

Abstract
Over the past two decades a plethora of crystal structures of molybdenum enzymes has appeared in the literature providing a clearer picture of the enzymatic active sites and increasing the challenge to chemists to develop accurate models for those sites. In this minireview we discuss the most recent model studies aimed to reproduce detailed features of the pterin-dithiolene ligand, both as the uncoordinated form and as a chelate coordinated to molybdenum.
Introduction
The catalytic sites of the molybdenum enzymes have long evoked curiosity. Accompanying articles in this issue address the importance of molybdenum enzymes, key players in the health of organisms spanning the range of bacteria to mammals and whose participation in global cycling of carbon, nitrogen and sulfur compounds impacts human health and the environment. The strategies of using synthetic analog or model compounds to define aspects of the molybdenum sites have been employed for more than a half-century. Early models revealed the coordination preferences of molybdenum while spectroscopic studies such as EPR1 and later, EXAFS,2 provided evidence for sulfur donor atoms in the first coordination sphere of Mo. The earliest models also indicated the likely role of oxo ligands on molybdenum in the catalytic cycle. The first extensive review to organize and contextualize early model work was Stiefel’s pivotal chapter in Progress in Inorganic Chemistry3 which set the stage for development of the first functional models to demonstrate reactivity characteristic of many molybdenum enzymes, and structural models to duplicate ever more closely the distinctive coordination environments found in the various enzyme families.4, 5 In subsequent years, numerous comprehensive review articles covered different aspects of model chemistry from functional models that incorporate oxygen atom transfer (OAT) reactivity 6–10 and coupled electron-proton transfer (CEPT),11, 12 to structural and spectroscopic models for the molybdenum catalytic site. 13–16 Readers are advised to consult those articles for more comprehensive and historical treatments, which are beyond the scope of this minireview.
Development of model chemistry has always progressed hand in hand with experimental data from molybdoenzymes. The notion that the catalytic moiety was a dissociable cofactor consisting of a molybdenum atom bound to a ligand of unknown structure arose from reconstitution of a mutant apoenzyme.17 More detailed information about the ligand (molybdopterin, MPT, or also called pyranopterin) on Moco came from careful degradation studies by Rajagopalan and coworkers, who established both the requirement of a pterin group in the ligand as well as the identity of the sulfur donor atoms within a dithiolene chelate (Fig. 1, top). 18, 19 The ligand itself was named molybdopterin (MPT) and when coordinated to molybdenum, it forms the molybdenum cofactor (Moco). At the time, the proposal for a dithiolene unit coordinating to a metal center in a biological system was novel. This biochemical work prompted the development of subsequent models incorporating dithiolene or pterin structures.6, 20–29 Spectroscopic studies coupled with computational methods suggested that the dithiolene unit was likely playing key electronic roles in modulating the Mo redox potentials through controlling the electronic structure, and in directing substrate reaction at a specific oxo ligand.30–33 Pterin molecules were established as potential redox partners.20–22, 34
Fig. 1.

Mo coordinated to the special ligand (top) as first proposed by Rajagopalan, (bottom) as observed in X-ray structures. R designates the phosphate substitution with several dinucleotides such as guanosine, adenosine or cytosine that is observed in some bacterial enzymes containing Moco.
The first X-ray crystal structure of the molybdenum cofactor bound to the protein was reported in 1995 and revealed the constituents of Moco as depicted in Fig. 1 (bottom).35 The presence of a reduced pterin was confirmed as well as the tethering of that pterin via a dithiolene to the Mo atom. Not predicted from previous studies was the presence of a pyran ring formed by the apparent cyclization of a side chain hydroxyl group with the pyrazine ring. Subsequent molybdoprotein structures, with nearly 50 now available, have confirmed this basic structure of the special ligand for Mo. Structures of some enzymes show the presence of two pterin units, thus highlighting the diverse nature of the molybdenum cofactor. This aspect was further established in Hille’s classification of molybdenum enzymes into three distinct families.4, 5 At the most fundamental level these families are distinguished by whether there is one or two pterin-dithiolene ligands on Mo, then these two groups are further differentiated by the number of attached oxygen or sulfur atoms, and whether there is an additional coordination from an amino acid residue.
With many X-ray structures of Moco providing the structural benchmarks, the quest for more faithful models continued. These studies now aim to synthesize compounds with as many components of the actual cofactor as possible in order to determine how they participate in the catalytic reactions. The quest to more closely replicate the structure of Moco in model compounds is challenging as models need to incorporate three redox active units (viz. the pterin, the dithiolene and the metal center) of the cofactor into one entity.34
The scope of this short minireview highlights recent model chemistry developed in our laboratories that are directed at understanding the role and impact of the pterin and the dithiolene chelate on Mo in the various forms of Moco in molybdoenzymes. The review begins with a general discussion on the atypical properties of the dithiolene ligands and pterin molecules that can be anticipated to play a part in the catalytic mechanism of Moco. Then model research directed at studying either the free pterin-dithiolene molecule or a Mo complex of such a ligand is placed in the context of prior models developed and discussed.
Properties That May Influence the Role of the Pterin-Dithiolene Ligand
Speculation regarding the role of the unique pterin dithiolene ligand in Moco can be found throughout the many reviews devoted to molybdenum enzymes and their models. 6–8, 10, 36–47 Certainly the dithiolene as a chelating ligand stabilizes bonding to the Mo atom but the versatility of the dithiolene ligand extends far beyond a simple bidentate sulfur chelate. The dithiolene also tunes the electronic environment at the metal center using several mechanisms. Conjugation within the dithiolene ligand mediated through the sulfur atoms to the molybdenum creates a highly covalent system that adjusts to variable electron density at the metal. The dithiolene can further influence the Mo electronic environment by a bending movement,31 that can change electron donation through pi interactions in response to Mo oxidation state so that larger dithiolene ‘fold angles’ are observed for the higher oxidation states of Mo.32 Both of these modes of modulating the electronic environment have led to the dithiolene ligand’s description as an “electronic buffer”.33 In addition to the electronic system tuning by the highly covalent bonding within dithiolenes, these ligands are redox active and can exist either in fully reduced (dithiolene, dianion), partially oxidized (radical anion) or fully oxidized (dithione, neutral) forms as shown in Fig. 2.
Fig. 2.
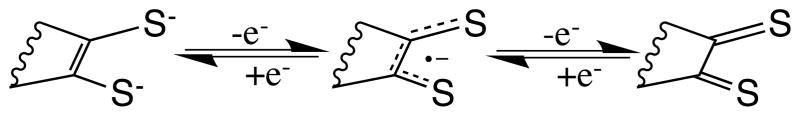
Different redox states of a dithiolene ligand.
A pterin substituent on the dithiolene chelate adds subtle electronic variation. Again, there are several mechanisms that allow the pterin to further tune the electronics through the dithiolene to the Mo atom. In the cofactor, the pterin is in conjugation with the dithiolene unit (Fig. 3). Insofar as the nitrogen-rich heterocyclic compounds such as a pterin is generally considered an electron-poor unit, this electron deficient nature is expected to exert an electron-withdrawing effect on the dithiolene chelate.
Fig. 3.
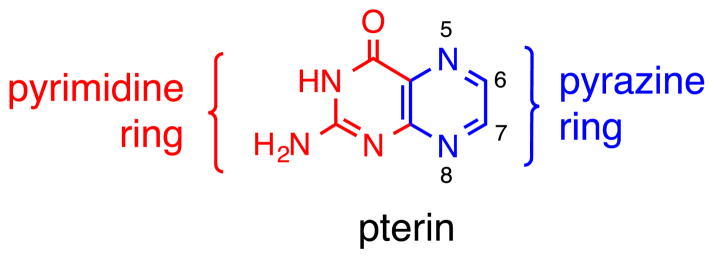
Numbering system for a simple pterin and its constituent N-heterocycles.
Electronic communication between the pterin and dithiolene can be further tuned by the redox capability of the pterin (Fig. 4), which can be reduced by a total of four electrons and protons. Additional electronic adjustment can occur through the multiple tautomeric forms of the semi-reduced, or dihydro-, pterin state (Fig. 5).
Fig. 4.
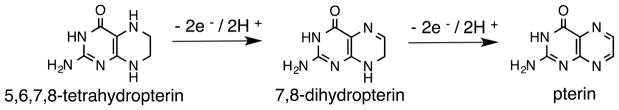
The three redox levels of the pterin heterocycle, where the semi-reduced form is illustrated in the most thermodynamically stable tautomer.
Fig. 5.

Several tautomers of the dihydropterin oxidation state.
Finally, the pterin in the vast majority, though not all, of the molybdoenzyme protein structures is observed with a third ring—the pyran ring—fused at positions C6 and C7 in the pterin. However, in two structures where the molybdenum is coordinated by two pterin-dithiolene ligands, one pterin-dithiolene does not have a pyran ring (Fig. 6).48, 49 The pyranopterin structure appears to be a consequence of the geometry imposed by the dithiolene at C6 and the presence of a hydroxyl substituent at the α-C of the dithiolene phosphate side chain (Fig. 6). The formation of the more rigid pyranopterin system can be expected to impose further constraints on the coordination geometry at Mo.
Fig. 6.

The two forms of the pterin-dithiolene ligand of Moco, a pyranopterin form and a non-cyclized form, occur simultaneously in certain bacterial molybdoproteins such as nitrate reductase and ethylbenzene dehydrogenase.
The multiple ways a pterin can influence the dithiolene interactions with the molybdenum atom must also be considered in light of the seat of the pterin unit within the protein. The many H-bonding acceptor and donor functional groups present in the pterin molecule are extensively used to tightly hold the pyranopterin within the protein scaffold. Indeed, it has been suggested that the strong H-bonding of the pterin to the protein may play a critical role in ensuring the appropriate geometry around the molybdenum, especially as these geometries are significantly different from ideal octahedral and trigonal prismatic polyhedra.50 Since pterin redox is necessarily a proton-coupled process, H-bonding to the protein can play a role in redox reactions of the pterin. A second way the protein may regulate pterin influence at the dithiolene can occur in the region of the pyran unit. Protein residues might trigger pyran ring opening and cyclization through interactions with the H-N8 and the hydroxyl group.
The above characteristics of dithiolene ligands and of pterin molecules suggest that together, the pair should create an incredibly redox rich and electronically nimble moiety. This expectation is one motivation behind the efforts to synthesize molybdenum dithiolene complexes having pterin substituents so that analytical methods can be used to provide detailed descriptions of electronic structure. The pursuit of synthesizing Mo-pterin-dithiolene complexes has been the focus of several laboratories since the 1990’s. In some cases, simplifying the model synthesis through replacing the complicated pterin system by a simpler quinoxaline group led to model compounds whose behavior contributed to understanding the pterin’s role. 51, 52 En route to successful preparation of Mo-pterin-dithiolene models, researchers have also learned from studies of simpler model compounds of the pterin-dithiolene ligand, MPT. Much of this model work first required the development of new pterin chemistry.26, 27, 29, 53, 54 The most recent results from our laboratories are described below and placed into the context of the question “what is the role of the pyranopterin dithiolene ligand in Moco?”
Models Illustrating the Role of Dithiolene Redox
As introduced above, dithiolenes are redox active ligands and can access partially oxidized (radical anion) or fully oxidized (dithione) forms (Fig. 2). While the majority of the model chemistry has focused on the fully reduced state, compounds with fully oxidized and neutral dithione ligands are rare. Dithione ligands are electron deficient and known to stabilize an electron rich, low valent metal center55 but it was not known whether dithiones could stabilize a higher valent oxo-Mo center.
An example of a dithione coordinated to high valent Mo was reported by Basu et al. and is shown in Fig. 7a; here, the use of a diisopropylpiperazine-2,3-dithione (iPr2Dt0) allowed the formation of an oxo-Mo(IV) complex, [(iPr2Dt0)2MoO][BF4]2.56 In this case, the Mo(IV) complex exhibits an unusual reactivity. In the presence of a base such as pyridine the terminal oxo-group is readily labilized forming a novel cluster of formula [(iPr2Dt0)Mo(BF4)]4. The Mo–Mo distance was determined to be 3.02 A, suggesting a considerable Mo…Mo bonding interaction. Each of the distorted trigonal bipyramid Mo centers is stabilized through coordination from two dithione ligands and one BF4 ligand. While the formation of the cluster has little biological consequence, such reactivity has not been observed in the case of Mo-complexes with reduced dithiolene ligands.56 The bonding pattern in [(iPr2Dt0)2MoOCl][PF6], has been investigated by resonance Raman spectroscopy. Acetonitrile solutions of [(iPr2Dt0)2MoOCl][PF6], exhibit a strong low energy absorption, unusual in mono- and bis-(dithiolene) Mo(IV) complexes. These low-spin complexes possess a (dx2 −y2)2 ground state HOMO that precludes LMCT transitions. In resonance pump probing of this band, several ligand backbone vibrational bands were enhanced. This led to the assignment of a valence bond description of the ligand architecture that includes a di-zwitterionic description (~37%) to the ground state (Fig. 7b).57 This was the first time a dithiolene backbone has been quantified in terms of different resonance contributions.
Fig. 7.

Examples of a Mo(IV)-dithione complexes: (A) an oxo-Mo(IV) center coordinated by two dithione and BF4− ligands; (B) an oxo-Mo(IV) center stabilized by two oxidized dithione ligands and chloride ligand (top); the dithione ligand exists as a resonance hybrid of dithione with its di-zweitterionic form (bottom).
Models of the MPT Ligand
In 1985 Joule and Garner14, 15 reported the first of many investigations of molecules related to MPT and Moco, where Fig. 8 provides several noteworthy accomplishments. They devised a successful route to a bis-(pterin-dithiolene)Mo(IV)-oxo complex (Fig 8, b) using a strategy of reacting a preassembled and protected pterin-dithiolene with K4[MoO2(CN)4] (Fig 8, a). 58 Also significant is their synthesis of the reduced pyranopterin dithiolene ligand in a protected form, the first reduced pyranopterin so closely related to the structure of MPT (Fig 8, c). 59–61
Fig. 8.

(a) dithiolene forming reaction, (b) a bis-(pterin-dithiolene)molybdenum complex, (c) a protected trithiolene incorporating a reduced pyranopterin.
More recently, the synthetic approach of using the trithiolene unit to protect the core structure of a pterin-dithiolene ligand was reported by Basu 29 et al (Fig. 9). In this work, retrosynthetic analysis led to a methodology based on a regioselective condensation reaction of an α-keto aldehyde (2), with 2,5,6-triamino-3,4-dihydropyrimidin-4-one (1) to obtain the desired 6-substituted regioisomers (3 and 4). In DMF, compound 4 is converted to the desired product, 3 under acidic conditions. The pterin functionality bears no protection, the only protection is in the dithiolene unit.
Fig. 9.

Synthetic route dephospho-MPT where the dithiolene chelate is protected as a trithiolene species.
The protected pterin-trithiolene 3 is a useful model since it has all the components of the dephospho MPT and can serve as a spectroscopic benchmark for future studies. Compound 3 in Fig. 9 will be reacted with suitable molybdenum reagents leading to Mo-pterin-dithiolene models.
Pterin-dithiolene Models of Moco
As an alternative to the approach developed by Garner and Joule in synthesizing Mo complexes of pterin-dithiolene, other researchers’ have used the synthetic strategy developed by Coucouvanis where a molybdenum tetrasulfide reacts with alkynes producing Mo dithiolene complexes (Eq. 1). 62–64
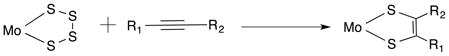 |
Eq. 1 |
The first such pterin-dithiolene complex Cp2Mo{S2(pterin)(COMe)} (where Cp = cyclopentadienide) (Fig. 10, A) was reported in 1991 but not further pursued. 65 A notable piece of data from this study is the 95,97Mo hyperfine value, whose small value (11 G) was interpreted as consistent with a highly covalent Mo-(pterin-dithiolene) system with a HOMO based on the C and S atoms of the dithiolene. 24, 66
Fig. 10.

Models incorporating a pterin substitution on a dithiolene chelate on Mo.
The tetrasulfide/alkyne coupling methodology to form dithiolenes was extended to a (scorpionate)Mo=X (X = O, S) scaffold, [Tp*Mo(=X)(S4)]−, (Tp* = hydrotris(3,5-dimethylpyrazolyl)borate) by Young and co-workers.67 Subsequently, Burgmayer and coworkers reported a generalized synthesis of (Tp*)Mo(=X)(pterin-dithiolene) complexes (Fig. 10, B), where the R-group can be a variety of hydroxyalkyl, aryl and, under current investigation, alkyl substituents.24 In the case of the Tp*MoX(pterin-dithiolenes) (X = S,O; R = aryl), it was concluded that the most significant effect of pterin substitution was to shift the Mo(5+/4+) redox potential considerably in the positive direction compared to simpler model complexes bearing 1,2-benzene dithiolate (bdt) or ethanedithiolate (edt).24 Surprisingly, the Magnetic Circular Dichroism and Electron Paramagnetic Resonance spectra exhibited by Tp*MoO(bdt) and Tp*MoO(pterin-dithiolene) complexes have nearly identical parameters suggesting the pterin substituent has little effect on the molybdenum environment.24
Burgmayer and co-workers found that when the pterinylalkyne in (Fig. 10, B) was substituted by an α-hydroxyl group, such as is observed in Moco, the resulting pterin-dithiolene complex readily cyclized to the pyranopterin-dithiolene form (Fig. 11).25 The pyranopterin structure was proven by X-ray crystal structures of the dithiolene complexes in both the 4+ and 5+ oxidation states.25 The formation of the pyran ring proceeds through a spontaneous intramolecular cyclization.
Fig. 11.
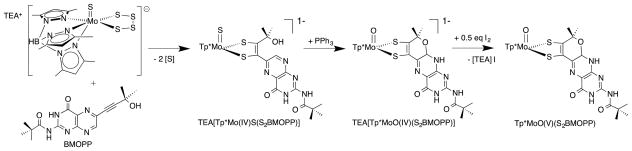
Synthetic route to Mo(IV) and Mo(V) complexes of a pterin-dithiolene ligand. TEA+ = tetraethylammonium cation.
The most intriguing feature of this model system is that the pyranopterin group can participate in an easily reversible pyran ring scission and recyclization (Fig. 12). 1H NMR in a variety of deuterated solvents revealed that the Mo(IV)-pterin-dithiolene complex exists in an equilibrium of cyclized and open species where solvent polarity controls the predominant species. Thus, the uncyclized pterin dithiolene complex predominates in the less polar (lower dielectric) chloroform while the pyranopterin dithiolene form predominates in more polar (higher dielectric) acetonitrile.25
Fig. 12.
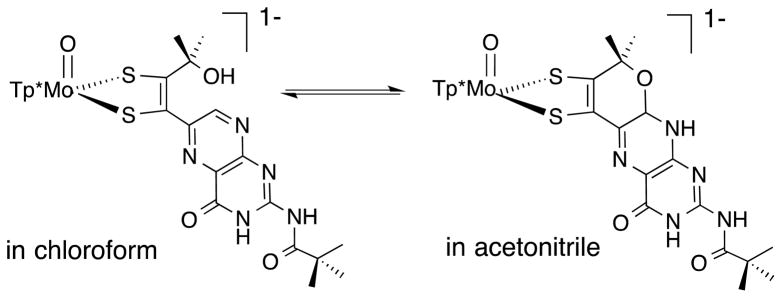
Reversible pyran ring formation observed in a pterin-dithiolene model system.
Pyran cyclization is facile and ring closure can apparently occur from both faces of the pterin. In the X-ray structure of (TEA) Tp*Mo(IV)O(S2BMOPP)] (where TEA+ = tetraethylammonium cation), two unique diastereomers are observed, that differ in terms of the chirality of the pyrano bridgehead carbon C7 directing H7 to either the side of the pterin plane (see the solid state structure of the anion [Tp*Mo(IV)O(S2BMOPP)]− in Fig. 13). In solution, 1H NMR spectra show distinct resonances for H7 depending on its position whether it is on the same side of the dithiolene as the Mo=O group or on the opposite side (Fig. 14). This has allowed determination of chemical shifts specific to the R,R and S,S conformations of the pyranopterin-dithiolene ligand. Thus, in solution the compound exists as a 1:1 mixture of diastereomers.
Fig. 13.
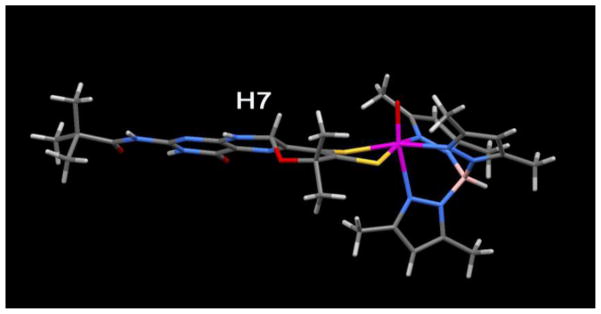
One of the two diastereomers observed in the X-ray structure for the pyranopterin-dithiolene model TEA[Tp*Mo(IV)O(S2-pyrano-BMOPP)] where H7 is oriented on the same side of the pterin-dithiolene ligand as the Mo=O group. The other diastereomer has H7 pointing down, on the opposite side of the pterin-dithiolene.
Fig. 14.

Pyran cyclization yields two diastereomers, depending on the orientation of hydroxyl oxygen atom attack at C7. Note the H atom of the –OH is not shown for clarity.
The results from this pterin-dithiolene model system are significant for two reasons. First, X-ray structures of several enzymes show Moco with one pterin-dithiolene ligand in the pyranopterin form and the other as an uncyclized pterin-dithiolene ligand, such as illustrated in Fig. 6.48, 49
Reversible pyran cyclization could be a possible mechanism for modulating the electronic environment of the dithiolene in molybdoenzymes. Second, the model system provides insight to understanding how reversible pyranopterin dithiolene formation is dependent on solvent polarity. This behavior implies that the pterin-dithiolene conformation in the molybdoenzymes could be similarly controlled by the protein environment around the pyran and pyrazine rings of the pterin.
During the evolution of the synthetic methodology for Mo-pterin-dithiolene models, studies were often initiated by employing a quinoxaline in place of pterin to serve as a simpler N-heterocyclic analog to test the synthetic strategy. It is worth noting the results from two cases directed at quinoxaline-dithiolene model systems. These are shown in Fig. 15 and serve to demonstrate the impact an N-heterocycle like pterin and quinoxaline can have on the electronic environment of the Mo-dithiolene moiety.52, 65, 68 In both model systems, the facile intramolecular cyclization producing pyrroloquinoxalyl-dithiolene ligands causes an asymmetric chelation to Mo, where the two Mo-S and C-S bonds have slightly different lengths (Fig. 16, left).
Fig. 15.
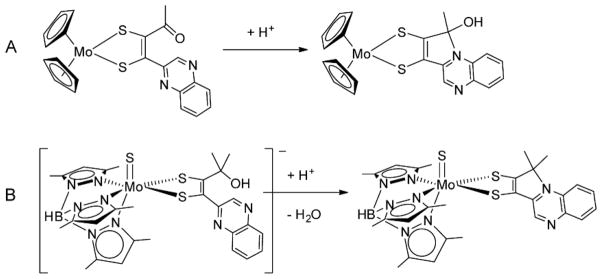
Electrophilic reactions of quinoxalyldithiolenes cause cyclization.
Fig. 16.
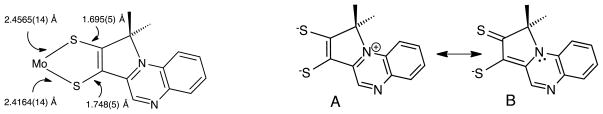
Resonance structures describing electronic delocalization in pyrroloquinoxalyl-dithiolene complexes.
For the product of reaction B in Fig. 15, this asymmetry has been correlated to an admixture of a thiolate-thione structure (Fig. 16, structure B) that is accessible because of the ability of the bridgehead N atom to accept a pair of electrons. Such behavior is characteristic of ligands now classified as “non-innocent” which have a high degree of covalency with the coordinated metal and which accommodate often unexpected electronic delocalization.
The above selection of recent and previous model studies of pterin- and quinoxaline-dithiolene Mo complexes provide examples of the rich possibility inherent in the pyranopterin-dithiolene MPT ligand of Moco. Fig. 17 brings together some of these features to shown how the pyran ring cleavage and electronic redistribution could mediate the immediate environment and hence, the reactivity of the Mo atom.
Figure 17.

Pyran ring-opening reaction in Moco, which can generate tautomers A-C by proton migration around the pyrazine ring of pterin. The bottom structure possessing the thione-thiolate chelate is a valence tautomer resulting from a formal two-electron intramolecular redox reaction yielding reduced Mo(IV) and an oxidized pterin dithiolene ligand.
It is instructive to compare the facile reversible pyran cyclization observed in the model system TEA[Tp*Mo(IV)O(S2-pyrano-BMOPP)] (Fig. 12) with the related quinoxalyl pyran system having a protected dithiolene group (Fig. 18). Only the pterin-dithiolene coordinated to Mo25 (Fig. 18, top) exhibits reversible pyran formation whereas the pyranoquinoxaline dithiolene molecule or simply pyran dithiolate27, 28 (Fig. 18, bottom) does not exhibit a similar reversible cyclization.
Fig. 18.

Comparison of two models possessing pyran rings and nitrogen heterocyclic substituents on a dithiolene or a protected dithiolene.
This difference in the pyran reactivity may point to a special structural feature of the pyranopterin-dithiolene portion of Moco that allows a specific mode of action required by the enzyme.
Unresolved Questions
We conclude this minireview with our perspective on the utility of pursuing model chemistry for the molybdenum enzymes. It might seem that the X-ray structural determinations of the molybdenum cofactor in a large number of molybdenum proteins would have brought to a close the benefit or even the need for model work. However, we argue that is not the case and there are fundamental aspects of Moco that have not been sufficiently answered by crystallographic studies. Despite the plethora of X-ray structures, which frequently present the molybdenum cofactor structure as definitively a fully reduced pyranopterin-dithiolene bound to Mo, there remains uncertainty in the exact structure of Moco in terms of the reduction state of the pterin as well as the dynamics available to the pyran ring.69, 70 Another related question is why does the pterin-dithiolene ligand appear in both a pyrano form and uncyclized in certain bacterial enzymes. Yet another structural issue is that no model has yet duplicated the geometrical parameters, specifically the inner coordination sphere angles, of Moco. The results from crystallography have in fact prompted more probing questions that require further detailed investigation.
Questions also remain in regards the mechanistic details operating in the enzymes, which suggest a need for further model studies. While a detailed discussion on reactivity is beyond the scope of this minireview, it has become increasingly clear that the OAT reactions, which are the modus operandi of many pterin-molybdenum enzymes, proceed at least via two steps,71–75 although details of the energetics of mechanistic steps and the electronic nature of the transition state may vary among different enzymes. In OAT model systems, the proposed steps are conceptually different from those in the enzymes and the enzymatic reactions are orders of magnitude faster, which argue that model systems developed to date are still inadequate mimics. Furthermore, since it has been shown that OAT model systems need not require the exact coordination about the metal nor specific ligand architecture as observed in the enzyme, it is provocative to ask why Nature chose such a complicated cofactor to carry out simple thermodynamic reactions. Since carrying out the stoichiometric reactions at a slow rate is not sufficient, it may be that the pterin-dithiolene ligand of the cofactor tunes the electronic structure such that it enhances the reaction rate and acts as a mediator for proton transfer to complete the catalytic cycle.
While our limited current knowledge does not provide a clear picture as to how the pterin cofactor may enhance the reaction rate, we offer here several suggestions. For example, a) the pterin located inside the protein milieu not only prevents the formation of unwanted dimolybdenum centers, but also reduces the reorganization energy to promote an efficient reaction. b) The extended structure of the pterin cofactor may poise the electronic states such that minimum reorganization energy is spent while the extended hydrogen bonding constraining the cofactor in a particular conformation influences the electronic state of the metal center. c) In addition, the redox state of the pterin might also influence the geometry at the metal center, in particular at the dithiolene, which again minimizes the reorganization of the metal center during electron transfer. d) A reversible pyran ring scission and cyclization at the pterin-dithiolene ligand, now documented in a model system, may also play a role in adjusting the electronic environment to control enzyme function. Clearly significant progress has been made in the development of new molecules as structural models, and understanding reactivity and electronic intricacies, but many questions are unresolved and more studies are needed.
Acknowledgments
We thank the National Institutes of Health for financial support of our research (NIH GM 061555 to PB and NIH GM 081848 to SB).
Contributor Information
Partha Basu, Email: basu@duq.edu, Department of Chemistry and Biochemistry, Duquesne University, Pittsburgh, PA 15282.
Sharon J. Nieter Burgmayer, Email: sburgmay@brynmawr.edu, Department of Chemistry, Bryn Mawr College, Bryn Mawr, PA 19010
References
- 1.Meriwether LS, Marzluff WF, Hodgson WG. Nature. 1966;212:465–467. doi: 10.1038/212465a0. [DOI] [PubMed] [Google Scholar]
- 2.Berg JM, Hodgson KO, Cramer SP, Corbin JL, Elsberry A, Pariyadath N, Stiefel EI. J Am Chem Soc. 1979;101:2774–2776. [Google Scholar]
- 3.Stiefel EI. Prog Inorg Chem. 1977;22:1–223. [Google Scholar]
- 4.Hille R. Chem Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- 5.Hille R, Hall J, Basu P. Chem Rev. 2014;114:3963–4038. doi: 10.1021/cr400443z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enemark JH, Cooney JJA, Wang JJ, Holm RH. Chem Rev. 2004;104:1175–1200. doi: 10.1021/cr020609d. [DOI] [PubMed] [Google Scholar]
- 7.Enemark JH, Young CG. Adv Inorg Chem. 1994;40:1–88. [Google Scholar]
- 8.Holm RH. Coord Chem Rev. 1990;100:183–221. [Google Scholar]
- 9.Holm RH. Chem Rev. 1987;87:1401–1449. [Google Scholar]
- 10.Majumdar A, Sarkar S. Coord Chem Rev. 2011;255:1039–1054. [Google Scholar]
- 11.Xiao Z, Bruck MA, Enemark JH, Young CG, Wedd AG. Inorg Chem. 1996;35:7508–7515. [Google Scholar]
- 12.Xiao Z, Bruck MA, Enemark JH, Young CG, Wedd AG. JBIC, J Biol Inorg Chem. 1996;1:415–423. doi: 10.1021/ic961983x. [DOI] [PubMed] [Google Scholar]
- 13.Stiefel EI, Pilato RS. In: Bioinorganic Catalysis. Reedijk J, editor. Marcel Dekker, Inc; 1993. [Google Scholar]
- 14.Collison D, Garner CD, Joule JA. Chem Soc Rev. 1996;25:25–32. [Google Scholar]
- 15.Hine FJ, Taylor AJ, Garner CD. Coord Chem Rev. 2010;254:1570–1579. [Google Scholar]
- 16.Pushie MJ, Cotelesage JJ, George GN. Metallomics. 2014;6:15–24. doi: 10.1039/c3mt00177f. [DOI] [PubMed] [Google Scholar]
- 17.Nason A, Lee KY, Pany SS, Ketchum PA, Lamberti A, DeVries J. Proc Natl Acad Sci USA. 1971;68:3242–3246. doi: 10.1073/pnas.68.12.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson JL, Rajagopalan KV. Proc Natl Acad Sci U S A. 1982;79:6856–6860. doi: 10.1073/pnas.79.22.6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajagopalan KV. Adv Enzymol Relat Areas Mol Biol. 1991;64:215–290. doi: 10.1002/9780470123102.ch5. [DOI] [PubMed] [Google Scholar]
- 20.Fischer B, Burgmayer SJN. Met Ions Biol Syst. 2002;39:265–316. [PubMed] [Google Scholar]
- 21.Kaufmann HL, Carroll PJ, Burgmayer SJN. Inorg Chem. 1999;38:2600–2606. [Google Scholar]
- 22.Fischer B, Schmalle HW, Baumgartner MR, Viscontini M. Helv Chim Acta. 1997;80:103–110. [Google Scholar]
- 23.Lim BS, Willer MW, Miao M, Holm RH. J Am Chem Soc. 2001;123:8343–8349. doi: 10.1021/ja010786g. [DOI] [PubMed] [Google Scholar]
- 24.Burgmayer SJN, Kim M, Petit R, Rothkopf A, Kim A, BelHamdounia S, Hou Y, Somogyi A, Habel-Rodriguez D, Williams A, Kirk ML. J Inorg Biochem. 2007;101:1601–1616. doi: 10.1016/j.jinorgbio.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams BR, Fu Y, Yap GPA, Burgmayer SJN. J Am Chem Soc. 2012;134:19584–19587. doi: 10.1021/ja310018e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bradshaw B, Collison D, Garner CD, Joule JA. Chem Commun. 2001:123–124. [Google Scholar]
- 27.Marbella L, Serli-Mitasev B, Basu P. Angew Chem, Int Ed. 2009;48:3996–3998. doi: 10.1002/anie.200806297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pimkov IV, Nigam A, Venna K, Fleming FF, Solntsev PV, Nemykin VN, Basu P. J Heterocycl Chem. 2013;50:879–886. doi: 10.1002/jhet.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pimkov IV, Peterson AA, Vaccarello DN, Basu P. RSC Adv. 2014;4:19072–19076. doi: 10.1039/C4RA02786H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirk ML, McNaughton RL, Helton ME. Prog Inorg Chem. 2003;52:111–212. [Google Scholar]
- 31.van Stipdonk MJ, Basu P, Dille SA, Gibson JK, Berden G, Oomens J. J Phys Chem A. 2014;118:5407–5418. doi: 10.1021/jp503222v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joshi HK, Cooney JJA, Inscore FE, Gruhn NE, Lichtenberger DL, Enemark JH. Proc Natl Acad Sci U S A. 2003;100:3719–3724. doi: 10.1073/pnas.0636832100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westcott BL, Gruhn NE, Enemark JH. J Am Chem Soc. 1998;120:3382–3386. [Google Scholar]
- 34.Fischer B, Enemark JH, Basu P. J Inorg Biochem. 1998;72:13–21. doi: 10.1016/s0162-0134(98)10054-5. [DOI] [PubMed] [Google Scholar]
- 35.Romao MJ, Archer M, Moura I, Moura JJG, LeGall J, Engh R, Schneider M, Hof P, Huber R. Science. 1995;270:1170–1176. doi: 10.1126/science.270.5239.1170. [DOI] [PubMed] [Google Scholar]
- 36.Young CG. J Biol Inorg Chem. 1997;2:810–816. [Google Scholar]
- 37.Young CG, Wedd AG. ACS Symp Ser. 1993;535:70–82. [Google Scholar]
- 38.Sugimoto H, Tsukube H. Chem Soc Rev. 2008;37:2609–2619. doi: 10.1039/b610235m. [DOI] [PubMed] [Google Scholar]
- 39.Stiefel EI. J Chem Soc, Dalton Trans. 1997:3915–3924. [Google Scholar]
- 40.Metz S, Thiel W. Coord Chem Rev. 2011;255:1085–1103. [Google Scholar]
- 41.McMaster J, Tunney JM, Garner CD. Prog Inorg Chem. 2003;52:539–583. [Google Scholar]
- 42.Majumdar A. Dalton Trans. 2014;43:8990–9003. doi: 10.1039/c4dt00631c. [DOI] [PubMed] [Google Scholar]
- 43.Kirk ML, Knottenbelt S, Habtegabre A. In: Computational Inorganic and Bioinorganic Chemistry. Solomon EI, Scott RA, King RB, editors. John Wiley & Sons Ltd; 2009. pp. 277–285. [Google Scholar]
- 44.Holm RH, Solomon EI, Majumdar A, Tenderholt A. Coord Chem Rev. 2011;255:993–1015. [Google Scholar]
- 45.Holm RH, Berg JM. Acc Chem Res. 1986;19:363–370. [Google Scholar]
- 46.Enemark JH, Cooney JJA, editors. Model complexes for molybdenum- and tungsten-containing enzymes. Wiley-VCH Verlag GmbH & Co. KGaA; 2006. [Google Scholar]
- 47.Basu P, Burgmayer SJN. Coord Chem Rev. 2011;255:1016–1038. doi: 10.1016/j.ccr.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bertero MG, Rothery RA, Palak M, Hou C, Lim D, Blasco F, Weiner JH, Strynadka NCJ. Nat Struct Biol. 2003;10:681–687. doi: 10.1038/nsb969. [DOI] [PubMed] [Google Scholar]
- 49.Kloer DP, Hagel C, Heider J, Schulz GE. Structure. 2006;14:1377–1388. doi: 10.1016/j.str.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 50.Schindelin H, Kisker C, Rees DC. JBIC. 1997;2:773–781. [Google Scholar]
- 51.Helton ME, Gebhart NL, Davies ES, McMaster J, Garner CD, Kirk ML. J Am Chem Soc. 2001;123:10389–10390. doi: 10.1021/ja0100470. [DOI] [PubMed] [Google Scholar]
- 52.Matz KG, Mtei RP, Leung B, Burgmayer SJN, Kirk ML. J Am Chem Soc. 2010;132:7830–7831. doi: 10.1021/ja100220x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taylor EC, Perlman KL, Kim YH, Sword IP, Jacobi PA. J Amer Chem Soc. 1973;95:6413–6418. [Google Scholar]
- 54.Soyka VR, Pfleiderer W, Prewo R. Helv Chim Acta. 1990;73:808–826. [Google Scholar]
- 55.Nemykin VN, Olsen JG, Perera E, Basu P. Inorg Chem. 2006;45:3557–3568. doi: 10.1021/ic051653p. [DOI] [PubMed] [Google Scholar]
- 56.Perera E, Basu P. Dalton Trans. 2009:5023–5028. doi: 10.1039/b904113c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mtei RP, Perera E, Mogesa B, Stein B, Basu P, Kirk ML. Eur J Inorg Chem. 2011;2011:5467–5470. doi: 10.1002/ejic.201101084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bradshaw B, Dinsmore A, Ajana W, Collison D, Garner CD, Joule JA. J Chem Soc, Perkin Trans. 2001;1:3239–3244. [Google Scholar]
- 59.Davies ES, Beddoes RL, Collison D, Dinsmore A, Docrat A, Joule JA, Wilson CR, Garner CD. J Chem Soc, Dalton Trans. 1997:3985–3996. [Google Scholar]
- 60.Dinsmore A, Birks JH, Garner CD, Joule JA. J Chem Soc, Perkin Trans. 1997;1:801–807. [Google Scholar]
- 61.Garner CD, Baugh PE, Collison D, Davies ES, Dinsmore A, Joule JA, Wilson CR. J Inorg Biochem. 1997;67:288. [Google Scholar]
- 62.Coucouvanis D, Hadjikyriacou A, Draganjac M, Kanatzidis MG, Ileperuma O. Polyhedron. 1986;5:349–356. [Google Scholar]
- 63.Coucouvanis D, Hadjikyriacou A, Toupadakis A, Koo SM, Ileperuma O, Draganjac M, Salifoglou A. Inorg Chem. 1991;30:754–767. [Google Scholar]
- 64.Draganjac M, Coucouvanis D. J Am Chem Soc. 1983;105:139–140. [Google Scholar]
- 65.Pilato RS, Eriksen KA, Greaney MA, Stiefel EI, Goswami S, Kilpatrick L, Spiro TG, Taylor EC, Rheingold AL. J Am Chem Soc. 1991;113:9372–9374. [Google Scholar]
- 66.Pilato RS, Eriksen K, Greaney MA, Gea Y, Taylor EC, Goswami S, Kilpatrick L, Spiro TG, Rheingold AL, Stiefel EI. ACS Symp Ser. 1993;535:83–97. [Google Scholar]
- 67.Sproules SA, Morgan HT, Doonan CJ, White JM, Young CG. Dalton Trans. 2005:3552–3557. doi: 10.1039/b506050h. [DOI] [PubMed] [Google Scholar]
- 68.Matz KG, Mtei RP, Rothstein R, Kirk ML, Burgmayer SJN. Inorg Chem. 2011;50:9804–9815. doi: 10.1021/ic200783a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rothery RA, Stein B, Solomonson M, Kirk ML, Weiner JH. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14773–14778. doi: 10.1073/pnas.1200671109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sparacino-Watkins C, Stolz JF, Basu P. Chem Soc Rev. 2014;43:676–706. doi: 10.1039/c3cs60249d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kail BW, Young CG, Johnson ME, Basu P. ACS Symp Ser. 2009;1012:199–217. [Google Scholar]
- 72.Kail BW, Perez LM, Zaric SD, Millar AJ, Young CG, Hall MB, Basu P. Chem-Eur J. 2006;12:7501–7509. doi: 10.1002/chem.200600269. [DOI] [PubMed] [Google Scholar]
- 73.Nemykin VN, Laskin J, Basu P. J Am Chem Soc. 2004;126:8604–8605. doi: 10.1021/ja049121f. [DOI] [PubMed] [Google Scholar]
- 74.Nemykin VN, Basu P. Inorg Chem. 2005;44:7494–7502. doi: 10.1021/ic0508324. [DOI] [PubMed] [Google Scholar]
- 75.Nemykin VN, Basu P. Dalton Trans. 2004:1928–1933. doi: 10.1039/b403964e. [DOI] [PubMed] [Google Scholar]