

Abstract
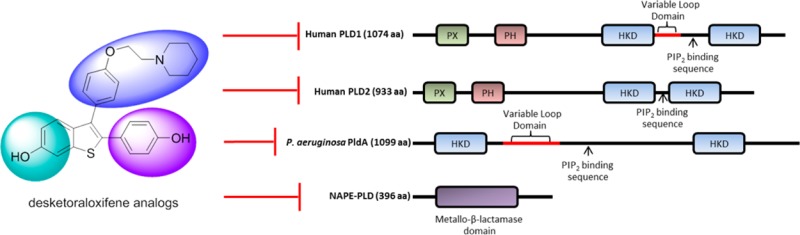
Phospholipase D (PLD) hydrolyses cellular lipids to produce the important lipid second messenger phosphatidic acid. A PLD enzyme expressed by Pseudomonas aeruginosa (PldA) has been shown to be important in bacterial infection, and NAPE-PLD has emerged as being key in the synthesis of endocannabinoids. In order to better understand the biology and therapeutic potential of these less explored PLD enzymes, small molecule tools are required. Selective estrogen receptor modulators (SERMs) have been previously shown to inhibit mammalian PLD (PLD1 and PLD2). By targeted screening of a library of SERM analogues, additional parallel synthesis, and evaluation in multiple PLD assays, we discovered a novel desketoraloxifene-based scaffold that inhibited not only the two mammalian PLDs but also structurally divergent PldA and NAPE-PLD. This finding represents an important first step toward the development of small molecules possessing universal inhibition of divergent PLD enzymes to advance the field.
Phospholipase D (PLD) is an important cellular enzyme that catalyzes the hydrolysis of phosphatidylcholine (PC) to phosphatidic acid (PA) and choline. PA is an essential signaling molecule and has been implicated in a plethora of cellular processes including cell growth, differentiation, and metastasis.1 There are over 4000 enzymes with PLD-like activity that hydrolyze phosphodiester bonds, neutral lipids, or polynucleotides. The largest PLD superfamily possesses two conserved histidine–lysine–aspartate (HKD) amino acid motifs that are thought to form the catalytic site; however, there are also non-HKD enzymes that exhibit distinct structures and mechanisms. Two mammalian isoforms have been identified, PLD1 and PLD2, with high sequence homology but disparate function. These isoforms share conserved phox homology (PX) and pleckstrin homology (PH) domains at the amino terminus, which are thought to be important for regulation of activity through lipid and protein binding (Figure 1).1 Several intracellular pathogens are known to secrete their own PLD enzymes to promote internalization or intracellular survival.2−8Pseudomonas aeruginosa is an opportunistic pathogen that infects immunocompromised patients and is a major cause of hospital-acquired infections. Cystic fibrosis patients are particularly susceptible to developing chronic P. aeruginosa infections that lead to severe lung damage and eventually respiratory failure. Recently, PldA was identified as a secreted effector of the Type VI secretion system of P. aeruginosa that targets human epithelial cells to promote bacterial internalization9 and was found to target bacterial cells to promote intra- and interbacterial species competition,10 both of which may be important components for establishing and maintaining infection. On the basis of phylogenetic analysis, PldA is closely related to eukaryotic PLD.9 In fact PldA has regions of high homology with the mammalian enzyme, including possessing two catalytic HKD motifs, but it does not share much homology with those enzymes from prokaryotes (Figure 1). Genetic examination even suggests that PldA may have been acquired through horizontal transfer by P. aeruginosa.11 NAPE-PLD, or N-acyl phosphatidylethanolamine phospholipase D, which hydrolyzes N-acyl phosphatidylethanolamine to generate N-acylethanolamines (NAE), such as anandamide, and phosphatidic acid is a representative non-HKD PLD of great interest (Figure 1).12 Anandamide is a major endocannabinoid shown to have antinociceptive and analgesic properties, to play a role in several neurodegenerative disorders, and to have antiproliferative, antimetastatic, and pro-apoptotic effects toward cancer cells in culture and in vivo.13
Figure 1.

Phospholipase D. (A) Sequences of HKD-containing mammalian PLD1 and PLD2, P. aeruginosa PldA, and the non-HKD containing NAPE-PLD, highlighting the divergent sequences and overall disparate homology. (B) Schematic of the enzyme-catalyzed reactions of these PLDs that result in phosphatidic acid (PA) production and diverse substrates.
Clearly, these four enzymes are divergent, yet the identification of a ligand that could bind to, and inhibit, with broad spectrum activity would be highly desirable to dissect their physiological roles and assess therapeutic potential. Very few published accounts of bacterial PLD inhibition currently exist. Early work on the non-HKD containing Streptomyces chromofuscus PLD utilized a nonhydrolyzable phosphoramidate substrate mimic to modestly inhibit enzymatic activity,14 and the literature does not contain any subsequent reports on small molecule inhibitor development for bacterial HKD or non-HKD PLDs. In the case of NAPE-PLD, most insights have been garnered through studies in NAPE-PLD–/– mice.15 In fact, only in the past decade has isoform-selective, direct inhibition of mammalian PLD1 and PLD2 been achieved with small molecules16−20 (Figure 2), and prior to that time, n-butanol was the primary tool to study PLD function. Compounds 1–6 elucidated new roles for PLD in oncology and viral infection, re-energizing the field; however, these potent and highly selective inhibitors of mammalian PLD have never been tested against PldA or NAPE-PLD.
Figure 2.

Structures of direct, allosteric mammalian PLD inhibitors 1–6. First-generation dual PLD1/2 inhibitor 1, PLD1-selective inhibitor 2, and PLD2-selective inhibitor 3 elucidated many important, and previously undefined, roles for PLD in oncology and virology. Second-generation PLD inhibitors 4–6 improved upon DMPK profiles and ancillary pharmacology as probes for the Molecular Library Probe Center Network (MLPCN).
Selective estrogen receptor modulators (SERMs) have long been used for the successful treatment of breast cancer (Figure 3). Two of the most commonly used SERMs are raloxifene (7)21 and tamoxifen (8).22 Both compounds were developed as antiestrogens acting at the estrogen receptor but have since been shown to have differential effects. Raloxifene is often used in the treatment of osteoporosis in postmenopausal women and is also used as a preventative measure for women at a high risk for breast cancer development, whereas tamoxifen has been used for more than 30 years for the treatment of ER+ breast cancer. Both 7 and 8 have been shown to have antiproliferative effects in ER negative breast cancer, suggesting that there is an alternative mechanism of action for these compounds.23 A previous report from our lab suggests that a potential ER-independent mechanism for the efficacy of these SERMs may be due to off-target inhibition of mammalian PLD.24 In that study, both 7 and 4-hydroxytamoxifen (9), the active cellular metabolite of 8, were shown to modestly inhibit mammalian PLD activity, both in cells and in vitro, whereas 8 stimulated PLD2 and showed no effect on PLD1. Desketoraloxifene (10) was developed by Eli Lilly in the late 90s as a raloxifene analogue that lacked the important carbonyl hinge in the parent molecule,25 thus leading to a SERM with unique, planar topology.26 This modification made the analogue a much stronger activator in the AP-1 site of ERα compared to that of ERβ, which is the opposite profile of 8. This new analogue had differential SERM efficacy with stimulatory activity in the uterus, similar to 9, but retained raloxifene-like character by remaining estrogenic in the bone.25 Analogue 10 was synthesized following the literature route, and its ability to inhibit PLDs was assessed; gratifyingly, in our exogenous PLD assays, 10 was a modest inhibitor of mammalian PLD1 (IC50 = 6.1 μM) and PLD2 (IC50 = 2.6 μM). However, as with the structurally distinct mammalian PLD inhibitors 1–6, the SERM-based PLD inhibitors 7–10 had also never been evaluated as inhibitors of either PldA or NAPE-PLD.
Figure 3.

Structures and PLD activities of known SERMS 7–10. These scaffolds represent a second chemotype, other than that of 1–6, that has been shown to inhibit mammalian PLD.
The use of either well-characterized 1–6 or SERMs 7–10 as starting points for inhibitor development aimed at PldA and NAPE-PLD is attractive due to the vast amount of preclinical and clinical information and pharmacokinetics already known about these compounds. Herein, we report a multipronged chemical biology effort that ultimately resulted in the identification of deketoraloxifene 10 and related analogues as the first small molecule inhibitors of PldA and NAPE-PLD; moreover, these ligands inhibited mammalian PLD1 and PLD2 as well. Thus, desketoraloxifene (10) represents the first example of a universal PLD inhibitor chemotype across structurally and phylogenetically distinct enzymes.
Results and Discussion
Evaluation of Known Mammalian PLD Inhibitors against PldA
The past decade witnessed significant progress in the discovery and development of mammalian PLD inhibitors 1–10.1,16−20 Although inhibitors based on halopemide (1–6) represented a major advance in our understanding of the physiology and therapeutic potential of individual isoenzymes PLD1 and PLD2, we were disappointed to find that they displayed no inhibitory activity against bacterial PldA (IC50’s > 20 μM), suggesting that the allosteric binding site occupied by 1–6 is not present in this PLD enzyme, thus requiring a search for novel chemotypes with alternate binding sites. Thus, our attention focused on the structurally distinct SERM ligands. With 7–10 previously being shown to inhibit mammalian PLD with mid-micromolar potency, we also assessed these against PldA. Gratifyingly (Table 1), 7–10 all displayed comparable potency at inhibition of PldA (IC50’s 4–18 μM). This unprecedented finding led us consider that other SERM analogues 10 may also exhibit broader spectrum PLD inhibitory activity, and it prompted a more thorough investigation of structure–activity relationships within scaffold 10. Here, we report a novel desketoraloxifene-based scaffold that inhibited not only the two mammalian PLDs but also PldA and NAPE-PLD by a multifaceted chemical biology approach.
Table 1. Activities of 1–10 at Mammalian PLD and P. aeruginosa PLD (PldA).
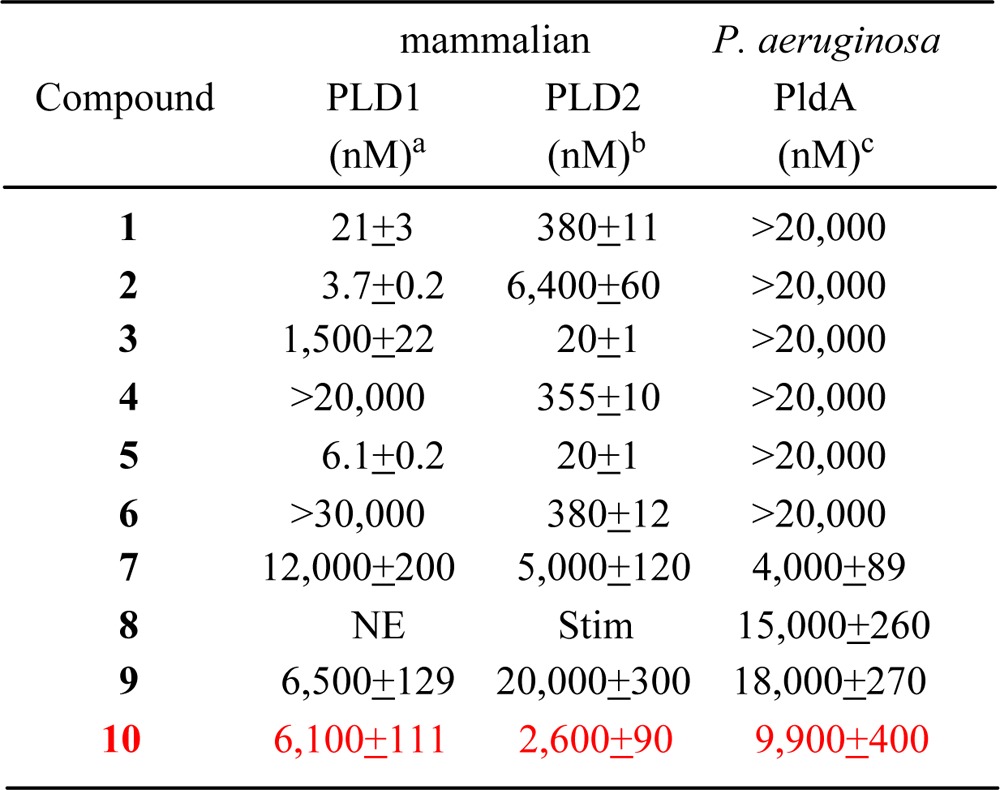
Cellular PLD1 assay in Calu-1 cells.
PLD2 cellular assay in HEK293-gfp-PLD2 cells.
Exogenous PldA assay. IC50 values are the average of n = 3 ± SEM. NE, no effect; Stim, stimulator of PLD2 activity.
Multiplatform PLD Screening
To follow-up on the unique PLD inhibitory profile of 10, we began to design analogue libraries; however, we took note of a library of analogues of 11 reported (Figure 4) by Larock and co-workers in conjunction with the Kansas University Center for Methodology and Library Development (CMLD) that surveyed three regions of the core.27 We were graciously provided 77 analogues 11, and we first screened the library (tested at 10 μg/mL) for inhibition of cellular mammalian PLD1 and PLD2. PLD activity was assessed utilizing a PLD transphosphatidylation reaction unique to this enzyme.16 Instead of the traditional biological nucleophile water, in the presence of a primary alcohol (n-butanol), PLD will alternatively use the alcohol to produce a metabolically stable transphosphatidylation product, phosphatidylbutanol (PtdBuOH), instead of producing PA. The PLD-specific lipid products (PtdBuOHs) were then isolated and detected using mass spectrometry as previously described (Figure 5). From the single-point data, a number of analogues 11 possessed PLD inhibitory activity, with a general preference toward inhibition of PLD1 over PLD2. In follow-up assays with full concentration–response curves (CRCs), several analogues displayed low micromolar potency (see Supporting Information Table S1) at PLD1 (IC50’s 2.7–13 μM) and no activity at PLD2 (IC50’s > 50 μM).
Figure 4.
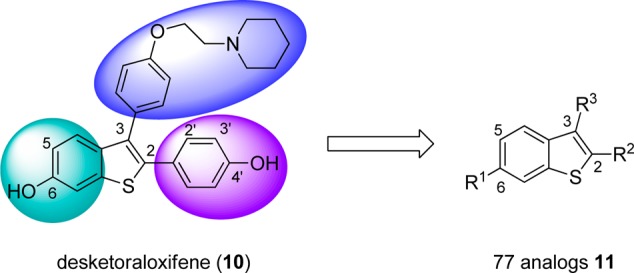
Three regions of 10 surveyed in a diversity library of desketoraloxifene analogues 11.
Figure 5.

Single-point screen of analogues 11. (A) Preliminary library screening approach for mammalian PLD inhibitor identification using a mass spectroscopy-based assay; (B) desketoraloxifene analogue 11 inhibition of cellular PLD1 (PMA stimulated Calu-1 cells) or (C) cellular PLD2 (HEK293-PLD2 cells) tested at 10 μg/mL.
To assess the ability of analogues 11 to inhibit PldA, PldA was first overexpressed in Escherichia coli and purified using immobilized metal-affinity chromatography and size-exclusion chromatography. PldA was determined to hydrolyze a wide array of phospholipid substrates, including PC. Analogues 11 were screened at a set 5 μg/mL concentration against purified PldA employing a modified version of the commercially available Amplex Red kit due to its convenience and high-throughput potential (Figure 6). The Amplex Red assay utilizes a three-step enzymatic process to produce the fluorescent compound, resorufin. Free choline is liberated from PC by a PLD that is then used by choline oxidase (CO) to generate H2O2. Horseradish peroxidase (HRP) in turn uses H2O2 to oxidize the Amplex Red compound to generate resorufin. The production of resorufin over time serves as a proxy for choline liberation and hence PLD activity. The most reproducible activity using the Amplex Red assay was with a synthetic short chain 1,2-diheptanoyl-sn-glycero-3-phosphocholine (7:0/7:0 PC) substrate below its critical micelle concentration, as opposed to a long chain PC in liposomes. Because 10 and analogues 11 are hydrophobic and could intercalate into the membrane, inhibition could be due to disruption of interaction with the lipid interface as opposed to directly altering catalytic activity, leading to false positives. The use of a monomeric, soluble substrate allows examination of the effects of the small molecules on PldA activity independent of protein binding to lipid membrane and enables selection of molecules that directly impact PldA activity. The strategy for addressing and identifying compounds with off-target effects during screening was to test each compound in the presence of choline chloride without PldA present to ensure that changes in the observed fluorescence were not attributable to inhibition of choline oxidase or horseradish peroxidase. The Amplex Red assay has been used in the past to screen large libraries of compounds for possible PLD inhibition; however, these reports tend to not fully investigate the possibility of false positives through inhibition of CO or HRP. For this reason, it is important to further validate leads using the previously published and well-established exogenous PLD assay as an alternative assay system.28,29 Excitingly, as shown in Figure 6, a number of analogues 11 displayed inhibition of PldA; however, we had exhausted our supply of the desketoraloxifene library 11 before we could acquire PldA CRCs. Still, the data from the three assays enabled the assessment of favorable SAR trends for inhibition of PLD1/2, PldA, or both (Figure 7).
Figure 6.

Single-point assay paradigm to assess PldA inhibition with analogues 11. (A) Modified Amplex Red high-throughput assay to assess PldA activity and (B) single-point desketoraloxifene analogue 11 inhibition of bacterial PldA tested at 5 μg/mL.
Figure 7.

Favorable desketoraloxifene analogue 11 SAR trends for (A) mammalian PLD and (B) P. aeruginosa PldA inhibition.
Chemistry
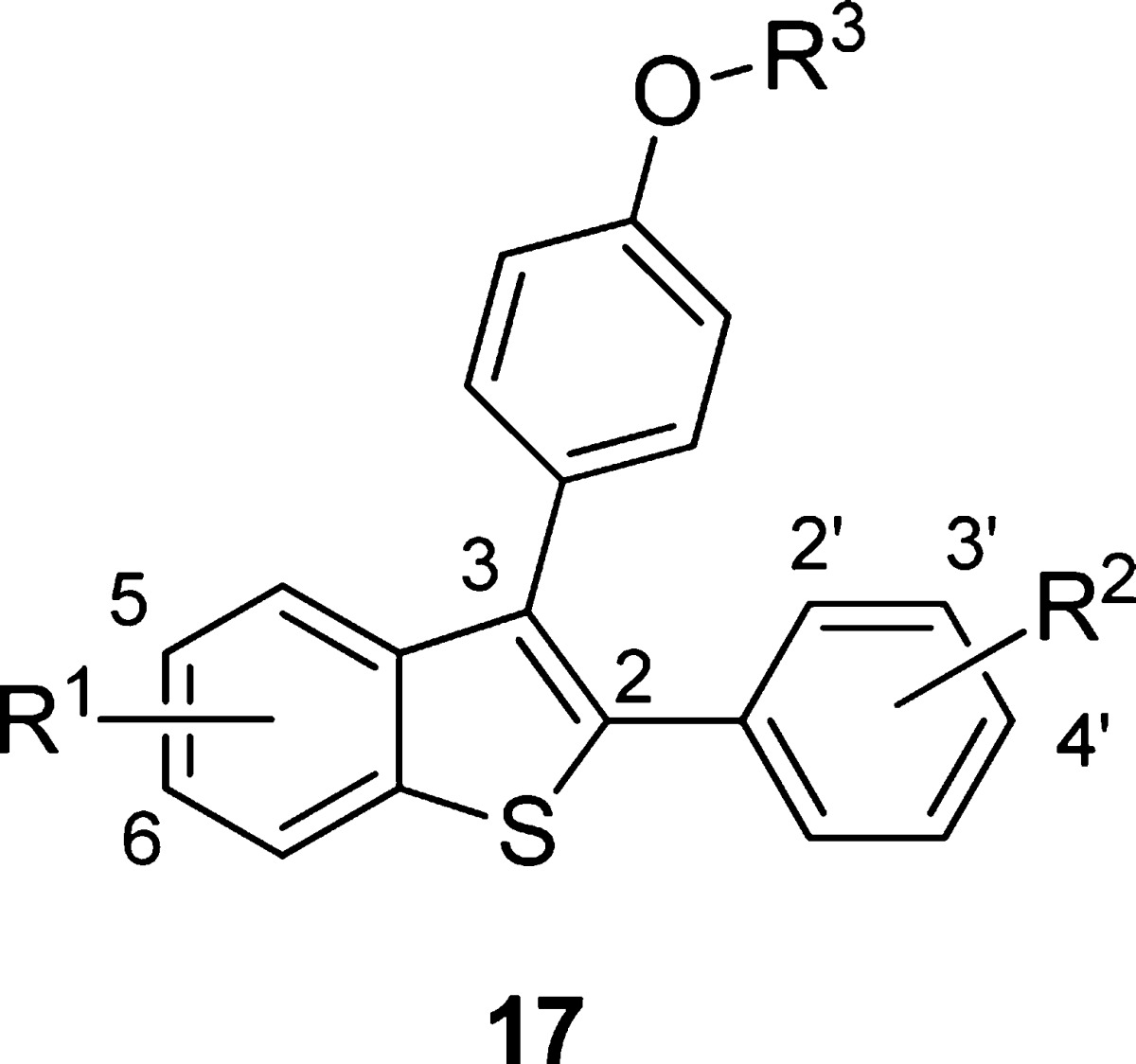
Using the SAR trends observed from the initial desketoraloxifene analogue library, additional targeted compounds were synthesized for the further development of small molecule mammalian and bacterial PLD inhibitors, but due to unfavorable DMPK properties and potential safety concerns, the acetylenic analogues were not pursued further; rather, synthesis was focused on aryl substitution patterns not represented in the provided library of analogues 11. Toward this goal, the original iodocyclization/palladium-catalyzed cross-coupling strategy employed by Larock in the preparation of analogues 11 was used with some modifications (Scheme 1).27,30 Here, dihaloarene 12 undergoes a Sonogashira coupling with ethynylanisole to provide 13. Conversion to the thioether and iodocyclization affords benzthiophene 14, and a subsequent Suzuki coupling delivers analogues 15. The aminoalkyl ether moiety is then installed through a Mitsunobu reaction to provide analogues 16, which were either assayed as the ethers, or deprotected under BBr3 conditions to yield the phenolic congeners 17. Overall yields for the six-step sequence ranged from 10 to 28%.
Scheme 1. Synthesis of Novel Desketoraloxifene Analogues 17.

Reagents and conditions: (a) ethynylanisole, 2 mol % PdCl2(PPh)3, 2 mol % CuI, Et3N, 50 °C, 56–89%; (b) n-BuLi, THF, −78 °C, then dimethyldisulfide, −78 °C to rt, 28–79%; (c) I2, DCM, rt, 80–99%; (d) (4-((tetrahydro-2H-pyran-yl)oxy)phenyl)boronic acid, 10 mol % Pd(dppf)2Cl2, K3PO4, dioxane/H2O, mw 140 °C, 20 min 72–90%; (e) HO(CH)2NR2, DIAD, PPh3, THF, rt, 45–85%; (f) 4.0 equiv. BBr3, DCM, 0 °C to rt, 37–90%.
Novel Analogue Screening and SAR
Novel analogues 15–17 were synthesized on the basis of the SAR trends observed in the initial published library and were assessed for their ability to inhibit human PLD1 and PLD2 and PldA. Moreover, we wanted to separate the SERM activity of 10 and analogues 11 from the PLD in inhibitory activity. The molecular determinants of SERM activity, i.e., the SERM pharmacophore, are well-established for both 7 and 10. The 6-OH (Figure 4) is essential for esterogen receptor binding and antiproliferative activity; in fact, deletion of the 6-OH leads to >100-fold loss in receptor affinity and antiproliferative action.31 In addition, replacement of the piperidinyl moiety in 10 with a N,N-dimethyl amine moiety similarly decreases SERM activity by >3-fold.31 Therefore, these clear implications from the SERM pharmacophore were incorporated into the design of new analogues. Human isoform inhibition was again assessed in the PLD1 and PLD2 model cellular systems at a single set 20 μM concentration. Inhibitory activity was determined by comparison with DMSO vehicle control, and hits were identified as compounds that inhibited >50% activity or showed an isoform-selective profile between PLD1 and PLD2 (Figure 8A,B). Compounds were also screened against PldA at 20 μM using the modified Amplex Red assay described above. Many compounds that significantly inhibited PldA activity were similar to those that inhibited the human enzymes (Figure 8C), and these compounds were further validated as hits using the exogenous PldA assay and found to inhibit PldA activity >50% at 20 μM. Thus, we were excited to find novel desketoraloxifene analogues that inhibited both mammalian PLD and bacterial PldA from this parallel library effort.
Figure 8.

Single-point screening of novel desketoraloxifene analogues 15–17 against mammalian (A) PLD1, (B) PLD2, and (C) PldA. Mammalian PLDs were screened via model cellular systems at 20 μM. PldA was screened using a modified Amplex Red assay at 20 μM. * denotes false positives in the modified Amplex Red assay.
Concentration–response curves of lead compounds were generated using the cellular model systems and exogenous assay for the human isoforms or using the modified Amplex Red assay and exogenous assay for PldA (Table 2). Each assay system has its own merits and limitations. The cellular model systems for human PLD1 and PLD2 require that the molecules be cell-penetrant, which is both a positive and a negative requirement, and are more complex, allowing for indirect effects on PLD activity. The exogenous PLD assay utilizes recombinant purified human PLD enzyme and reconstituted liposomes in an in vitro system. Cell penetrance is no longer an issue, and effects are likely to be more direct. As for PldA, the Amplex Red assay uses monomeric substrate, whereas substrate is presented as a liposome in the exogenous assay. These compounds are highly hydrophobic with limited water solubility. Decrease in potency in the exogenous assay may be attributable to inhibitor sequestration in liposomal membranes or within vesicles that alter the concentration of inhibitor accessible to PldA. A similar phenomenon seems to occur for the human isoforms. There is a selective inhibition of PLD1 over PLD2 using the cellular screening assay; however, analysis with recombinant protein using the exogenous assay demonstrates that the compounds do directly affect the activity of both PLD isoforms. Differences in the subcellular localization, and therefore accessibility, of these compounds to PLD1 and PLD2 within these cells may also explain this discrepancy.
Table 2. Activities of Desketoraloxifene Analogues 17 at Mammalian PLD and P. aeruginosa PLD (PldA).
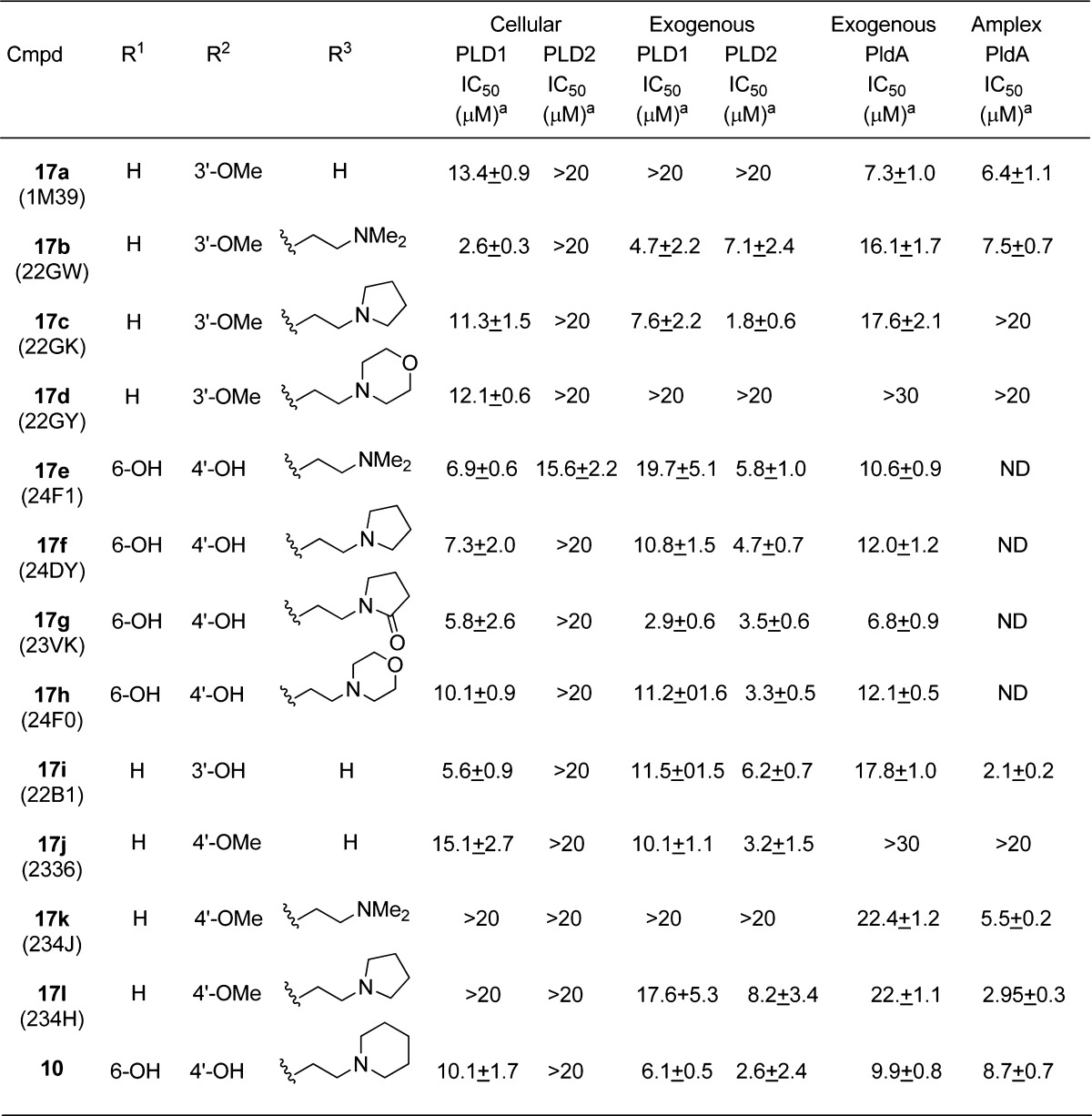
Each IC50 is the average of three experiments expressed ± SEM; ND, not determined.
In general, compounds closely related to the desketoraloxifene parent structure 10 were the most favorable pan-PLD inhibitors. These compounds contained modest modifications to the C ring of the scaffold but retained hydroxyl substitutions in the A and B rings. Many compounds that inhibited mammalian PLDs also inhibited the bacterial enzyme (17a–c, 17e–i, 17k, and 17l), although the overall SAR trends were not completely conserved. Several compounds, e.g., 17a and 17l, displayed enhanced inhibition of PldA compared to that toward mammalian PLD1 and PLD2. Alternatively, 17c and 17j preferentially inhibited mammalian PLD1 and PLD2 over PldA (Table 2). However, the SAR was far from robust and highlights the power of preparing libraries of analogues to capture serendipitous bioactivity.
Alternative Inhibitor Characterization against PldA
Potent small molecule inhibitors of mammalian PLD1 and PLD2 containing benzimidazole and triazaspirone scaffolds 1–6 were previously reported (Figure 2).16−18 Single-point 20 μM concentration comparisons and CRCs of representative PLD1-, PLD2-, or dual PLD1/2-selective mammalian PLD inhibitors 1–3 were performed alongside raloxifene (7) to evaluate their potency against PldA (Figure 9A). The raloxifene IC50 (3.9 μM) was 5- to 10-fold more potent than that of the PLD1-selective, PLD2-selective, and dual PLD1/2 inhibitors, 2 (26.1 μM), 3 (19.1 μM), and 1 (40 μM), respectively. Hence, a major discrepancy exists between potency against PldA relative to the human PLD isoforms of 1000- to 10 000-fold. This data further suggests that although PldA is evolutionarily similar to the mammalian PLDs there is a differential conservation of inhibitor binding sites for these varied scaffolds. Unlike the raloxifene (7), desketoraloxifene (10), and analogues 11 and 17, which displayed similar SAR trends and potency among PldA, PLD1, and PLD2, the benzimidazole and triazaspirone scaffolds 1–6, which potently inhibit human PLD1 and PLD2 in the low nanomolar range, are comparably poor inhibitors of PldA, suggesting that these compounds bind to a region of the mammalian enzymes that is lacking or is not well conserved in the bacterial PldA. The identification of two variable lead scaffolds with differential binding sites supports the possibility of developing specific inhibitors of P. aeruginosa phospholipase D, PldA.
Figure 9.

Screening of unique structural PLD inhibitors against (A) bacterial PldA and (B) mammalian NAPE-PLD. Compounds were screened against NAPE-PLD via a mass spectrometry assay at 20 μM, except for MAFP, which was used at 500 μM. PldA was screened using the exogenous assay at 20 μM. (C) Full CRC for 17b on mammalian NAPE-PLD (IC50 = 67 μM).
Characterization of Alternative NAPE-PLD Enzymes
As previously mentioned, other enzymes, such as NAPE-PLD, exhibit phospholipase D-like phosphodiesterase activity; however, they are not structurally related to the HKD-containing PLD family, which includes human PLD1 and PLD2 and PldA. The potency of various structural classes of PLD inhibitors developed against HKD-containing PLD proteins was evaluated on recombinant human NAPE-PLD. Each compound was assayed at a set 20 μM final concentration, with the exception of methyl arachidonyl fluorophosphonate (MAFP), a published NAPE-PLD inhibitor,32 which was used at 500 μM (Figure 9B). PA production by NAPE-PLD was assessed utilizing mass spectrometry.33 MAFP has been shown to inhibit NAPE-PLD activity in vitro with an IC50 of 1 mM and served as a positive control. The PLD1-selective, PLD2-selective, and dual PLD1/2 inhibitors 1–3 have poor potency against NAPE-PLD under these conditions (Figure 9B), which, like the data for PldA, suggests that the mammalian allosteric site is absent in this PLD enzyme as well. Raloxifene (7) was also observed not to inhibit NAPE-PLD activity (at 20 μM), although it was capable of inhibiting PLD1, PLD2, and PldA. Inhibition of NAPE-PLD by MAFP is likely due to perturbations of the lipid–detergent micelles disrupting protein–lipid interactions attributable to the high concentrations required for inhibition. As 10 was the most potent across the three PLD enzymes, we also evaluated it as an inhibitor of the highly structurally divergent NAPE-PLD and discovered that desketoraloxifene also inhibited this PLD (IC50 = 58 μM), albeit weakly. Moreover, the desketoraloxifene analogue 17b also displayed inhibition of NAPE-PLD, with an IC50 of 67 μM (Figure 9C), and was also active against mammalian PLD1 and PLd2 and PldA. Moreover, 17b was devoid of the 6-OH moiety, which is critical for estrogen receptor binding and antiproliferative action, and also contained an N,N-diemthylamino moiety, also known to greatly reduced SERM activity in this core. On the basis of the well-established SERM pharmacophore model for 7 and 10,31 it is reasonable to presume that 17b separates PLD activity from SERM activity. Although weak, 10 and 17b are rare examples of NAPE-PLD inhibitors and are the first examples of a chemotype that can universally inhibit mammalian PLD1/2, bacterial PldA, and NAPE-PLD, suggesting a potentially common binding site among structurally and phylogenetically distinct PLD enzymes.
Conclusions
A new class of PLD inhibitors has been classified on the basis of the raloxifene–tamoxifen hybrid SERM desketoraloxifene 10. Herein, we identify the first published inhibitors of an important bacterial PLD, P. aeruginosa, PldA, as well as weak, yet improved, inhibitors of NAPE-PLD. The compounds are modest dual mammalian PLD1/2 inhibitors but are the first to inhibit a diverse range of structurally and phylogentically diverse PLDs. PldA has been implicated in promoting chronic infection of the opportunistic pathogen P. aeruginosa.11 Therapeutics targeting this bacterial PLD may serve as viable targets for treating infections in view of the fact that it is a secreted protein that targets human epithelial cells to promote bacterial internalization. The identification of the first submillimolar inhibitor of NAPE-PLD will help to better define the role of this enzyme in cellular processes. SERM compounds are often well-tolerated in patients, have good pharmacokinetics, and have been used for decades in the clinic, which makes this class of analogues an ideal starting point for the development of novel, universal inhibitors of mammalian (PLD1/2, NAPE-PLD) and bacterial PLD (PldA). These are significant findings that have provided a lead series for further, focused optimization efforts toward potent pan-PLD inhibitors for in vivo studies. Further structural refinements are in progress and will be reported in due course.
Experimental Section
Cell Culture
Calu-1 cells were purchased from American Type Culture Collection (Manassas, VA). Calu-1 cells were maintained in DMEM supplemented with 10% FBS, 100 μg/mL penicillin–streptomycin, and 0.25 μg/mL amphotericin. HEK293 cells stably expressing GFP-tagged human PLD2A were generated in the lab. To sustain selection pressure, low-passage-number HEK293-gfpPLD2 cells were maintained in DMEM supplemented with 10% FBS, 100 μg/mL penicillin–streptomycin, 2 μg/mL puromycin, and 600 μg/mL G418. All HEK293-gfpPLD2 experiments were done on tissue culture plates that had been coated with low levels of polylysine. All cells were maintained in a humidified 5% CO2 incubator at 37 °C.
Endogenous PLD Activity Assay of Cell Lines Using Deuterated 1-Butanol Incorporation
Endogenous PLD activity was determined using a modified in vivo deuterated 1-butanol (1-butanol-d10) PLD assay.16,28 Cells were seeded into 12-well tissue culture plates to reach 90% confluence at the time of assay. All cell types, aside from the HEK293-gfpPLD2 cells, were serum-starved 18 h prior to experiment in DMEM, 0.5% FBS, 1% AA. Cells were pretreated in the presence of test compound (50 μM to 5 nM) or DMSO (vehicle control) in DMEM for 5 min at rt. After pretreatment, media was removed, and cells were treated with DMEM + 1 μM PMA + 0.3% 1-butanol-d10, and either test compound, DMSO vehicle control, or DMEM alone for 30 min at 37 °C. HEK293-gfpPLD2 cells were treated in the presence of DMEM + 0.3% 1-butanol-d10 and either test compound or DMSO but were not stimulated with PMA. After treatment, samples were extracted, and 1,2-dihexanoyl-sn-glycero-3-phosphomethanol (32:0 PtdMeOH) was added as an internal standard. The lipids were isolated, the solvent was evaporated, and the resulting lipid film was resuspended in MS solvent (9:1 methanol/chloroform + 1 μL of NH4OH). Samples were directly injected into a Finnigan TSQ Quantum triple quadrupole mass spectrometer, and data was collected in negative ionization mode. Data was analyzed by plotting the ratio of the major phosphatidylbutanol-d9 lipid products of PLD stimulation to the standard 32:0 PtdMeOH. Background signal was subtracted using cells not treated with 1-butanol-d10 as a negative control. The data was compared to vehicle control samples and is expressed as percent of PMA-stimulated PLD activity in the case of the Calu-1 cell line or as percent of basal PLD activity in HEK293-gfpPLD2 cells. Experiments were performed once in triplicate. IC50 values were determined using a concentration–response curve, and nonlinear curve fitting and statistical analysis were performed using GraphPad Prism, v. 4.0. Data are plotted as mean ± SEM unless otherwise stated.
Modified Amplex Red Assay of PldA
Amplex Red assay kit was purchased from Invitrogen (cat. no. A12219) and used with modifications detailed below. In a 96-black well plate, in a final reaction volume of 200 μL, the following components were combined on ice to yield final concentrations of 50 mM Tris (pH 7.5), 80 mM KCl, 5 mM MgCl2, 5 mM CaCl2, 50 μM Amplex Red reagent, 0.1 U/mL choline oxidase, 1 U/mL horseradish peroxidase, 1.9 nM PldA, 20 μM (or 5 μg/mL) test compound, and 0.5 mM 7:0/7:0 PC. Lipid in chloroform was dried under a N2 stream in a glass test tube and resuspended in 50 mM Tris (pH 7.5) and 80 mM KCl buffer. All components were combined on ice, and the reaction was initiated by incubating at 37 °C and reading fluorescence emission at 590 nm (excitation = 530 nm) continuously at 90 s intervals for 60 min using a fluorescence plate reader. The signal was normalized by subtracting the background signal produced in the presence of DMSO and absence of PldA. Initial velocities were determined from the linear component of the fluorescence signal. Data are presented as percent total activity normalized to vehicle control. All experiments were performed once in triplicate.
To detect false positive hits, each compound was tested again under identical conditions described above except that PldA and PC substrate were removed and substituted with 50 μM choline chloride. This provides a way of assessing the ability of the compounds to inhibit either the choline oxidase or horseradish peroxidase enzyme independent of their effect on PldA. Compounds that were potential false positives were screened at 20 μM in the secondary method (exogenous PLD assay) to confirm PldA inhibition.
To generate concentration–response curves for each hit, the assay as described above was used except that a range of concentrations (0.1–50 μM) was assayed for each compound.
Exogenous Measurement of Human PLD Activity
In vitro PLD activity was measured with an exogenous substrate assay as previously described.28,29 Briefly, PLD activity was measured as the release of free [3H]-choline from [choline-methyl-3H] dipalmitoyl-phosphatidylcholine ([3H]-DPPC). 3–50 nM PLD1 or PLD2 was reconstituted with phospholipid vesicle substrates composed of 10 μM dipalmitoyl-PC, 100 μM PE (bovine liver), 6.2 μM PIP2 (porcine brain), 1.4 μM cholesterol, and 2.5 μCi [3H]-DPPC. Lipid solutions were dried under a gentle stream of nitrogen and resuspended in 100 mM HEPES (pH 7.5), 160 mM KCl, 6 mM EGTA, and 0.2 mM DTT. Small unilamellar vesicles were prepared by bath sonication (2 × 2 min intervals at 80 W). All assays were performed at 37 °C on agitation for 30 min in 50 mM HEPES, pH 7.5, 80 mM KCl, 3 mM EGTA, 0.1 mM DTT, 3.6 mM MgCl2, 3.6 mM CaCl2, and 10 μM GTPγS. Reactions were stopped with the addition of trichloroacetic acid and bovine serum albumin. Free [3H]-choline was separated from precipitated lipids and proteins by centrifugation and was analyzed by liquid scintillation counting. Raw data was normalized by subtracting background radioactivity levels and is presented as percent total activity. Experiments were performed once in triplicate. IC50 values were determined using a concentration–response curve for each compound against each protein, and the data were analyzed using GraphPad Prism, v 4.0. Nonlinear curve fitting and statistical analysis were done using built-in functions to determine IC50 values. Data are plotted as mean ± SEM unless otherwise stated. Nonspecific effects of compounds by disruption of vesicles were addressed by cross-checking effects in the modified Amplex Red assay described above.
Exogenous Measurement of PldA Activity
Phospholipase D activity was measured using a modified exogenous assay.28,29 In brief, 6 nM purified PldA was incubated with liposomes containing 90 μM 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol), 10 μM 1,2-dipalmitoyl-sn-glycero-3-phosphocholine, and 2.5 μCi [3H]-DPPC for 10 min at 37 °C in buffer containing 50 mM HEPES, pH 7.5, 100 mM KCl, and 3 mM MgCl2. The reaction was quenched with 10% trichloroacetic acid and bovine serum albumin on ice. Protein and lipid were removed by centrifugation. Free [3H]-choline was measured by scintillation counting. Experiments were performed once in triplicate. Data are presented as percent total activity normalized to vehicle control. Data was analyzed by nonlinear regression, and IC50 values were generated using GraphPad Prism, v 4.0. Data are plotted as mean ± SEM unless otherwise stated.
Recombinant Human NAPE-PLD Expression and Purification
E. coli expressing the plasmid pQE-80-PLDHis encoding C-terminus 6× His-tagged human NAPE-PLD was provided by Dr. Sean Davies (Vanderbilt University). Protein purification was performed as described previously.34 Protein was concentrated, and buffer was exchanged into 10 mM Na2HPO4 and 500 mM NaCl.
NAPE-PLD Activity Assay
In a final reaction volume of 100 μL, the following components were combined: 50 mM Tris, pH 7.5, 10 μM diC18:1, C19 NAPE, 1% N-octylglucoside or 0.1% Triton X-100, and 0.5 μg of partially purified NAPE-PLD, and the reaction was incubated at 37 °C for 30 min to 1 h. At the end of the reaction, the lipid products were extracted with a 2:1 chloroform/methanol solution. The lipids were analyzed using previously described glycerophospholipid mass spectrometric methodology.28,32
Chemistry Experimental
All commercial chemicals and solvents were reagent grade and were used without further purification unless otherwise specified. All reactions were carried out employing standard chemical techniques under an inert atmosphere. Analytical thin-layer chromatography performed on 250 μm silica gel plates from Sorbent Technologies was employed routinely to follow the course of reactions. NMR spectra were recorded on a 400 MHz Bruker AV-400, 500 MHz Bruker DRX-500, and 600 MHz AV-II instruments. 1H chemical shifts are reported as δ values in ppm relative to the residual solvent peak (DMSO-d6 = 2.50, CDCl3 = 7.26). Data are reported as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, dd = double of doublet, t = triplet, q = quartet, m = multiplet), and coupling constant (Hz). 13C chemical shifts are reported as δ values in ppm relative to the residual solvent peak (DMSO-d6 = 39.52, CDCl3 = 77.16). Analytical HPLC was performed on an Agilent 1200 LCMS with UV detection at 214 and 254 nm along with ELSD detection. The purity of all tested compounds was greater than 98% based on analytical HPLC at 214 and 254 nm and ELSD. Preparative purification of library compounds was performed on a Gilson 215 preparative LC system. Low-resolution mass spectra were obtained on an Agilent 1200 LCMS with electrospray ionization. High-resolution mass spectra were recorded on a Waters QToF-API-US plus Acquity system with electrospray ionization.
4-(2-(3-Methoxyphenyl)benzo[b]thiophen-3-yl)phenol (15)
Starting material iodide 14 (0.400 g, 1.09 mmol, 1.00 equiv), 4-hydroxyphenyl boronic acid (0.485 g, 2.18 mmol, 2.00 equiv), K3PO4 (0.302 g, 2.18 mmol, 2.00 equiv), and Pd(dppf)2Cl2 (0.089 g, 0.11 mmol, 0.10 equiv) were added to a microwave vial. The solvent was added (6.1 mL dioxane/1.7 mL H2O), and it was heated in a microwave reactor at 130 °C for 40 min. The reaction was then filtered through a pad of Celite, eluting with ethyl acetate (2 × 20 mL). The mixture was concentrated under reduced pressure, and the concentrated material was resuspended in THF (11 mL). Aqueous HCl (10% w/w) (11 mL) was added to the mixture at rt, and the reaction was vigorously stirred for 1 h at rt. The mixture was then transferred to a separatory funnel and partitioned between additional water (10 mL) and ethyl acetate (25 mL). The crude mixture was then extracted with ethyl acetate (3 × 25 mL), and the organic phase was dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The material was purified by flash column chromatography (25–35% ethyl acetate in hexanes, Rf = 0.15 in 25% ethyl acetate in hexanes) to afford product 15 as a white solid (0.281 g, 78% yield). 1H NMR (400.1 MHz, CDCl3) δ 7.90–7.85 (m, 1H), 7.62–7.57 (m, 1H), 7.39–7.32 (m, 2H), 7.25–7.16 (m, 3H), 6.99–6.96 (m, 1H), 6.91–6.86 (m, 3H), 6.80 (ddd, J1 = 8.3 Hz, J2 = 2.6 Hz, J3 = 0.7 Hz, 1H), 3.65 (s, 3H). 13C NMR (100.6 MHz, CDCl3) δ 159.37, 155.04, 141.15, 139.11, 138.84, 135.74, 133.08, 131.86, 129.53, 128.14, 124.70, 124.56, 123.47, 122.22, 115.80 (×2), 114.81, 113.99, 55.21. HRMS (TOF, ES+): C21H17O2S [M + H]+ calcd, 333.0949; found, 333.0950.
4-(4-(2-(3-Methoxyphenyl)benzo[b]thiophen-3-yl)phenoxy)-N,N-dimethylethan-1-amine (17b)
To an oven-dried assimilation vial equipped with a magnetic stir bar under argon containing phenol 15 (0.046 g, 0.14 mmol, 1.00 equiv), triphenylphosphine (0.073 g, 0.28 mmol, 2.00 equiv), and dimethylamino ethanol (0.024 g, 0.28 mmol, 2.00 equiv) stirring in THF (2.00 mL) was added diisopropyl azodicarboxylate (0.042 g, 0.021 mmol, 1.50 equiv) at 0 °C. The resulting solution was allowed to warm to rt over the next hour and was stirred for 48 h. The reaction was then concentrated under reduced pressure and purified by reverse-phase HPLC (35–80% gradient acetonitrile in acidic water (0.1% trifluoromethanesulfonic acid), monitoring product elution at 230 nm) to provide product 17b (0.037 g, 65% yield). 1H NMR (400.1 MHz, CDCl3) δ 7.89–7.84 (m, 1H), 7.58–7.53 (m, 1H), 7.39–7.31 (m, 2H), 7.31–7.26 (m, 2H), 7.18 (t, J = 7.9 Hz, 1H), 6.96–6.89 (m, 3H), 6.88–6.85 (m, 1H), 6.82–6.78 (m, 1H), 4.40 (t, J = 4.3 Hz, 2H), 3.67 (s, 3H), 3.53 (t, J = 4.3 Hz, 2H), 2.97 (s, 6H). 13C NMR (100.6 MHz, CDCl3) δ 159.48, 156.61, 140.96, 139.44, 138.86, 135.58, 132.62, 131.97, 129.57, 129.48, 124.76, 124.63, 123.33, 122.24, 122.23, 115.07, 114.79, 113.79, 62.88, 56.56, 55.22, 43.77. HRMS (TOF, ES+): C25H26NO2S [M + H]+ calcd, 404.1684; found, 404.1685.
Acknowledgments
We would like to thank Dr. Ronald Bruntz for the production and purification of mammalian PLD2 protein for in vitro characterization. Benjamin Neuenswander is thanked for the purification of the SERM screening library, and we sincerely thank the University of Kansas NIH Center of Excellence in Chemical Methodology and Library Development (CMLD) for providing a copy of the SERM library for our PLD screens.
Glossary
Abbreviations
- PLD
phospholipase D
- PldA
Pseudomonas aeruginosa
- SERMs
selective estrogen receptor modulators
- CRCs
concentration–response curves
- SAR
structure–activity relationship
Supporting Information Available
Experimental procedures and spectral data for new compounds. Figure S1: Structures and identification codes for analogue library 11. Table S1: Cellular PLD1 and PLD2 IC50 values. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
# S.A.S. and C.T.S. contributed equally to this work. S.A.S characterized all compound in both cellular and biochemical assays for human PLD1 and PLD2 and prepared the manuscript. C.T.S. developed the in vitro Amplex Red PLD assay, performed all PldA inhibitor characterization, and contributed to the preparation of the manuscript. C.-H.C., D.I.J., and R.C.L. designed and synthesized the SERM screening library. M.C.O. synthesized novel desketoraloxifene analogues. K.A.B. was an undergraduate researcher who helped in resynthesis of hit compounds. R.R.L. expressed and purified mammalian PLD1. H.A.B. oversaw the project and contributed to manuscript preparation. C.W.L. oversaw synthesis, provided SAR comments, and prepared the final version of the manuscript.
We thank the McDonnell Foundation and Voices Against Brain Cancer for partial financial support of this work as well as the NIH (MLPCN U54084679) and Warren Family and Foundation for support of our programs.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Selvy P. E.; Lavieri R. R.; Lindsley C. W.; Brown H. A. (2011) Phospholipase D: enzymology, functionality, and chemical modulation. Chem. Rev. 111, 6064–6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viner R.; Chetrit D.; Ehrlich M.; Segal G. (2012) Identification of two Legionella pneumophila effectors that manipulate host phospholipids biosynthesis. PLoS Pathol. 8, e1002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darville T.; Welter-Stahl L.; Cruz C.; Sater A. A.; Andrews C. W. Jr.; Ojcius D. M. (2007) Effect of the purinergic receptor P2X7 on Chlamydia infection in cervical epithelial cells and vaginally infected mice. J. Immunol. 179, 3707–3714. [DOI] [PubMed] [Google Scholar]
- Renesto P.; Dehoux P.; Gouin E.; Touqui L.; Cossart P.; Raoult D. (2003) Identification and characterization of a phospholipase D-superfamily gene in Rickettsiae. J. Infect. Dis. 188, 1276–1283. [DOI] [PubMed] [Google Scholar]
- Edwards J. L.; Entz D. D.; Apicella M. A. (2003) Gonococcal phospholipase d modulates the expression and function of complement receptor 3 in primary cervical epithelial cells. Infect. Immun. 71, 6381–6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards J. L.; Apicella M. A. (2006) Neisseria gonorrhoeae PLD directly interacts with Akt kinase upon infection of primary, human, cervical epithelial cells. Cell. Microbiol. 8, 1253–1271. [DOI] [PubMed] [Google Scholar]
- Zhu W.; Banga S.; Tan Y.; Zheng C.; Stephenson R.; Gately J.; Luo Z. Q. (2011) Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6, e17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs A. C.; Hood I.; Boyd K. L.; Olson P. D.; Morrison J. M.; Carson S.; Sayood K.; Iwen P. C.; Skaar E. P.; Dunman P. M. (2010) Inactivation of phospholipase D diminishes Acinetobacter baumannii pathogenesis. Infect. Immun. 78, 1952–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F.; Waterfield N. R.; Yang J.; Yang G.; Jin Q. (2014) A Pseudomonas aeruginosa type VI secretion phospholipase D effector targets both prokaryotic and eukaryotic cells. Cell Host Microbe 15, 600–610. [DOI] [PubMed] [Google Scholar]
- Russell A. B.; LeRoux M.; Hathazi K.; Agnello D. M.; Ishikawa T.; Wiggins P. A.; Wai S. N.; Mougous J. D. (2013) Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature 496, 508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilderman P. J.; Vasil A. I.; Johnson Z.; Vasil M. L. (2001) Genetic and biochemical analyses of a eukaryotic-like phospholipase D of Pseudomonas aeruginosa suggest horizontal acquisition and a role for persistence in a chronic pulmonary infection model. Mol. Microbiol. 39, 291–303. [DOI] [PubMed] [Google Scholar]
- Okamoto Y.; Morishita J.; Tsuboi K.; Tonai T.; Ueda N. (2004) Molecular characterization of a phospholipase D generating anandamide and its congeners. J. Biol. Chem. 279, 5298–5305. [DOI] [PubMed] [Google Scholar]
- Pacher P.; Kunos G. (2013) Modulating the endocannabinoid system in human health and disease—successes and failures. FEBS J. 280, 1918–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh M. K.; Yang H.; Roberts M. F. (2003) Using O-(n-alkyl)-N-(N,N′-dimethylethyl)phosphoramidates to investigate the role of Ca2+ and interfacial binding in a bacterial phospholipase D. Biochim. Biophys. Acta 1649, 146–153. [DOI] [PubMed] [Google Scholar]
- Egertova M.; Simon G. M.; Cravatt B. F.; Elphick M. R. (2008) Localization of N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD) expression in mouse brain: a new perspective on N-acylethanolamines as neural signaling molecules. J. Comp. Neurol. 506, 604–615. [DOI] [PubMed] [Google Scholar]
- Scott S. A.; Selvy P. E.; Buck J. R.; Cho H. P.; Criswell T. L.; Thomas A. L.; Armstrong M. D.; Arteaga C. L.; Lindsley C. W.; Brown H. A. (2009) Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat. Chem. Biol. 5, 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavieri R.; Scott S. A.; Lewis J. A.; Selvy P. E.; Armstrong M. D.; Alex Brown H.; Lindsley C. W. (2009) Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part II. Identification of the 1,3,8-triazaspiro[4,5]decan-4-one privileged structure that engenders PLD2 selectivity. Bioorg. Med. Chem. Lett. 19, 2240–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavieri R. R.; Scott S. A.; Selvy P. E.; Kim K.; Jadhav S.; Morrison R. D.; Daniels J. S.; Brown H. A.; Lindsley C. W. (2010) Design, synthesis, and biological evaluation of halogenated N-(2-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)benzamides: discovery of an isoform-selective small molecule phospholipase D2 inhibitor. J. Med. Chem. 53, 6706–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. A.; Scott S. A.; Lavieri R.; Buck J. R.; Selvy P. E.; Stoops S. L.; Armstrong M. D.; Brown H. A.; Lindsley C. W. (2009) Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I: impact of alternative halogenated privileged structures for PLD1 specificity. Bioorg. Med. Chem. Lett. 19, 1916–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monovich L.; Mugrage B.; Quadros E.; Toscano K.; Tommasi R.; LaVoie S.; Liu E.; Du Z.; LaSala D.; Boyar W.; Steed P. (2007) Optimization of halopemide for phospholipase D2 inhibition. Bioorg. Med. Chem. Lett. 17, 2310–2311. [DOI] [PubMed] [Google Scholar]
- Cummings S. R.; Eckert S.; Krueger K. A.; Grady D.; Powles T. J.; Cauley J. A.; Norton L.; Nickelsen T.; Bjarnason N. H.; Morrow M.; Lippman M. E.; Black D.; Glusman J. E.; Costa A.; Jordan V. C. (1999) The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA 281, 2189–2197. [DOI] [PubMed] [Google Scholar]
- Fisher B.; Costantino J. P.; Wickerham D. L.; Redmond C. K.; Kavanah M.; Cronin W. M.; Vogel V.; Robidoux A.; Dimitrov N.; Atkins J.; Daly M.; Wieand S.; Tan-Chiu E.; Ford L.; Wolmark N. (1998) Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J. Natl. Cancer. Inst. 90, 1371–1388. [DOI] [PubMed] [Google Scholar]
- Plowman P. N. (1993) Tamoxifen as adjuvant therapy in breast cancer. Current status. Drugs 46, 819–833. [DOI] [PubMed] [Google Scholar]
- Eisen S. F.; Brown H. A. (2002) Selective estrogen receptor (ER) modulators differentially regulate phospholipase D catalytic activity in ER-negative breast cancer cells. Mol. Pharmacol. 62, 911–920. [DOI] [PubMed] [Google Scholar]
- Grese T. A.; Sluka J. P.; Bryant H. U.; Cullinan G. J.; Glasebrook A. L.; Jones C. D.; Matsumoto K.; Palkowitz A. D.; Sato M.; Termine J. D.; Winter M. A.; Yang N. N.; Dodge J. A. (1997) Molecular determinants of tissue selectivity in estrogen receptor modulators. Proc. Natl. Acad. Sci. U.S.A. 94, 14105–14110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherman R. V.; Carroll D. C.; Scanlan T. S. (2001) Activity of a tamoxifen-raloxifene hybrid ligand for estrogen receptors at an AP-1 site. Bioorg. Med. Chem. Lett. 11, 3129–3131. [DOI] [PubMed] [Google Scholar]
- Cho C. H.; Jung D. I.; Neuenswander B.; Larock R. C. (2011) Parallel synthesis of a desketoraloxifene analogue library via iodocyclization/palladium-catalyzed coupling. ACS Comb. Sci. 13, 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown H. A.; Henage L. G.; Preininger A. M.; Xiang Y.; Exton J. H. (2007) Biochemical analysis of phospholipase D. Methods Enzymol. 434, 49–87. [DOI] [PubMed] [Google Scholar]
- Henage L. G.; Exton J. H.; Brown H. A. (2006) Kinetic analysis of a mammalian phospholipase D: allosteric modulation by monomeric GTPases, protein kinase C, and polyphosphoinositides. J. Biol. Chem. 281, 3408–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S.; Larock R. C. (2010) Iodine/palladium approaches to the synthesis of polyheterocyclic compounds. J. Org. Chem. 75, 1652–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grese T. A.; Sluka J. P.; Bryant H. U.; Cullinan G. J.; Glasebrook A. L.; Jones C. D.; Matsumoto K.; Palkowitz A. D.; Sato M.; Termine J. D.; Winter M. A.; Yang N. N.; Dodge J. A. (1997) Molecular determinants of tissue selectivity in estrogen receptor modulators. Proc. Natl. Acad. Sci. U.S.A. 94, 14105–14110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen G.; Hansen H. S. (1999) N-Acylphosphatidylethanolamine-hydrolysing phospholipase D lacks the ability to transphosphatidylate. FEBS Lett. 455, 41–44. [DOI] [PubMed] [Google Scholar]
- Ivanova P. T.; Milne S. B.; Byrne M. O.; Xiang Y.; Brown H. A. (2007) Glycerophospholipid identification and quantitation by electrospray ionization mass spectrometry. Methods Enzymol. 432, 21–57. [DOI] [PubMed] [Google Scholar]
- Guo L.; Gragg S. D.; Chen Z.; Zhang Y.; Amarnath V.; Davies S. S. (2013) Isolevuglandin-modified phosphatidylethanolamine is metabolized by NAPE-hydrolyzing phospholipase D. J. Lipid Res. 54, 3151–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.