

Abstract

The histone proteins in nucleosome core particles are known to catalyze DNA cleavage at abasic and oxidized abasic sites, which are produced by antitumor antibiotics and as a consequence of other modalities of DNA damage. The lysine rich histone tails whose post-translational modifications regulate genetic expression in cells are mainly responsible for this chemistry. Cleavage at a C4′-oxidized abasic site (C4-AP) concomitantly results in modification of lysine residues in histone tails. Using LC-MS/MS, we demonstrate here that that Lys8, -12, -16, and -20 of histone H4 were modified when C4-AP was incorporated at a hot spot (superhelical location 1.5) for DNA damage within a nucleosome core particle. A new DNA–protein cross-linking method that provides a more quantitative analysis of individual amino acid reactivity is also described. DNA–protein cross-links were produced by an irreversible reaction between a nucleic acid electrophile that was produced following oxidatively induced rearrangement of a phenyl selenide derivative of thymidine (3) and nucleophilic residues within proteins. In addition to providing high yields of DNA–protein cross-links, kinetic analysis of the cross-linking reaction yielded rate constants that enabled ranking the contributions by individual or groups of amino acids. Cross-linking from 3 at superhelical location 1.5 revealed the following order of reactivity for the nucleophilic amino acids in the histone H4 tail: His18 > Lys16 > Lys20 ≈ Lys8, Lys12 > Lys5. Cross-linking via 3 will be generally useful for investigating DNA–protein interactions.
Chromatin
is comprised of monomeric
nucleosome core particles (NCPs). The NCPs (Figure 1A) consist of 145–147 bp of DNA wrapped ∼1.6–1.7
times around a highly positively charged octameric core of histone
proteins (H2A, H2B, H3, and H4).1,2 The central base pair
of the DNA sequence (N73 of a 145 bp duplex) sits at the
dyad axis of the octameric core (superhelical location (SHL) 0) and
extends approximately seven helical turns in each direction (SHL ±
1–7) (Figure 1A,B). The accessibility
of nucleosomal DNA to other DNA binding proteins depends upon the
dynamics of the nucleic acid’s interactions with histone proteins,
which are regulated by the lysine rich tails that protrude through
the NCP.3 For instance, histone H4 contains
five lysine residues in its amino terminal tail (Figure 1C). These lysines are often post-translationally methylated,
acetylated, or otherwise modified by an increasing variety of groups
in cells.4−6 Lysine (and other) modifications affect chromatin
structure and a variety of biochemical processes, including transcription.7,8 The effects of specific amino acid modifications on biochemistry
are hypothesized to be part of an incompletely understood histone
code.9,10 In addition, the histone tails are believed
to directly interact with DNA.11 This is
also supported by recent discoveries that histone proteins in NCPs
catalyze DNA cleavage at the sites at which three alkali-labile lesions
(AP, L, C4-AP) are commonly formed by therapeutic agents and other
DNA damaging agents.12−16 Strand scission at these abasic sites is accelerated as much as
450-fold compared with that observed in free DNA under identical solvent
conditions. For instance, the half-life of C4-AP in a NCP is as short
as 14 min, significantly less than the expected lifetime of this lesion
in cells.13,17 Mutagenesis studies revealed that amino
terminal histone tails are involved in this chemistry, but it has
been difficult to ascertain the role of individual amino acids.13−15,18 This in turn makes it difficult
to postulate possible connections between abasic site mediated histone
modification and genetic regulation. Given the importance of specific
amino acids in the histone code, we sought to determine their roles
in catalyzing strand scission of damaged DNA.
Figure 1.

Nucleosome core particle structure. (A) X-ray crystal structure of NCP (taken from PDB 1kx5); nucleotide positions modified and superhelical locations (SHL) 0–7 are noted (see text). For clarity, a single DNA gyre is shown. (B) Cartoon showing nucleotides (N) modified in this study and their approximate positioning within the NCP (gray oval) with respect to SHL. (C) Peptide sequence of lysine rich histone H4 tail.
NCP catalyzed strand scission of AP and C4-AP (Scheme 1) proceeds via Schiff-base intermediates. Mutation of the five lysines in the H4 tail to arginine and a single histidine to alanine (Figure 1C) accounts for 90% of the acceleration of cleavage at AP89.13,14,18 Mutating individual lysine residues and the single histidine present in the H4 tail to alanines revealed that the side chains of lysine and histidine residues are involved in Schiff-base formation and contribute to the subsequent β-elimination.14 However, these experiments failed to determine the relative contributions of individual lysine residues. For instance, mutating lysines 16 and 20 to alanine only reduced the rate of AP disappearance ∼2-fold, accounting for a small fraction of the ∼100-fold rate acceleration in the NCP.14 Mutating individual lysines in H4 had a similarly small effect on the decomposition of L or C4-AP at the same position within the core particle.13,15 The lack of an effect of individual lysine residues in the H4 tail on decomposition rates of AP and C4-AP lesions in NCPs was attributed at least in part to the reversible nature of Schiff-base formation (Scheme 1) and the conformational flexibility of the histone tails. Although the histone tails are present in X-ray crystal structures (Figure 1A), the positions shown are not the result of the observed electron density, but rather what the authors believe they could be. The histone tails that protrude from the core particle are sufficiently long and flexible that they can interact with distal portions of DNA. For instance, the H4 tail exits through the space between the DNA gyres between SHLs 2 and 3 (Figure 1A) but can form a Schiff base with AP at SHL 0.18 Herein, we describe alternative approaches for identifying contributions of individual lysine residues, including a new, generally useful method for probing DNA–protein interactions.
 |
1 |
Scheme 1.

Results and Discussion
Preparation of NCPs Containing C4-AP and 3
NCPs containing stable precursors to C4-AP and 4 were prepared by synthesizing the 145 bp DNA containing 2 or 3, respectively, at specific sites. The DNA sequence employed in the NCPs was based upon the strong positioning “601” sequence discovered by Widom.19,20 Modifications 2 and 3 were introduced into chemically synthesized oligonucleotides that were then ligated using T4 DNA ligase, as previously described (see Supporting Information).15,21 The denaturing PAGE purified 145 nt products were then hybridized with their complements, and NCPs were reconstituted with purified octamer produced from histone proteins expressed in Escherichia coli using established methods (see Supporting Information).23 The modified nucleotides were introduced in the vicinity of SHL 0 (N73, N218) where the DNA is held tightly by the octamer but is not close to the exit point of any of the histone tails (Figure 1A,B). Nucleotides 2 and 3 were also incorporated in the region of SHL 1.5 (N89, N202, N205) and SHL 4.5 (N119, N172) where the DNA is bent and stretched, respectively. SHL 1.5 and 4.5 are also close to where histone tails pass between the DNA gyres (Figure 1A). DNase I cleavage on reconstituted NCPs confirmed that the incorporation of either modified nucleotide did not affect the register of the 601 DNA with respect to the histone octameric core (see Figures S11 and S12, Supporting Information). The absence of cleavage at the sites where 3 was incorporated suggested that the minor groove at SHLs 0, 1.5, and 4.5 is pointed toward the histone octameric core (inward).
C4-AP is generated from 2 upon short irradiation at 350 nm (eq 1) following NCP reconstitution.24,25 Mild oxidation of 3 (e.g., NaIO4) produces nucleic acid electrophile 4, which reacts with an opposing dA in free DNA to form an interstrand cross-link (ICL, 5) (Scheme 2).26,27 DNA and subsequently NCPs containing 3 at specific positions were prepared via a similar manner described for 2 (see Supporting Information).
Scheme 2.

Mass Spectral Analysis of Histone H4 Modification by C4-AP
C4-AP reactivity in nucleosome core particles is distinct from that of AP and L in that the lesion is transferred to the lysine side chain(s) of the histone protein(s) in the form of a lactam concomitantly with strand scission (1, Scheme 1).13 The symmetry of the octameric histone core (two copies each of four histone proteins) provides two equivalent nucleotide positions in each NCP. We took advantage of the NCP symmetry to maximize the yield of modified histone protein containing 1 (Scheme 1) and incorporated C4-AP at equivalent positions within the two gyres. MALDI-TOF MS analysis was carried out following incubation of a NCP containing C4-AP at positions 89 (C4-AP89) and 234 (C4-AP234) (Figure 1B).13 The amino terminal tail was the only region of the histone H4 protein where modification was detected. The masses of peptides digested by trypsin, Glu C plus Asp N, or thermolysin were consistent with formation of 1 from reaction of C4-AP with a lysine residue but did not enable identification of which lysines were modified or their relative amounts. Specific modified sites were identified via LC-MS/MS analysis of digested histone H4 (Figure 2). After incubation of the C4-AP containing NCP, the DNA was digested and the individual proteins were purified by reverse phase HPLC. Histone H4 was treated separately with trypsin or thermolysin. The protein was acetylated prior to digestion with trypsin to limit hydrolysis at lysine residues. Three peptides containing the lactam modification (1) were detected (Figure 2). The shortest fragment contained amino acids 20–23 and its fragmentation pattern revealed that Lys20 was modified (Figure 2A). The two other fragments contained amino acids 4–17 but differed from one another with respect to which lysine was modified. Fragmentation analysis indicated that Lys16 was modified in the more rapidly eluting peptide (Figure 2B) and Lys12 in the peptide with later retention time (Figure 2C). LC-MS/MS analysis of the thermolysin digest affirmed lactam modification of Lys12 and Lys16 (see Supporting Information). Moreover, treatment of H4 with thermolysin revealed that Lys8 is modified (1) in the peptide fragment consisting of amino acids 1–9 (Figure 3).
Figure 2.

LC-MS/MS identification of modified lysines in the histone H4 tail following trypsin digestion. (A) Lys20 modification in the Lys20–Arg23 peptide. (B) Lys16 modification in the Gly4–Arg17 peptide. (C) Lys12 modification in the Gly4–Arg17 peptide. Fragments containing modified amino acids, either 1 or an acetylated lysine, are indicated by ∗. See Supporting Information for calculated and observed m/z.
Figure 3.

LC-MS/MS identification of modified Lys8 in the Ser1–Gly9 peptide of the histone H4 tail following thermolysin digestion. Fragments containing 1 are indicated by ∗. See Supporting Information for calculated and observed m/z.
Overall, adducts with C4-AP89 or this lesion at the equivalent position in the other gyre of the NCP (C4-AP234) were detected at four of the five lysines in the histone H4 tail. Only modification at Lys5 and the amino terminus of the protein was not observed. The lack of evidence for adduct formation at these positions can be rationalized based on their greater distance from C4-AP89 and C4-AP234. However, these data do not provide quantitative insight into the relative reactivity of the individual lysines with C4-AP.
Detecting DNA–Protein Interactions Using an Irreversible Trap
The preceding LC-MS/MS analysis and previously reported kinetic experiments using mutant histone proteins provide qualitative evidence and inferential support, respectively, for the involvement of individual lysines.13 Given the importance of modified histone lysines in regulating genetic expression in cells, we sought a more quantitative method for detecting interactions between protein nucleophiles and DNA.
Reactive species, such as carbenes and nitrenes, have been used to detect biomacromolecular interactions, including DNA–protein interactions, by forming cross-links.28,29 However, with a small number of exceptions, the yields of such processes are often low.30−34 Other methods can be slow or require modifying Watson–Crick base pairing.35−37 We postulated that readily generated 4 might provide the right balance between kinetic reactivity and selectivity (Scheme 2). Phenyl selenide 3 possesses a number of properties that make it an attractive candidate for probing DNA–protein interactions.26 As mentioned above, the phenyl selenide (3) is compatible with solid-phase oligonucleotide synthesis, and the corresponding deoxynucleotide triphosphate is a substrate for DNA polymerase.21,38 Electrophile 4 is produced from 3 under mild oxidative conditions (e.g., NaIO4, H2O2, 1O2). Furthermore, the modified nucleotide does not alter the Watson–Crick hydrogen-bonding pattern of the native nucleotide, and the electrophilic species is situated in the major groove when 4 is in the anti conformation. Under these circumstances, 4 is well positioned to react with nucleophilic amino acid side chains. In duplex DNA, 4 forms interstrand cross-links (ICLs) with an opposing dA in competition with trapping by exogenous nucleophiles (e.g., azide) via population of the syn conformational isomer.39 Furthermore, 4 reacts slowly with H2O.
The viability of 4 as a trap of DNA–protein interactions was examined using the respective nucleoside (3; please note that for convenience comparable compounds in DNA or as monomers are assigned the same number) and the amides of N-acetylated amino acids (Scheme 3). LC-MS analysis of the oxidation of 3 by NaIO4 in the presence of lysine (6a), histidine (6b), or cysteine (6c) produced the anticipated adducts (7a–c), as determined by their masses (see Supporting Information). However, adducts were not observed from arginine, tyrosine, alanine, aspartic acid, serine, or tryptophan.
Scheme 3.

The reactivity of 4 was examined at several positions within a NCP by introducing 3 at specific sites within both strands of the 145 bp DNA (see Figure 1A,B for locations at which 3 was incorporated). The phenyl selenide (3) was introduced at the dyad axis (SHL 0, N73, N218) of the NCP, SHL 1.2 (N205), and two regions where the DNA is strongly kinked, SHL 1.5 (N89, N202) and SHL 4.5 (N119, N172). SHL 1.5 is a hot spot for DNA binding molecules, and nucleotides at this site are accessible to histone H4 and to a lesser extent to the histone H3 tail.40−42 SHL 4.5 is more proximal to the H2A and H2B histone tails that protrude through the core particle. The dyad region is not closely positioned to the lysine rich tail of any of the histone proteins, but the DNA is tightly held in this region.3 In separate experiments, 3 was incorporated in opposing positions within the duplex.
Reactivity of 4 in NCPs Containing Wild-type Histones
Generating 4 in NCPs results in considerably lower yields of ICLs (5) at most positions tested compared with that in free DNA (Figure 4). ICL yields were reduced by more than 70% when 4 was produced in either strand at SHL 4.5 and ∼60% at SHL 1.5. The smallest percentage change was observed when 4 was generated at the dyad position (SHL 0, N73) of the NCP, where the histone tails are further away and the DNA helix is less perturbed than at SHL 1.5 and 4.5 where it is kinked. In fact, no difference in ICL yield was observed from 4 at N218 (SHL 0) in the NCP or in free DNA. Phenyl selenide placed at the dyad location (SHL 0, N73) is flanked by different nucleotides (5′-dA, 3′-dC, 5′-dA4C) in the 601 DNA sequence than the other sites. For instance, positions dN89 and dN119, which are in the same strand as N73 are flanked by 5′-T and 3′-dA (5′-dT4A). However, changing the sequence flanking 4 at position 73 (73* in Figure 4) to match those at N89 (SHL 1.5) and N119 (SHL 4.5) has little effect on its reactivity in free DNA or the NCP.
Figure 4.

DNA ICL yield from 4 in free DNA and within NCPs as a function of position. The numbers correspond to the nucleotide positions as shown in Figure 1A,B. Each yield is an average of three independent experiments ± SD; 73* indicates the T3(4)A precursor sequence.
The decrease in DNA ICLs is compensated by the formation of DNA–protein cross-links (DPCs) (Figure 5). DPC yields were considerably greater at SHLs 1.5 and 4.5 than at SHL 0, indicative of their closer proximity to the lysine rich histone tails. Reaction at N205 is the one exception to this trend. However, DPC yields did not depend upon whether 4 pointed outward or inward at a given SHL. The importance of proximity between 4 and the histone proteins was more evident when the protein(s) with which the electrophile reacted was identified. This was accomplished by adapting a previous assay in which the 5′-phosphate of the nucleotide involved in cross-linking was 32P-labeled.16 Following incubation of 4 generated in a NCP in which the 5′-phosphate of 3 was radiolabeled, the DNA was digested, and the histone proteins were separated by either SDS gel or triton-acid-urea polyacrylamide gel. Coomassie staining was used to verify the presence of each histone, and the amount of radiation associated with each protein (Table 1) was quantified using phosphorimaging analysis.
Figure 5.

DNA–protein cross-links are formed from 4 in NCPs: DPC yield as a function of position. The numbers correspond to the nucleotide positions as shown in Figure 1A,B. Each yield is an average of three independent experiments ± SD; 73* indicates the precursor T3(4)A sequence.
Table 1. Identification of Histone Proteins Involved in DPCs with 4 as a Function of Its Position in NCPs.
| % of total
DNA–protein cross-linksa |
||||
|---|---|---|---|---|
| SHL position of 4 (nt no.)b | H2A | H2B | H3 | H4 |
| 1.5 (89) | d | <1 | 6.9 ± 0.1 | 92.6 ± 0.3 |
| 1.5 (202) | d | d | 69.2 ± 1.6 | 33.4 ± 2.4 |
| 1.2 (205) | 4.3 ± 0.5 | 8.0 ± 0.3 | 67.1 ± 5.3 | 20.7 ± 6.4 |
| 4.5 (119) | 62.5 ± 3.5 | 37.5 ± 3.5 | d | d |
| 4.5 (172) | 3.5 ± 2.1 | 93.9 ± 3.7 | 2.5 ± 4.5 | d |
| 0 (73)c | 40.8 | d | 48.8 | 10.4 |
| 0 (218)c | 56.3 | 3.8 | 27.1 | 12.8 |
Values are the average of three independent reactions ± SD.
nt no. refers to the position of 4 within the 601 DNA sequence. See Figure 1A,B.
Values are from a single experiment.
Not detected.
Protein cross-linking to nucleotides at the dyad region (SHL 0) was fairly nonselective. This is consistent with their greater distance from any of the histone lysine rich tails compared with the other SHLs examined. In contrast, >97% of the cross-links involving 4 at SHL 4.5 (4119, 4172) were with H2A and H2B. The preference for either of these proteins depended upon which DNA strand 4 was incorporated (Figure 6). Examination of a NCP crystal structure that contains the histone tails suggests that the H2B tail is closer to 4172. Similarly, the opposing position (4119) favors reaction with H2A whose shorter tail exits the core particle in the vicinity of the strand containing this nucleotide but must traverse the major groove to react with 4172 (Figure 6).
Figure 6.
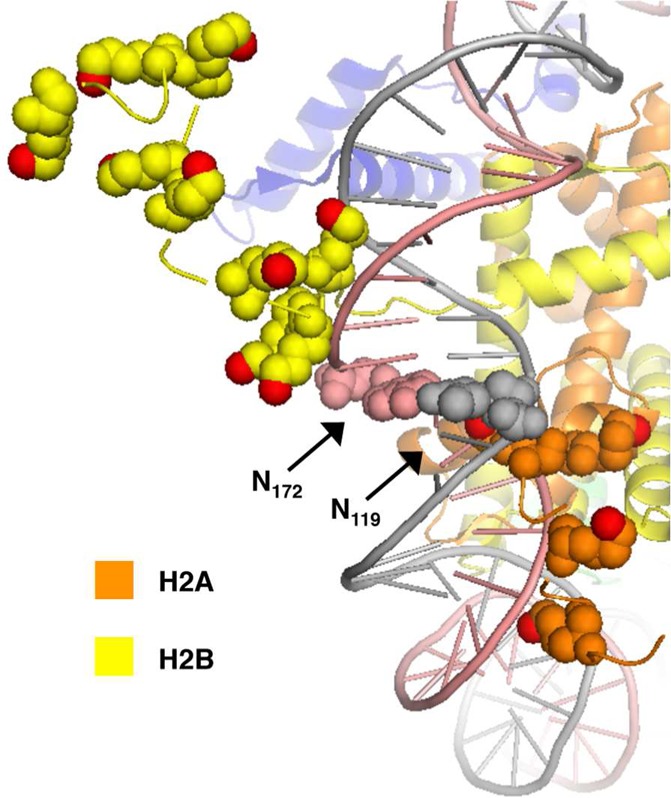
Proximity of histone H2A and H2B tails to nucleotides 119 and 172 in the vicinity of SHL 4.5. Proximal lysine residues are shown as spheres with ε-nitrogen atoms in red. Image taken from PDB 1kx5.
Reactivity at SHL 1.2–1.5 parallels that of 4 at SHL 4.5. DPCs are predominantly distributed between histones H3 and H4, whose lysine rich tails protrude through the DNA in this region. The preference for cross-linking to histone H3 or H4 is consistent with the proximity of the different proteins’ tails to opposite DNA strands (Figure 7). In this region of the 601 DNA, the strand containing 489 forms a minor groove that faces (“inward”) the octameric core. Moreover, the major groove containing 489 is well positioned to react with the H4 tail. Reaction with the H3 tail requires the protein to traverse the minor groove to react with 489 in its anti conformation. The change in the rotational orientation of 4205 resulted in different reactivity. The electrophilic carbon in the major groove is well positioned to react with histone H3 and is consistent with the protein’s major contribution to DPC formation (67.1% ± 5.3%, Table 1). The reactivity of 4205 with histone H4 (20.7% ± 6.4%) may be attributed to the syn conformation resulting from the rotation around the glycosidic bond that also yields DNA–DNA interstrand cross-links (ICLs).
Figure 7.

Proximity of histone H3 and H4 tails to nucleotides 89, 202, and 205 in the vicinity of SHL 1.2–1.5. Proximal lysine residues and H4 His18 are shown as spheres with ε- and imidazole nitrogen atoms in red. Image taken from PDB 1kx5.
Product Analysis of 4 at SHL 1.5 in NCPs Containing Histone H4 Variants
Although the preceding assay identified which protein(s) reacts with 4, the specific amino acids involved were unidentified. To shed light on the amino acids that form DPCs by reacting with 489, we examined the electrophile’s reactivity in NCPs containing histone H4 variants (Table 2). Mutating one or more lysines in the H4 tail to alanine or deleting the 20 amino acid tail in its entirety had an insignificant effect on the yield of DNA interstrand cross-links (ICLs). Similarly, substituting one or more lysines with alanine had no effect on the DPC yield (Table 2). Although we were unable to express the H4 protein in which all five lysines in the amino tail region were mutated to alanines, the K5R, K8R, K12R, K16R, K20R variant was obtained. NCPs containing this H4 variant produced DPCs in slightly lower yield than those with wild-type protein. However, NCPs containing H4 protein in which H18 was mutated to alanine in addition to substituting arginines for the five lysine residues reduced the DPC yield by approximately 50%. This is approximately the same yield as when the entire H4 tail was deleted (H4 deletion, Table 2).
Table 2. Reactivity of 489 in NCPs Containing Histone H4 Variants.
| histone H4 variant | % ICL yielda | % DPC yielda |
|---|---|---|
| WT | 23.8 ± 2.9 | 49.7 ± 3.1 |
| K16A | 24.7 ± 0.5 | 50.4 ± 0.6 |
| K20A | 24.8 ± 0.5 | 50.3 ± 1.0 |
| K16A, K20A | 24.8 ± 2.1 | 51.5 ± 1.5 |
| K5A, K8A, K12A | 29.1 ± 1.5 | 50.7 ± 0.7 |
| H18A | b | 43.5 ± 3.7 |
| K5R, K8R, K12R, K16R, K20R | b | 43.8 ± 1.1 |
| K5R, K8R, K12R, K16R, K20R, H18A | b | 24.2 ± 1.5 |
| H4 deletion | 21.2 ± 2.1 | 27.3 ± 3.0 |
Values are the average of three independent reactions ± SD.
Not determined.
The proteins responsible for cross-linking with 489 in the NCPs containing various histone H4 variants correlated with the effects (or lack thereof) on DPC yields (Table 3). The electrophile reacted solely with histone H4 when lysines 16 and 20 were mutated to alanines (K16A, K20A). In addition, only 6% of the DPCs produced when lysines 5, 8, and 12 were substituted with alanine involved histone H3. Histone H4 was still the major protein cross-linked with 489 when either histidine 18 was mutated to alanine or all five lysines within this protein’s tail were substituted by arginine. However, the involvement of H3 increased significantly when all of the nucleophilic side chains in the tail were replaced by arginine or alanine, leaving only H4’s amino terminus. H3 was entirely responsible for DPCs when the H4 tail was deleted. The increased involvement of H3 correlated with reduced DPC yield at position 89 and is consistent with its anticipated poorer access of the nucleophilic amino acids in its tail to 489 (Figure 7).
Table 3. Effect of Histone H4 Mutations on Proteins Involved in DPCs with 489.
| % of DNA–protein
cross-linksa |
|||
|---|---|---|---|
| histone H4 variant | H2A/H2B | H3 | H4 |
| WT | 0.6 ± 0.8 | 6.9 ± 0.1 | 92.6 ± 0.9 |
| K16A, K20A | 0 | 0 | 100 |
| K5A, K8A, K12A | 0 | 5.5 ± 2.7 | 94.7 ± 3.8 |
| H18A | 3.2 ± 1.8 | 10.2 ± 4.5 | 86.7 ± 6.2 |
| K5R, K8R, K12R, K16R, K20R | 3.7 ± 1.3 | 10.9 ± 0.6 | 85.4 ± 1.8 |
| K5R, K8R, K12R, K16R, K20R, H18A | 19.5 ± 7.1 | 63.3 ± 5.2 | 17.3 ± 1.9 |
| H4 deletion | 0 | 100 | 0 |
Values are the average of three independent reactions ± SD.
A similar examination of the reactivity of 4 produced at the position opposite N89 (4202) (Figure 1A,B) revealed similarities but important distinctions as well (Table 4). Various mutations in histone H4 gave rise to a slightly larger fractional decrease in ICLs than was observed for 489. However, the histone H4 mutations, including deleting the lysine rich tail in its entirety, had little effect on DPC yield. Moreover, reducing the lysine content in the H4 tail resulted in greater reactivity between 4202 and histone H3. For instance, substituting alanine at positions 16 and 20 of H4 gave rise to a small increase in cross-linking to H3 (73.3% ± 0.4% versus 69.7% ± 1.6% in the presence of wt histone H4). However, DPCs between 4202 and H3 increased to 94.3% ± 0.5% when the K5A, K8A, K12A H4 variant was used, and 100% of the cross-links involved H3 when the entire H4 tail was deleted. These data suggest that lysines 5, 8, and 12 in histone H4 are more likely to react with 4202 than are lysines 16 or 20. The NCP structure suggests that the accessibility of lysines 16 and 20 in histone H4 to 4202 is partially blocked by the opposing strand, whereas the tail’s flexibility provides amino acids closer to the amino terminus of H4 better access the electrophile at this site (Figure 7).
Table 4. Reactivity of 4202 in NCPs Containing Histone H4 Variants.
| histone H4 variant | % ICL yielda | % DPC yielda |
|---|---|---|
| WT | 17.8 ± 0.2 | 43.5 ± 1.9 |
| K16A | 12.9 ± 0.9 | 43.9 ± 1.5 |
| K20A | 13.1 ± 0.2 | 42.8 ± 1.3 |
| K16A, K20A | 14.0 ± 0.2 | 44.8 ± 1.3 |
| K5A, K8A, K12A | 10.4 ± 1.3 | 38.5 ± 1.1 |
| H4 deletion | 11.6 ± 1.1 | 42.8 ± 1.0 |
Values are the average of three independent reactions ± SD.
Kinetic Analysis of 4 at SHL 1.5 in NCPs Containing Histone H4 Variants
The preceding experiments indicate that nucleophilic amino acids (e.g., lysines, histidines) and possibly even the H4 protein’s amino terminus can compensate for one another in reactions with electrophilic 4. However, measuring yields alone does not address how facile the reactions are. Consequently, we measured the kinetics for DPC formation by 489 (SHL 1.5). To do this, the appropriate NCP was treated with NaIO4 (3 mM) for 5 min, at which time oxidation was quenched by the addition of Na2SO3 (30 mM). Independent experiments on duplex DNA (data not shown) indicated that this concentration of Na2SO3 was sufficient to prevent any further oxidation of the phenyl selenide by NaIO4. The quench was required to enable monitoring product growth under conditions in which 4 is not still being formed. DPC growth was measured from this point forward by removing aliquots at various times. DPC formation followed first-order growth (Figure 8). The kinetic analysis of DPC formation revealed that although product yield (Table 2) was not strongly influenced by all histone H4 variants, the rate constants were (Table 5). For instance, substituting alanine for lysines 16, 20, or 16 and 20 in histone H4 had no effect on DPC yield or the level of this protein’s involvement in cross-linking (Table 3). However, the rate constant for DPC formation was reduced by more than 60% in the double mutant and >45% when the NCPs contained a single lysine mutation in H4. Similarly, histone H3 partially compensates for the absence of all nucleophilic residues (K5R, K8R, K12R, K16R, K20R, H18A) on histone H4 tail and yields ∼1/2 as much DPCs as that measured in NCP containing wild-type histone H4. However, the rate constants (Table 5) for the reaction (with H3 mostly, Table 3) were significantly slower. The kinetic experiments indicate that histidine 18 reacts relatively rapidly with 489. The relatively high rate constant for reaction of 489 with histidine 18 could be attributed to the imidazole ring’s nucleophilicity and the access of the amino acid’s side chain to 489. The kinetic experiments also indicate that lysines 16 and 20 react more readily with 489 than do lysines 5, 8, and 12, which is not evident from measuring product yields (Table 2). The rate constant for DPC formation from 489 in NCP containing H4 variant K8R, K12R, K16R, K20R was indistinguishable from that containing K5R, K8R, K12R, K16R, K20R. Similarly, the rate constants for DPC formation in NCPs containing H4 K8R, K12R, K16R, K20R, H18A or H4 K5R, K8R, K12R, K16R, K20R, H18A were also indistinguishable from one another. These results suggest that lysine 5 does not react with 489 and corroborate the absence of lactam modification on lysine 5 by C4-AP in the LC-MS/MS experiments (Figures 2 and 3). Finally, the kDPC for NCPs containing the K5R, K8R, K12R, K16R, K20R, H18A histone H4 protein is more than 13-fold slower than that when wild-type protein is present (Table 5). Since the H3 protein is responsible for the majority of DPCs formed in this NCP (Table 3), these data suggest that this protein reacts at least an order of magnitude more slowly with 489 than does the H4 protein.
Figure 8.

Representative plot of DPC growth involving 489 in NCP containing wild-type histone H4.
Table 5. Rate Constant for DPC Formation by 489 in NCPs Containing Histone H4 Variants.
| histone H4 variant | kDPC (min–1)a |
|---|---|
| WT | 0.65 ± 0.06 |
| K16A | 0.31 ± 0.01 |
| K20A | 0.36 ± 0.03 |
| K16A, K20A | 0.25 ± 0.03 |
| K5A, K8A, K12A | 0.36 ± 0.05 |
| H18A | 0.19 ± 0.01 |
| K8R, K12R, K16R, K20R | 0.19 ± 0.02 |
| K5R, K8R, K12R, K16R, K20R | 0.19 ± 0.02 |
| K8R, K12R, K16R, K20R, H18A | <0.05 |
| K5R, K8R, K12R, K16R, K20R, H18A | <0.05 |
Values are the average of three independent reactions ± SD.
Conclusions
This study provides specific details about a unique chemical process that occurs in nucleosome core particles and introduces a generally applicable method for probing nucleic acid–protein interactions.13 Using LC-MS/MS, we identified multiple lysines in the histone H4 tail that are modified upon catalyzed cleavage of a suitably positioned C4′-oxidized abasic lesion within a NCP. This is the first reaction ever characterized in which an oxidized abasic lesion that is produced by therapeutic agents results in the modification of histone amino acids that are involved in genetic regulation. However, product analysis does not provide information on the relative contributions of histone tail lysines. While mass spectrometry generally enables identification of individual interactions in biomacromolecular ensembles, quantitation is difficult without isotopically enriched samples.43,44 Consequently, we developed a method for probing DNA–protein interactions that utilizes an irreversible reaction, much like an affinity tagging reagent. Kinetic analysis is infrequently used as a tool in conjunction with covalent trapping for probing biomacromolecular interactions.45 However, it is the use of kinetics that provides more quantitative insight into the interactions between 4 and specific amino acids.
Unlike other electrophilic reagents, such as carbenes and nitrenes, that are used in affinity labeling experiments, 4 only reacts with the most nucleophilic native amino acids (lysine, cysteine, and histidine) and reacts much more slowly with water. The selective nature of 4 contributes to the high DNA–protein cross-link yields. Product studies with 4 reinforce previous experiments on NCP catalyzed DNA strand scission at abasic sites.13−15,18 Specifically, reaction of 4 reveals that multiple nucleophilic amino acid side chains in histone tails react with the DNA and that reactive sites vary when one or more of these are substituted with non-nucleophilic amino acids. More importantly, kinetic experiments provide a qualitative ranking of amino acids in terms of reactivity. Kinetic analysis indicates that 489 reacts with His18 > Lys16 > Lys20 ≈ Lys8, Lys12 > Lys5. A more quantitative determination of dependence on any one nucleophile is not possible in this system. This may be attributed to multiple reasons, including different conformations of the protein tail in the various histone variants that affect the effective molarity of specific amino acids with respect to 4 and possible differences in DNA–protein interactions as the charge varies.
Protein tagging by 4 will be useful for obtaining information regarding DNA–protein interactions between abasic sites at other locations in nucleosome core particles and consequently will provide insight into possible connections between NCP catalysis of DNA cleavage and post-translational histone modification. This electrophilic probe exhibits a higher degree of selectivity and yields than most examples of electron deficient species (e.g., carbenes, nitrenes), which rarely provide yields comparable to those observed here.28−31,34 Electrophile 4 will be a generally useful tool for probing DNA–protein interactions, especially because in addition to its precursor (3) being compatible with chemical oligonucleotide synthesis, DNA polymerase accepts the corresponding triphosphate as a substrate.38
Methods
General Procedure for the Oxidation Reaction of NCPs Containing 3
The ligation of 145-mer 601 DNA containing 3 and the reconstitution of 32P-labeled NCPs were carried out as described previously using the appropriate oligonucleotides (Figure S2, Supporting Information).16 To the reconstituted NCP solution was added 5 mM NaIO4. Following incubation at 37 °C for 2 h, the samples were divided into two portions. One portion was treated with proteinase K (0.1 μg) for 5 min at RT and analyzed by 8% denaturing PAGE (40 × 32 × 0.04 cm3) to detect DNA interstrand cross-links. To the second portion was added 4 × SDS loading buffer (400 mM Tris·HCl, 400 mM DTT, 8% SDS, 40% glycerol), and the mixture was separated by SDS–PAGE (10% resolving acrylamide/bis(acrylamide) = 29:1, 5% stacking layer, 20 × 16 × 0.1 cm3). The gel was run at 250 V until the bromophenol blue band migrated to the bottom. The products in the gels were quantified using a Storm 840 phosphorimager and ImageQuant TL software.
General Procedure for the Kinetic Studies
NCPs containing 5′-32P-labeled DNA were mixed with 3 mM NaIO4 without additional buffer. Na2SO3 (30 mM) was added 5 min afterward to quench excess NaIO4. An aliquot was immediately withdrawn from the reaction and thereafter at each indicated time point and stored at −80 °C. The individual aliquots were mixed with 4 × SDS loading buffer and subjected to SDS–PAGE separation (10% resolving layer, acrylamide/bis(acrylamide) 29:1, 5% stacking layer, 20 × 16 × 0.1 cm3). The products in the gels were quantified using a Storm 840 phosphorimager and ImageQuant TL software.
Acknowledgments
We are grateful for generous financial support from the National Institute of General Medical Sciences (Grants GM-063028 and GM-054996). L.Weng is grateful for the Ada Sinz Hill Fellowship from Johns Hopkins University.
Supporting Information Available
Experimental procedures, representative autoradiograms, kinetic plots, LC-MS/MS data (and table of observed and calculated m/z of fragment ions), ESI-MS and MALDI-TOF MS of modified oligonucleotides, and chromatograms for reactions of amino acids with 4. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Davey C. A.; Sargent D. F.; Luger K.; Maeder A. W.; Richmond T. J. (2002) Solvent mediated interactions in the solution of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 319, 1097–1113. [DOI] [PubMed] [Google Scholar]
- Luger K.; Mader A. W.; Richmond R. K.; Sargent D. F.; Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- Hall M. A.; Shundrovsky A.; Bai L.; Fulbright R. M.; Lis J. T.; Wang M. D. (2009) High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat. Struct. Mol. Biol. 16, 124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L.; Peng C.; Montellier E.; Lu Z.; Chen Y.; Ishii H.; Debernardi A.; Buchou T.; Rousseaux S.; Jin F.; Sabari B. R.; Deng Z.; Allis C. D.; Ren B.; Khochbin S.; Zhao Y. (2014) Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 10, 365–370. [DOI] [PubMed] [Google Scholar]
- Patel D. J.; Wang Z. (2013) Readout of epigenetic modifications. Annu. Rev. Biochem. 82, 81–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganuma T.; Workman J. L. (2011) Signals and combinatorial functions of histone modifications. Annu. Rev. Biochem. 80, 473–499. [DOI] [PubMed] [Google Scholar]
- Jorgensen S.; Schotta G.; Sorensen C. S. (2013) Histone H4 lysine 20 methylation: Key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 41, 2797–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Carey M.; Workman J. L. (2007) The role of chromatin during transcription. Cell 128, 707–719. [DOI] [PubMed] [Google Scholar]
- Strahl B. D.; Allis D. (2000) The language of covalent histone modifications. Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- van Rossum B.; Fischle W.; Selenko P. (2012) Asymmetrically modified nucleosomes expand the histone code. Nat. Struct. Mol. Biol. 19, 1064–1066. [DOI] [PubMed] [Google Scholar]
- Zheng C.; Hayes J. J. (2003) Structures and interactions of the core histone tail domains. Biopolymers 68, 539–546. [DOI] [PubMed] [Google Scholar]
- Greenberg M. M. (2014) Abasic and oxidized abasic site reactivity in DNA: Enzyme inhibition, crosslinking, and nucleosome catalyzed reactions. Acc. Chem. Res. 47, 646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C.; Sczepanski J. T.; Greenberg M. M. (2013) Histone modification via rapid cleavage of C4′-oxidized abasic sites in nucleosome core particles. J. Am. Chem. Soc. 135, 5274–5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C.; Sczepanski J. T.; Greenberg M. M. (2012) Mechanistic studies on histone catalyzed cleavage of apyrimidinic/apurinic sites in nucleosome core particles. J. Am. Chem. Soc. 134, 16734–16741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C.; Greenberg M. M. (2012) Histone-catalyzed cleavage of nucleosomal DNA containing 2-deoxyribonolactone. J. Am. Chem. Soc. 134, 8090–8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczepanski J. T.; Wong R. S.; McKnight J. N.; Bowman G. D.; Greenberg M. M. (2010) Rapid DNA-protein cross-linking and strand scission by an abasic site in a nucleosome core particle. Proc. Natl. Acad. Sci. U. S. A. 107, 22475–22480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamna H.; Cheung I.; Ames B. N. (2000) A method for detecting abasic sites in living cells: Age-dependent changes in base excision repair. Proc. Natl. Acad. Sci. U.S.A. 97, 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczepanski J. T.; Zhou C.; Greenberg M. M. (2013) Nucleosome core particle-catalyzed strand scission at abasic sites. Biochemistry 52, 2157–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary P. T.; Widom J. (1998) New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 276, 19–42. [DOI] [PubMed] [Google Scholar]
- Vasudevan D.; Chua E. Y. D.; Davey C. A. (2010) Crystal structures of nucleosome core particles containing the ‘601’ strong positioning sequence. J. Mol. Biol. 403, 1–10. [DOI] [PubMed] [Google Scholar]
- Hong I. S.; Greenberg M. M. (2005) Efficient DNA interstrand cross-link formation from a nucleotide radical. J. Am. Chem. Soc. 127, 3692–3693. [DOI] [PubMed] [Google Scholar]
- Dyer P. N.; Edayathumangalam R. S.; White C. L. B.; Yunhe; Chakravarthy S.; Muthurajan U. M.; Luger K. (2004) Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 375, 23–44. [DOI] [PubMed] [Google Scholar]
- Sczepanski J. T.; Jacobs A. C.; Greenberg M. M. (2008) Self-promoted DNA interstrand cross-link formation by an abasic site. J. Am. Chem. Soc. 130, 9646–9647. [DOI] [PubMed] [Google Scholar]
- Kim J.; Gil J. M.; Greenberg M. M. (2003) Synthesis and characterization of oligonucleotides containing the C4′-oxidized abasic site produced by bleomycin and other DNA damaging agents. Angew. Chem., Int. Ed. 42, 5882–5885. [DOI] [PubMed] [Google Scholar]
- Hong I. S.; Greenberg M. M. (2005) DNA interstrand cross-link formation initiated by reaction between singlet oxygen and a modified nucleotide. J. Am. Chem. Soc. 127, 10510–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H.; Majumdar A.; Tolman J. R.; Greenberg M. M. (2008) Multinuclear nmr and kinetic analysis of DNA interstrand cross-link formation. J. Am. Chem. Soc. 130, 17981–17987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persinger J.; Bartholomew B. (2009) Site-directed DNA crosslinking of large multisubunit protein-DNA complexes. Methods Mol. Biol. 543, 453–474. [DOI] [PubMed] [Google Scholar]
- Winnacker M.; Breeger S.; Strasser R.; Carell T. (2009) Novel diazirine-containing DNA photoaffinity probes for the investigation of DNA-protein-interactions. ChemBioChem 10, 109–118. [DOI] [PubMed] [Google Scholar]
- Dechassa M. L.; Zhang B.; Horowitz-Scherer R.; Persinger J.; Woodcock C. L.; Peterson C. L.; Bartholomew B. (2008) Architecture of the SWI/SNF-nucleosome complex. Mol. Cell. Biol. 28, 6010–6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai K.; Ozawa S.; Yamada R.; Yasui T.; Mizuno S. (2014) Comparison of the reactivity of carbohydrate photoaffinity probes with different photoreactive groups. ChemBioChem 15, 1399–1403. [DOI] [PubMed] [Google Scholar]
- Qiu Z.; Lu L.; Jian X.; He C. (2008) A diazirine-based nucleoside analogue for efficient DNA interstrand photocross-linking. J. Am. Chem. Soc. 130, 14398–14399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatsepin T. S.; Dolinnaya N. G.; Kubareva E. A.; Ivanosvskaya M. G.; Metelev V. G.; Oretskaya T. S. (2005) Covalent binding of modified nucleic acids to proteins as a method for investigation of specific protein-nucleic acid interactions. Russ. Chem. Rev. 74, 77–95. [Google Scholar]
- Shigdel U. K.; Zhang J.; He C. (2008) Diazirine-based DNA photo-cross-linking probes for the study of protein-DNA interactions. Angew. Chem., Int. Ed. 47, 90–93. [DOI] [PubMed] [Google Scholar]
- Fromme J. C.; Banerjee A.; Huang S. J.; Verdine G. L. (2004) Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature 427, 652–656. [DOI] [PubMed] [Google Scholar]
- Dadová J.; Orság P.; Pohl R.; Brázdová M.; Fojta M.; Hocek M. (2013) Vinylsulfonamide and acrylamide modification of DNA for cross-linking with proteins. Angew. Chem., Int. Ed. 52, 10515–10518. [DOI] [PubMed] [Google Scholar]
- Nagatsugi F.; Kawasaki T.; Usui D.; Maeda M.; Sasaki S. (1999) Highly efficient and selective cross-linking to cytidine based on a new strategy for auto-activation within a duplex. J. Am. Chem. Soc. 121, 6753–6754. [Google Scholar]
- Hong I. S.; Ding H.; Greenberg M. M. (2006) Radiosensitization by a modified nucleotide that produces DNA interstrand cross-links under hypoxic conditions. J. Am. Chem. Soc. 128, 2230–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong I. S.; Ding H.; Greenberg M. M. (2006) Oxygen independent DNA interstrand cross-link formation by a nucleotide radical. J. Am. Chem. Soc. 128, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K.; Rechsteiner T. J.; Flaus A. J.; Waye M. M. Y.; Richmond T. J. (1997) Characterization of nucleosome core particles containing histone proteins made in bacteria. J. Mol. Biol. 272, 301–311. [DOI] [PubMed] [Google Scholar]
- Davey G.; Wu B.; Dong Y.; Surana U.; Davey C. A. (2010) DNA stretching in the nucleosome facilitates alkylation by an intercalating antitumor agent. Nucleic Acids Res. 38, 2081–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuduvalli P. N.; Townsend C. A.; Tullius T. D. (1995) Cleavage by calicheamicin γ1 of DNA in a nucleosome formed on the 5S RNA gene of xenopus borealis. Biochemistry 34, 3899–3906. [DOI] [PubMed] [Google Scholar]
- Herzog F.; Kahraman A.; Boehringer D.; Mak R.; Bracher A.; Walzthoeni T.; Leitner A.; Beck M.; Hartl F.-U.; Ban N.; Malmström L.; Aebersold R. (2012) Structural probing of a protein phosphatase 2a network by chemical cross-linking and mass spectrometry. Science 337, 1348–1352. [DOI] [PubMed] [Google Scholar]
- Rappsilber J. (2011) The beginning of a beautiful friendship: Cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J. Struct. Biol. 173, 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock H.; Bachu R.; Beal P. A. (2011) Covalent stabilization of a small molecule-rna complex. Bioorg. Med. Chem. Lett. 21, 5002–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.