

Abstract
B-cell chronic lymphocytic leukemia (CLL) is the most common human leukemia occurring as indolent or aggressive form. CLL clinical features and genetic abnormalities are well documented, but molecular details are still under investigation. MicroRNAs are small non-coding RNAs involved in several cellular processes and expressed in a tissue-specific manner. MicroRNAs regulate gene expression, and their deregulation can alter expression levels of genes involved in development/progression of tumors. In CLL, microRNAs can function as oncogenes or tumor suppressors and can also serve as markers for CLL onset/progression. Here, we discuss the most recent findings about the role of microRNAs in CLL and how this knowledge can be used to identify new biomarkers and treatment approaches.
Keywords: microRNA, CLL
CLL: characteristics and outcomes
Chronic lymphocytic leukemia (CLL) is the most common human leukemia. This disease can arise in two forms, aggressive and indolent, both characterized by the accumulation of incompetent CD5+ B lymphocytes [1]. Unfavorable prognosis is associated with the expression of unmutated immunoglobulin heavy variable genes (UM-IgH-VH) and high level of 70 kD zeta-associated protein (ZAP-70). Chromosomal alterations are detected in >80% of cases and can discriminate patients with different outcomes [2,3]: (i) low risk, normal karyotype or 13q deletion; (ii) intermediate risk, 11q deletion or trisomy 12; and (iii) high risk, 17p deletion or complex karyotype [4]. The ability of malignant cells to respond to microenvironmental stimuli via B-cell receptor (BCR) signaling, interaction with accessory cells or microvescicles identified in plasma has recently emerged as another factor in the clinical course of CLL [5,6].
Signatures of microRNAs in CLL cells and microenvironment
MicroRNA profiles can distinguish normal B cells from malignant CLL cells and have been associated with prognosis, progression, and drug resistance [7]. A signature profile describing 13 microRNAs able to differentiate aggressive and indolent CLL was reported [8]. Patients with high miR-21 and miR-155 had a higher risk of death compared to patients with low expression of these microRNAs [9,10], and miR-181b can predict time to treatment, acting as a biomarker of progression [11]. MicroRNA signatures can also predict refractoriness to fludarabine [7]. MiR-148a, miR-222, and miR-21 exhibited a higher expression in non-responders while lower levels of miR-34a were observed in resistant subjects even in absence of p53 aberrations [12]. Recent studies demonstrated that microRNAs are involved in an intricate interplay with BCR signaling and microenvironmental stimuli. Indeed, BCR signaling and immunoglobulin production can be regulated by microRNAs [13] while the expression of certain microRNAs can be altered via BCR stimulation [5]. A signature of 39 differentially expressed miRNAs was found upon BCR stimulation [14]. BCR activation can lead to reduced levels of miR-29c, miR-150, miR-181b, or miR-223 [15], and low expression of these microRNAs was observed in patients with shorter survival and/or time to treatment [16]. MiR-155 is instead upregulated in response to BCR ligation and appears to play a role during T- and B-cell development [5,13]. Recent reports proposed that miRNAs are released by donor cells through circulating microvescicles (MVs) [6]. Extracellular miRNAs are present in the plasma of CLL patients at different levels from healthy controls. Since therapy often reduced the pool of malignant cells but does not affect plasma features, it is likely that MVs continue to induce abnormal gene profiles. Therefore, miRNA profiling in plasma could reflect another mechanism of malignant B-cell proliferation [17].
Role of microRNAs in CLL
Recently, researchers have focused on the molecular impact of deregulation of microRNA expression in CLL. MiR-15/16 cluster, miR-34b/c, miR-29, miR-181b, miR-17/92, miR-150, and miR-155 family members, the most deregulated microRNAs in CLL, were found to regulate important genes, helping to clarify molecular steps of disease onset/progression.
MicroRNA-15a/16-1
In 2002, a cluster of two microRNA genes, miR-15a and miR-16-1, was located within the 13q14.3 deleted region [18]. 13q14.3 deletion in CLL, the most frequent genomic aberration, associates with the longest treatment-free interval [19]. Accordingly, miR-15a/16-1 expression was found downregulated in ~66% of CLL cases [18]. The importance of miR-15a/16-1 was confirmed in a study in New Zealand black (NZB) mice, the only mouse strain that naturally develops CLL [20]. A point mutation causing a decrease of miR-16-1 expression in NZB lymphoid tissues and elevated levels of Bcl-2 was found in the miR-15a/16-1 precursor (located in the mouse genomic region homologous to 13q14) [20]. B-cell lymphoma 2 (BCL2) is a central player in the genetic program of eukaryotic cells, promoting survival by inhibiting apoptosis [21], and it is overexpressed in many human cancers [22]. MiR-15a/16-1 and BCL2 expression levels were found inversely correlated in CLL [23], and downregulation of these microRNAs in leukemic cell lines resulted in an increase of Bcl2 expression with consequent inhibition of apoptosis [23]. Moreover, microarray experiments performed on CLL patients with high vs low levels of miR-15a/16-1 identified a gene signature which also contains MCL1, an antiapoptotic BCL-2 family member associated with B-CLL cell survival and chemotherapy resistance [24].
The first genetic manipulation in mice that confirmed the importance of miR-15a/16-1 deletion in CLL was carried out by Dr. Dalla-Favera and colleagues [25]. These authors designed a model with conditional alleles that either resembled the loss of the minimal deleted region (Mdr), spanning entirely the host gene Dleu2 gene [26], or the specific miR-15a/16-1 cluster deletion, without altering the expression of Dleu2 [25]. Mdr knockout (KO) animals lived less than wild-type (WT) siblings and succumbed to leukemia, while the differential survival between miR-15a/16-1 KO and their WT littermates was not statistically significant, indicating that the latter were affected by a milder phenotype than the former [25].
Additional factors regulate miR-15a/16-1 expression besides chromosomal deletion. Veronese et al. identified an allele-specific transcription mechanism. Normally, one allele of miR-15a/16-1 is transcribed by RNA polymerase II together with DLEU2 and the other by RNA polymerase III independently of the host gene. In 13q14 deleted patients, exclusive RNA polymerase III-driven transcription was observed and found to associate with high expression of ZAP70. Indeed, in a CLL case of monozygotic twins that differed in ZAP70 status and clinical features, transcription of primiR-15a/16-1 was driven by RPIII in the aggressive ZAP70-positive patient and by RPII in the indolent ZAP70-negative case [27]. Furthermore, CLL cells show a reduced amount of processed intermediates pre-miR-15a/16-1, while the precursor pri-miR-15a/16-a was not decreased, indicating a block of miRNA maturation at the DROSHA processing step. Interestingly, the mRNA levels of pri-miRNA processing cofactors were not decreased, and CLL cells retained the ability to cleave other microRNA precursors, suggesting that DROSHA processing is specifically impaired for mir-15a/16-1 and possibly other specific miRNAs [28]. Lastly, the two copies of the critical region were found to replicate asynchronously. Differential replication timing represents an early epigenetic mark resulting in different chromatin packaging and monoallelic expression. These findings support a model of consecutive inactivation of both alleles by a series of different mechanisms, resulting in a final complete inactivation. Indeed, in the majority of CLLs, monoallelic deletion is sufficient for complete loss of mir-15a/16-1 function rather than a twofold downregulation. Inactivation of the active chromosome copy by epigenetic mechanisms could therefore be a tumor-initiating event [29].
MicroRNA-34b/c and microRNA-34a
11q deleted region includes the miR-34b/c cluster locus [30], while deletion of 17p leads to abrogation of the p53 tumor suppressor [31], and 13q deletion involves miR15a/16-1 downregulation. To verify possible interactions between these chromosomal alterations, we investigated if the miR-15a/16-1 cluster, tumor protein p53, and miR-34b/c cluster are connected in a molecular pathway that could explain the prognostic implications of 11q, 17p, and 13q deletions in CLL [32]. Several TP53 binding sites were found upstream miR-15a/16-1 on chromosome 13, miR-34b/c on chromosome 11, and miR-34a on chromosome 1. Thus, TP53 could induce the expression of these microRNAs [32]. Besides, miR-15a/16-1 target TP53 while miR-34 targets ZAP-70 mRNA expression [32]. In 13q deleted patients, the loss of miR-15a/16-1 expression shifts the balance not only toward higher levels of anti-apoptotic Bcl2 [10,23] but also toward higher levels of pro-apoptotic p53. Consequently, the number of apoptotic cells decreases because of the lower level of Bcl2, but the intact p53 pathway keeps the tumor growth relatively low. This finding could explain how 13q deletions associate with indolent CLL. Moreover, increased p53 levels in patients with 13q deletions associate with transactivation of miR-34b/c leading to reduced levels of ZAP-70, positively correlating with survival [3]. CLL patients with 11q deletion, instead, show lower levels of miR-34b/c and higher levels of ZAP-70. In these patients, TP53 is not upregulated because miR-15a/16-1 are not deleted and this condition is associated with lower control on apoptosis. TP53 transactivation of miR-34b/c is ineffective, since the locus is deleted, leading to a higher expression of ZAP-70 which correlates with poor prognosis [32] (Figure 1). 17p deletion and TP53 mutation identifies the majority of chemotherapy-resistant patients. However, almost half of the refractory cases cannot be explained by a direct defect of p53. Zenz et al. studied miR-34a and miR-34b/c expression in refractory CLL with and without 17p deletion or TP53 mutation [33]. While no expression of miR-34b/c was detected, downregulation of miR-34a was observed in 17p deleted and/or TP53 mutated cases and in fludarabine-refractory cases even in the absence of 17p deletion/TP53 mutation. Therefore, low expression of miR-34a in CLL is associated not only with p53 inactivation but also with chemotherapy-refractory disease, impaired DNA damage response, and apoptosis resistance, regardless of 17p deletion/TP53 mutation [33].
Figure 1.
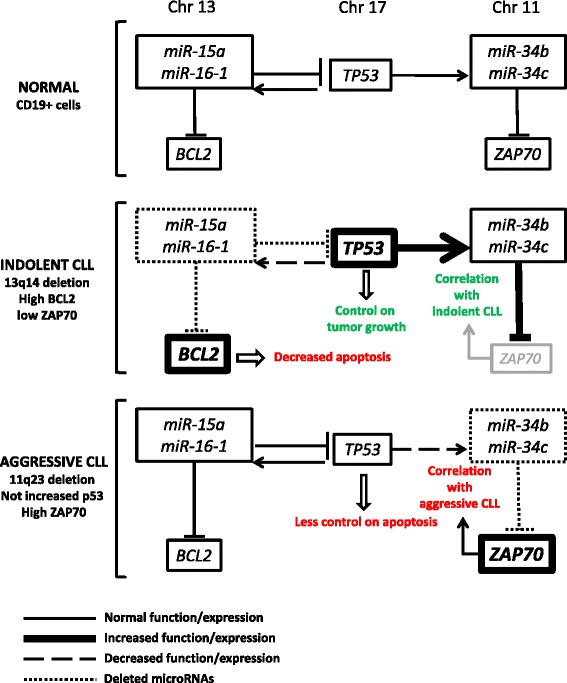
Interplay between microRNA expression and chromosomal aberration in CLL.
MicroRNA-181b and microRNA-29
In both indolent and aggressive CLLs, miR-29 is overexpressed when compared to normal B cells while miR-181b is downregulated when compared to normal B cells. Although, when comparing indolent and aggressive CLLs, both mir-29 and mir-181b show higher expression levels in indolent cases [34-36]. Moreover, miR-181b expression is decreased during CLL progression when sequential samples from the same patients are compared [11]. In the same study, miR-181b is also reported to target MCL1 in CLL, while in another study on human gastric and lung cancer cell lines, it was found able to target BCL2 and involved in the development of multidrug resistance [37]. To clarify the role of miR-29 in CLL, we designed a transgenic mouse model and mice overexpressing miR-29 developed a disease similar to the indolent form of CLL. The role of downregulation of miR-29 and miR-181b in aggressive CLLs appears to correlate with Tcl1 overexpression [35]. Activation of TCL1 oncogene (T cell leukemia/lymphoma 1) is a central event in the initiation of aggressive CLL, and high Tcl1 expression correlates with aggressive phenotype [38]. We verified that coexpression of TCL1 with miR-29 and miR-181 decreased its expression and found inverse correlation between miR-29b, miR-181b, and Tcl1 expression in CLL samples [35].
MicroRNA-17/92
MiR-17/92 is a polycistronic microRNA cluster overexpressed in several lymphoid malignancies. In 2008, Xiao et al. [39] generated mice with higher expression of miR-17/92 in lymphocytes. These mice developed lymphoproliferative disease and autoimmunity and died prematurely. Lymphocytes from these mice showed more proliferation and less apoptosis. MiR-17/92 inhibited the expression of the tumor suppressor PTEN and the proapoptotic protein Bim. These data indicate that PTEN and Bim were direct targets of miR-17/92 cluster molecules [39].
A recent study demonstrated that BCR signals can modulate expression of microRNAs. Evidence that BCR response in aggressive UM-IgH VH CLL is accomplished through upregulation of miR-17/92 has been described. In this study, the BCR-driven induction of miR-17/92 in UM-IgH VH CLLs was proposed as possible regulator of B-cell proliferation/survival by downregulating anti-proliferative and/or pro-apoptotic genes [40].
To determine whether miR-17/92 overexpression induces lymphomagenesis, we generated a transgenic mouse overexpressing miR-17/92 in B cells [41]. Eighty percent of mir-17/92 transgenics developed a B-cell malignancy characterized by expansion of CD19+ B cells. Forty-four microRNAs and 680 genes were differentially expressed in these malignant B cells compared to controls. Eight upregulated miRs were correlated with 101 downregulated target mRNAs, and 11 downregulated miRs were correlated with 66 upregulated target mRNAs. ABCC3, a member of the ABC transporter family involved in anticancer agents transport, phosphoinositide 3-kinase (PI3K), growth arrest, and DNA damage 45 (GADD45), IL-4 were among downregulated mRNAs in miR-17/92 mice [41].
MicroRNA-155
MiR-155 and BIC, its host gene, have been reported to accumulate in human B-cell lymphomas, especially in diffuse large B-cell lymphomas, Hodgkin lymphomas, and certain types of Burkitt lymphomas [42]. To study its role in B-cell development and lymphomagenesis, we generated a transgenic mouse expressing mmu-miR-155 in B cells [42]. We showed that Eμ-mmu-miR-155 mice exhibit initially a preleukemic pre-B-cell proliferation evident in the spleen and bone marrow, followed by frank B-cell malignancy resembling the human diseases. These findings indicate that miR-155 is able to induce polyclonal expansion, favoring the capture of secondary genetic changes for full transformation and suggests that miR-155 is directly implicated in the initiation and/or progression of these diseases [42].
It has been reported that expression of miR-155 can alter the signaling pathways triggered by BCR response by modulating the expression of SHIP1 in CLL. SHIP1 encodes for Src homology-2 domain containing inositol 5-phosphatase 1, a phosphatase that may suppress surface immunoglobulin and BCR signaling. SHIP1 is a predicted target of miR-155, and transfection experiments confirmed the targeting by reducing its expression and enhancing cell sensitivity to BCR stimulation. Survival signals from auxiliary cells in the microenvironment might contribute to the upregulation of miR-155. Indeed, T cells or accessory cells of the lymphoid tissue can enhance expression of miR-155 by stimulating the BCR response via CD154/CD40 or BAFF/APRIL interaction and in vitro CLL cells stimulated with CD154 or BAFF/APRIL show reduced expression of SHIP1 and enhanced responsiveness to BCR ligation. Similar effects were also noted in normal B cells, indicating that miR-155 might play a physiologic role in regulating the B-cell response to BCR ligation [13].
Another microenvironmental system that has recently been studied is represented by the role of microvescicles (MVs) in intercellular communication [6]. Recently, Ferrajoli et al. [17] hypothesized that some miRNAs might be involved in the transition from monoclonal B-cell lymphocytosis (MBL) to CLL and that these miRNAs may also predict response to therapy. They found miR-155 increasingly overexpressed both in cells and in circulating microvesicles (MVs) from normal to MBL and CLL samples. Furthermore, miR-155 expression was higher in non-responder than responder patients and the plasma level of miR-155 was lower in patients who achieved complete remission than in patients who experienced other responses. These data validate the ability of miR-155 to predict response to treatment and suggest that increasingly higher expression of miR-155 from normal B cells to MBL to CLL and its overexpression in the plasma can be used as a predictor for poor response to treatment and disease progression.
Conclusions
The ability of microRNAs to modulate gene expression is essential to provide fine control on several cell processes and its deficiency can be involved in CLL development/progression. Indeed, their deregulation can affect downstream pathways on cell cycle and proliferation. MiR-15a/16-1 deletion is an initializing step in CLL, eliciting the control on Bcl2 expression. MiR-34 family members are involved in a fine-regulated feedback circuitry with p53 and miR-15a/16-1 in 13q deleted CLL, suggesting bidirectional interplay between microRNAs and genes. Moreover, restoring expression of miR-15a/16-1 indirectly affects expression of miR-34 family by modulating p53 expression and downregulation of miR-29 and miR-181b in aggressive CLL contributes to overexpression of Tcl1 [43]. Furthermore, study of miR-17/92 and miR-155 may provide useful insights into drug design, delivery, resistance mechanisms, and microenvironmental responses [17,41]. BCR stimulation can alter the expression of certain microRNAs, and BCR-regulated microRNAs might affect B-cell proliferation and apoptosis [14]. Further studies could aim to develop new therapies that disrupt the capacity of CLL cells to home to tissue microenvironments or that inhibit the signaling from accessory cells within the microenvironment. For example, agents that block the capacity of CLL cells to engage accessory cells might be expected to decrease the levels of miR-155, which increases cell sensitivity to BCR [13]. Lastly, deregulation of microRNAs can originate from chromosomal alteration, epigenetic modulation, allele selection, aberrant precursor processing, or interaction with other genes. Given these results, we can underline that microRNAs have a deep impact on CLL development/progression.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VB, YP, and CMC wrote the manuscript and designed the figure. All authors read and approved the final manuscript.
Contributor Information
Veronica Balatti, Email: Veronica.Balatti@osumc.edu.
Yuri Pekarky, Email: Yuri.Pekarsky@osumc.edu.
Carlo M Croce, Email: Carlo.Croce@osumc.edu.
References
- 1.Bullrich F, Croce C. Molecular biology of chronic lymphocytic leukemia, Chronic lymphocytic leukemias, second edition, revised and expanded, Bruce Cheson, Ed Marcel Dekker, Inc New York. 2001. p. 9–32
- 2.Orchard JA, Ibbotson RE, Davis Z, Wiestner A, Rosenwald A, Thomas PW, et al. ZAP-70 expression and prognosis in chronic lymphocytic leukaemia. Lancet. 2004;363:105–11. doi: 10.1016/S0140-6736(03)15260-9. [DOI] [PubMed] [Google Scholar]
- 3.Rassenti LZ, Huynh L, Toy TL, Chen L, Keating MJ, Gribben JG, et al. ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med. 2004;351:893–901. doi: 10.1056/NEJMoa040857. [DOI] [PubMed] [Google Scholar]
- 4.Moreno C, Montserrat E. Genetic lesions in chronic lymphocytic leukemia: what’s ready for prime time use? Haematologica. 2010;95:12–5. doi: 10.3324/haematol.2009.016873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;27:847–59. doi: 10.1016/j.immuni.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cortez MA, Bueso-Ramos C, Ferdin J, Lopez-Berestein G, Sood AK, Calin GA. MicroRNAs in body fluids—the mix of hormones and biomarkers. Nat Rev Clin Oncol. 2011;8:467–77. doi: 10.1038/nrclinonc.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferracin M, Zagatti B, Rizzotto L, Cavazzini F, Veronese A, Ciccone M, et al. MicroRNAs involvement in fludarabine refractory chronic lymphocytic leukemia. Mol Cancer. 2010;9:123. doi: 10.1186/1476-4598-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, Dumitru CD, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci U S A. 2004;101:11755–60. doi: 10.1073/pnas.0404432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rossi S, Shimizu M, Barbarotto E, Nicoloso MS, Dimitri F, Sampath D, et al. microRNA fingerprinting of CLL patients with chromosome 17p deletion identify a miR-21 score that stratifies early survival. Blood. 2010;116:945–52. doi: 10.1182/blood-2010-01-263889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calin GA, Pekarsky Y, Croce CM. The role of microRNA and other non-coding RNA in the pathogenesis of chronic lymphocytic leukemia. Best Pract Res Clin Haematol. 2007;20:425–37. doi: 10.1016/j.beha.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 11.Visone R, Veronese A, Rassenti LZ, Balatti V, Pearl DK, Acunzo M, et al. miR-181b is a biomarker of disease progression in chronic lymphocytic leukemia. Blood. 2011;118:3072–9. doi: 10.1182/blood-2011-01-333484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zenz T, Mohr J, Eldering E, Kater AP, Buhler A, Kienle D, et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood. 2009;113:3801–8. doi: 10.1182/blood-2008-08-172254. [DOI] [PubMed] [Google Scholar]
- 13.Cui B, Chen L, Zhang S, Mraz M, Fecteau JF, Yu J, et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood. 2014;124:546–54. doi: 10.1182/blood-2014-03-559690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kluiver JL, Chen CZ. MicroRNAs regulate B-cell receptor signaling-induced apoptosis. Genes Immun. 2012;13:239–44. doi: 10.1038/gene.2012.1. [DOI] [PubMed] [Google Scholar]
- 15.Mraz M, Kipps TJ. MicroRNAs and B cell receptor signaling in chronic lymphocytic leukemia. Leuk Lymphoma. 2013;54:1836–9. doi: 10.3109/10428194.2013.796055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li S, Moffett HF, Lu J, Werner L, Zhang H, Ritz J, et al. MicroRNA expression profiling identifies activated B cell status in chronic lymphocytic leukemia cells. PLoS One. 2011;6:e16956. doi: 10.1371/journal.pone.0016956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrajoli A, Shanafelt TD, Ivan C, Shimizu M, Rabe KG, Nouraee N, et al. Prognostic value of miR-155 in individuals with monoclonal B-cell lymphocytosis and patients with B chronic lymphocytic leukemia. Blood. 2013;122:1891–9. doi: 10.1182/blood-2013-01-478222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–6. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 20.Raveche ES, Salerno E, Scaglione BJ, Manohar V, Abbasi F, Lin YC, et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood. 2007;109:5079–86. doi: 10.1182/blood-2007-02-071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 22.Sanchez-Beato M, Sanchez-Aguilera A, Piris MA. Cell cycle deregulation in B-cell lymphomas. Blood. 2003;101:1220–35. doi: 10.1182/blood-2002-07-2009. [DOI] [PubMed] [Google Scholar]
- 23.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–9. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calin GA, Cimmino A, Fabbri M, Ferracin M, Wojcik SE, Shimizu M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–71. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17:28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 26.Migliazza A, Bosch F, Komatsu H, Cayanis E, Martinotti S, Toniato E, et al. Nucleotide sequence, transcription map, and mutation analysis of the 13q14 chromosomal region deleted in B-cell chronic lymphocytic leukemia. Blood. 2001;97:2098–104. doi: 10.1182/blood.V97.7.2098. [DOI] [PubMed] [Google Scholar]
- 27.Veronese A, Pepe F, Chiacchia J, Pagotto S, Lanuti P, Veschi S, et al. Allele-specific loss and transcription of the miR-15a/16-1 cluster in chronic lymphocytic leukemia. Leukemia. 2015;29:86–95. doi: 10.1038/leu.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allegra D, Bilan V, Garding A, Dohner H, Stilgenbauer S, Kuchenbauer F, et al. Defective DROSHA processing contributes to downregulation of MiR-15/-16 in chronic lymphocytic leukemia. Leukemia. 2014;28:98–107. doi: 10.1038/leu.2013.246. [DOI] [PubMed] [Google Scholar]
- 29.Mertens D, Wolf S, Tschuch C, Mund C, Kienle D, Ohl S, et al. Allelic silencing at the tumor-suppressor locus 13q14.3 suggests an epigenetic tumor-suppressor mechanism. Proc Natl Acad Sci U S A. 2006;103:7741–6. doi: 10.1073/pnas.0600494103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Auer RL, Riaz S, Cotter FE. The 13q and 11q B-cell chronic lymphocytic leukaemia-associated regions derive from a common ancestral region in the zebrafish. Br J Haematol. 2007;137:443–53. doi: 10.1111/j.1365-2141.2007.06600.x. [DOI] [PubMed] [Google Scholar]
- 31.Merkel O, Asslaber D, Pinon JD, Egle A, Greil R. Interdependent regulation of p53 and miR-34a in chronic lymphocytic leukemia. Cell Cycle. 2010;9:2764–8. doi: 10.4161/cc.9.14.12267. [DOI] [PubMed] [Google Scholar]
- 32.Fabbri M, Bottoni A, Shimizu M, Spizzo R, Nicoloso MS, Rossi S, et al. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA. 2011;305:59–67. doi: 10.1001/jama.2010.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zenz T, Habe S, Denzel T, Mohr J, Winkler D, Buhler A, et al. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood. 2009;114:2589–97. doi: 10.1182/blood-2009-05-224071. [DOI] [PubMed] [Google Scholar]
- 34.Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, et al. A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 35.Pekarsky Y, Santanam U, Cimmino A, Palamarchuk A, Efanov A, Maximov V, et al. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res. 2006;66:11590–3. doi: 10.1158/0008-5472.CAN-06-3613. [DOI] [PubMed] [Google Scholar]
- 36.Santanam U, Zanesi N, Efanov A, Costinean S, Palamarchuk A, Hagan JP, et al. Chronic lymphocytic leukemia modeled in mouse by targeted miR-29 expression. Proc Natl Acad Sci U S A. 2010;107:12210–5. doi: 10.1073/pnas.1007186107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu W, Shan X, Wang T, Shu Y, Liu P. miR-181b modulates multidrug resistance by targeting BCL2 in human cancer cell lines. Int J Cancer. 2010;127:2520–9. doi: 10.1002/ijc.25260. [DOI] [PubMed] [Google Scholar]
- 38.Herling M, Patel KA, Khalili J, Schlette E, Kobayashi R, Medeiros LJ, et al. TCL1 shows a regulated expression pattern in chronic lymphocytic leukemia that correlates with molecular subtypes and proliferative state. Leukemia. 2006;20:280–5. doi: 10.1038/sj.leu.2404017. [DOI] [PubMed] [Google Scholar]
- 39.Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–14. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bomben R, Gobessi S, Dal Bo M, Volinia S, Marconi D, Tissino E, et al. The miR-17 approximately 92 family regulates the response to Toll-like receptor 9 triggering of CLL cells with unmutated IGHV genes. Leukemia. 2012;26:1584–93. doi: 10.1038/leu.2012.44. [DOI] [PubMed] [Google Scholar]
- 41.Sandhu SK, Fassan M, Volinia S, Lovat F, Balatti V, Pekarsky Y, et al. B-cell malignancies in microRNA Emu-miR-17 ~ 92 transgenic mice. Proc Natl Acad Sci U S A. 2013;110:18208–13. doi: 10.1073/pnas.1315365110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006;103:7024–9. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pekarsky Y, Croce CM. Is miR-29 an oncogene or tumor suppressor in CLL? Oncotarget. 2010;1:224–7. doi: 10.18632/oncotarget.129. [DOI] [PMC free article] [PubMed] [Google Scholar]