/mL streptomycin, or in androgen-deprived media i.e. phenol red-free RPMI1640 medium (Biosource) supplemented with 10% charcoal stripped FBS (CSS, Gemini, West Sacramento, CA), 50 units/mL penicillin and 50 μg/mL streptomycin. Cultures treated with EGF were starved for at least 3 hours in serum-free medium (SFM) because this eliminates the background effect of endogenous EGF that may be present in the serum, in order to optimize the effects of endogenously added EGF.
RNA preparation and Quantitative RT-PCR (qPCR)
Cells were plated at low density (3×105/per well) in 60 mm petri dishes. After overnight starvation in SFM, the cells were treated with or without EGF at a concentration of 50 ng/mL in SFM for 6 hours. Total mRNA was then extracted using Trizol (Life Technology, Carlsbad, CA) and isolated with RNA mini-prep columns (Qiagen, Valencia, CA). The mRNA was reverse transcribed to cDNA with M-MLV reverse transcriptase (Roche Diagnostics, Basel, Switzerland). 10-100 ng of the reverse-transcribed cDNA was then PCR-amplified in real-time with IQ SYBR green mix (Bio-Rad, Hercules, CA) using the MyiQ Single-Color Real-Time PCR Detection System (Bio-Rad). Each reaction was performed in duplicate. Total BCRP, CREB and CRTC2 mRNA were amplified using appropriate primers and normalized with β-actin mRNA. The sequences of BCRP and β-actin primers were described previously [12]. The sequences of CREB and CRTC2 primers used for qPCR are provided in Table 1.
Table 1.
| Point mutant primers | |
| Sense | 5′- CGGCGCTCCGGCCAGTGAAAGCGACCAAACCC -3′ |
| Antisense | 5′- GGGTTTGGTCGCTTTCACTGGCCGGAGCGCCG -3′ |
| ChiP assay of CRE-site primers for qPCR (amplifies BP –504 to –403 of the BCRP promoter) | |
| Forward | 5′-CCCTTTCCTTCCTTGGGTTA-3′ |
| Reverse | 5′-AAATGGGTGGTTTCTGGTGA-3′ |
| CREB primers for qPCR: | |
| Forward: | 5′-AAGCGGAGTGTTGGTGAGTG-3′ |
| Reverse: | 5′-GCTCCTCCGTCACTGCTTTC-3′; |
| CRTC2 primers for qPCR: | |
| Forward: | 5′- TCAGAGCCTGTTGGAAAGCA-3′ |
| Reverse: | 5′-CAAGGGGAAGAGTGGTGAGG-3′ |
Western Blot Analysis
1 × 105 cells/mL were cultured in 60 mm plates. Following overnight starvation in SFM, some plates were treated with EGF (50 ng/mL) and others with vehicle for 24 hours, then lysed in radioimmune precipitation buffer (RIPA buffer: 50 mM Tris–HCl, pH 7.4; 1% NP-40; 0.25% sodium deoxycholate; 150 mM NaCl; 1 mM EDTA; 1× protease inhibitor cocktail and 1× phosphatase inhibitor cocktail (EMD, Millipore) for 30 min on ice with occasional vortexing. The clarified lysates were separated by 4-12% SDS-polyacrylamide gel electrophoresis, transferred onto polyvinylidene difluoride membranes, and analyzed by Western blotting with primary antibodies against BCRP, p-AKT, p-ERK, p-CREB, p-EGFR and GAPDH, respectively (see “Materials” for source). The bands were visualized by enhanced chemiluminescence (GE Healthcare Bio-Sciences Corp., Piscataway, NJ) and quantified by densitometric analysis (Visionworks LS image acquisition and analysis software, UVP, Upland, CA).
Isolation of nuclear and cytosolic fractions
LNCaP cells were seeded in 60 mm dishes and cultured with 10% FBS or CSS, respectively, for 7 days. Nuclear extracts from the cells were prepared using the NE-PER extraction kit (Pierce Thermo Scientific Inc., Rockford, IL) and protein was quantified using the BCA assay kit (Pierce Thermo Scientific Inc.).
Cytotoxicity Assay
Cytotoxicity was assessed by the method of Vichai and Kirtikara [13]. Briefly, IGROV1 cells were plated in 96-well plates at a density of 1 × 104 cells/well and cultivated in SFM overnight. Sensitivity to mitoxantrone was measured in four different groups: untreated (control), 10 μM FTC, (a specific inhibitor of BCRP), 10 ng/mL EGF, or 10 ng/mL EGF plus 10 μM FTC. Following incubation of these groups for 24 hours in complete medium, cells in each group were continuously exposed to the indicated concentrations of mitoxantrone for 2 days. Then, cells were stained with 0.02% sulforhodamine B (SRB, Sigma-Aldrich, Milwaukee, WI) according to the SRB assay protocol [13].
Reporter gene assay
Cells were seeded in 24-well plates at a density of 5 × 104 cells/well and grown in complete growth media for 6 hours. The pGL4/1285 reporter construct containing the BCRP promoter (-1285/+362) upstream of firefly luciferase, the pGL4/1285Mut construct containing a mutated CRE site in the BCRP promoter, the pGL4-Basic empty vector (negative control), and an internal control vector pRL-TK (Promega, Madison, WI) which expresses Renilla luciferase, were used for various co-transfection experiments using the X-tremeGENE HP DNA transfection reagent (Roche Diagnostics). Following transfection, the cells were cultured for an additional 48 h in complete medium, then starved overnight in SFM followed by treatment with EGF (50 ng/mL) or 8Br-cAMP (2 mM) for 6 hours. Cells were lysed in passive lysing buffer (Promega) and dual luciferase assay performed. Luminescence was quantified in a TD-20/20 luminometer (Turner Designs, Inc., Sunnyvale, CA). Results are expressed as the ratio of firefly luciferase luminescence in the BCRP promoter construct relative to the Renilla luciferase luminescence in the internal control.
Identification of a CRE in the BCRP promoter and CRE site mutation
The BCRP genomic sequence spanning -668 to +529 bps around the E1b/c human promoter was interrogated for CREB binding sites (CRE) using the Matinspector promoter/transcription factor scan program [14, 15].
Point mutation within the CRE site was obtained using the QuickChange Site-Directed Mutagenesis Kit (Agilent Technologies Inc., Santa Clara, CA), performed according to the manufacturer's instructions. The sequences of the primers used in making the point mutation are provided in Table 1.
Chromatin immunoprecipitation (ChiP) assay
IGROV1 cells were cultured in 100 mm dishes at a density of 1×106 cells/per well. After starvation overnight in SFM, cells were stimulated with 50 nM EGF for 60 min then subjected to ChiP assay as per the manufacturer's instructions (Millipore, Temecula, CA). Briefly, cells were cross-linked with 1% formaldehyde for 10 min at room temperature. Cells were then harvested in sodium dodecyl sulfate lysis buffer and the DNA sheared to 200–1000 base pairs by sonication. The sonicated cell lysates were pre-cleared with salmon sperm DNA blocked protein A agarose beads (Millipore). Immunoprecipitation of protein-DNA complexes was performed on the pre-cleared lysates with anti-rabbit p-CREB antibody. Simultaneously, pre-cleared lysates were also immunoprecipitated with rabbit IgG (Cell Signaling Technology) as control for potential non-specific co-immunoprecipitations. The bound DNA was de-crosslinked with high salt and the purified DNA was quantified by real time PCR using promoter-specific primers for the CRE site in the BCRP promoter. The sequences of the primers used for real time PCR are given in Table 1. Results are expressed as percentage of immunoprecipitated p-CREB-DNA to total DNA input (input).
RNA interference
CREB or CRTC2 mRNA interference was performed using pre-designed CREB or CRTC2 siRNA (final concentration 10 nM, Ambion, Austin, TX) or non-specific siRNA (10 nM, negative control, Ambion). Briefly, cells were seeded in 60 mm Petri dishes and transfected with siRNA using HiPerfect reagent (Qiagen) according to the manufacturer's protocol. After 24 hours of incubation, the cells were transferred to new 60 mm dishes at a cell density of 3 × 105 cells/well, and incubated for another 24 hours. After overnight starvation in SFM, the cells were treated with EGF (50 ng/mL) or vehicle for the designated times before cell harvest. In experiments with LNCaP cells, the cells were incubated in either 10% FBS medium or 10% CSS medium for the indicated number of days after transfection, then harvested for analysis.
Statistical methods
Student's t-test was used for statistical comparisons (as detailed in the figure and table legends).
Results
In silico prediction of CREB binding sites in the BCRP promoter
Matinspector identified several putative CREB response elements (CREs) within the BCRP genomic sequence spanning -668 to +529 bps in the human E1b/c promoter (Figure 1). Two CREB binding sites with matrix similarity >0.8 were identified in this region located at bps -329 and -123 relative to the published transcriptional start site [16]. The matrix similarity for each CRE is given in Figure 1.
Figure 1.
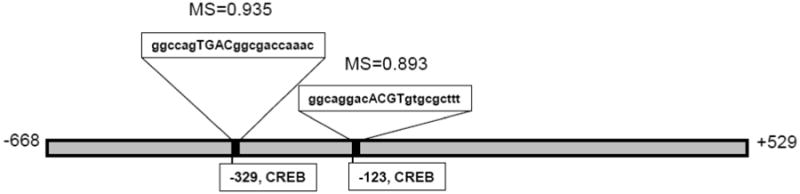
Putative cAMP-response elements (CRE) in the BCRP/ABCG2 promoter, identified by the MatInspector program. The “matrix similarity” (MS) for each site is indicated in the figure. Matrix similarity is a measure of goodness of fit of the putative response element identified with a known functional response element. A perfect fit has an MS of 1.0; MS scores >0.8 are considered to indicate good matches.
Effects of 8Br-cAMP on BCRP expression
To establish whether CREB is involved in regulation of human BCRP expression, the effects of the PKA pathway activator 8-Br cAMP were measured in cells transfected with a luciferase reporter construct (pGL4/1285) containing the BCRP promoter region (-1285 to +362). Treatment with 8-Br cAMP significantly (P<0.05) increased the luciferase reporter activity of the pGL4/1285 transfected cells in human ovarian carcinoma IGROV1 cells (Figure 2A) and breast carcinoma MDAMB468 cells (Figure 2B). BCRP mRNA levels also increased 2-fold in IGROV1 cells following 8Br-cAMP treatment (Figure 2C) for 6 hours.
Figure 2.
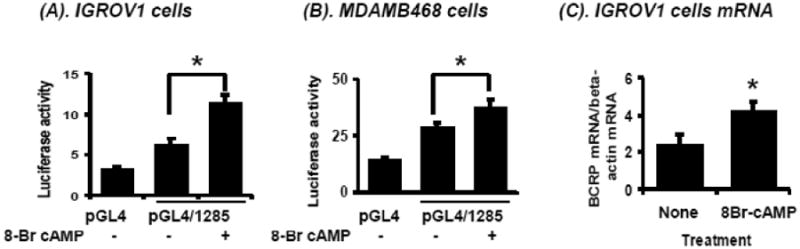
Effects of the c-AMP analogue 8Br-cAMP on BCRP promoter activity and BCRP mRNA expression. Cells were cultured in 24-well plates and transfected with pGL4 empty vector or pGL4/1285, a reporter construct containing the promoter region (-1285/+362) of BCRP, respectively. After culture overnight in SFM, cells were treated with 8Br-cAMP (2 mM, final concentration) for 6 hours, then luciferase activity was determined for IGROV1 cells (A) and MDAMB468 cells (B). IGROV1 cells were treated with 2 mM 8Br-cAMP for 6 hours after culture overnight in SFM, then total RNA was isolated and BCRP mRNA expression was determined by real time qPCR (C). Results represent the means ± S.D. from three separate experiments, each done with triplicate determinations per data point. *, significantly (P<0.05) different vs. untreated, Student's t-test.
Effects of EGF on CREB phosphorylation and BCRP mRNA and protein expression in IGROV1 and MDAMB468 cells
The two major downstream pathways of EGFR signaling – MAPK and PI3K – ultimately can lead to CREB phosphorylation [17-20]. We examined the phosphorylation state of several downstream proteins following treatment of IGROV1 cells for up to 24 hours or MDAMB468 cells for up to 6 hours with EGF (50 ng/mL). EGF induced EGFR, CREB, AKT, and ERK phosphorylation starting at 30 minutes. Maximal phosphorylation of CREB occurred at 30 minutes, and of EGFR between 30 minutes and 3 hours, while elevated p-AKT and p-ERK levels were sustained for at least 6 hours following treatment with EGF in both IGROV1 (Figure 3A) and MDAMB468 cells (Figure 3B). The findings in each cell line are consistent with previous observations that EGF induces CREB phosphorylation via either the AKT or ERK pathways [4, 5]. Interestingly, EGF treatment resulted in EGFR degradation in IGROV1 cells, but not in MDAMB468 cells, as was reported previously [21, 22].
Figure 3.
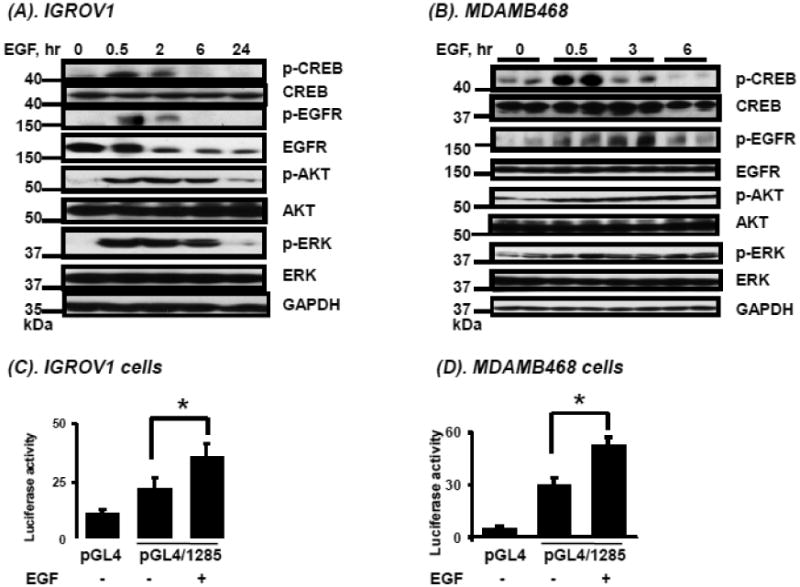
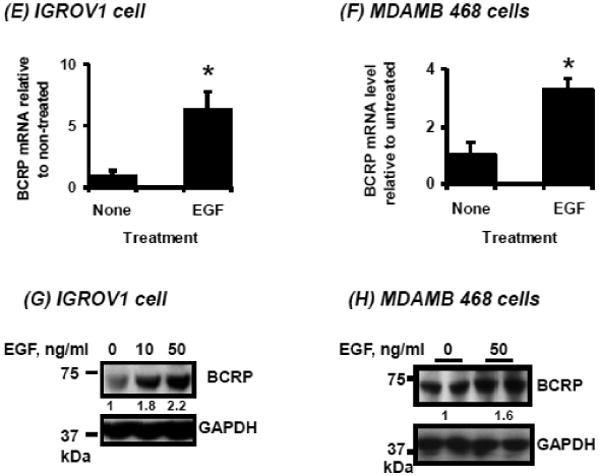
Effects of EGF on pathways downstream of EGFR, CREB activation, and on BCRP promoter activity, mRNA and protein expression. A, B. Time-course of phosphorylated and unphosphorylated EGFR, AKT, ERK and CREB following EGF treatment (see Methods) in IGROV1 human ovarian carcinoma cells (A) and MDAMB468 human breast carcinoma cells (B) determined by Western blotting as described in “Methods.” C, D. The numbers on the left of the blot reflect the molecular size of marker proteins (kDa). Effects of EGF on activity of a BCRP promoter reporter construct transfected into human carcinoma cells. Cells were cultured, transfected with reporter constructs and exposed to EGF as described in Methods, then reporter activities were determined for IGROV1 ovarian (C) and MDAMB468 breast cancer cells (D). E, F. Effects of EGF on BCRP mRNA expression in human carcinoma cells. IGROV1 or MDAMB468 cells were treated with and without EGF as described in Methods, then total RNA was isolated and BCRP mRNA expression was were determined by real time qPCR for IGROV1 (E) and MDAMB468 cells (F). G, H. Effects of EGF on BCRP protein expression in human carcinoma cells. Cells were exposed to EGF as described in Methods, then harvested and BCRP expression was determined by Western blot in IGROV1 (G) and MDAMB468 cells (H). Western blots for GAPDH were used as a loading control. The data shown represent the mean and standard deviation of 3 different experiments, done on different days. Each individual assay was run in duplicate. *, significantly (P<0.05) different vs. untreated by student's t-test. Western blots shown represent one of three independent blots done on different days, with similar results obtained.
Treatment of pGL4/1285-transfected MDAMB468 and IGROV1 cells with EGF resulted in increased luciferase activity (p<0.05) in both cell lines (Figures 3C, D). The EGF-induced BCRP promoter activity in these cell lines correlated with enhanced BCRP mRNA and protein expression in these cells, as shown in Figures 3E-H.
EGF reduces cellular sensitivity to mitoxantrone in IGROV1 cells
To determine whether the EGF-induced BCRP protein is functional, the sensitivity of IGROV1 cells to the cytotoxic BCRP-substrate drug mitoxantrone was measured. Mitoxantrone caused a dose-dependent decrease in survival (Figure 4, -EGF/-FTC group); however, cell survival was significantly higher (P<0.01) in the EGF-treated group (+EGF/-FTC) compared to the non-EGF treated control group at mitoxantrone concentrations of 0.1, 0.3, and 1 μM (Figure 4). Furthermore, this EGF-induced resistance to mitoxantrone cytotoxicity was reduced in the presence of FTC, a specific inhibitor of the BCRP transporter (Figure 4, +EGF/+FTC group, 0.3 μM mitoxantrone, P=0.037), suggesting that BCRP is contributing to the mitoxantrone resistance observed in the EGF-treated cells.
Figure 4.
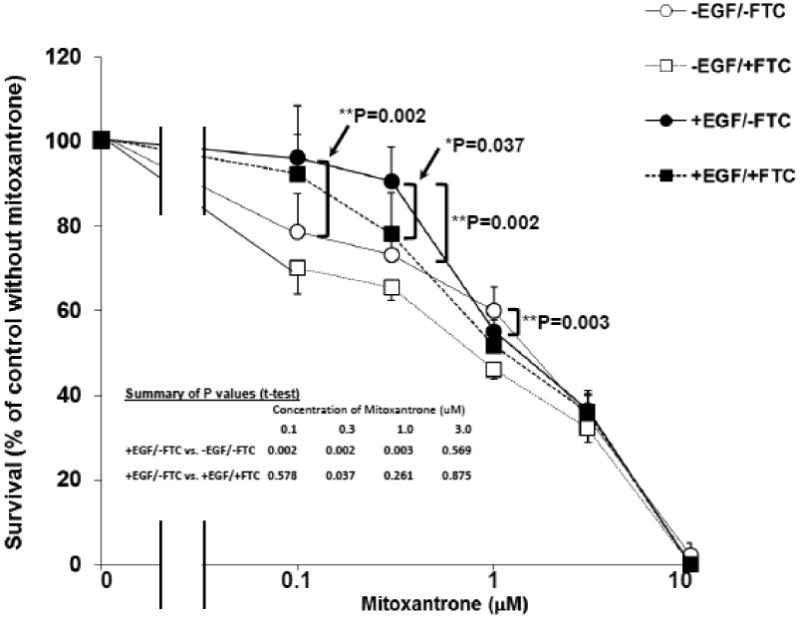
Effects of EGF on cellular sensitivity to mitoxantrone in IGROV1 cells. IGROV1 cells were plated at 2 × 104 cells/mL in 96-well plates in complete medium. Following starvation overnight in SFM, the cells were divided to four different groups: untreated (control), Fumitremorgin C (FTC, 10 μM, a specific inhibitor of BCRP), EGF (10 ng/mL), or EGF (10 ng/mL) plus FTC (10 μM). After 24 hours of EGF treatment, cells in each treatment group were continuously exposed to the indicated concentrations of mitoxantrone for 48 hours, after which survival was assessed by staining with SRB as described in Materials and Methods. Results are expressed as percent survival compared to cultures without mitoxantrone, presented as the mean of six individual values ± SD. *: Significantly different compared to no FTC by student's t-test, P=0.037; **: Significantly different compared to no EGF by Student's t-test, P<0.01.
Effect of EGFR and its downstream pathways on EGF-induced BCRP transcription
There are two major signaling pathways downstream of EGFR: RAS/MAPK and PI3K/AKT [23]. To identify which signaling pathways are involved in EGF-induced BCRP gene transcription, cells transfected with pGL4/1285 or basic pGL4 vector were pre-treated with selective inhibitors of EGFR (ZD1839), PI3K (LY294002), or MEK (PD98059) prior to exposure to EGF (50 ng/mL, 6 hours). Cells were lysed and reporter activities determined. All of the inhibitors reduced the pGL4/1285 reporter activity to that approximating the activity in IGROV1 cells without EGF stimulation (Figure 5A), suggesting that both pathways downstream of EGFR contribute to EGF-mediated BCRP transcriptional regulation.
Figure 5.
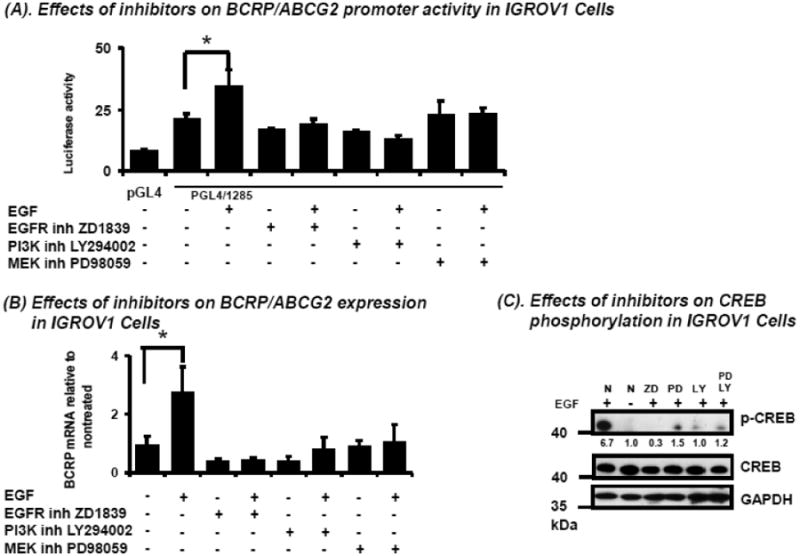
Effects of EGFR pathway inhibitors on EGF-induced human BCRP or p-CREB expression in IGROV1 cells. A. Effects of EGFR pathway inhibitors on EGF-induced BCRP promoter activity. Cells were transfected with pGL4 or pGL4/1285 respectively. After 24-hours of culture in complete medium, then overnight starvation in SFM, cells were pretreated with the EGFR selective inhibitor ZD1839 (10 μM), the PI3K inhibitor LY294002 (15 μM), or the MEK inhibitor PD98059 (30 μM) respectively for 1 hour, followed by treatment with EGF (50 ng/mL) for 6 hours. Then, BCRP promoter reporter gene activities were determined by measuring luciferase luminescence as described in Methods. B. Effects of EGFR pathway inhibitors on EGF-induced BCRP mRNA expression. Following treatment with EGF and inhibitors as described for (A) above, total mRNA was isolated and BCRP mRNA expression were determined by real-time qPCR and normalized for the expression of β-actin mRNA (B). Results shown in A and B represent the means ± S.D. from duplicate measurements and three independent experiments for promoter activity and mRNA, respectively. *, significantly (P<0.05) different vs. untreated by student's t-test. C. Effects of EGFR pathway inhibitors on EGF-induced p-CREB expression. Following treatment with inhibitors as described for (A) above, then 30 min treatment with EGF (50 ng/mL), the expression of p-CREB, CREB, and GAPDH (done as loading control) was determined by Western blot. Bands represent results typical for three independent experiments, done on different days.
Similar effects of these inhibitors were observed with respect to BCRP mRNA expression; the 3-fold induction of BCRP mRNA by EGF was completely abolished after inhibition of EGFR or either of its downstream pathways (Figure 5B). Further, inhibition of EGFR by ZD1839 or PI3K by LY294002 or MAPK by PD98059 significantly reduced EGF-induced CREB phosphorylation in IGROV1 cells after 30 minutes of drug exposure (Figure 5C). However, inhibiting both PI3K/AKT and MAPK pathways did not completely abolish EGF-induced CREB phosphorylation (Figure 5C), suggesting that other EGF-dependent mechanisms may also be involved in the generation of p-CREB. Taken together, these data demonstrate that EGF can regulate BCRP by phosphorylating CREB via the PI3K and MAPK pathways.
CREB knockdown diminishes constitutive BCRP mRNA expression and abolishes EGF-induced BCRP mRNA expression in IGROV1 cells
To confirm further the role of p-CREB in the regulation of BCRP mRNA expression by EGF, CREB silencing was performed using CREB-specific siRNA. IGROV1 cells were transfected with either negative control siRNA (NC) or CREB siRNA. After 48 hours, cells were starved in SFM for 3 hours, and then treated with EGF or vehicle for 16 hours before cell harvest. CREB mRNA levels decreased by approximately two-thirds in the CREB siRNA-transfected IGROV1 cells when compared to NC siRNA transfected cells (Figure 6A). CREB silencing significantly reduced the basal BCRP mRNA expression in IGROV1 cells compared to control siRNA transfected cells, and completely abolished the EGF-stimulated enhancement of BCRP mRNA expression seen in the control siRNA transfected cells (Figure 6B). Protein analysis confirmed that CREB protein was reduced by CREB siRNA (Figure 6C). Although EGF induced CREB phosphorylation in both CREB silenced or CREB non-silenced cells, the induction of CREB phosphorylation was much lower in CREB silenced cells under stimulation with EGF for 30 min (Figure 6C). Interestingly, the total BCRP protein level did not change significantly between the CREB silenced and the non-silenced cells (data not shown); this in part could be due to the relatively long half-life of BCRP of approximately 39 hours [24].
Figure 6.
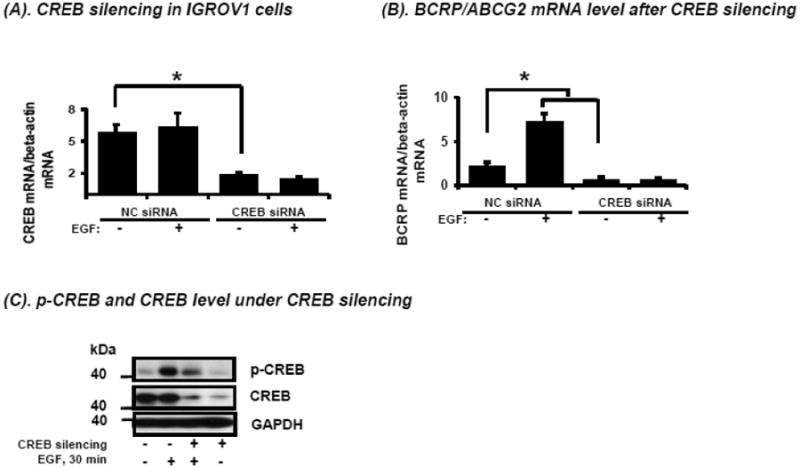
Effects of CREB silencing on CREB mRNA expression (A), BCRP mRNA expression (B) and CREB and p-CREB protein expression (C) in IGROV1 cells. Cells were transfected with CREB siRNA or negative control siRNA under culture conditions as described in Materials and Methods, followed by treatment with or without EGF (50 ng/mL) for 12 hours. CREB mRNA expression (A) and BCRP mRNA expression (B) were determined by real-time qPCR and normalized for the expression of β-actin mRNA. Expression of p-CREB and CREB protein was determined by Western blot following transfection with CREB siRNA or negative control (NC) siRNA in the presence or absence of EGF treatment (50 ng/mL) for 30 minutes. An immunoblot for GAPDH is used as loading control.
The CRE site at -329 binds to p-CREB, and is critical to EGF-induced BCRP mRNA expression
Amongst the CREB binding sites identified by Matinspector, the -329 site had the highest matrix similarity (Figure 1). Hence, a point mutation of this putative CRE was made in the -1285/+362 BCRP promoter construct (designated pGL4/1285Mut). IGROV1 cells were then transfected and reporter assays carried out in the absence or presence of EGF (50 ng/mL, 6 hours). Interestingly, upon transfection the pGL4/1285Mut construct demonstrated higher basal reporter activity than the parental pGL4/1285 construct. Although the basis for this is presently not clear, importantly EGF treatment did not induce a further increase in pGL4/1285Mut reporter gene activity in contrast to the wild-type pGL4/1285 vector (Figure 7A), underscoring the relevance of this CRE site in EGF-mediated regulation of BCRP expression.
Figure 7.
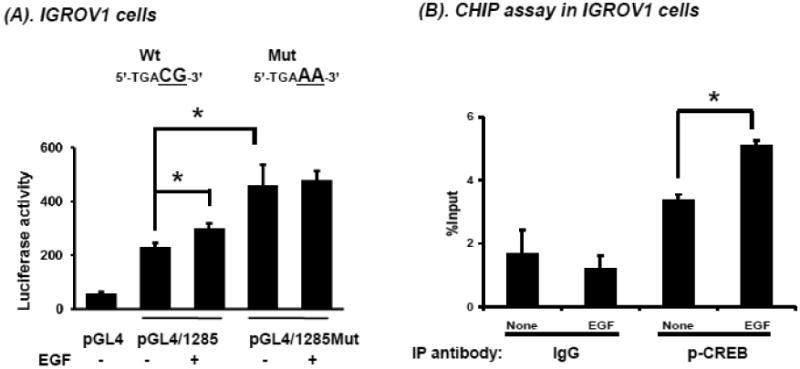
Validation of the putative CRE site at -329 in the BCRP promoter. A. IGROV1 cells were cultured in 24-well plates and transfected with pGL4 empty vector or pGL4/1285 or a construct containing a point mutation of the CRE located at BP -329 (pGL4/1285Mut). After 48 hours, cells were transferred to SFM for 3 hours then treated overnight with EGF (50 ng/mL). Reporter gene activities were determined in cell lysate. B. ChiP analysis of p-CREB interaction with the CRE located at -329 in the BCRP promoter was performed in IGROV1 cells in the presence or absence of EGF (50 ng/mL) for 1 hour. A ChiP assay with IgG isotype control was also performed to exclude non-specific interactions. The data shown are the mean and standard deviation of 3 different experiments, done on different days. Each individual assay was run in duplicate. *, significantly different (P< 0.05) compared to no EGF control group.
To determine whether direct interaction exists between p-CREB and the CRE-site in the BCRP promoter, ChiP assays were performed in IGROV1 cells in the presence or absence of EGF. EGF treatment significantly increased p-CREB binding to the BCRP promoter -329 CRE site compared to no EGF treatment or to IgG isotype control immunoprecipitations (Figure 7B), strengthening the notion of a direct interaction of p-CREB (Ser-133) with the BCRP promoter.
Androgen deprivation in LNCaP human prostate cancer cells enhances AKT and CREB phosphorylation, nuclear translocation of CRTC2, and BCRP transcription
In prostate cancer, androgen deprivation therapy usually produces initial tumor regression; however, with time the cancer usually recurs despite hormone withdrawal. It has been shown recently that androgen deprivation induces the phosphorylation of AKT [25]. Therefore, we sought to determine whether androgen deprivation would also induce CREB phosphorylation and subsequently BCRP up-regulation via the AKT pathway in prostate cancer cells. Androgen deprivation can also result in down-regulation of Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2), the upstream activating kinase for AMPK [26, 27]. Because p-AMPK is required for phosphorylation and hence cytoplasmic sequestration of the CREB co-activator CRTC2, we postulated that dephosphorylation and nuclear translocation of CRTC2 should also occur under androgen-depleted conditions.
To test our hypothesis, we carried out studies with human LNCaP prostate cancer cells, which express androgen receptor and are sensitive to androgens. LNCaP cells were cultured in ‘normal’ medium (RPMI1640 + 10% FBS) or androgen-depleted medium (phenol red-free RPMI1640 + 10% CSS) for 7 days, and then p-AKT, p-CREB and BCRP protein levels determined by Western blot. Figure 8A shows that p-AKT, p-CREB and BCRP increase approximately 2-fold when LNCaP cells are cultured in CSS-containing medium. Androgen-depleted conditions also enhance BCRP mRNA expression in the LNCaP cells (Figure 8B).
Figure 8.
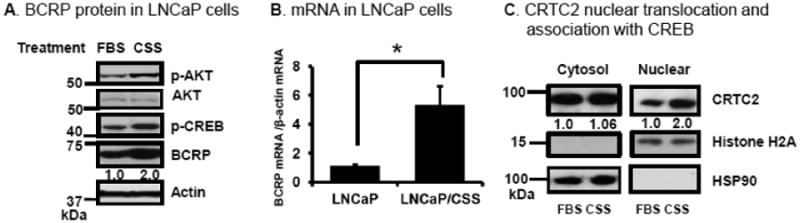
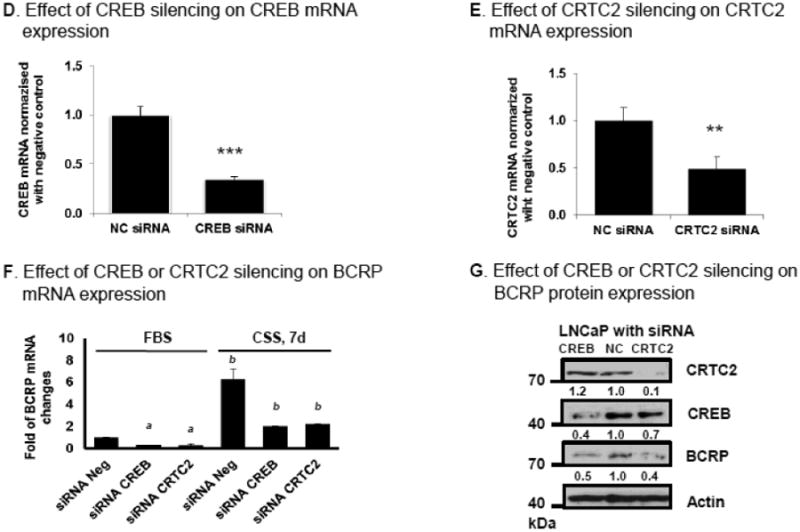
Effects of CREB and the CREB co-activator CRTC2 on BCRP mRNA and protein expression in human LNCaP prostate cancer cells. (A) p-AKT, p-CREB and BCRP protein expression in LNCaP cells cultured in medium containing either 10% FBS or 10% charcoal stripped (CSS) medium for 7 days, as determined by Western blot. A Western blot for actin is used as loading control. (B) BCRP mRNA expression in LNCaP or LNCaP/CSS cells (LNCaP cells were cultured in medium containing 10% CSS for 7 days) as measured by real time qPCR, normalized for expression of β-actin mRNA. (C) Distribution of CRTC2 between cytosol and nucleus analyzed by Western blot, as described in Methods. (D-F) Effects of CREB or CRTC2 silencing on CREB, CRTC2 and BCRP mRNA expression in LNCaP cells. Twenty four hours after transfection of LNCaP cells with CREB siRNA, CRTC2 siRNA, or negative control siRNA were cultured in medium containing 10% FBS or 10% CSS respectively for 7 days, then total RNA was isolated and mRNA expression was determined by real-time qPCR for CREB (D), CRTC2 (E) or BCRP (F); the mRNA expression data shown were normalized for the mRNA expression by the normal control (NC) siRNA transfected cells, which was given a value of 1.0. The data shown are the mean and standard deviation of 3 different experiments, done on different days. Each individual assay was run in duplicate. *, P< 0.05 compared to untreated control group. a, P< 0.05 compared to negative control siRNA transfected FBS group. b, P< 0.05 compared to control siRNA transfected CSS group. (G) Effects of CREB or CRTC2 silencing on BCRP protein expression in LNCaP cells. LNCaP cells in complete medium were transfected with CREB siRNA or CRTC2 siRNA, or negative control siRNA as described in Materials and Methods; after 72 hours, cells were lysed in RIPA buffer and CREB, CRTC2 and BCRP protein expression was determined by Western blot. A Western blot for actin is used as loading control. The Western blot results shown are representative of the results of three independent replicate experiments, done on different days.
To test whether androgen deprivation increases CRTC2 nuclear translocation, nuclei were isolated after LNCaP cells were cultured in CSS medium for 7 days. It is apparent that this treatment results in a two-fold increase in CRTC2 nuclear content (Figure 8C).
Treatment of LNCaP cells with CREB or CRTC2 siRNA decreases the expression of the respective mRNAs in the LNCaP cells (Figures 8D, 8E). These siRNAs also decrease the expression of BCRP mRNA in LNCaP cells under both ‘normal’ growth conditions (FBS media) as well as androgen-deprived conditions (CSS media) (Figure 8F). No further decrease in BCRP mRNA expression was noted in cultures treated with siRNA to both CREB and CRTC2 (data not shown). CREB and CRTC2 siRNA also decreased the respective protein levels of CREB and CRTC2 in the LNCaP cells, which in parallel are associated with a decrease in BCRP protein levels by 50-60% compared to NC siRNA transfected cells (Figure 8G). Taken together, these data demonstrate that both CREB and CRTC2 are involved in modulating BCRP gene expression in LNCaP cells.
Discussion
We have previously demonstrated that CREB can modulate BCRP expression via a CRE site within the Bcrp1 promoter in the mouse intestine. While the structural organization of the mouse and human BCRP promoters are different [16, 28], in the present study we have now also identified CRE sites within the human BCRP promoter, and show that at least one of these sites binds to p-CREB and contributes to the complex transcriptional regulation of BCRP in several cancer cell lines of different lineages, including ovarian, breast, and prostate cancers. We show that a commonly dysregulated growth factor in human cancers – EGF – can regulate BCRP expression via phosphorylation of CREB; furthermore, androgen withdrawal in hormone sensitive LNCaP prostate cancer cells increases their content of p-CREB and nuclear content of the CREB co-activator CRCT2.
Although CREB was originally reported to be activated via the PKA pathway, CREB is now known to be regulated by a variety of extracellular signals, including growth factors, osmotic stress and ultraviolet irradiation [17, 29-33]. For example, EGF activates EGFR and its two major downstream signaling pathways, PI3K and MAPK, resulting in CREB phosphorylation [4, 17]. Our data show that inhibition of EGFR activity by ZD1839 abolished EGF-induced CREB phosphorylation. In addition, inhibition of either the PI3K or the MAPK pathway abolished EGF-induced CREB phosphorylation, as well as EGF-induced BCRP expression. These data demonstrate that the EGF-EGFR axis can regulate CREB phosphorylation and subsequent BCRP expression via its two major downstream signaling pathways, PI3K and MAPK, in human cancer cells. Given BCRP's role as a transporter of xenobiotics and anti-neoplastic drugs, modulation of its expression by a frequently dysregulated growth promoting pathway such as EGFR in a number of human cancers would not only provide a growth advantage but also potentially make the cancer cells resistant to certain cytotoxic agents. Targeting EGFR and/or its downstream signaling axis, including under conditions where BCRP has also been recruited, could be particularly useful in rendering the cancer cells more susceptible cytotoxic therapies.
EGFR, which is frequently overexpressed or activated in cancer cells, plays a critical role in cell survival, growth, maintenance, and function [34, 35]. Inhibitors of EGFR tyrosine kinase activity such as gefitinib and erlotinib (which are also substrates for BCRP) have been used clinically as a treatment for non-small-cell lung cancer, pancreatic cancer, breast cancer, colon cancer and other cancers that are associated with EGFR up regulation. Unfortunately, cancers that initially respond to EGFR-targeted chemotherapy frequently develop resistance to the therapy [36], which often involves autonomous activation of the PI3K/AKT or MAPK pathway. Understanding the mechanisms by which EGFR and its two major downstream pathways affect BCRP expression via p-CREB may aid in developing novel strategies for treatment of acquired or innate resistance. EGFR mutations are frequently associated with drug resistance, often occurring concomitantly with PI3K mutations [37]. The PI3K/AKT signaling cascade is frequently dysregulated in many human malignancies, including pancreatic, colon, ovarian and breast cancers [38]. Up-regulation of the PI3K pathway in relation to multidrug resistance has been described in some tumors, especially in acquired drug resistance to tyrosine kinase inhibitors (TKIs, e.g., gefitinib) [39]. Inactivation of AKT prevented TKI-induced up-regulation of BCRP [23]. These observations further support the notion that both EGFR and its downstream signaling PI3K/AKT pathway are involved with the induction of BCRP expression.
Previously, our laboratory observed the involvement of the PI3K pathway alone in maintaining BCRP expression in human K562 chronic myelogenous leukemia cells [40]. PI3K/AKT-dependent BCRP up-regulation was also reported by Huang et. al.; however, this study demonstrated that PI3K/AKT activation caused EGFR to be translocated to the nucleus, where it bound to an EGFR DNA binding site on the BCRP promoter, revealing a novel role for EGFR as a transcription factor [7]. Although relatively rare, mutations in the MAPK pathway (e.g. BRAF) were also observed in cells resistant to EGFR inhibitors by Ohashi and co-workers [41], suggesting that the MAPK pathway may also be involved in the induction of drug resistance involving BCRP. Thus, current evidence suggests that the EGFR/PI3K/AKT/CREB and EGFR/MAPK/CREB pathways may mediate BCRP expression and hence may be involved in acquired drug resistance.
As a transcription factor, CREB signaling is implicated in tumor progression. CREB is overexpressed and constitutively phosphorylated in a number of forms of human cancer including prostate cancer (see [42] for review). An increase in p-CREB has been observed in bone metastases obtained from patients with prostate cancer [43]. Prostate cancer patients may experience recurrent tumors after irradiation treatment or hormone deprivation treatment. It was reported that prostate cancer cells that survived irradiation treatment had an increased nuclear content of CREB [44]. In our current study, we observed that androgen withdrawal enhanced CREB phosphorylation and the nuclear content of the CREB co-activator CRTC2, associated with enhanced expression of BCRP, strengthening the notion that CREB and its co-activator may regulate BCRP expression, and that CREB may be a promising cancer chemotherapeutic target [42].
Androgen deprivation therapy such as orchiectomy and/or luteinizing hormone-releasing agonists is currently the mainstay of treating not only metastatic prostate cancer but also locally advanced and recurrent prostate cancers [45]. Our findings demonstrate that androgen withdrawal can up-regulate BCRP in the androgen-sensitive human prostate cancer cell line LNCaP. This in part occurs through enhanced phosphorylation of CREB. In addition, another underlying mechanism appears to be androgen withdrawal-dependent translocation of the CREB co-activator CRTC2 from the cytoplasm to the nucleus. CRTC2 is a downstream target of AMP-dependent protein kinase (AMPK) and the phosphorylation of CRTC2 by AMPK and AMPK-related kinases, salt-inducible-kinase-1 (SIK-1) and -2, promotes CRTC2 binding to 14-3-3 proteins in the cytoplasm and prevents the translocation of CRTC2 to the nucleus, thereby reducing CREB-dependent mRNA expression [9]. Hormone deprivation is known to inactivate CaMKK2, the upstream kinase of AMPK, which results in inactivation of AMPK [26, 27]. However, it has not been reported previously that androgen deprivation can decrease CRTC2 phosphorylation as a consequence of AMPK inactivation. Hence, our demonstration that an increase in CRTC2 nuclear translocation occurs in LNCaP cells cultured for 7 days in hormone-depleted medium is novel, and supports the notion that androgen deprivation decreases CRTC2 phosphorylation. Furthermore, either CREB or CRTC2 silencing reduces BCRP expression in LNCaP cells deprived of androgens. Thus, BCRP expression appears to be at least in part under the regulation of CREB, as well as the CREB co-activator CRTC2, in prostate cancer cells. This finding is of particular interest with regard to androgen deprivation therapy: recruitment of BCRP upon androgen withdrawal could potentially render the prostate cancer cells undergoing androgen deprivation relatively resistant to further therapies. Further, although highly effective initially, androgen deprivation therapy fails in most patients, particularly those maintained on androgen deprivation for long periods of time, resulting in castration resistant prostate cancer (CRPC). Castration resistance can lead to a relatively chemotherapy-resistant state, and can be associated with enhanced expression of anti-apoptotic proteins such as Bcl-2 [46]. It will also be of interest to determine the rate of expression and role of BCRP in patients undergoing long term androgen deprivation therapy and among those who develop CRPC.
In conclusion, our study demonstrates for the first time that the human BCRP promoter region contains a functional CRE-site that can integrate diverse signals such as those triggered by growth factors like EGF in ovarian and breast cancers, or withdrawal of androgens in prostate cancer, to regulate BCRP promoter activity. These results reveal a novel mechanism of BCRP regulation and emphasize that signaling pathways that ultimately result in CREB phosphorylation or nuclear translocation of its co-activator may be responsible for inducing BCRP expression in human cancer cells. Enhanced BCRP expression under such conditions may help protect the cancer cells from xenobiotics and cytotoxic agents, and contribute to innate or acquired resistance to anti-tumor therapies ranging from cytotoxic agents to androgen deprivation therapy.
Figure 9.
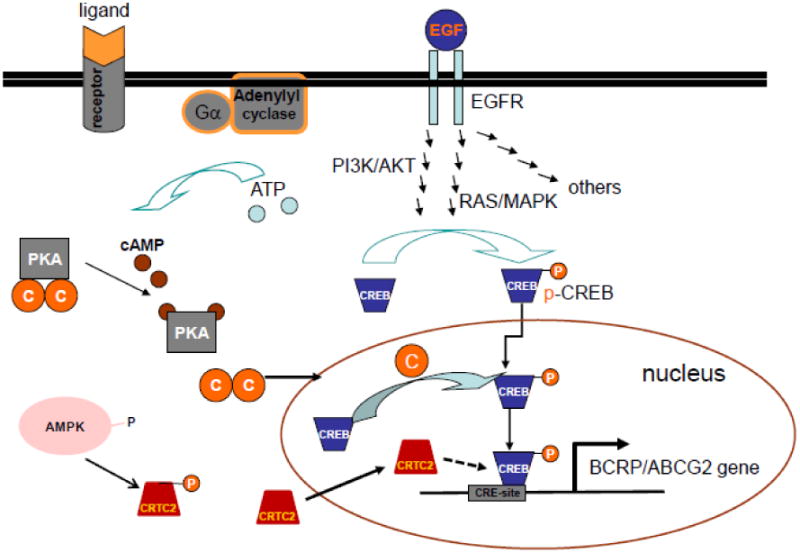
Schematic of how p-CREB acts as a common downstream effector for multiple signaling pathways that regulate BCRP mRNA expression Xie, et.al.
Highlights.
A novel cyclic AMP response element (CRE) in the BCRP promoter binds p-CREB
This CRE plays a central role in stimulating BCRP transcription
EGFR signaling enhances BCRP transcription via this CRE
Androgen withdrawal in prostate cancer cells increases p-CREB and nuclear CRTC2
Androgen withdrawal in prostate cancer cells activates BCRP transcription
Acknowledgments
Dr. Ross' and Dr. Hussain's research is supported by Department of Veterans' Affairs Merit Review Awards. Dr. Hamburger's research is supported by grant 1R01CA138583 from the National Cancer Institute, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Natarajan K, Xie Y, Baer MR, Ross DD. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem Pharmacol. 2012;83:1084–1103. doi: 10.1016/j.bcp.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakanishi T, Ross DD. Breast cancer resistance protein (BCRP/ABCG2): its role in multidrug resistance and regulation of its gene expression. Chin J Cancer. 2012;31:73–99. doi: 10.5732/cjc.011.10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Natarajan K, Xie Y, Nakanishi T, Beck WT, Bauer KS, Ross DD. Identification and characterization of the major alternative promoter regulating Bcrp1/Abcg2 expression in the mouse intestine. Biochim Biophys Acta. 2011;1809:295–305. doi: 10.1016/j.bbagrm.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang S, Dong S, Guo A, Ruan H, Lonial S, Khoury HJ, Gu TL, Chen J. Epidermal growth factor stimulates RSK2 activation through activation of the MEK/ERK pathway and src-dependent tyrosine phosphorylation of RSK2 at Tyr-529. J Biol Chem. 2008;283:4652–4657. doi: 10.1074/jbc.M709673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gan Y, Shi C, Inge L, Hibner M, Balducci J, Huang Y. Differential roles of ERK and Akt pathways in regulation of EGFR-mediated signaling and motility in prostate cancer cells. Oncogene. 2010;29:4947–4958. doi: 10.1038/onc.2010.240. [DOI] [PubMed] [Google Scholar]
- 6.Meyer zu Schwabedissen HE, Grube M, Dreisbach A, Jedlitschky G, Meissner K, Linnemann K, Fusch C, Ritter CA, Volker U, Kroemer HK. Epidermal growth factor-mediated activation of the map kinase cascade results in altered expression and function of ABCG2 (BCRP) Drug Metab Dispos. 2006;34:524–533. doi: 10.1124/dmd.105.007591. [DOI] [PubMed] [Google Scholar]
- 7.Huang WC, Chen YJ, Li LY, Wei YL, Hsu SC, Tsai SL, Chiu PC, Huang WP, Wang YN, Chen CH, Chang WC, Chen AJ, Tsai CH, Hung MC. Nuclear Translocation of Epidermal Growth Factor Receptor by Akt-dependent Phosphorylation Enhances Breast Cancer-resistant Protein Expression in Gefitinib-resistant Cells. J Biol Chem. 2011;286:20558–20568. doi: 10.1074/jbc.M111.240796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, Takemori H, Montminy M. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- 9.Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 10.Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, Montminy M. TORCs: transducers of regulated CREB activity. Molecular cell. 2003;12:413–423. doi: 10.1016/j.molcel.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 11.Katoh Y, Takemori H, Lin XZ, Tamura M, Muraoka M, Satoh T, Tsuchiya Y, Min L, Doi J, Miyauchi A, Witters LA, Nakamura H, Okamoto M. Silencing the constitutive active transcription factor CREB by the LKB1-SIK signaling cascade. FEBS J. 2006;273:2730–2748. doi: 10.1111/j.1742-4658.2006.05291.x. [DOI] [PubMed] [Google Scholar]
- 12.Nakanishi T, Karp JE, Tan M, Doyle LA, Peters T, Yang W, Wei D, Ross DD. Quantitative analysis of breast cancer resistance protein and cellular resistance to flavopiridol in acute leukemia patients. Clin Cancer Res. 2003;9:3320–3328. [PubMed] [Google Scholar]
- 13.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 14.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 15.Quandt K, Frech K, Karas H, Wingender E, Werner T. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bailey-Dell KJ, Hassel B, Doyle LA, Ross DD. Promoter characterization and genomic organization of the human breast cancer resistance protein (ATP-binding cassette transporter G2) gene. Biochim Biophys Acta. 2001;1520:234–241. doi: 10.1016/s0167-4781(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 17.Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- 18.Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi YH, Lee SN, Aoyagi H, Yamasaki Y, Yoo JY, Park B, Shin DM, Yoon HG, Yoon JH. The extracellular signal-regulated kinase mitogen-activated protein kinase/ribosomal S6 protein kinase 1 cascade phosphorylates cAMP response element-binding protein to induce MUC5B gene expression via D-prostanoid receptor signaling. J Biol Chem. 2011;286:34199–34214. doi: 10.1074/jbc.M111.247684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hisaoka K, Tsuchioka M, Yano R, Maeda N, Kajitani N, Morioka N, Nakata Y, Takebayashi M. Tricyclic antidepressant amitriptyline activates fibroblast growth factor receptor signaling in glial cells: involvement in glial cell line-derived neurotrophic factor production. J Biol Chem. 2011;286:21118–21128. doi: 10.1074/jbc.M111.224683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bianchi F, Magnifico A, Olgiati C, Zanesi N, Pekarsky Y, Tagliabue E, Croce CM, Menard S, Campiglio M. FHIT-proteasome degradation caused by mitogenic stimulation of the EGF receptor family in cancer cells. Proc Natl Acad Sci U S A. 2006;103:18981–18986. doi: 10.1073/pnas.0605821103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rush JS, Quinalty LM, Engelman L, Sherry DM, Ceresa BP. Endosomal accumulation of the activated epidermal growth factor receptor (EGFR) induces apoptosis. J Biol Chem. 2012;287:712–722. doi: 10.1074/jbc.M111.294470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 24.Imai Y, Ishikawa E, Asada S, Sugimoto Y. Estrogen-mediated post transcriptional down-regulation of breast cancer resistance protein/ABCG2. Cancer Res. 2005;65:596–604. [PubMed] [Google Scholar]
- 25.Mikhailova M, Wang Y, Bedolla R, Lu XH, Kreisberg JI, Ghosh PM. AKT regulates androgen receptor-dependent growth and PSA expression in prostate cancer. Adv Exp Med Biol. 2008;617:397–405. doi: 10.1007/978-0-387-69080-3_38. [DOI] [PubMed] [Google Scholar]
- 26.Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, Warren A, Scott H, Madhu B, Sharma N, Bon H, Zecchini V, Smith DM, Denicola GM, Mathews N, Osborne M, Hadfield J, Macarthur S, Adryan B, Lyons SK, Brindle KM, Griffiths J, Gleave ME, Rennie PS, Neal DE, Mills IG. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. The EMBO journal. 2011;30:2719–2733. doi: 10.1038/emboj.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karacosta LG, Foster BA, Azabdaftari G, Feliciano DM, Edelman AM. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J Biol Chem. 2012;287:24832–24843. doi: 10.1074/jbc.M112.370783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie Y, Natarajan K, Bauer KS, Nakanishi T, Beck WT, Moreci RS, Jeyasuria P, Hussain A, Ross DD. Bcrp1 transcription in mouse testis is controlled by a promoter upstream of a novel first exon (E1U) regulated by steroidogenic factor-1. Biochim Biophys Acta. 2013;1829:1288–1299. doi: 10.1016/j.bbagrm.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. The EMBO journal. 1998;17:4426–4441. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dash PK, Karl KA, Colicos MA, Prywes R, Kandel ER. cAMP response element-binding protein is activated by Ca2+/calmodulin- as well as cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1991;88:5061–5065. doi: 10.1073/pnas.88.11.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheng M, Thompson MA, Greenberg ME. CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- 32.Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb MJ. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. The EMBO journal. 1996;15:4629–4642. [PMC free article] [PubMed] [Google Scholar]
- 33.Iordanov M, Bender K, Ade T, Schmid W, Sachsenmaier C, Engel K, Gaestel M, Rahmsdorf HJ, Herrlich P. CREB is activated by UVC through a p38/HOG-1-dependent protein kinase. The EMBO journal. 1997;16:1009–1022. doi: 10.1093/emboj/16.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foley J, Nickerson NK, Nam S, Allen KT, Gilmore JL, Nephew KP, Riese DJ., 2nd EGFR signaling in breast cancer: bad to the bone. Seminars in cell & developmental biology. 2010;21:951–960. doi: 10.1016/j.semcdb.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soeda A, Inagaki A, Oka N, Ikegame Y, Aoki H, Yoshimura S, Nakashima S, Kunisada T, Iwama T. Epidermal growth factor plays a crucial role in mitogenic regulation of human brain tumor stem cells. J Biol Chem. 2008;283:10958–10966. doi: 10.1074/jbc.M704205200. [DOI] [PubMed] [Google Scholar]
- 36.Morgillo F, Bareschino MA, Bianco R, Tortora G, Ciardiello F. Primary and acquired resistance to anti-EGFR targeted drugs in cancer therapy. Differentiation. 2007;75:788–799. doi: 10.1111/j.1432-0436.2007.00200.x. [DOI] [PubMed] [Google Scholar]
- 37.Chaft JE, Arcila ME, Paik PK, Lau C, Riely GJ, Pietanza MC, Zakowski MF, Rusch V, Sima CS, Ladanyi M, Kris MG. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther. 2012;11:485–491. doi: 10.1158/1535-7163.MCT-11-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rychahou PG, Jackson LN, Silva SR, Rajaraman S, Evers BM. Targeted molecular therapy of the PI3K pathway: therapeutic significance of PI3K subunit targeting in colorectal carcinoma. Ann Surg. 2006;243:833–842. doi: 10.1097/01.sla.0000220040.66012.a9. discussion 843-834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, Rinehart C, Seidel B, Yee D, Arteaga CL, Engelman JA. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. 2008;118:2609–2619. doi: 10.1172/JCI34588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakanishi T, Shiozawa K, Hassel BA, Ross DD. Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood. 2006;108:678–684. doi: 10.1182/blood-2005-10-4020. [DOI] [PubMed] [Google Scholar]
- 41.Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL, Pan Y, Wang L, de Stanchina E, Shien K, Aoe K, Toyooka S, Kiura K, Fernandez-Cuesta L, Fidias P, Yang JC, Miller VA, Riely GJ, Kris MG, Engelman JA, Vnencak-Jones CL, Dias-Santagata D, Ladanyi M, Pao W. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109:E2127–2133. doi: 10.1073/pnas.1203530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiao X, Li BX, Mitton B, Ikeda A, Sakamoto KM. Targeting CREB for cancer therapy: friend or foe. Current cancer drug targets. 2010;10:384–391. doi: 10.2174/156800910791208535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu D, Zhau HE, Huang WC, Iqbal S, Habib FK, Sartor O, Cvitanovic L, Marshall FF, Xu Z, Chung LW. cAMP-responsive element-binding protein regulates vascular endothelial growth factor expression: implication in human prostate cancer bone metastasis. Oncogene. 2007;26:5070–5077. doi: 10.1038/sj.onc.1210316. [DOI] [PubMed] [Google Scholar]
- 44.Deng X, Liu H, Huang J, Cheng L, Keller ET, Parsons SJ, Hu CD. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: implications for disease progression. Cancer Res. 2008;68:9663–9670. doi: 10.1158/0008-5472.CAN-08-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roach M., 3rd Current trends for the use of androgen deprivation therapy in conjunction with radiotherapy for patients with unfavorable intermediate-risk, high-risk, localized, and locally advanced prostate cancer. Cancer. 2014;120:1620–1629. doi: 10.1002/cncr.28594. [DOI] [PubMed] [Google Scholar]
- 46.Tang Y, Khan MA, Goloubeva O, Lee DI, Jelovac D, Brodie AM, Hussain A. Docetaxel followed by castration improves outcomes in LNCaP prostate cancer-bearing severe combined immunodeficient mice. Clin Cancer Res. 2006;12:169–174. doi: 10.1158/1078-0432.CCR-05-1388. [DOI] [PubMed] [Google Scholar]