

Abstract
The field of metabolomics has witnessed an exponential growth in the last decade driven by important applications spanning a wide range of areas in the basic and life sciences and beyond. Mass spectrometry in combination with chromatography and nuclear magnetic resonance are the two major analytical avenues for the analysis of metabolic species in complex biological mixtures. Owing to its inherent significantly higher sensitivity and fast data acquisition, MS plays an increasingly dominant role in the metabolomics field. Propelled by the need to develop simple methods to diagnose and manage the numerous and widespread human diseases, mass spectrometry has witnessed tremendous growth with advances in instrumentation, experimental methods, software, and databases. In response, the metabolomics field has moved far beyond qualitative methods and simple pattern recognition approaches to a range of global and targeted quantitative approaches that are now routinely used and provide reliable data, which instill greater confidence in the derived inferences. Powerful isotope labeling and tracing methods have become very popular. The newly emerging ambient ionization techniques such as desorption ionization and rapid evaporative ionization have allowed direct MS analysis in real time, as well as new MS imaging approaches. While the MS-based metabolomics has provided insights into metabolic pathways and fluxes, and metabolite biomarkers associated with numerous diseases, the increasing realization of the extremely high complexity of biological mixtures underscores numerous challenges including unknown metabolite identification, biomarker validation, and interlaboratory reproducibility that need to be dealt with for realization of the full potential of MS-based metabolomics. This chapter provides a glimpse at the current status of the mass spectrometry-based metabolomics field highlighting the opportunities and challenges.
Keywords: Mass spectrometry, Ionization methods, Quantitative metabolomics, Mass analyzers, Ambient ionization, MS-imaging, Chromatography, Capillary electrophoresis
1 Brief History of Metabolic Profiling
Although metabolic profiling or metabolomics is considered a relatively new field in systems biology, the first reports of metabolic studies can be traced to the ancient China, where ants were used to detect diabetes by evaluating the levels of glucose in urine samples [1]. “Urine charts” correlating smell, taste, and color of urine were employed in the Middle Ages to diagnose various medical conditions that are metabolic in origin [2]. The idea that individuals might have a distinctive “metabolic pattern” that could be “fingerprinted” by studying their biological fluids was proposed and tested by Roger Williams and his co-workers in late 1940s [3]. They utilized paper chromatography to determine that metabolic patterns significantly varied among different subjects, but were relatively constant for a given individual. Their studies of a variety of subjects including alcoholics and schizophrenics produced the evidence that each of these groups has a particular metabolic pattern. The technological advances in gas chromatography (GC), liquid chromatography (LC), and mass spectrometry (MS) in the 1960s and 1970s allowed quantitative metabolic profiling studies. Horning and co-workers in 1971 successfully used GC-MS to measure metabolites in human urine and tissue extracts [4, 5]. Horning, along with Pauling and Robinson and their research groups, led the development of GC-MS-based techniques for the metabolic measurements in biological fluids through the 1970s to early 1980s [6]. Later, developments of high-resolution/sensitivity MS and NMR instrumentation in combination with multivariate statistical analysis have allowed metabolomics to become a fast-growing field in system biology over the past decade.
Metabolomics is having major impacts on numerous disciplines including the life, food and plant sciences, drug development, toxicology, environmental science, and medicine. Metabolites, being the downstream products of cellular function, represent a sensitive measure of the actions of upstream molecular species such as genes, transcripts, and enzymes, including the effects of disease, drugs, toxicity, and the environment. Hence, identification and quantitative analysis of metabolites in humans and animal and cell models of numerous human diseases offer avenues for understanding, diagnosing, and managing human diseases; assessing disease risk factors associated with drugs, toxins, and the environment; and ultimately developing personalized treatment options.
2 Analytical Techniques
The analytical tools of choice for small-molecule analysis in metabolomics are mass spectrometry (MS) and nuclear magnetic resonance (NMR). MS and NMR methods are both supplementary and complementary to one another. Numerous techniques within MS and NMR offer multifaceted approaches to detect and identify a variety of metabolites, and measure their concentrations accurately. MS is intrinsically a highly sensitive method for detection, quantitation, and structure elucidation of upwards of several hundred metabolites in a single measurement. The sensitivity and accuracy of detection by MS are dependent on the nature of the experimental conditions and the instrumental settings; major factors that contribute include the nature of metabolite extraction, separation, ionization (and possibly ion suppression), and detection approaches. Because of the complexity of biological matrices, it is often necessary to separate metabolites of interest prior to MS acquisition. Thus, hyphenated analytical techniques combining separation technology with MS have become highly effective tools for small-molecule analysis. The main chromatography methods that are typically coupled with MS are high-performance liquid chromatography (HPLC), gas chromatography (GS), and capillary electrophoresis (CE). In the last decade or so, each of these approaches has seen tremendous growth. Such advances combined with the development of new software packages and databases now enable quantitative analysis of several hundreds of metabolites in automation mode.
Among the three separation methods, LC and GC methods are most widely used, while CE is gaining increased interest in the field [7]. GC-MS achieves better metabolite separation than LC and generally avoids ion suppression, due to its use of the gaseous phase and the nature of its MS ionization. However, unlike LC, GC typically requires chemical derivatization of the metabolic species prior to the GC-MS analysis [8–10]. LC-MS has seen a major uptake in the field as it detects a larger pool of intact metabolites with no need for chemical modification. The traditional reverse-phase chromatography is used in the separation of nonpolar to slightly polar molecules [11] whereas HILIC (hydrophilic interaction liquid chromatography) mode is becoming a technique of choice for strongly to slightly polar metabolites [12].
Ionization is one of the most critical steps in MS-based metabolite measurements. The degree of its ionization determines the ability to detect and quantify a metabolite. The most often used ionization methods in the field of metabolomics are electrospray ionization (ESI) and electron impact (EI) ionization. ESI is the favorite ionization technique for HPLC-MS for multiple reasons. It adequately ionizes molecules in the liquid phase and can universally be used for small molecules (<1,000 amu) as well as for large molecules such as peptides and proteins. Moreover, ESI is a soft ionization technique, so it does not induce a significant fragmentation of the molecular ions. A drawback in using ESI is that its ionization efficiency is deleteriously affected by the presence of salts, so the chromatography methods are limited to the use of only volatile buffers such as ammonium acetate or ammonium formate. In addition, ion suppression can occur when co-eluting metabolites compete for a limited number of molecular ions with low electron or proton affinity metabolites are obscured, or not detected at all. EI is the ionization method of choice for GC-MS analysis. EI is a hard ionization method as it causes fragmentation of metabolites and it enables detection with minimal matrix effects due to co-eluting metabolites. Recently, atmospheric pressure chemical ionization (APCI) and atmospheric pressure photoionization (APPI) are gaining traction in the field of LC-MS-based metabolic analysis [13].
Metabolite detection with high resolution and sensitivity is generally desired. However, achieving both goals in a single MS detection mode is challenging because as a general rule higher sensitivity leads to lower resolution and vice versa. There are various options including single (MS) or tandem (MS/MS) mass analyzers to choose from, each of which has different sensitivity and resolution performance. The single-configuration mass analyzers include the quadrupole (Q), linear ion trap (LIT), quadrupole ion trap (QIT), time of flight (TOF), Fourier transform ion cyclotron resonance (FTICR), and Orbitrap. Quadrupole and ion trap analyzers offer high sensitivity, but limited resolution whereas TOF, FTICR, and Orbitrap offer high mass resolution. Mass analyzers arranged in a tandem configuration include triple-quadrupole ion trap (QTrap), triple quadrupole (TQ), quadrupole-TOF (Q-TOF), and linear-quadrupole ion trap-Orbitrap (LTQ-Orbitrap). Because of their high sensitivity and selectivity, TQ and Qtrap analyzers are the most common MS spectrometers hyphenated to LC and employed in targeted metabolic studies, while Q-TOF, LTQ-Orbitrap, and FTICR analyzers are more suitable for global profiling and metabolite identification (including isotopomer analysis) due to their higher mass-resolving power. Mass analyzers used with GC are usually single quadrupoles or TOFs, but some recent GC-MS instruments are now equipped with QTOF or TQ mass spectrometers.
3 MS-Based Metabolomics Studies
The number of MS-based metabolomics studies has increased exponentially over the last decade (see Fig. 1). To date MS-based metabolomics approaches have been applied to study, among others, the effect of drugs, toxins, and various diseases on metabolite levels, to trace metabolic pathways and measure fluxes. Numerous reviews provide accounts of MS-based metabolomics methods development and applications. For example, investigations detailing a number of diseases including breast cancer [14], colorectal cancer [15], prostate cancer [16], esophageal and gastric cancer [17], cardiovascular diseases [18,19], kidney diseases [20], inborn errors of metabolism [21], effects of toxicology [22] and nutrition [23], and metabolic fluxes [24,25] reviewed recently provide a partial account of the advances in the field.
Fig. 1.
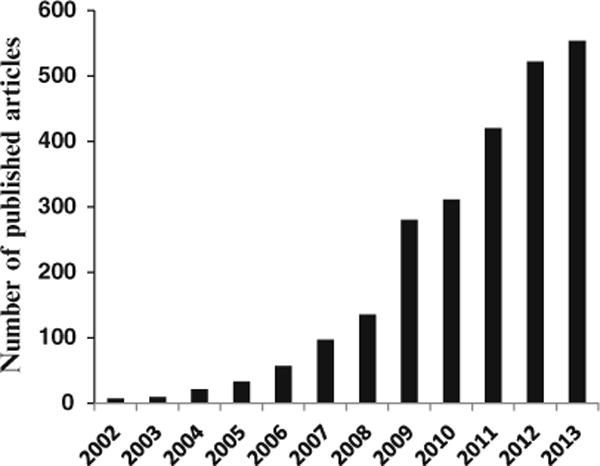
A qualitative representation of the number of MS-based metabolomics research studies published during the past 12 years. The numbers were obtained from the PubMed search using the keywords “mass spectrometry” and “metabolomics”
The metabolomics field has steadily progressed from conventional chemometrics approach that combines global metabolite profiles and statistical methods to a more robust and reliable quantitative approach (quantitative metabolomics). Currently, a major emphasis in the field is being placed on the quantitative analysis of metabolites. A major benefit of the quantitative metabolomics approach is that it potentially reduces errors arising from numerous factors including background distortions, matrix interferences, and peak misalignments. Quantitative metabolomics generally involves targeted tandem MS approaches in which the combined use of precursor and product ions, along with robust chromatography, imparts a high degree of reliability to the data. Utilization of isotope-labeled internal standards in targeted quantitative approaches enables determination of absolute metabolite concentrations. The high costs or unavailability of isotope-labeled standards for many compounds, however, pose a significant challenge for enhancing the pool of quantified metabolites. An approach focused on circumventing this problem utilizes mass production of labeled compounds in vivo through microbial metabolism [26, 27]. In this approach, using a single uniformly 13C-labeled substrate, for example, several hundred labeled compounds can be obtained using microorganisms such as yeast or bacteria. Although potentially attractive, this approach involves tedious steps to make and calibrate the labeled compounds before they can be used for quantitative applications, and challenges exist in utilizing such extracts for studying mammalian systems. It remains to be seen if such approaches find widespread use in metabolomics.
4 Fast MS Methods
The development of ambient ionization methods in mass spectrometry altogether bypasses the use of chromatography separation and shows significant promise for screening applications because such methods are fast and involve little or no sample preparation. DESI-MS (desorption electrospray atmospheric ionization-mass spectrometry) [28], DART-MS (direct analysis in real-time-mass spectrometry) [29], and EESI-MS (extractive electrospray ionization-mass spectrometry) [30] are a few of the early methods with demonstrated potential for real-time analysis of complex mixtures. In DESI-MS the charged and nebulized solvent is directed towards a biological specimen, such as a tissue slice deposited on a surface, for example. In DART-MS, a stream of excited metastable helium gas and hot nitrogen gas are used to volatilize and ionize analytes of interest and obtain real-time metabolite data. In, EESI, two colliding spray sources are used for ionization and introduction into the mass spectrometer. Since then the number of ambient ionization methods have grown rapidly focusing applications to both medical and basic sciences with more than 30 different methods available currently [31,32]. Importantly, the advent of ambient ionization has opened avenues for in situ analysis of tissue specimens, which promises real-time diagnostic information and accurate surgical resection of tumors [33].
5 MS Imaging
An important corollary of advances in ambient ionization methods is the further development of MS imaging. MS imaging has opened new avenues for real-time tissue characterization, in two and three dimensions, of biological specimens such as tissue samples and promises immense utility for clinical applications and numerous other areas [34]. High-quality images that can be obtained with high sensitivity and resolution represent important molecular characterization tools in vivo as well as ex vivo, and promise diagnostic applications in clinical settings. Numerous investigations demonstrate disease diagnostic potential of MS imaging for diseases such as bladder, kidney, prostate, and brain cancers [35–38]. Comparable performance to the current gold standard, histopathology, was shown based on DESI-MS imaging of lipids for the diagnosis of human brain tumors, which highlights the potential of MS imaging for guiding surgeries in real time [39].
6 Statistical Analysis
Metabolite data generated by mass spectrometry are generally complex, and hence multivariate statistical methods are often needed to extract information from such complex datasets. The metabolomics field has witnessed the development of a large number of statistical methods. Of these, commonly used workhorse approaches are principal component analysis (PCA), logistic regression, and partial least squares discriminant analysis (PLS-DA). PCA, an unsupervised method, clusters samples without the prior knowledge of the sample class based on the variance of the signals in the metabolite profiles [40,41] and is often used as a starting point in the analysis. PLS-DA, a supervised method, offers the construction of predictive models based on the regression of the data matrix (X) against a matrix (Y) that contains class information, such as disease or healthy control, for each sample [42]. Logistic regression analysis enables selection of the highly ranked metabolites that contribute most to the classification. The validity of the derived statistical models is often tested internally using leave-one-out or leave 1/3rd, 1/5th, or 1/7th out cross-validation procedures [43]. Further, the robustness and accuracy of PLS-DA models are also tested using Monte Carlo cross validation (MCCV) [44]. The most stringent test for the validity of models is testing data from an independent sample set. Such a test is challenging as it needs large patient cohorts, but is important to assess and eliminate the deleterious effects of confounding factors, and develop robust statistical models.
7 Opportunities, Challenges, and Future Directions
Mass spectrometry continues to play a dominant role as an important analytical tool in the metabolomics field. Advances in the field enable discovery of numerous putative disease biomarkers and provide insights into the pathogeneses of many diseases. Numerous findings have also been tested for translational applications including early disease detection, therapy prediction and prognosis, monitoring treatment, and recurrence detection [45].
With consideration to the high complexity of biological mixtures, the vast majority of MS analysis methods involve prior separation using LC, GC, or CE. However, the fast-growing number of applications using continuously evolving separation methods and protocols presents opportunities and challenges. Clearly, advances in chromatography methods enabled efficient separation of metabolites and enhanced the number of detected metabolites. A major challenge, however, is the inability to compare and correlate the results of such studies on the same or similar samples obtained by independent research groups. This is a major bottleneck to the growth of the field. Other factors such as sample preparation, sample matrix, and carryover effects also contribute to the variability of the data. To circumvent these challenges, there is a need to move away from the conventionally used relative metabolite concentration measurements to more reliable absolute concentration determinations, which will then be independent of the analytical platforms, methods, and protocols used. This approach is not straightforward for MS, as it requires suitable internal standards and proper calibration, but is very important. Such efforts not only enable comparison and correlation of the vast data accumulated in the literature, but the results are more meaningful as, ultimately, clinical translation of metabolite-based biomarker technology requires biomarkers to be measured and validated in absolute concentration. The evolutions in the fast MS methods that have emerged relatively more recently promise altogether different types of applications in the biomedicine. The ambient ionization methods used in these approaches enable real-time analysis of tissue in situ potentially under intraoperative environments. However, unlike the chromatography-based MS methods, fast MS methods are even more challenging to calibrate on an absolute basis. A major focus is therefore needed to test the reproducibility of these methods for real clinical utility.
Acknowledgments
We acknowledge financial support from NIH (National Institute of General Medical Sciences NIH 2R01GM085291).
References
- 1.Van der Greef J, Smilde AK. Symbiosis of chemometrics and metabolomics: past, present, and future. J Chemometr. 2005;19:376–386. [Google Scholar]
- 2.Nicholson JK, Lindon JC. Systems biology: metabolomics. Nature. 2008;455:1054–1056. doi: 10.1038/4551054a. [DOI] [PubMed] [Google Scholar]
- 3.Gates SC, Sweeley CC. Quantitative metabolic profiling based on gas chromatography. Clin Med. 1978;24:1663–1673. [PubMed] [Google Scholar]
- 4.Horning EC, Horning MG. Human metabolic profiles obtained by GC and GC/MS. J Chromatogr Sci. 1971;9:129–140. [Google Scholar]
- 5.Horning EC, Horning MG. Metabolic profiles: gas-phase methods for analysis of metabolites. Clin Chem. 1971;17:802–809. [PubMed] [Google Scholar]
- 6.Griffiths WJ, Wang Y. Mass spectrometry: from proteomics to metabolomics and lipidomics. Chem Soc Rev. 2009;38:1882–1896. doi: 10.1039/b618553n. [DOI] [PubMed] [Google Scholar]
- 7.Ramautar R, Somsen GW, de Jong GJ. CE-MS for metabolomics: developments and applications in the period 2010–2012. Electrophoresis. 2013;34:86–98. doi: 10.1002/elps.201200390. [DOI] [PubMed] [Google Scholar]
- 8.Fiehn O, Kopka J, Dörmann P, Altmann T, Trethewey RN, Willmitzer L. Metabolite profiling for plant functional genomics. Nat Biotechnol. 2000;18:1157–61. doi: 10.1038/81137. [DOI] [PubMed] [Google Scholar]
- 9.Sparkman OD, Penton Z, Kitson FG. Gas chromatography and mass spectrometry: a practical guide. 2. Academic; Burlington, MA: 2011. [Google Scholar]
- 10.Dunn WB, Broadhurst D, Ellis DI, Brown M, Halsall A, O’Hagan S, Spasic I, Tseng A, Kell DB. A GC-TOF-MS study of the stability of serum and urine metabolomes during the UK Biobank sample collection and preparation protocols. Int J Epidemiol. 2008;37:23–30. doi: 10.1093/ije/dym281. [DOI] [PubMed] [Google Scholar]
- 11.Roberts LD, Souza AL, Gerszten RE, Clish CB. Targeted metabolomics. Curr Protoc Mol Biol. 2012;30:1–24. doi: 10.1002/0471142727.mb3002s98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bajad SU, Lu W, Kimball EH, Yuan J, Peterson C, Rabinowitz JD. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J Chromatogr A. 2006;1125:76–88. doi: 10.1016/j.chroma.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 13.Tian H, Bai J, An Z, Chen Y, Zhang R, He J, Bi X, Song Y, Abliz Z. Plasma metabolome analysis by integrated ionization rapid-resolution liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2013;27:2071–2080. doi: 10.1002/rcm.6666. [DOI] [PubMed] [Google Scholar]
- 14.O’Connell TM. Recent advances in metabolomics in oncology. Bioanalysis. 2012;4:431–451. doi: 10.4155/bio.11.326. [DOI] [PubMed] [Google Scholar]
- 15.Williams MD, Reeves R, Resar LS, Hill HH., Jr Metabolomics of colorectal cancer: past and current analytical platforms. Anal Bioanal Chem. 2013;405:5013–5030. doi: 10.1007/s00216-013-6777-5. [DOI] [PubMed] [Google Scholar]
- 16.Trock BJ. Application of metabolomics to prostate cancer. Urol Oncol. 2011;29:572–581. doi: 10.1016/j.urolonc.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abbassi-Ghadi N, Kumar S, Huang J, Goldin R, Takats Z, Hanna GB. Metabolomic profiling of esophago-gastric cancer: a systematic review. Eur J Cancer. 2013;49:3625–3637. doi: 10.1016/j.ejca.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Shah SH, Kraus WE, Newgard CB. Metabolomic profiling for the identification of novel biomarkers and mechanisms related to common cardiovascular diseases: form and function. Circulation. 2012;126:1110–1120. doi: 10.1161/CIRCULATIONAHA.111.060368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhee EP, Gerszten RE. Metabolomics and cardiovascular biomarker discovery. Clin Chem. 2012;58:139–147. doi: 10.1373/clinchem.2011.169573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weiss RH, Kim K. Metabolomics in the study of kidney diseases. Nat Rev Nephrol. 2011;8:22–33. doi: 10.1038/nrneph.2011.152. [DOI] [PubMed] [Google Scholar]
- 21.Mamas M, Dunn WB, Neyses L, Goodacre R. The role of metabolites and metabolomics in clinically applicable biomarkers of disease. Arch Toxico. 2011;85:5–17. doi: 10.1007/s00204-010-0609-6. [DOI] [PubMed] [Google Scholar]
- 22.Robertson DG, Watkins PB, Reily MD. Metabolomics in toxicology: preclinical and clinical applications. Toxicol Sci Suppl. 2011;1:S146–S170. doi: 10.1093/toxsci/kfq358. [DOI] [PubMed] [Google Scholar]
- 23.Scalbert A, Brennan L, Fiehn O, Hankemeier T, Kristal BS, van Ommen B, Pujos-Guillot E, Verheij E, Wishart D, Wopereis S. Mass-spectrometry-based metabolomics: limitations and recommendations for future progress with particular focus on nutrition research. Metabolomics. 2009;5:435–458. doi: 10.1007/s11306-009-0168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reaves ML, Rabinowitz JD. Metabolomics in systems microbiology. Curr Opin Biotechnol. 2011;22:17–25. doi: 10.1016/j.copbio.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zamboni N, Fendt SM, Rühl M, Sauer U. (13)C-based metabolic flux analysis. Nat Protoc. 2009;4:878–892. doi: 10.1038/nprot.2009.58. [DOI] [PubMed] [Google Scholar]
- 26.Birkemeyer C, Luedemann A, Wagner C, Erban A, Kopka J. Metabolome analysis: the potential of in vivo labeling with stable isotopes for metabolite profiling. Trends Biotechnol. 2005;23:28–33. doi: 10.1016/j.tibtech.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 27.Mashego MR, Wu L, van Dam JC, Ras C, Vinke JL, Van Winden WA, Van Gulik WM, Heijnen JJ. MIRACLE: mass isotopomer ratio analysis of U-13C-labeled extracts. A new method for accurate quantification of changes in concentrations of intracellular metabolites. Biotechnol Bioeng. 2004;85:620–628. doi: 10.1002/bit.10907. [DOI] [PubMed] [Google Scholar]
- 28.Takats Z, Wiseman JM, Gologan B, Cooks RG. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science. 2004;306:471–473. doi: 10.1126/science.1104404. [DOI] [PubMed] [Google Scholar]
- 29.Cody RB, Laramee JA, Durst HD. Versatile new ion source for the analysis of materials in open air under ambient conditions. Anal Chem. 2005;77:2297–2302. doi: 10.1021/ac050162j. [DOI] [PubMed] [Google Scholar]
- 30.Chen H, Venter A, Cooks RG. Extractive electrospray ionization for direct analysis of undiluted urine, milk and other complex mixtures without sample preparation. Chem Commun. 2006;19:2042–2044. doi: 10.1039/b602614a. [DOI] [PubMed] [Google Scholar]
- 31.Wu C, Dill AL, Eberlin LS, Cooks RG. Mass spectrometry imaging under ambient conditions. Mass Spectrom Rev. 2013;32:218–243. doi: 10.1002/mas.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang MZ, Cheng SC, Cho YT, Shiea J. Ambient ionization mass spectrometry: a tutorial. Anal Chim Acta. 2011;702:1–15. doi: 10.1016/j.aca.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 33.Balog J, Sasi-Szabó L, Kinross J, Lewis MR, Muirhead LJ, Veselkov K, Mirnezami R, Dezső B, Damjanovich L, Darzi A, Nicholson JK, Takáts Z. Intraoperative tissue identification using rapid evaporative ionization mass spectrometry. Sci Transl Med. 2013;5:194ra93. doi: 10.1126/scitranslmed.3005623. [DOI] [PubMed] [Google Scholar]
- 34.Nemes P, Vertes A. Ambient mass spectrometry for in vivo local analysis and in situ molecular tissue imaging. TrAC Trends Anal Chem. 2012;34:22–34. [Google Scholar]
- 35.Dill AL, Eberlin LS, Zheng C, Costa AB, Ifa DR, Cheng L, Masterson TA, Koch MO, Vitek O, Cooks RG. Multivariate statistical differentiation of renal cell carcinomas based on lipidomic analysis by ambient ionization imaging mass spectrometry. Anal Bioanal Chem. 2010;398:2969–2978. doi: 10.1007/s00216-010-4259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dill AL, Eberlin LS, Costa AB, Zheng C, Ifa DR, Cheng L, Masterson TA, Koch MO, Vitek O, Cooks RG. Multivariate statistical identification of human bladder carcinomas using ambient ionization imaging mass spectrometry. Chem A Eur J. 2011;17:2897–2902. doi: 10.1002/chem.201001692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eberlin LS, Dill AL, Costa AB, Ifa DR, Cheng L, Masterson T, Koch M, Ratliff TL, Cooks RG. Cholesterol sulfate imaging in human prostate cancer tissue by desorption electrospray ionization mass spectrometry. Anal Chem. 2010;82:3430–3434. doi: 10.1021/ac9029482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eberlin LS, Norton I, Dill AL, Golby AJ, Ligon KL, Santagata S, Cooks RG, Agar NY. Classifying human brain tumors by lipid imaging with mass spectrometry. Cancer Res. 2012;72:645–654. doi: 10.1158/0008-5472.CAN-11-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eberlin LS, Norton I, Orringer D, Dunn IF, Liu X, Ide JL, Jarmusch AK, Ligon KL, Jolesz FA, Golby AJ, Santagata S, Agar NY, Cooks RG. Ambient mass spectrometry for the intraoperative molecular diagnosis of human brain tumors. Proc Natl Acad Sci USA. 2013;110:1611–1616. doi: 10.1073/pnas.1215687110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holmes E, Antti H. Chemometric contributions to the evolution of metabonomics: mathematical solutions to characterising and interpreting complex biological NMR spectra. Analyst. 2002;127:1549–1557. doi: 10.1039/b208254n. [DOI] [PubMed] [Google Scholar]
- 41.Griffin JL. Metabolic profiles to define the genome: can we hear the phenotypes? Philos Trans Royal Soc Lond B Biol Sci. 2004;359:857–871. doi: 10.1098/rstb.2003.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barker M, Rayens W. Partial least squares for discrimination. J Chemometr. 2003;17:166–173. [Google Scholar]
- 43.Heather LC, Wang X, West JA, Griffin JL. A practical guide to metabolomic profiling as a discovery tool for human heart disease. J Mol Cell Cardiol. 2013;55:2–11. doi: 10.1016/j.yjmcc.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 44.Girard DA. A fast “Monte-Carlo cross validation” procedure for large least squares problems with noisy data. Num Math. 1989;56:1–23. [Google Scholar]
- 45.Nagana Gowda GA, Raftery D. Biomarker discovery and translation in metabolomics. Curr Metabolom. 2013;1:227–240. doi: 10.2174/2213235X113019990005. [DOI] [PMC free article] [PubMed] [Google Scholar]