

Abstract
Myeloperoxidase (MPO) catalyzes the breakdown of hydrogen peroxide and the formation of the potent oxidant hypochlorous acid. We present the application of the fluorogenic peroxidase substrate 10-acetyl-3,7-dihydroxyphenoxazine (ADHP) in steady-state and transient kinetic studies of MPO function. Using initial kinetic parameters for the MPO system, we characterized under the same conditions a number of gold standards for MPO inhibition, namely 4-amino benzoic acid hydrazide (4-ABAH), isoniazid and NaN3 before expanding our focus to isomers of 4-ABAH and benzoic acid hydrazide analogs. We determined that in the presence of hydrogen peroxide that 4-ABAH and its isomer 2-ABAH are both slow-tight binding inhibitors of MPO requiring at least two steps, whereas NaN3 and isoniazid-based inhibition has a single observable step. We also determined that MPO inhibition by benzoic acid hydrazide and 4-(trifluoromethyl) benzoic acid hydrazide was due to hydrolysis of the ester bond between MPO heavy chain Glu 242 residue and the heme pyrrole A ring, freeing the light chain and heme b fragment from the larger remaining MPO heavy chain. This new mechanism would essentially indicate that the benzoic acid hydrazide analogs impart inhibition through initial ejection of the heme catalytic moiety without prior loss of the active site iron.
Keywords: Myeloperoxidase, Inflammation, Reactive Oxygen Species
INTRODUCTION
Myeloperoxidase (MPO) is the only peroxidase capable of consuming hydrogen peroxide (H2O2), in order to catalyze formation of hypochlorous acid (HOCl). Besides MPO-dependent generation of HOCl and hydroxyl radials (•OH), MPO can accommodate and oxidize a number of small molecule substrates that bind to the active site displacing water molecules that are used in the electron transport chain along the heme ring surface [1]. Here, we sought to better understand the mechanism of MPO-mediated oxidation reactions by use of rapid reaction kinetic analysis of a previous uncharacterized fluorescence substrate to determine the overall kinetic steps from a substrate oxidation vantage point and to use this methodology for rapid characterization of MPO inhibitors for relative potency and mechanism of action.
Enzymatically, MPO follows the classical peroxidase cycle (Eq. 1-3) and is tolerant of diverse pH environments with peak activity in the absence of Cl− exhibited at pH 5.5 [2, 3]. MPO is a ferric iron enzyme (MPO-Fe(III)), which becomes oxidized in the presence of H2O2 to a short-lived redox intermediate termed compound I (half life ~100 ms; [4]), which contains a ferryl π cation radical (MPO-Fe(IV)=O ·+π) (Eq. 1) [5, 6]. Compound I can oxidize halides (e.g. Cl−, Br−, I−) with Cl−, presumably being its main physiological substrate [7]. The MPO-dependent reactions follow the classic peroxidases catalytic cycle shown in Equations 1-3.
| (Eq. 1) |
| (Eq. 2) |
| (Eq. 3) |
| (Eq. 4) |
| (Eq. 5) |
Reactive halide (eg. Cl−) generate HOCl through interaction with Compound I (Eq. 2) but not with Compound II (Eq. 2) or the superoxide inactivated Compound III (Eq. 5).
The active site Fe molecule drives these reactions (Eq. 1-5) through octahedral coordination between the central protoporphyrin IX heme and the proximal histidine 336 on the MPO heavy chain (HCHis336), with redox chemistry occurring on the distal surface through displacement of 5 water molecules. There are hydroxylated methyl groups on pyrrole rings A and C that form two ester bonds with evolutionarily conserved peroxidase residues, namely a distal aspartate residue of the light chain (LCAsp94) and a distal glutamate residue of the heavy chain (HCGlu242). Interestingly, the active site of MPO differs from other peroxidases in that the heme moiety is slightly bowed cradling the five water molecules and these two ester bond linkages are in this distal heme environment. The heme has in-plane rings B and D and the asymmetry is caused by the aforementioned separate ester bonds with LCAsp94 and HCGlu242, as well as a sulfonium ion linkage between the heme 2-vinyl group on ring A and methionine 243 on the heavy chain (HCMet243) [8]. Due to the unique spectral signature of MPO, some distinct intermediates are produced by Equations 1-5, which can be observed through standard and stopped flow UV-Vis spectroscopy [8] and by magnetic circular dichroism [9, 10]. The MPO signature is exemplified by the Soret band at 430 nm representing the heme moiety with additional lower energy bands that arise from π–π* transitions in the four frontier orbital bands denoted as charge transfer band 1 (CT1; 496nm), α (570nm), β (620nm) and CT2 (690 nm) [11, 12]. Covalent modification or destruction of the heme can be monitored through rapid reaction kinetic analysis using stopped flow rapid sequential mixing techniques where changes to these absorbance peaks can be assigned and monitored for changes. Given the tremendous spectral and magnetic properties associational with the catalytic activity of MPO, it is a rich source with which to study enzyme inhibition and structure activity relationships.
It has been reported that certain reactive nitrogen containing compounds such as sodium azide (NaN3) [13, 14], salicylhydroxamic acid [15, 16], 4-aminobenzoic acid hydrazide (4-ABAH) [17], isonicotinylhydrazine (isoniazid) [18], N-acetyl-5-methoxytryptamine (melatonin) [19] and 4-hydroxy-2,2,6,6-tetra-methyl-1-piperidinyloxy (tempol) [20], can inhibit MPO activity. Other sulfur containing MPO inhibitors have also been reported such as diamino-diphenyl sulfone (dapsone) [21], and propylthiouracil [22]. Recently, AstraZeneca has gained FDA-approval for the use of an irreversible MPO suicide substrate (2-thioxanthine; AZD5904) in the treatment of chronic obstructive pulmonary disease [23]. Despite the identification of these inhibitors there is no study currently in the literature that has taken on the challenge to analysis their inhibitor parameters for MPO in an attempt to define kinetic steps that govern the inhibitor potential under identical conditions and in parallel. It should be emphasized that despite recent advances in solving MPO-inhibitor crystal structures, there is no current structural data available for the inhibitors examined here.
Our study describes the use of steady state equilibrium and rapid-reaction kinetics investigation of the fluorogenic substrate 10-acetyl-3, 7-dihydroxyphenoxazine (ADHP) for studies of MPO catalysis, characterization of the global dependency of ADHP oxidation by the MPO-H2O2 system and its use to define the mechanism of the widely-used MPO inhibitors namely 4-ABAH, isoniazid and NaN3 before expanding our focus to isomers and analogs of 4-ABAH compounds. The proposed mechanism by which ADHP is oxidized to fluorescent resorufin is shown in Fig. 1a, whereby an initial ADHP radicals are formed and then interaction with another AHDP is needed to generate one molecule of resorufin [24].
Fig. 1. Characterization of the ADHP oxidation by MPO-H2O2 complex.

A, General mechanism of ADHP oxidation to resorufin by a proposed two step reaction, whereby the MPO-H2O2 complex generates two ADHP radicals that undergo a subsequent enzyme independent dismutation reaction to complete formation of one resurofin and one ADHP molecule [33]. B, The H2O2 dependency for ADHP oxidation by Compound I assessed by initial rates of fluorescence change. Reactions of MPO (2.8 nM) and ADHP (20 μM) were initiated by different concentrations of H2O2. C, Michaelis-Menten plot of initial rates versus given ADHP concentrations, progress curves of ADHP oxidation to resorufin generated by MPO-H2O2 complex over ADHP concentrations ranging from 6.25, 12.5, 25, 50, 100, 200, 400, and 800 μM. Reactions of MPO (23 nM) and given ADHP concentrations were initiated by H2O2 (22 μM) auto-injection. D, Stopped-flow progress curves of resorufin generation by MPO (23 nM) initiated by addition of H2O2 (22 μM) for a series of given ADHP concentrations. E, the original fluorescence data was converted into produced resorufin and a set of produced resorufin progress curves was fitted to one phase exponential function using the non-linear regression. Observed rate constant (Kobs) increases with increasing concentration of ADHP in Panel F and MPO-H2O2 complex in Panel G. Determination of apparent first order rate constant from the slope of Kobs as a function of ADHP concentrations. Single exponential function was used to obtain the observed rate constant (Kobs). Titrations were performed and analyzed as described under “Experimental Procedures.”
For our studies presented here, we chose to focus on hydrazide analogs prior to moving to other classes of MPO inhibitors. Our results support the hypothesis that 4-ABAH and its isomer 2-ABAH are slow-tight binding inhibitors of MPO requiring at least two independent steps, whereas NaN3 and isoniazid-based inhibition have a single observable kinetic / inhibitory step. Covalent modification of MPO active site heme group was also readily apparent by use of ADHP oxidation following equilibrium dialysis of mixtures of the enzyme and its inhibitor, in this case for NaN3 or 4-ABAH. We determined that 4-ABAH covalently binds to the MPO enzyme active site, but that inhibition of peroxidase activity is not initiated until after H2O2 is added. Analysis of the state of the heme moiety and whether it was destroyed by the benzoic acid hydrazide itself lead us to discover that this compound does not destroy the heme but rather mediates the preferentially cleavage of the HCGlu242 ester linkage causing a chemical rearrangement resulting in loss of the absorbance signature for the MPO sulfonium ion linkage at HCMet243. Mass spectroscopic analysis confirmed the oxylated heme protoporphyrin ring is released through hydrolysis of the ester linkage between LCAsp93 and pyrrol ring C generating free heme in solution and MPO light chain. This study collectively demonstrates a new method for comparing MPO inhibitors reliability against one and other with regard to their overall efficacy and assessment of the minimal kinetic steps observed in the ADHP oxidation reactions, and the covalent nature of inhibition and their H2O2–dependency. We have used this methodology to define a new mechanism that explains H2O2 catalyzed hydrazide-based inhibition of MPO.
EXPERIMENTAL PROCEDURES
Reagents, Proteins and Chemicals
Ultra-pure myeloperoxidase (MPO) was purchased from Lee Biosolutions Inc. (St. Louis, MO) and 10-acetyl-3, 7-dihydroxyphenoxazine (ADHP) was purchased from ABD Bioquest Inc. (Sunnyvale, CA). Sodium acetate, 3,3’, 5,5’-tetramethylbenzidine (TMB), H2O2, 2-aminobenzoic acid hydrazide (2-ABAH), 4-(trifluoromethyl) benzoic acid hydrazide (4-TFMBAH), 3-(dimethylamino) benzoic acid hydrazide (3-DMABAH) were obtained from Sigma-Aldrich (St. Louis, MO, USA). 4-aminobenzoic acid hydrazide (4-ABAH), benzoic acid hydrazide (BAH), 4-fluorobenzoic acid hydrazide (4-FBAH), 4-nitrobenzoic acid hydrazide (4-NBAH), sodium azide (NaN3), Dimethyl sulfoxide (DMSO) were obtained from Alfa Aesar (Ward Hill, MA). For gel staining reagent Gelcode Blue and enhanced chemiluminescence reagent Western Lightning ultra was purchased from Pierce (Rochford, IL) and Perkin Elmer (Waltham, MA), respectively.
Sodium acetate buffer was adjusted with acetic acid to pH 5.6 (assay buffer). H2O2 stock of 30% from obtained from BDH Chemicals (London, UK) and diluted to concentration on the day of the experiment with concentration verified by absorbance at λ240nm using an extinction coefficient of 39.4 M−1cm−1[25]. ADHP, 4-ABAH, 2-ABAH, 4-BAH, 4-FBAH, 4-NBAH, 4-TFMBAH, 3-DMABAH, NaN3 and isoniazid were dissolved in DMSO and subsequently diluted into assay buffer. The final concentration of DMSO in the reaction was less than 0.5 % (v/v), which did not affect fluorescence of the oxidized ADHP product 7-hydroxyl-3H-phenoxazin-3-one (resorufin; data not shown).
Steady State Analysis of MPO activity Kinetics
ADHP oxidation (Fig. 1A) was measured as a function of H2O2 by use of Varioskan Flash plate reader with excitation of λ530nm and emission of λ590nm. Reactions of ADHP (20μM) were incubated with MPO (2.8 nm) in assay buffer and initiated by the addition of 1/ 10th volume H2O2 from a serial dilution basin. Initial rates for the velocity of these reactions plotted against H2O2 concentration and kinetic parameters (Fig. 1B). Due to the rapid rate of the reaction initial rates were obtained using the auto-injection functionality built into the reader. SkanIt software 2.4.3 parameters included an interval of 0.1 s for each of 1000 reads. Data was timed to remove the substrate depletion exhibited by the high rate of the substrate turnover and used to obtain approximation of the kinetic parameters of Compound I of the MPO-H2O2 system. These results were confirmed by analysis by simultaneous fits of the progress curves obtained in the stopped flow data as an ADHP dependency.
Transient State Analysis by Stopped Flow Kinetics
ADHP oxidation (Fig. 1A) was measured as a function of fluorescence intensity over time using QuantaMaster 80 (Photon Technology International, Birmingham, NJ) with a Xe - Arc lamp. Mixtures of MPO and ADHP were rapidly mixed with H2O2 in acetate buffer (assay buffer) using RX2000 rapid mixing stopped-flow accessory (Applied Photophysics Ltd, Leatherhead, UK) under constant 25°C reaction conditions using isotemp 3016D (Fisher Scientific, Pittsburgh, PA). In addition, the mixture of MPO with ADHP and H2O2 in assay buffer were loaded into two different drive syringes of the stopped-flow apparatus and maintained at 25°C for at least 10 min prior to triggering the pneumatic driver. Experiments were initiated with the optimized concentration of H2O2 (22 μM; refer to Fig. 1B) and performed in a total final volume of 500 μL. After initiation by H2O2, we measured the time-dependent fluorescence change of produced resorufin using an excitation wavelength of 530 nm and an emission wavelength of 590 nm to follow the reaction. Felix GX4.2.2 captured 10 reads per 1 s for 120 s. For all experiments, the slit widths of the spectrofluorometer were set to 8 nm excitation and 2 nm emissions. The fidelity of the kinetic experiments was maintained by RX pneumatic drive of stopped-flow rapid mixing accessory and recordings started prior to triggering rapid mixing of the reactants. The kinetic traces were trimmed to eliminate the delay in observed fluorescence change following mixing and each curve represents an average of at least 4 independently triggered event traces.
Fluorescence Data Analysis
Raw fluorescence intensities measurements were converted into molar resorufin production using a standard curve. Sets of progress curves were initially fit with either a single or double exponential function using GraphPad Prism 5.0. The apparent first order rate constant (Kappon) was obtained by fitting kobs independently versus the concentration for ADHP and MPO (see Fig. 2);
| (Eq. 6) |
where y is the molar product formed, t is time, a is amplitude of the decay, c is the signal at infinite time and kobs is the exponential rate constant.
Fig. 2. NaN3 inhibition of Compound I.

A, Stopped flow analysis of MPO (23 nM) without inhibitor (a), in the presence of NaN3 (30 μM) after pre-incubation for 1 hr prior to triggering H2O2 addition (b) and after equilibrium dialysis (c). B, Lineweaver-Burk plots of for NaN3 inhibition on MPO calculated from resorufin production data. Titrations of 0, 7.5, 30 μM NaN3 are shown for the different concentration of ADHP from 25, 50, 100, 200 and 400 μM. Titrations were performed and analyzed as described under “Experimental Procedures.”
Simultaneous Fits of Progress Curves to Determine the Specificity Constant of MPO-H2O2System for ADHP
To obtain Km and kcat, Scientist software (MicroMath Inc., Saint Louis, Missouri) was used to fit simultaneously the produced resorufin progress curve as a function of ADHP concentration for the MPO-H2O2 system (23 nM/ 22 μM) using the integrated Michaelis-Menten equation;
| (Eq. 7) |
where P is the concentration of product formed, T is time, E0 is the total concentration of enzyme, S0 is the initial concentration of substrate ADHP, Km is the Michaelis-Menten constant, and kcat is the rate of substrate turnover.
Global Analysis of the MPO Inhibition
For the inhibitory effect of ABAH on MPO activity, a set of the time-dependent fluorescence progress curves was fit simultaneously by a slow-tight binding model using DynaFit 3 software ([26]; Biokine, Ltd. Watertown, MA, USA). Kinetic models of a one-step or a two-step mechanism are shown as follows: The inhibitory efficiencies for the inhibitory effects of 4-ABAH and its analogs on MPO activity are obtained from DynaFit software. The overall inhibition constant (Ki) associated with the formation of the reversible EI complex was determined when it met the model of a one-step slow tight-binding inhibitor, such as:
| (Eq. 8) |
When the inhibitory pattern met the requirement of a two-step slow tight-binding inhibitor model, the overall inhibition constant was determined as given by the following equation:
| (Eq. 9) |
k+3, k-3, k+4, k-4 were obtained from simulating a set of fluorescence progress curves inhibited by different inhibitors using DynaFit 3, where Ki =k-3/ k+3.
SDS-PAGE Analysis
To determine the effect that the simplest benzoic acid hydrazide inhibitor or its analog 4-TFMBAH has on the heme catalytic ability of MPO, we incubated MPO (1.2 μM) for 10 min with different concentrations of BAH inhibitor (0, 0.025, 0.25, 2.5, 12.5 and 25 mM) with ADHP (40 μM) and timing of the reaction was measured following addition of H2O2 (20 μM). ADHP All reactions were measured in assay buffer at room temperature. Samples of 20 μL were added to non-reducing sample loading buffers, and then loaded without prior heating and resolved by 4-15% gradient SDS-polyacrylamide gel electrophoresis. Note that heat treatment was avoided due to the established autocatalytic cleavage of MPO heavy chain that results in the appearance of a 39 and 20 kDa band by SDS gel electrophoresis through specific cleavage of the HCMet243-HCPro244 bond [27, 28]. Total protein stained by GelCode Blue reagent (Pierce) and the location and status of the heme moiety of MPO was probed by use of Western Lightning reagent (Perkin Elmer) directly on the gel with images collected with a Fujifilm LAS-1000 luminescence imager.
MALDI-TOF Method
Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectroscopy was used to analyze reaction conditions that were determined to maximized light chain-heme formation products generation as determined by protein electrophoresis, namely MPO (1.2 μM) with BAH (2.5 mM) pre-incubated for 10 min prior to the addition of H2O2 (20 μM). Protein samples were diluted to optimal concentration and spotted onto a 96-well plate using a sandwiched method where the protein solution was diluted 1 to 1 with sinapinic acid matrix (30 mg matrix in 1 ml 33% acetonitrile, 0.1% TFA, freshly prepared) were let air dry to complete at room temperature, and rinsed with H2O and dried prior to sinapinic acid matrix addition (1 μL) to allow for crystal growth. The spectra were acquired using a MALDI-TOF (Microflex, Burker Daltonics) with flexControl (version 3.3) and data analysis by flexAnalysis software (version 3.3). Mass calibration used Insulin and BSA for instrument and for external spectrum. Laser power and voltages were tuned so an optimum signal to noise ratio was achieved. MALDI spectrum was calibrated against external standard, smooth and subtracted background. MPO parent and cleaved fragments with and without heme were identified in the MALDI spectrum range from 10,000 to 150,000 Da.
Liquid Chromatography with Tandem Mass Spectroscopy Method
Protein samples were prepared identical to the MALDI-TOF measurements but instead analyzed using an Ultra Performance LC Systems (ACQUITY, Waters Corp., Milford, MA) coupled with a quadrupole time-of-flight mass spectrometer (Q-TOF Premier, Waters) with electrospray ionization (ESI) detector in positive ion mode operated by the Masslynx software (V4.1). Each sample in H2O were injected into a C18 column (ACQUITY UPLC® BEH C18, 1.7 μm, 2.1 × 50 mm, Waters) with a 150 μL/min flow rate of mobile phase of solution A (95% H2O, 5% acetonitrile, 0.1% formic acid) and solution B (95% acetonitrile, 5% H2O, 0.1% formic acid) in a 10 min gradient starting at 95% A to 5% A in 6 min and back to 95% in 8 min.
The ion source voltages were set at 3 KV, sampling cone at 37 V and the extraction cone at 3 V. The source and desolvation temperature were maintained at 120°C and 225°C, respectively, with the desolvation gas flow at 200 L/h. The TOF MS scan was from 500 to 3500 m/z at 1 s with 0.1 s inter-scan delay. For real time mass calibration, direct infusion of sodium formate solution (10% formic acid/0.1M NaOH/isopropanol at a ratio of 1:1:8) at 1 sec/10 sec to ion source at 1μL/min was used. Scans at 4 min peak (data not shown) of 10 min LC chromatogram were combined, the multiply charged protein envelop from 800 to 2100 m/z were processed using MaxEnt1 (Masslynx) to deconvolute to the molecular ion with iterations that converged. The spectrum was then smooth and centered to obtain the molecular mass. Result from LC-ESI+-MS measurement agrees with those by MALDI-TOF. For clarity, the MALDI-TOF results are shown in this paper.
RESULTS
Effect of H2O2on Oxidation of ADHP by MPO
Our initial attempts at characterization of the MPO-H2O2 system involved use of guaiacol and 3, 3’, 5, 5’-tetramethylbenzidine (TMB)-based absorbance assays because of our previous success in the use of these substrates in tissue homogenate assays of MPO activity [29-32]. The oxidation of ADHP by MPO in the presence of H2O2 is an ordered two-substrate (a.k.a. Ping-Pong) reaction (Fig. 1A). Solubility issues of TMB under the reaction conditions over the range necessary for accurate Km determination limited its utility. Similar problems were seen for guaiacol, which when added to buffer is a suspension not a true solution but these issue did not occur with ADHP. For oxidation of ADHP by MPO, reactions reached a plateau after 20 s following H2O2 addition. It has been well documented that H2O2 concentrations have a profound impact on the catalytic activity of MPO [3], but there is still uncertainty regarding the cause of this phenomena. During the preparation of this manuscript, Kettle et al. reported that H2O2 at high concentrations resulted in suicide inhibition of MPO by degradation of the heme linkage due to modification of methionine residues [33]. Interestingly, in that study HCMet243 was not one of the methionine groups oxidized by H2O2. Furthermore, it was confirmed that our ratio of MPO to H2O2 concentrations used here would represent Compound I (Fig. 1B) and validates our contention that the kinetic properties would represent a defined species in the MPO reaction mechanism. Initially, we wanted to ensure that assessment of any of the MPO inhibitors was performed at the pinnacle of the H2O2 dependency on the specificity constant for the enzyme (kcat/Km) for ADHP, as such the relationship between velocity of ADHP oxidation at various H2O2 concentrations was determined using the integrated Michaelis-Menten equation including H2O2 substrate inhibition driven by the modification of methionine residues by the H2O2. Our results indicated that H2O2 dependence of MPO activity was fit well to a model that included substrate inhibition step with an apparent inhibitory rate constant Ki of 10.8 nM of this H2O2 effect (Fig. 1B) with data fitting demonstrated a maximal velocity of 26 μM s−1. Interpretation of the significance of this Ki value is problematic and not pursued further as it may represent a collective effect resulting from oxidation of several MPO residues.
Michaelis-Menten Analysis of the ADHP Substrate by MPO-H2O2 System
To obtain the Michaelis-Menten parameters Km and kcat for Compound I (Eq. 1), we chose two independent methods. Initially, we studied the oxidation of ADHP using the injector functionality built-in to the VarioSkan Flash fluorescence plate reader. The auto-injector dispensed the H2O2 to initiate the reaction, as a means of generating a set of progress curves. Plots of initial velocities as a function of different concentrations of substrate ADHP are shown in Fig. 1C. Michaelis-Menten analysis for MPO-mediated oxidation of ADHP gave a Km of 31 ± 4 μM and the kcat of 186 ± 6 s−1.
Our second method for determination of these Michaelis-Menten parameters was to use ADHP in stopped flow fluorescence spectroscopy studies. This method involved collection of the full substrate depletion / progress curves for molar product formed (also known as resorufin) and simultaneous fitting to the integrated Michaelis-Menten equation (Eq. 7) using Scientist software (Fig. 1D). A residual plot stands for the goodness-of-fit of the model to the data (data not shown). The integrated Michaelis-Menten analysis determined the Km to be 39 ± 11 μM with a kcat of 224 ± 50 s−1. These two independent methods gave overlapping values and provided confidence as to initial values needed to begin analysis of the hydrazide analogs as MPO inhibitors. All errors are reported as 2 times standard deviations.
Characterization of ADHP conversion to resorufin by the MPO-H2O2 System
Initially, we sought to determine the behavior of the ADHP substrate as a reporter of Compound I (Eq. 1), specifically how reaction Vmax varied as a function of either altered substrate concentration (Fig. 1D) or varied enzyme concentration (Fig. 1E). Titrations demonstrate increasing reaction velocity over the range of ADHP substrate concentration assayed, namely 1, 2, 3, 10, 20, 40, 60, 70, and 80 μM ADHP with MPO at 23 nM and H2O2 at 22 μM (Fig. 1D). The enzyme dependency is shown in Fig. 1E with constant ADHP concentration of 40 μM with similar behavior exhibited. Observed rate constant (kobs) for both dependencies was calculated from single exponential function using the non-linear regression function built into GraphPad Prism 5.0. As expected, the kobs also increased over the experimental range of ADHP concentrations from 1 to 80 μM (Fig. 1F) and for the converse experiment holding substrate constant over 3 to 45 nM MPO (Fig. 1G). The apparent second order rate constant (Kappon) obtained from the slope of kobs against ADHP concentration Kappon was 2.1 ± 0.2 mM −1 s−1.
Lineweaver-Burk Analysis of 4-ABAH and NaN3-based Inhibition of the MPO-H2O2 System
To determine the mechanism by which 4-ABAH inhibits MPO; we initially attempted to generate the classic Lineweaver-Burk profile to determine competitive, noncompetitive and mixed-type inhibition for 4-ABAH. In general, multi-substrate systems are typically not amendable to this analysis [34], due presumably to the multiple velocities observed. The reaction of MPO with 4-ABAH has been reported to be irreversible [17] and as such permanently disabled and was not competitive in nature. Unlike reversible inhibitors these types of mechanisms of inactivation are time-dependent as formation of a covalent bond can be generally slow, so we further sought to determine the number of steps that could be observed using ADHP as a reporter.
To determine the mechanism by which NaN3, inhibits MPO, we initially wanted to confirm whether NaN3 was a reversible inhibitor in the absence of H2O2. ADHP oxidation was measured for a control reaction with NaN3 and before and after equilibrium dialysis (Fig. 2A). Results indicated that NaN3 binding to the MPO enzyme was itself reversible as near complete fluorogenic potential was recovered (see the difference between progress curves b and c in Fig. 2A). From there, we sought to conduct a full Lineweaver-Burk profile of NaN3 binding / inhibition of Compound I. Fig. 2B shows the profile produced via auto-injection of substrate H2O2 with a Varioskan Flash plate reader. The results of this experiment showed that NaN3 inhibits MPO by a competitive inhibition or mixed-type inhibition with ADHP (Fig. 2B). From this, kinetic parameters (kcat of 207 s−1 and Km of 28.8 μM) for MPO oxidation of ADHP were determined from curves in the absence of inhibitor. These values were identical to values determined initially determined (Fig. 1 and data not show). From interpretation of the Lineweaver-Burk plots, the addition of NaN3 (7.5 μM) decreased the apparent kcat to 161 s−1 and the apparent Km to 111 μM. This result was extended when 30 μM NaN3 was added further decreasing the kcat (108 s−1) and the apparent Km (167 μM). This represents a 11-fold reduction in the apparent specificity constant (kcat/Km) for the native system as compared to the highest concentration of NaN3 tested.
Global Analysis of 4-ABAH on Oxidation of the ADHP Substrate by the MPO-H2O2 system by Use of DynaFit
Modeling Software
Here we explored whether hydrazide analogs are indeed a slow-tight binding inhibitors of MPO. Initially, we analyzed sets of 4-ABAH-dependent inhibition of MPO activity and altered 4-ABAH under constant ADHP concentration and compiled them in the DynaFit software. Simultaneous modeling of these curves with initial Michaelis-Menten parameters determined for the simplified MPO-H2O2 system (Fig. 1) allowed us to discriminate between the one step model (Scheme I) and a two-step model (Scheme II) of slow binding inhibition. DynaFit results are shown in Table 1, where appropriateness of the model is assessed by automatic Akaike information criterion [35, 36]. The inhibitory efficiencies measured by DynaFit modeling for the inhibitory effects of 4-ABAH on MPO activity are shown in Table 2. Our results of NaN3 for the one-step model fit of slow binding inhibition can be found in Fig. 3A and results for the two step model of the slow binding inhibition mechanism for 4-ABAH are shown in Fig. 3B. The corresponding residual plots from Dynafit 3 are shown in their bottom panels, respectively.
Scheme I.
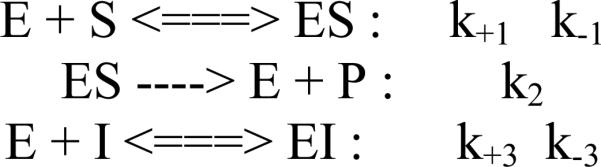
The one-step model tested.
Scheme II.
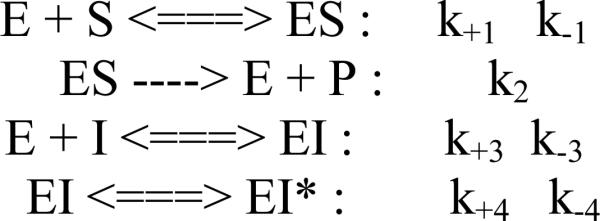
The two-step model tested.
Table 1.
Discrimination of MPO inhibition by Dynafit 3 analysis such that the lowest change in Akaike's information criterion (ΔAICc) and highest Akaike weight.
| Inhibitor | Model | ΔAICc | Weight |
|---|---|---|---|
| 2-ABAH | 1 step | 8.9 | 0.01127 |
| 2 step | 0 | 0.98873 | |
| 4-ABAH | 1 step | 6.9 | 0.03093 |
| 2 step | 0 | 0.96907 | |
| 3-DMABAH | 1 step | 0 | 0.88393 |
| 2 step | 4.1 | 0.11607 | |
| BAH | 1 step | 0 | 0.99685 |
| 2 step | 11.5 | 0.00315 | |
| 4-FBAH | 1 step | 0 | 0.9196 |
| 2 step | 4.9 | 0.0804 | |
| NaN3 | 1 step | 0 | 0.88144 |
| 2 step | 4.0 | 0.11856 | |
| 4-TFMBAH | 1 step | 0 | 0.90896 |
| 2 step | 4.6 | 0.09104 | |
| 4-NBAH | 1 step | 0 | 0.82051 |
| 2 step | 3.0 | 0.17949 | |
| Isoniazid | 1 step | 0 | 0.88569 |
| 2 step | 4.1 | 0.11431 |
Table 2.
Inhibitory efficiencies of hydrazine analogs on MPO activity, as measured in the MPO fluorescence stopped flow assay and simultaneous fit with either a 1 or 2-step slow tight-binding mechanism using DynaFit 3. Fixed constants k+1 = 10 μM−1s−1, k−1 = 80 s−1, k+2 = 224 s−1.
| Inhibitors | k+3 (μM−1s−1) | k−3 (s−1) | k+4 (s−1) | k−4 (s−1) | Ki (μM) | Ki* (μM) |
|---|---|---|---|---|---|---|
| 2-ABAH | 0.10 ± 0.0014 | 0.026 ± 0.0003 | 0.0067 ± 0.0004 | 0.0094 ± 0.002 | 0.26 | 0.16 |
| 4-ABAH | 0.13 ± 0.0007 | 0.031 ± 0.0001 | 0.038 ± 0.003 | 0.73 ± 0.06 | 0.24 | 0.23 |
| 3-DMABAH | 0.09 ± 0.0009 | 0.05 ± 0.0005 | - | - | 0.56 | - |
| BAH | 0.0098 ± 5.47E-05 | 0.0068 ± 9.5E-05 | - | - | 0.69 | - |
| 4-FBAH | 0.0075 ± 4.93E-05 | 0.0062 ± 0.0001 | - | - | 0.83 | - |
| NaN3 | 0.185 ± 0.0018 | 0.156 ± 0.001 | - | - | 0.84 | - |
| 4-TFMBAH | 0.0013 ± 1.36E-05 | 0.004 ± 0.0002 | - | - | 3.08 | - |
| 4-NBAH | 0.0027 ± 1.65E-05 | 0.01 ± 0.0001 | - | - | 3.70 | - |
| Isoniazid | 0.0011 ± 7.31E-06 | 0.011 ± 0.0001 | - | - | 10.00 | - |
Fig. 3. Comparison of Compound I inhibition by NaN3 and 4-ABAH using simultaneous reaction modeling.

A, Fluorescence progress curves presented in arbitrary units (au) for various indicated concentration of NaN3 were globally fit to a one-step slow-tight binding model detailed in Scheme I. Representative goodness-of-fit for the 30 μM ABAH with 15 μM ADHP reaction is shown as a residual plot. B, Similar plots for 4-ABAH analyzed by a two-step slow-tight binding model (Scheme II) for indicated concentrations of 4-ABAH to accommodate the biphasic phenotypic of the reaction. Representative goodness-of-fit for 0.64 μM ABAH with 16 μM ADHP are also shown. Titrations were performed and analyzed as described under “Experimental Procedures.”
Effect of 4-ABAH on Oxidation of the ADHP Substrate by MPO-H2O2 system
Due to the fact that individual progress curves for 4-ABAH can be modeled well using a simple double exponential expression, we hypothesized that 4-ABAH could be acting as a slow-tight binding irreversible inhibitor of MPO. To test this, we first had to confirm that 4-ABAH was an irreversible inhibitor of Compound I by equilibrium dialysis and reanalysis by stopped flow fluorescence spectroscopy. Our dialysis result showed no increase in activity post-dialysis indicating a permanent modification to the MPO active site (Fig. 4A and Fig. 4B). Phenotypically, there was loss of one step involved in 4-ABAH mediated inhibition of MPO, presumably due to pre-incubation of the reactants. It is worth noting that the presence of H2O2 is required for this irreversible inhibition of MPO by 4-ABAH, consistent with the literature [17].
Fig. 4. Reversibility of 4-ABAH-mediated MPO inhibition.

A, Stopped flow analysis of MPO (23 nM) and ABAH were mixed and incubated in assay buffer for 1 hr prior to triggering H2O2 addition. B, Exhaustive dialysis of identical mixtures into assay buffer followed addition with H2O2. Titrations were performed and analyzed as described under “Experimental Procedures.”
Comparison of Hydrazide Analogs and Their Effect on Oxidation of ADHP by the MPO-H2O2 system
We sought to compare the inhibition of Compound I by hydrazide-containing compounds with various electron withdrawing and electron donating groups. The IC50s for different inhibitors using the half maximum of ADHP oxidation are shown along with their respective structures in Table 3. For comparison, kinetic parameters were also determined for isoniazid and NaN3. Akaike information criterion based discrimination analysis is shown in Table 1, with 2-ABAH fitting best with a two-step slow binding inhibitor K *i of 0.16 μM, which has a similar effect as 4-ABAH (Fig. 3B). 3-DMABAH, NaN3, 4-TFMBAH, and isoniazid are all best represented by a one-step slow binding inhibition model as shown in Table 2 with Ki of 0.56 μM, 0.84 μM, 3.08 μM, 10 μM, respectively. These results indicate that 2-ABAH is a 62-fold higher affinity inhibitor of Compound I as compared to isoniazid under the conditions tested. To evaluate this phenomenon further, the relationship between the overall inhibitory rate constants Ki for the meta- and para-substituted compounds with respect to the appropriate Hammett coefficients (σ) were examined as shown in Fig. 5A. It is linear between the σ value and its corresponding Ki. Further examination of the dependency of the σ value with respect to the phenomenological IC50 values showed a linear but less precise relationship (Fig. 5B). There was weak linear correlation between the inhibitor constants and the hydrophobicity of the substituent under our conditions as shown in Fig. 5C.
Table 3.
Profile of IC50 values for MPO inhibition. Inhibition of MPO activity (%) was determined from progress curves for resorufin generation by mixtures of MPO (23 nM) with ADHP (40 μM) initiated with H2O2 (22 μM) in the presence of varied concentrations of 2-ABAH, 4-ABAH, 3-DMABM, NaN3, 4-FBAH, BAH, 4-NBAH, 4-TFMBH and Isoniazid. Columns presented are abbreviate compound names (inhibitor), compound structure with varied R groups shown in red, and 50% inhibitory concentration with standard error of the mean (IC50 ± SEM).
| Inhibitor | Structure | IC50 ± SEM (μM) |
|---|---|---|
| 2-ABAH |
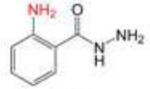
|
0.50 ± 0.03 |
| 4-ABAH |
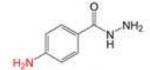
|
0.71 ± 0.01 |
| 3-DMABAH |
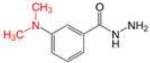
|
1.27 ± 0.07 |
| NaN3 |
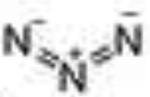
|
6.57 ± 0.32 |
| 4-FBAH |
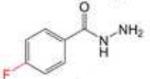
|
22.45 ± 2.19 |
| BAH |
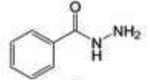
|
40.83 ± 4.00 |
| 4-NBAH |
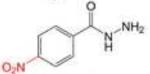
|
54.62 ± 14.06 |
| 4-TFMBAH |
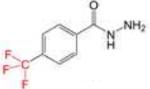
|
142.70 ± 10.98 |
| Isoniazid |
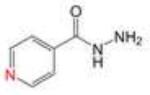
|
277.10 ± 6.65 |
Fig. 5. Linear free-energy relationship of substituted benzoic acid hydrazide compounds tested on inactivation of Compound I.

A, Correlation of the log of overall inhibitory rate constant (Ki) for the compounds tested with their respective Hammett's constants (σ). A positive value of σ indicates electron-withdrawing group and a negative value an electron-donating group. B, Correlation of the log of the half maximal inhibitory concentration (IC50) versus σ. C, Correlation of Log(Ki) versus hydrophobicity parameters. Titrations were performed and analyzed as described under “Experimental Procedures.”
Benzoic acid hydrazide and analog mediated cleavage of MPO
It has been reported that the heme group linked to the MPO active site can be degraded by H2O2 when it was added to MPO at a 1000:1 ratio and incubated for 2 hrs [33]. As is shown in Fig. 6A, the light chain of MPO is cleaved from MPO as we increase the concentration of BAH because the heavy chain (58.5 kDa) and light chain (14.5kDa) are linked via their covalent bound with the heme prosthetic group [33]. MPO is a dimer of heterodimers (146 kDa) consists of two identical glycosylated heavy and two light chains with the heavy chains linked by a single disulfide bond. Interestingly, once cleavage of the heme ring occurs the iron containing active site heme migrates with the light chain as evident by its ability to oxidize the chemiluminescent substrate (Fig. 6B). Examination of the concentration dependence of this cleavage also shows a progressive loss of one and then both of the MPO light chains converting the MPO heavy chain into a specific product without peroxidase potential. To determine whether this observation was an isolated BAH phenomena, we tested in the same manner 4-TMFBAH, which showed a similar results, but with slightly altered concentration dependency (Supplementary Fig. 1). These results indicate that the heme moiety was not degraded and remained intact with the Fe remaining coordinated in such a way as to still allow for peroxidase activity.
Fig. 6. Benzoic acid hydrazide-mediated cleavage of MPO active site heme.

A-B, MPO (1.2 μM), ADHP (40 μM) with different concentrations of BAH inhibitor (0, 0.025, 0.25, 2.5, 12.5 and 25 mM) were incubated with H2O2 (20 μM) at room temperature for 10 min. Samples of 20 μL were added to non-reducing sample loading buffers, and then loaded without prior heating and resolved by 4-15% gradient SDS-polyacrylamide gel electrophoresis. Concentration dependence of BAH-mediated cleavage of heme moiety freeing the MPO light chain shown by SDS gel for protein (panel A) and by chemiluminescence (panel B). Note the progressive oxidative cleavage of the MPO protein marked by loss of one and then both of the MPO light chains (~14kD). The active site heme is attach to light chain as evident by the chemiluminescence band.
Analysis of Hydrazide induced Cleavage product by Mass Spectroscopy
We were interested in determining the exact mass of the fragments of MPO generated in reaction mixtures of MPO, BAH and H2O2. To address these we used parallel techniques of MALDI-TOF and tandem liquid chromatography and electrospray mass spectroscopy. Our results are shown in Fig. 7, where a reference active site of MPO and linkage between ester of the hydroxylated methyl groups with (LCAsp94) and a distal HCGlu242 are shown by the blue arrows in Panel A and the MALDI-TOF data is shown in Panel B. For clarity, the heme group in MPO is a protoporphyrin IX group or also referred to as heme b, which is comprised of a tetra-pyrrole structure with rings denoted as A, B, C and D (Fig. 7A). Although we noted peaks associated with the MPO dimer representing MPO (HC-heme b-LC), monomer of MPO (HC-heme b-LC), and cleavage products, we honed in on peaks of interest critical for our question coinciding with the range between 8,000 to 20,000 Da (Fig. 7B). Our results indicate peaks in this range with m/z 13,056.1 Da, which is converted overtime into a m/z 12,407.1 Da peak representing a mass difference of 649.00 Da (data not shown regarding time course). The calculated average mass of heme b (C34H32FeN4O4) is 616.48 Da and the addition of 2 oxygen molecules would render a theoretical mass nearly identical to the results we obtained (C34H32FeN4O6; 648.49 Da). The appearance of the second cleavage product is important as it also explains why we also saw diminished chemiluminescence for the peroxidase activity in band roughly correlating to the LC-heme b reaction product at high concentration of the BAH (data not shown) and for 4-TFMBAH (Supplementary Fig. 1). It should be emphasized that reactions without BAH or with high level of H2O2 compared to MPO (1000 to 1 molar ratio) did not result in these ester cleavage products. Presumably, hydrolysis of the second ester is also by a nucleophilic acyl substitution to generate a free form of heme b with any modifications made to the rings by these cleavage reactions and as it turns out in our gel experiments it results in a LC band devoid of catalytic potential. As a confirmation that this result was not artifact of the MALDI-TOF technique and preparation, we performed also a parallel liquid chromatograph and electrospray mass spectroscopy experiment and that showed a parent monomer MPO (i.e. LCHeme b-HC) peak at m/z 77,368 Da and a cleaved HC corresponding to the m/z 64,470 Da peak. This results in difference of 12,898 Da, which is in good agreement the value of seen for the electrospray mass of the LC-heme b intermediate of m/z 13,049.5 Da.
Fig. 7. Sequential hydrolysis of ester bonds results in cleavage of the catalytic heme from MPO active site.

A, Structure and nomenclature of the tetrapyrrole ring of the heme in the MPO active site. Figure adapted from (Kooter IM, 1999). B, Maldi-TOF mass spectroscopy analysis of reactions under conditions that maximize light chain-heme product formation, namely MPO (1.2 μM) with BAH (2.5 mM) pre-incubated for 10 min prior to the addition of H2O2 (20 μM). These reactions are identical to those in lane 6 in Fig. 6. C, Spectroscopic analysis of MPO heme signature following addition of BAH. Curves represent MPO (black line), MPO with hydrazide (dotted), MPO with hydrazide post H2O2 addition after 10 min (dashed) and again after 1hr (dot-dashed). D, Schematic representation of the release of the active site heme from MPO by reaction with BAH and H2O2. MPO subunits are shown as light chain (red), heme (green) and heavy chain (gray).
Analysis of the Spectral Changes that occur during MPO based hydrazide inhibition
Mechanistic studies on the interaction of MPO with potential active site ligands have been based primarily, in recent years, on measurements of spectral changes that accompany formation of reactive heme intermediates (Eq. 1-5) during the myeloperoxidase catalytic cycle [17, 19, 20, 23, 37]. The spectral signature displays the classic Soret peak and four lower energy Q bands (Fig. 7C). One of there bands is formed by the sulfonium linkage that heme makes with HCMet243. Of note, Kettle et al. recently reported that the HCMet243 was not one of the methionine groups oxidized by high concentrations of H2O2 leading to MPO inactivation [33]. Here, we sought to identify whether the sulfonium linkage is maintained in the BAH-mediated cleavage products or whether this unique bond is lost. We noted a blue shift of the 430 nm Soret peak to 418 nm peak when H2O2 was added. There was also loss of both the α (570nm) and CT2 (690 nm) peaks (Fig. 7C). Collectively, these results indicate that the sulfonium linkage with HCMet243 was lost with incubation of MPO with BAH and H2O2.
DISCUSSION
Mechanistic studies on the interaction of MPO with potential active site ligands have been based primarily, in recent years, on measurements of spectral changes that accompany formation of reactive heme intermediates (Eq. 1-3) during the classic peroxidase catalytic cycle [17, 19, 20, 23, 37]. This is a powerful technique that allows correlates of various agents or conditions on the formation of this short-lived Compound I and conversion to Compound II defined by Equation 1 & 2, respectively. Other studies have sought to characterize the potential role of the chlorination cycle (Eq. 4) and its interplay with the peroxidase activity of enzyme and the potential added complexity of superoxide intermediate reactions (Eq. 5) and the superoxide dismutase activity of the MPO active site [38, 39]. Here we sought initially to provide a reliable platform to compare MPO inhibitors in the broadest sense without introducing questions of experimental bias (i.e. comparisons of potency across platforms). Furthermore we wanted to utilize a system or assay that was amendable to future high through put screening efforts. We chose here to study the fluorogenic peroxidase substrate ADHP [40, 41] as it has properties amendable to this goal. Furthermore, we wanted to study this substrate under conditions that maximize MPO function (i.e. pH 5.6) and in the absence of Cl− ions, thereby eliminating the 2-electron transfer in Equation 5. Using this strategy, we determined the Michaelis-Menten parameters for MPO oxidation of ADHP (Fig. 1). Once we confirmed the reliability of this new method and appropriate substrate / enzyme relationship occurred, we tackled the problem of examining the classic MPO inhibitors like NaN3 (Fig. 2 & 3) and 4-ABAH (Fig. 3 & 4). Keeping in mind that we were focused on a systematic approach to examining these inhibitors, we collected transient kinetic datasets for each inhibitor (Table 3) and then batch-wise analyzed them with DynaFit modeling software to assure consistent handling of the data. We were able to validate the obtained Ki and Ki* values by these methods and compare them to the original phenotypic IC50 values (Table 3). From our results, we determine that both 2-ABAH and 4-ABAH were the most potent MPO inhibitors causing their action through a slow-tight binding mechanism with respective Ki* formation rates of 0.16 and 0.23 μM, which is in good agreement with the respective observed IC50 values of 0.71 and 0.50 μM (Table 2). Furthermore 2-ABAH and 4-ABAH inhibited by a two-step mechanism as opposed to the other BAH and NaN3 inhibitors tested (Table 1) suggesting that the mere presence of the amino group at either position on the BAH would be favorable; leading to this second step interaction.
A simple yet powerful benefit to the use of ADHP was our ability to discriminate between covalent and non-covalent interaction of the inhibitors with MPO in the absence of H2O2 providing direct insight into the overall mechanism of action of a given compound. We examined the reversibility of the compound inhibitor by dialyzing mixes of the MPO and, for this experiment, either with NaN3 shown in Fig. 2 and 4-ABAH in shown in Fig. 4. It is established that NaN3 inhibits peroxidases by modification to the heme ring through formation of a δ-meso-azidoheme adduct rather than direct interaction of the azide with the chelated Fe molecule [42]. In MPO, the A, B, C and D rings are bridged by four faces referred to as the α-heme edge between ring A and B, β-heme edge between ring B and C, γ-heme edge between ring C and D, and δ-heme edge between ring D and back to the A ring. The δ-meso-azidoheme adduct at this site is a result of solvent accessibility of the substrate to the distal heme cavity on peroxidases [43]. Our studies here (Fig. 2A) confirm that this inhibition of MPO by NaN3 is reversible and the interaction of 4-ABAH with MPO was covalent, both under conditions lacking H2O2. Furthermore, one step we observed for 4-ABAH inhibition of Compound I corresponds presumably to the slow binding step or covalent modification of the MPO protein is lost upon exhaustive dialysis (Fig. 4). It should be emphasized that if modification of MPO active site were competitive or non-covalent there would be release of the weaker binding compound overtime by this equilibrium dialysis, essentially returning the MPO to its native state. Interestingly, this step of potential high affinity interaction or covalent modification is markedly absent in the other compounds tested with the exception of 2-ABAH, indicating that a like mechanism may occur.
Our report is not the first to study inhibition of peroxidases by hydrazide containing compounds as it has been described for MPO and others peroxidase, such as horseradish peroxidase by alkyl hydrazine [44, 45] and phenylhydrazine [44, 45]; not to mention, microperoxidase-11 by 4-ABAH [46]. To evaluate this phenomenon further, we also wanted to validate that ADHP was accurately reporting on catalytic events within the active site. To do this we examined the relationship between the overall inhibitory rate constants, namely Ki, for the meta- and para-substituted compounds used here with respect to the appropriate Hammett coefficients (σ) (Fig. 5A). Given this linear relationship of this plot and high degree of correlation, it is likely that the same general mechanism is operating throughout the series [47]. Keeping in mind that a positive value of σ indicates an electron-withdrawing group and a negative value an electron-donating group, so –CF3 substitution would have an overall withdrawing function with a σ value of + 0.54 and be contrasted by the –NH2 substitution that carries a σ value of -0.66. Further examination of the dependency of the σ value with respect to the phenomenological IC50 values (Fig. 5B) showed a similar but less precise relationship was indicated. Under our conditions there was extremely weak correlation with between the inhibitor constants and the hydrophobicity of the substituent added (Fig. 5C). It should be noted that due to limitations of the Hammett's coefficient interpretations, the 2-ABAH could not be examined in this way due to the ortho-effects resulting in potential interference between the moieties [47]. Taken together, there was clear evidence that use of ADHP oxidation is a powerful approach to study inhibitory aspects related to targeting MPO function and our handling of the batch data was appropriate given the high correlation of the Hammett plots (Fig. 5A).
Spurred on by our findings that there was two distinct observable kinetic events occurring in 4-ABAH-mediated inhibition of Compound I, we decided to better understand the nature of inhibition for the simple un-substituted BAH, as a means to ascribe one step to a physical event. Our initial hypothesis was that the heme b was being destroyed as was reported for potential carbene Fe-porphyrin complex described for cytochrome P450 [48] and more recently for H2O2-oxidative inhibition of MPO [33]. It is speculated that a phenol radical would add to the heme and cause a rearrangement that would free the Fe molecule, similar to the inactivation of myoglobin with phenylhydrazine. Contrary to this, we observed no evidence that Fe was released and that the heme b group was being destroyed. Moreover, Fig. 6 showed convincingly that there is a LC-heme b band that remained active towards the luminol-based chemiluminescence substrate. This cleavage event could partially explain the problem in obtaining a structure of MPO with BAH, given BAH-mediated inhibition showed a release of heme b group, adding significant disorder to the forming unit cell due to a conversion of the covalent linked ester into an equilibrium interaction. One simple analogy would be that overtime the BAH essentially promotes ejection of the heme b payload from the peroxidase into the surrounding environment. To prove that the LC-heme b inhibition product was indeed formed, we setup two parallel and independent experiments to determine the actual mass of the LC-heme b reaction product. To confirm that this ester hydrolysis mechanism was broadly used by other BAH analog we also showed that 4-TFMBAH was capable of generating the similar reaction products (Supplementary Fig. 1), though at a slightly altered concentration profile suggesting that there maybe potential in the future to modulate hydrolysis of the MPO ester bonds based on chemical properties of the modifications to the BAH platform. Our UV-Vis spectroscopic data (Fig. 7C) also demonstrated the loss of the sulfonium ion linkage between HCMet243 and the pyrrole A ring, which would be necessary to have the LC-heme b moiety release from the HC of MPO.
It should be emphasized that simplification of the overall number of parameters has aided in the overall evaluation of these inhibitors tested here and conditions will be examine in the future as layers of complexity are added (e.g. increasing the pH to more neutral conditions and addition of Cl− ions to the system). Studies will also be necessary to expand the structure and activity relationship to analogous compounds to those tested to gain a greater understanding of the constraints of the MPO active site environment and the chemical influence analogous substitution have on MPO ester hydrolysis in the presence of H2O2. It is our contention that application of this new methodology (i.e. use of ADHP) will aid in rapid assessment of the MPO peroxidase function making high through put screening possible and leading to rationale design application of inhibitors targeting MPO specifically. Furthermore, our observation here that BAH promotes the release of the heme b from the active site of MPO through cleavage of the ester linkages is novel and may promote a better understanding of the mechanism of action of this widely used inhibitor.
Supplementary Material
Supplemental Fig. 1. 4-(trifluoromethyl) benzoic acid hydrazide-mediated cleavage of MPO active site heme. A-B, MPO (1.2 μM), ADHP (40 μM) with different concentrations of 4-TFMBAH inhibitor (0, 0.025, 0.25, 2.5, 12.5 and 25 mM) were incubated with H2O2 (20 μM) at room temperature for 10 min. Samples of 20 μL were added to non-reducing sample loading buffers, and then loaded without prior heating and resolved by 4-15% gradient SDS-polyacrylamide gel electrophoresis. Concentration dependence of 4-TFMBAH-mediated cleavage of heme moiety freeing the MPO light chain shown by SDS gel for protein (panel A) and by chemiluminescence (panel B).
Highlights.
ADHP fluorogenic substrate as a reporter of rapid MPO-H2O2 reaction kinetics
Comparative analysis of MPO-H2O2 inhibition by benzoic acid hydrazide analogs
MPO-H2O2 inhibition by 2- or 4- amino benzoic acid hydrazide occurs in 2 steps
Benzoic acid hydrazide inhibits MPO by ejecting of heme by ordered ester cleavage
Benzoic acid hydrazide inhibition of MPO-H2O2 does not require iron release
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Yonnie Wu of the Auburn University Mass Spectroscopy facility in the Department of Chemistry and Biochemistry for collection of the MALDI-TOF and electrospray mass spectrometric data. The authors would also like to thank Dr. Doug Goodwin for helpful suggestions.
The abbreviations used are
- MPO
myeloperoxidase
- LC
light chain of myeloperoxidase
- HC
heavy chain of myeloperoxidase
- ADHP
10-acetyl-3, 7-dihydroxyphenoxazine
- ABAH
aminobenzoic acid hydrazide, 2-ABAH, 2-aminobenzoic acid hydrazide
- 4-ABAH
4-aminobenzoic acid hydrazide
- BAH
benzoic acid hydrazide
- 4-FBAH
4-fluorobenzoic acid hydrazide
- 4-NBAH
4-nitrobenzoic acid hydrazide
- 4-TFMBAH
4-(trifluoromethyl) benzoic acid hydrazide
- 3-DMABAH
3-(dimethylamino) benzoic acid hydrazide
- HOCl
hypochlorous acid
- H2O2
hydrogen peroxide
- TMB
3,3’, 5,5’-tetramethylbenzidine
- MALDI-TOF
Matrix-assisted laser desorption/ionization time-of-flight
- au
arbitrary units
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supported by the National Institutes of Health through grants provided by the National Heart, Lung, and Blood Institute, R00HL094533 (to P.P.) & R01HL114477 (to P.P.), and the National Institute of Allergy and Infectious Diseases grant 2R44AI085840-02 (to P.P.).
REFERENCES
- 1.Fiedler TJ, Davey CA, Fenna RE. J. Biol. Chem. 2000;275:11964–11971. doi: 10.1074/jbc.275.16.11964. [DOI] [PubMed] [Google Scholar]
- 2.Andrews PC, Krinsky NI. Anal. Biochem. 1982;127:346–350. doi: 10.1016/0003-2697(82)90185-3. [DOI] [PubMed] [Google Scholar]
- 3.Andrews PC, Krinsky NI. J. Biol. Chem. 1982;257:13240–13245. [PubMed] [Google Scholar]
- 4.Harrison JE, Araiso T, Palcic MM, Dunford HB. Biochem. Biophys. Res. Commun. 1980;94:34–40. doi: 10.1016/s0006-291x(80)80183-5. [DOI] [PubMed] [Google Scholar]
- 5.Everse J. Free Radic. Biol. Med. 1998;24:1338–1346. doi: 10.1016/s0891-5849(97)00451-6. [DOI] [PubMed] [Google Scholar]
- 6.Kettle AJ, van Dalen CJ, Winterbourn CC. Redox Rep. 1997;3:257–258. doi: 10.1080/13510002.1997.11747120. [DOI] [PubMed] [Google Scholar]
- 7.Fliss H. Mol. Cell. Biochem. 1988;84:177–188. doi: 10.1007/BF00421053. [DOI] [PubMed] [Google Scholar]
- 8.Furtmuller PG, Zederbauer M, Jantschko W, Helm J, Bogner M, Jakopitsch C, Obinger C. Arch. Biochem. Biophys. 2006;445:199–213. doi: 10.1016/j.abb.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 9.Kooter IM, Koehler BP, Moguilevsky N, Bollen A, Wever R, Johnson MK. J. Biol. Inorg. Chem. 1999;4:684–691. doi: 10.1007/s007750050340. [DOI] [PubMed] [Google Scholar]
- 10.Kooter IM, Moguilevsky N, Bollen A, Sijtsema NM, Otto C, Dekker HL, Wever R. Eur. J. Biochem. 1999;264:211–217. doi: 10.1046/j.1432-1327.1999.00606.x. [DOI] [PubMed] [Google Scholar]
- 11.Andrews PC, Krinsky NI. J. Biol. Chem. 1981;256:4211–4218. [PubMed] [Google Scholar]
- 12.Wever R, Plat H. Biochim. Biophys. Acta. 1981;661:235–239. doi: 10.1016/0005-2744(81)90009-7. [DOI] [PubMed] [Google Scholar]
- 13.Vissers MC, Winterbourn CC. Biochem. J. 1987;245:277–280. doi: 10.1042/bj2450277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hampton MB, Kettle AJ, Winterbourn CC. Infect. Immun. 1996;64:3512–3517. doi: 10.1128/iai.64.9.3512-3517.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikeda-Saito M, Shelley DA, Lu L, Booth KS, Caughey WS, Kimura S. J. Biol. Chem. 1991;266:3611–3616. [PubMed] [Google Scholar]
- 16.Forbes LV, Sjogren T, Auchere F, Jenkins DW, Thong B, Laughton D, Hemsley P, Pairaudeau G, Turner R, Eriksson H, Unitt JF, Kettle AJ. J. Biol. Chem. 2013 doi: 10.1074/jbc.M113.507756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kettle AJ, Gedye CA, Winterbourn CC. Biochem. J. 1997;321(Pt 2):503–508. doi: 10.1042/bj3210503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forbes LV, Furtmuller PG, Khalilova I, Turner R, Obinger C, Kettle AJ. Biochem. Pharmacol. 2012;84:949–960. doi: 10.1016/j.bcp.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 19.Galijasevic S, Abdulhamid I, Abu-Soud HM. Biochemistry. 2008;47:2668–2677. doi: 10.1021/bi702016q. [DOI] [PubMed] [Google Scholar]
- 20.Vaz SM, Augusto O. Proc. Natl. Acad. Sci. USA. 2008;105:8191–8196. doi: 10.1073/pnas.0708211105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bozeman PM, Learn DB, Thomas EL. Biochem. Pharmacol. 1992;44:553–563. doi: 10.1016/0006-2952(92)90449-s. [DOI] [PubMed] [Google Scholar]
- 22.Malle E, Furtmuller PG, Sattler W, Obinger C. Br. J. Pharmacol. 2007;152:838–854. doi: 10.1038/sj.bjp.0707358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tiden AK, Sjogren T, Svensson M, Bernlind A, Senthilmohan R, Auchere F, Norman H, Markgren PO, Gustavsson S, Schmidt S, Lundquist S, Forbes LV, Magon NJ, Paton LN, Jameson GN, Eriksson H, Kettle AJ. J. Biol. Chem. 2011;286:37578–37589. doi: 10.1074/jbc.M111.266981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorris HH, Walt DR. J. Am. Chem. Soc. 2009;131:6277–6282. doi: 10.1021/ja9008858. [DOI] [PubMed] [Google Scholar]
- 25.Nelson DP, Kiesow LA. Anal. Biochem. 1972;49:474–478. doi: 10.1016/0003-2697(72)90451-4. [DOI] [PubMed] [Google Scholar]
- 26.Kuzmic P. Anal. Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 27.Taylor KL, Strobel F, Yue KT, Ram P, Pohl J, Woods AS, Kinkade JM., Jr. Arch. Biochem. Biophys. 1995;316:635–642. doi: 10.1006/abbi.1995.1083. [DOI] [PubMed] [Google Scholar]
- 28.Taylor KL, Pohl J, Kinkade JM., Jr. J. Biol. Chem. 1992;267:25282–25288. [PubMed] [Google Scholar]
- 29.Nahrendorf M, Sosnovik D, Chen JW, Panizzi P, Figueiredo JL, Aikawa E, Libby P, Swirski FK, Weissleder R. Circulation. 2008;117:1153–1160. doi: 10.1161/CIRCULATIONAHA.107.756510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swirski FK, Wildgruber M, Ueno T, Figueiredo JL, Panizzi P, Iwamoto Y, Zhang E, Stone JR, Rodriguez E, Chen JW, Pittet MJ, Weissleder R, Nahrendorf M. J. Clin. Invest. 2010;120:2627–2634. doi: 10.1172/JCI42304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su HS, Nahrendorf M, Panizzi P, Breckwoldt MO, Rodriguez E, Iwamoto Y, Aikawa E, Weissleder R, Chen JW. Radiology. 2012;262:181–190. doi: 10.1148/radiol.11110040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cortez-Retamozo V, Etzrodt M, Newton A, Ryan R, Pucci F, Sio SW, Kuswanto W, Rauch PJ, Chudnovskiy A, Iwamoto Y, Kohler R, Marinelli B, Gorbatov R, Wojtkiewicz G, Panizzi P, Mino-Kenudson M, Forghani R, Figueiredo JL, Chen JW, Xavier R, Swirski FK, Nahrendorf M, Weissleder R, Pittet MJ. Immunity. 2013;38:296–308. doi: 10.1016/j.immuni.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paumann-Page M, Furtmuller PG, Hofbauer S, Paton LN, Obinger C, Kettle AJ. Arch. Biochem. Biophys. 2013;539:51–62. doi: 10.1016/j.abb.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marquez LA, Dunford HB. J. Biol. Chem. 1995;270:30434–30440. doi: 10.1074/jbc.270.51.30434. [DOI] [PubMed] [Google Scholar]
- 35.Wagenmakers EJ, Farrell S. Psychon. Bull Rev. 2004;11:192–196. doi: 10.3758/bf03206482. [DOI] [PubMed] [Google Scholar]
- 36.Burnham KP, Anderson DR. Model selection and Multimodel inference: a practical information-theroretic approach. second edition Springer; 2002. [Google Scholar]
- 37.Chapman AL, Mocatta TJ, Shiva S, Seidel A, Chen B, Khalilova I, Paumann-Page ME, Jameson GN, Winterbourn CC, Kettle AJ. J. Biol. Chem. 2013;288:6465–6477. doi: 10.1074/jbc.M112.418970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winterbourn CC, Kettle AJ. Jpn. J. Infect. Dis. 2004;57:S31–33. [PubMed] [Google Scholar]
- 39.Winterbourn CC, Hampton MB, Livesey JH, Kettle AJ. J. Biol. Chem. 2006;281:39860–39869. doi: 10.1074/jbc.M605898200. [DOI] [PubMed] [Google Scholar]
- 40.Meng Y, High K, Antonello J, Washabaugh MW, Zhao Q. Anal. Biochem. 2005;345:227–236. doi: 10.1016/j.ab.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 41.Pulli B, Ali M, Forghani R, Schob S, Hsieh KL, Wojtkiewicz G, Linnoila JJ, Chen JW. PloS one. 2013;8:e67976. doi: 10.1371/journal.pone.0067976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilks A, Black SM, Miller WL, Ortiz de Montellano PR. Biochemistry. 1995;34:4421–4427. doi: 10.1021/bi00013a034. [DOI] [PubMed] [Google Scholar]
- 43.Sharma S, Singh AK, Kaushik S, Sinha M, Singh RP, Sharma P, Sirohi H, Kaur P, Singh TP. Int. J. Biochem. Mol. Biol. 2013;4:108–128. [PMC free article] [PubMed] [Google Scholar]
- 44.Ator MA, David SK, Ortiz de Montellano PR. J. Biol. Chem. 1987;262:14954–14960. [PubMed] [Google Scholar]
- 45.Ator MA, Ortiz de Montellano PR. J. Biol. Chem. 1987;262:1542–1551. [PubMed] [Google Scholar]
- 46.Arvadia P, Narwaley M, Whittal RM, Siraki AG. Arch. Biochem. Biophys. 2011;515:120–126. doi: 10.1016/j.abb.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 47.Smith MB, March J. March Advanced Organic Chemistry: Reaction, Mechanisms, and Structure. Sixth edition John Wiley & Sons, Inc.; 2006. [Google Scholar]
- 48.Lange M, Mansuy D. Tetrahedron Lett. 1981;22:2561–2564. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. 4-(trifluoromethyl) benzoic acid hydrazide-mediated cleavage of MPO active site heme. A-B, MPO (1.2 μM), ADHP (40 μM) with different concentrations of 4-TFMBAH inhibitor (0, 0.025, 0.25, 2.5, 12.5 and 25 mM) were incubated with H2O2 (20 μM) at room temperature for 10 min. Samples of 20 μL were added to non-reducing sample loading buffers, and then loaded without prior heating and resolved by 4-15% gradient SDS-polyacrylamide gel electrophoresis. Concentration dependence of 4-TFMBAH-mediated cleavage of heme moiety freeing the MPO light chain shown by SDS gel for protein (panel A) and by chemiluminescence (panel B).