

Abstract
Obesity is reaching epidemic proportions so there is growing interest in the mechanisms that regulates adipose tissue development and function. Although murine adipose cell lines are useful for many mechanistic studies, primary human adipose stromal cells (ASCs), which can be isolated from distinct adipose depots and cultured in vitro, have clear translational relevance. We describe the methods to isolate, culture, and differentiate human ASCs to adipocytes that respond to physiologically relevant hormones, such as insulin and β-adrenergic agonists. We also describe methods for assaying hormonal effects on glucose transport and lipolysis.
1. INTRODUCTION
Obesity, defined as accumulation of excess adipose tissue (AT), is increasing worldwide. AT mass expands by increases in the size (hypertrophy) and number of adipocytes (hyperplasia). There is a growing interest in understanding mechanisms of adipocyte differentiation and function as alterations in AT metabolic and endocrine function are thought to play key roles in the pathogenesis of obesity and related metabolic diseases. The life span of subcutaneous (sc) adipocytes in humans is estimated as between ~2–3 (Strawford, Antelo, Christiansen, & Hellerstein, 2004) and 10 years (Spalding et al., 2008). Thus, significant numbers of adipocytes are replaced though the recruitment of adipose precursors and mechanisms that regulate preadipocyte differentiation are important for understanding the maintenance of a healthy AT. Although mouse embryonic fibroblast cell lines, 3T3-L1 and 3T3-F422A, among others, have provided invaluable model systems for mechanistic studies of proliferation and adipogenesis, there is a clear need for cultures of human adipose precursors. Several human adipose cell strains are available including SGBS and hMAD (Bordicchia et al., 2012; Fischer-Posovszky, Newell, Wabitsch, & Tornqvist, 2008), and the use of primary adipose stromal cells (ASCs) that prepared from subjects with fat distribution and/or obesity phenotypes are essential for translational research.
Large number of ASCs, often called AT stem cells or preadipocytes, can be isolated with collagenase digestion of AT. AT specimens can be easily obtained through needle aspiration or during elective surgeries with only minimal risk to the volunteers. ASCs are multipotent cells (adipogenic, chondrogenic, osteogenic, and myogenic; Cawthorn, Scheller, & Macdougald, 2012b), are capable of expansion in vitro, and can be cryopreserved for long periods without significant loss in proliferation and differentiation capacity. The differentiated adipocytes display phenotypic characteristics of genuine adipocytes, that is, freshly isolated ones. Specially, they respond to physiologically relevant concentrations of hormones, including insulin and β-adrenergic agonists (Fried et al., 2010; Lee & Fried, 2012; Lee, Wu, & Fried, 2012; Lystedt et al., 2005; Zierath et al., 1998). Further, ASCs are also useful for assessing donor- and origin-dependent effects (depot, sex, age, obesity, etc.) on cell proliferation and differentiation capacity, which is not possible with cell lines. Visceral adipose depots contain fewer adipogenic precursors and they exhibit lower proliferation and differentiation capacity (Hauner, Wabitsch, & Pfeiffer, 1988; Tchkonia et al., 2002, 2006). Femoral compared to abdominal sc preadipocytes exhibit lower differentiation capacity (Hauner & Entenmann, 1991; Tchoukalova et al., 2010). Obesity- and aging-related declines in ASC proliferation and differentiation capacity have also been reported (Isakson, Hammarstedt, Gustafson, & Smith, 2009; Perez et al., 2013; Sepe, Tchkonia, Thomou, Zamboni, & Kirkland, 2011). Taken together, available data show that cultures of human ASCs reflect the depot-specific biology of human ATs and are therefore valuable models for understanding the underlying mechanisms regulating fat distribution and its influence on metabolic disease.
Conditions to proliferate and differentiate ASCs (e.g., seeding density, culture media and conditions, and subculturing) vary among published studies. Standardization of growth and differentiation conditions is critical as they affect the degree to which the culture will differentiate. We describe methods to proliferate and differentiate ASCs and discuss potential confounders in the processes. We also describe methods to use differentiated human adipocytes for analyses of lipolysis and glucose transport.
2. ISOLATION OF ASCs FROM AT
2.1. Materials
Most chemicals are purchased from Sigma-Aldrich (St Louis, MO), cell culture media and FBS are from Life Technologies (Carlsbad, CA), and cell culture plates and plasticware are from Corning (Corning, NY) or BD Biosciences (Franklin Lakes, NJ). Surgical grade scissors, forceps, and perforated spoons can be purchased from Fine Scientific Tools (Foster City, CA):
AT, obtained from surgical resection or sc fat aspiration.
Laminar flow hood.
Tissue culture incubator, 5% CO2 atmosphere.
Sterile scissors, dissecting forceps, and perforated spoons.
Funnels with 250 μm nylon mesh affixed on top and autoclaved.
Tissue culture plates and 50 ml tubes.
Sterile PBS or saline.
Collagenase (type 1, Worthington Biochemical, Lakewood, NJ) or Liberase (Roche, Indianapolis, IN) solution can be prepared in advance (1 mg/ml HBSS), filtered through 0.2 μm filter, and frozen in aliquots at −20 °C.
Red blood cell (RBC) lysis buffer (0.154 mM NH4Cl, 10 mM K2HPO4, and 0.1 mM EDTA, pH 7.3).
Culture media (growth media): alpha-MEM (5 mM glucose, 11900-024, Life Technologies, Carlsbad, CA) with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin (pen/strep) (15140, Life Technologies, Carlsbad, CA).
2.2. Procedures for ASC isolation
All procedures are done under sterile conditions.
-
AT is brought to the lab in room temperature PBS or Medium 199 (12340-030, Life Technologies, Carlsbad, CA). From most fat aspirations and surgical resection, 0.3–10 g AT is obtained. With panniculectomy, a large chunk of AT, often with skin attached, is obtained and thus, it is necessary to dissect AT using scissors and forceps.
Note: AT can be stored 4°C overnight and ASCs can be isolated at 4 the next day. The yields, however, may be lower.
Mince AT into small pieces, approximately 5–10 mg pieces (1–2 mm3) using sharp scissors. Tissues from fat aspirations are already fragmented and do not require mincing.
Pour the minced tissue through a 250 μm mesh. The mesh is formed into the shape of a funnel, stapled to hold this shape, inserted into a funnel, affixed with tape, and autoclaved. Place this apparatus on top of an empty 500 ml bottle to capture the waste.
Rinse AT with saline or PBS. Remove any visible blood clots and obvious connective tissue using sterile forceps.
Estimate tissue amount (weigh an empty sterile container such as a petri dish, add AT to the container, and weigh again the container with the tissue, and the difference is tissue weight).
Transfer the tissue into 50 ml tubes and add collagenase solution, 1–2 ml/g of AT.
Digest AT by incubating in a water bath with shaking (100 rpm) at 37 °C for 2 h until there are few intact pieces of AT and the mixture becomes a homogeneous soupy consistency. Vortexing or rapid swirling by hand will facilitate digestion.
Filter the digested AT through a 250 μm mesh affixed to a funnel and capture the flow through into a 50 ml tube. The flow through contains the ASCs.
Wash the mesh with culture media. When a large amount of tissue is used, undigested or connective tissue will clog the mesh. Scraping the mesh with a spoon or forceps will facilitate the filtering process and increase yield. Repeat wash as needed.
Centrifuge the 50 ml tubes at 500 × g for 10 mins at room temperature. After centrifugation, three layers are visible: a fat cake at the top, an intermediate layer of medium, and a cell pellet at the bottom.
Carefully aspirate off the upper fat cake and the internatant above the cell pellet.
-
Add 1–5 ml of RBC lysis buffer (depending on the amount of RBC) and pipette up and down to resuspend cells. Incubate in RBC lysis buffer no more than 15 min at room temp or 10 min at 37 °C.
Note: RBCs are denser than ASCs and are thought to block cell attachment. When a very small amount of tissue is used (e.g., less than 0.5 g), cells can be plated without RBC lysis.
Centrifuge the tubes at 500 × g for 5 mins.
Aspirate off the supernatant and resuspend cells in growth media.
Count cell number using a hemocytometer and plate accordingly.
-
After overnight attachment, gently wash the plates with PBS to remove residual RBC and cell debris and refeed.
Note: Washing and refeeding after just several hours of attachment may enrich preadipocytes/fibroblasts (personal communication—JL Kirkland). Based on cell surface markers (Cawthorn, Scheller, & Macdougald, 2012a), an adipocyte progenitor population can be isolated using FACS or antibody conjugated magnetic beads.
3. PROTOCOLS FOR PROLIFERATION, SUBCULTURE, AND FREEZING DOWN
10% FBS-supplemented DMEM, DMEM/F12, or alpha-MEM is mostly used during ASC culture. FBS, however, is known to decrease the proliferation and differentiation capacity (Hauner, Schmid, & Pfeiffer, 1987; Skurk, Ecklebe, & Hauner, 2007). Human serum or growth factors can substitute for FBS with preserved proliferation and differentiation capacity (Bieback et al., 2012; Chieregato et al., 2011; Im, Chung, Kim, & Kim, 2011; Tunaitis et al., 2011).
3.1. Materials
Growth media (GM): alpha-MEM with 10% FBS and antibiotics (usually pen/strep).
Freezing media: 10% dimethyl sulfoxide (DMSO) supplemented GM.
0.25% trypsin/EDTA (25200, Life Technologies, Carlsbad, CA).
Cryogenic tubes.
Freezing container.
Culture plates and plasticware.
3.2. Proliferation of cells
Grow and proliferate cells in the GM, with refeeding every 2–3 days.
When cells reach 70–80% confluence, subculture or freeze down cells. Cells may lift off from the plates if they are kept too long after reaching confluence.
3.3. Subculturing
When cells are 70–80% confluent, aspirate off the media and wash once with PBS.
Aspirate off the PBS, and add ~1.5 ml of 0.25% trypsin to 10 cm dishes (less volume for smaller dishes).
Swirl the plates to distribute the trypsin and then aspirate off most of the liquid, leaving about 0.3 ml per dish.
Incubate dishes 2–3 min in the incubator at 37 °C. Check whether cells are lifted off from the plates by examining under a microscope. If cells have not lifted off, incubate longer.
Add 5 ml GM to each dish to inactivate the trypsin. Pipette up and down, washing the plate thoroughly, to collect all of the cells off the plate.
Transfer the media and cells into 15 or 50 ml tubes and centrifuge at 500×g for 5 min.
Remove the supernatant and resuspend the cells.
-
Count cell numbers and subculture cells accordingly. With 1–4 splitting, it generally takes 3–5 days until they reach 70–80% confluence.
Note: Cells derived from visceral depots grow slower than sc depots. In addition, there is also subject-dependant variation in proliferation rates.
3.4. Freezing and storage
When cells have grown for several days, they survive freezing/thawing cycle better.
At 70–80% confluency, trypsinize cells following steps 1–6 described in the previous section.
Aspirate off the supernatant, add 1–2 ml freezing media, and resuspend cells.
Transfer the cell suspension to prelabeled cryogenic vials. Place the vials into a freezing container and store at −80 °C for several days.
-
Transfer cell vials to a cryostorage system for long-term storage.
Note: In our experience, ASCs can be stored in a cryostorage system without any significant loss in proliferation or differentiation capacity for more than 20 years.
3.5. Protocol for thawing cells
Remove vial(s) from cryostorage system and immediately place in 37 °C water bath for 60–90 s. Remove as soon as cells are thawed.
Add 1 ml of GM in the cell vial, pipette up and down to mix, and transfer the contents of the vial to centrifuge tubes containing GM.
Centrifuge for 5 min at 500×g.
Aspirate off the supernatant and resuspend cells in GM.
Count the cells for plating or just plate into 10 cm dishes overnight and split on the following day.
4. PROTOCOLS TO DIFFERENTIATE HUMAN ASCs TO ADIPOCYTES
This is a modified protocol from Hauner’s (Hauner, Skurk, & Wabitsch, 2001) and Kirkland’s (Tchkonia et al., 2005), in which differentiation is carried out in a serum-free, chemically defined media. In standard protocols, adipogenic induction cocktail contains insulin, glucocorticoids, and 1-methyl-3-isobutylxanthine (IBMX) (Hauner et al., 2001; Yu et al., 2011). Thiazolidinediones (TZD), which are PPAR-γ agonists, are often used to improve the degree of differentiation as human ASCs do not differentiate well in the absence of PPAR-γ ligands (Gustafson & Smith, 2012; Nimitphong, Holick, Fried, & Lee, 2012).
4.1. Materials
-
Complete differentiation media (CDM): make up stock solutions, prepare the CDM by adding the components and stock solutions, filter through a 0.2 micron filters, and store at 4 °C up to 1 month.
Final concentrations Stock concentrations Amount/l DMEM/F12, 18.5 mM glucose 1 pack HEPES 15 mM 3.57 g NaHCO3 25 mM 2.22 g Penicillin/streptomycin 100 units/ml (pen) and 100 μg/ml (strep) 10,000 units/ml (pen) and 10,000 μg/ml (strep) 10 ml d-Biotin 33 μM 3.3 mM 10 ml Pantothenate 17 μM 1.7 mM 10 ml Dexamethasone 100 nM 10 mM 10 μl Insulin 100 nM 600 μM 167 μl Rosiglitazone or other TZDs 1 μM 10 mM 100 μl IBMX 0.5 mM 110 mg T3 2 nM 20 μM 100 μl Transferrin 10 μg/ml 10 mg/ml 1 ml Maintenance media (MM): DMEM/F12 with pen/strep, HEPES, NaHCO3, d-biotin, pantothenate, 10 nM insulin and 10 nM dexamethasone.
4.2. Preparation and storage of stock solutions
d-Biotin (3.3 mM): dissolve in DMEM/F12, sterilize by filtering through a 0.2-micron filter, and store at 4 °C.
Pantothenate (1.7 mM): dissolve in DMEM/F12, sterilize by filtering through a 0.2-micron filter, and store at 4 °C.
Transferrin (10 mg/ml): reconstitute in water, aliquot, and store at −80 °C.
T3 (20 μM): make 2 mM stock solution in 1 N NaOH, then dilute into ethanol to make 20 μM, and store at −20 °C.
IBMX: weigh out the amount needed, add H2O followed by addition of 1 N KOH dropwise, and vortex until it is dissolved. Once it is reconstituted, immediately transfer the solution into the media.
Rosiglitazone (10 mM, ALX-350-125, Enzo, Farmingdale, NY): make 10 mM solution in DMSO, and store at −80 °C in aliquots.
Insulin (Humulin R, Lilly, Indianapolis, IN): recombinant human insulin from pharmacy.
Dexamethasone (10 mM): make stock solutions in ethanol and store at −20 °C.
4.3. Procedures for differentiating ASCs
Count and plate cells depending on the experimental design with a plating density of 5000–15,000 cells/cm2.
Grow cells in the GM until 2 days after confluence. With a plating density of 5000 cells/cm2, it generally takes 5–7 days until they are ready for differentiation.
-
Once cells are ready for differentiation, aspirate off the GM and refeed with the CDM.
Note: Both serum-free (Deslex, Negrel, Vannier, Etienne, & Ailhaud, 1987; Entenmann & Hauner, 1996; Shahparaki, Grunder, & Sorisky, 2002; Tchkonia et al., 2002) and supplemented (Bunnell, Flaat, Gagliardi, Patel, & Ripoll, 2008; Yu et al., 2011) conditions are used during differentiation, the two most commonly used conditions being serum-free or 3% FBS-supplemented conditions. Although up to 3% FBS does not significantly affect differentiation, metabolic properties of adipocytes, however, are different between serum-free and 3% FBS protocols (Lee et al., 2012).
Replenish the CDM every 2 to 3 days. Lipid droplet accumulation is usually visible after 3 days of differentiation.
-
After 3–7-day induction, remove the CDM and feed cells with the MM.
Note: In commonly used protocols, IBMX and TZD are present only during the initial 3 days (Hauner et al., 2001; Yu et al., 2011). Longer periods of induction are also used and may actually improve extent of differentiation (Dicker et al., 2004; Fischer-Posovszky et al., 2008; Skurk & Hauner, 2012; Yu et al., 2011), especially for cells with poor differentiation capacity under standard conditions (Lee et al., 2012).
-
Keep cells in the MM and replenish every 2 to 3 days until fully differentiated. We generally consider cells as having matured into adipocytes after at least 12 days of differentiation. Adipocytes can be maintained in the culture>40 days and the size of lipid droplets grows until they have several enlarged droplets per cell or even become unilocular.
Note: The MM contains insulin (10 nM) and dexamethasone (10 nM), as the combination of these two hormones best maintains high expression levels of adipocyte genes such as GLUT4 and adiponectin. Extra caution is required during refeeding well-differentiated adipocytes as lipid-filled adipocytes tend to lift off the plates. The use of 1 ml pipetter to remove and refeed media can be considered to minimize disturbing the cells. Unless required, leaving ~20% of the media during refeeding is also helpful.
4.4. Determination of differentiation degree
Substantial changes in cell morphology occur during differentiation (Fig. 4.1). Starting from day 3 of differentiation, a small amount of lipid accumulation is observed. By day 8, cells should be rounding up to a more spherical shape from a more elongated fibroblast shape. Well-differentiated cultures will have over 80% of the cells containing lipid droplets.
Figure 4.1.

Microscopic images of human adipose stromal cells at various stages. Microscopic images are taken at preconfluence (A), 2 days postconfluence (B), d3 (C), d7 (D), d14 (E), and d35 (F) of differentiation.
The formation of lipid droplets is a hallmark of adipogenesis and can be easily observed by phase-contrast microscope. The lipid droplets can be stained with Oil Red O or Nile red. The amount of triacylglycerol in cell lysates can be measured for quantification.
-
Expression levels of adipogenic markers can be determined at the mRNA and protein levels. Typical markers include
adipogenic transcription factors: PPAR γ, CEBP β, and CEBP α;
adipocyte genes: fatty acid binding protein 4, lipoprotein lipase, adiponectin, leptin, perilipin, adipose triglyceride lipase, etc.
Note: When measuring the expression levels of lipid droplet proteins, especially perilipin, add 5–10% SDS to cell lysis buffer and incubate cell lysates at 37 °C for 1 h with vortexing every 5–10 min. This procedure is required to release the tight association of these hydrophobic proteins with the lipid droplet surface.
Note: Differentiated adipocytes also express significant amount of adiponectin and leptin that can be easily measured in culture media with commercially available ELISA kits.
5. METHODS TO USE NEWLY DIFFERENTIATED ADIPOCYTES FOR METABOLIC STUDIES
Newly differentiated adipocytes provide a valuable adipocyte system for metabolic and endocrine functions of adipocytes. In this section, we will describe the protocols for lipolysis and glucose transport assays. The procedures described are based on 12-well plates and adjustment is needed when different formats are used. The results of lipolysis and glucose transport assay performed in sc adipocytes derived from five independent subjects are presented in Fig. 4.2.
Figure 4.2.
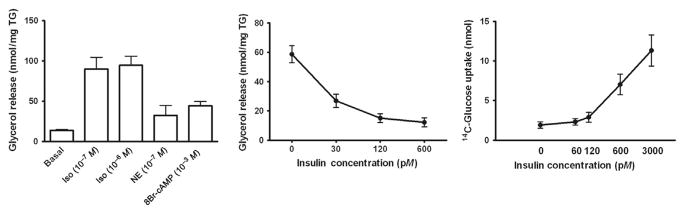
Metabolic phenotypes of newly differentiated human adipocytes. Lipolytic rates, basal and in response to isoproterenol (Iso), norepinephrine (NE), or 8-bromo-cAMP (A) and in response to insulin (B), are measured on d14 of differentiation. Basal lipolysis was measured in the conditions of 1 units/ml ADA +20 nM PIA. Inhibition of lipolysis by insulin was measured against 8-bromo-cAMP (1 mM)-stimulated conditions. (C) The rates of 14C-2-deoxy-glucose uptake were measured after 3–4 h of starvation at 0, 60, 120, 600, and 3000 pM insulin concentrations. Data are obtained from sc ASCs derived from five independent subjects. Some of these data have been published (Lee & Fried, 2012; Lee et al., 2012).
5.1. Lipolysis
Newly differentiated adipocytes in culture respond to β-adrenergic agonists or cAMP analogs that stimulate lipolysis and respond to physiological concentrations of insulin that inhibit lipolysis (Figure 4).
5.1.1 Materials
DMEM/F12
-
Krebs Ringer bicarbonate (KRB) buffer with 5 mM glucose and 4% bovine serum albumin (BSA, fatty acid free, EMD Millipore, Darmstadt, Germany).
10 × mixed salts (MS)
-
Combine the following salts in approximately 800 ml of deionized and distilled H2O:
76.14 g NaCl.
3.51 g KCl.
3.06 g MgSO4.
3.63 g CaCl2.
Bring up to 1 l with H2O and store at 4 °C.
10 × bicarbonate/phosphate buffer
-
Combine:
20.66 g NaHCO3.
1.51 g KH2PO4.
Dissolve the salts in H2O, adjust to pH 7.6, bring to 1 l with H2O, and store at 4 °C.
KRB with 5 mM glucose, 200 nM adenosine, and 4% BSA: prepare on the day of the experiment
-
In a clean flask, combine
80 parts of H2O.
10 parts of 10× MS solution.
10 parts of 10× bicarbonate/phosphate buffer.
Gas for 15 min with 95% O2/5% CO2.
Add BSA and glucose and 100 μl of adenosine (200 μM stock, prepared in H2O and stored in −80 °C) per 100 ml buffer.
Stir with a stir bar until dissolved.
-
Adjust pH to 7.4.
Note: There are lot-to-lot variations in BSA that can affect metabolic and secretion properties of adipocytes. Pretesting and buying in bulk are recommended.
-
-
Reagents for lipolysis assay: adenosine, adenosine deaminase (ADA, 10102121001, Roche, Indianapolis, IN), phenylisopropyl adenosine (PIA), β-adrenergic agonists, cAMP analogs, and insulin.
Note: ADA and PIA are added to standardize any potential variations in adenosine levels (Honnor, Dhillon, & Londos, 1985).
5.1.2 Procedure for lipolysis
Plate and differentiate cells according to experimental design. Plate extra wells for cell number and triglyceride quantification.
On the day of assay, wash cells with warm basal DMEM/F12 or PBS several times to remove any residual hormones, especially when measuring inhibition of lipolysis by insulin.
Add basal DMEM/F12 to the cells and incubate for 3–4 h for hormone starvation.
Prepare KRB buffer with 5 mM glucose and 4% BSA, pH 7.4.
-
Prepare various conditions for lipolysis assay, hormones, and lipolytic reagents in KRB buffer with 4% BSA, 0.5 ml per well of 12-well cell culture plates.
Note: Basal lipolytic rates are defined as ADA (1 units/ml)+PIA (20 nM) (Fried et al., 2010). Stimulated lipolytic rates are measured with isoproterenol (10−8–10−6 M), norepinephrine (10−7–10−6 M), and 8-bromo-cAMP (1 mM). Inhibition of lipolysis by insulin can be measured under 8-bromo-cAMP-stimulated condition (1 mM). The effective concentration 50 (EC50) of insulin for well-differentiated sc adipocytes is 10–30 pM (Lee & Fried, 2012).
After preincubation, remove media and add the prepared KRB buffer with different reagents (step 5), 0.5 ml per well.
Incubate cells in the incubator at 37 °C for 2 h (rates are linear within this time).
After incubation, place cell culture plates on ice directly to stop the reaction. Collect and save incubation media for measurement of glycerol and FFA.
Wash cells with ice-cold PBS several times to remove BSA and scrape cells in cell lysis buffer for future use for quantifying cell numbers or tri-glyceride or immunoblotting. Data are expressed the amount of glycerol released per cell number or triacylglycerol amount.
5.2. Glucose uptake
Uptake of glucose is measured at different insulin concentrations (0–10 nM) to establish concentration dependency. The following protocol is optimized for 12-well cell culture plates in total incubation volume of 300 μl per well. Total incubation volume, the amount, and specific activity of labeled glucose can be adjusted.
5.2.1 Materials
-
Krebs Ringer HEPES (KRH) buffer with 0.01% BSA, with or without 5 mM glucose, 200 nM adenosine, pH 7.4.
10 × Mixed Salts: same as previously described.
-
10 × HEPES/phosphate buffer:
23.8 g HEPES.
-
3.42 g NaH2PO4H2O.
Dissolve the salts in H2O, adjust pH 7.6, bring to 1 l with H2O, and store at 4 °C.
KRH buffer with 0.01% BSA, with or without 5 mM glucose, 200 nM adenosine, pH 7.4: prepare on the day of experiment.
-
In a clean flask, combine:
80 parts of H2O
10 parts of 10 × MS
10 parts of 10 × HEPES buffer
Add BSA and adenosine to the buffer. Divide the buffer into two parts, one without glucose and one with glucose. Then add glucose to KRH buffer with glucose (5 mM).
Stir with a stir bar until dissolved.
Adjust pH to 7.4.
2-Deoxyglucose (2-DOG), unlabeled and 14C-labeled
Insulin
Lysis buffer: Triton X-100 lysis buffer (1% Triton X-100, 20 mM Tris, and 150 mM NaCl)
Cytochalasin B: 0.96 mg/ml (2 mM) dissolved in ethanol and stored at −20 °C
5.2.2 Procedure for glucose transport assay
Plate and differentiate cells for glucose transport assay. Plate extra wells for cell number counting and nonspecific uptake measurement.
On the day of assay, prepare KRH buffer plus 0.01% BSA, with or without 5 mM glucose.
Wash cells times with warm PBS, being careful not to disturb cells.
Add KRH buffer with 5 mM glucose into each well and incubate cells for 3–4 h for insulin starvation.
Prepare varying incubation conditions, containing different concentrations of insulin in the KRH buffer without glucose (0.27 ml per well of 12-well plates). EC50 for insulin-stimulated glucose transport is 100–300 pM (Lee et al., 2012).
Wash the preincubated cells with warm KRH buffer without glucose several times to remove any residual glucose.
Add the KRH buffer prepared in step 5 into cells and incubate cells for 30 min. For nonspecific uptake, treat separate wells with 1.5 μl cyto-chalasin B stock.
-
Prepare 10 × 2-DOG mixture (final concentrations 0.25–1 μCi/ml [14C]2-DOG and 0.05–0.2 mM unlabeled 2-DOG).
Note: The amount of 2-DOG (both cold and radioactive) can be adjusted. Be sure to count a known volume of the 2-DOG mixture for specific activity.
After preincubation with insulin, add 10 × 2-DOG mixture (30 μl per 300 μl total volume) to each well as quickly as possible, and incubate for a further 10 min in the incubator.
Immediately place the cells on ice to stop the reaction after incubation and wash three times with ice-cold PBS to remove unincorporated 2-DOG.
Remove PBS, add 0.5 ml of Triton X-100 lysis buffer, and shake the plates on a rocker for 1 h.
Transfer the cell lysates to scintillation vials and count radioactivity. Also count the specific activity of the 2-DOG mixture.
Subtract cytochalasin B values from each condition to determine the specific uptake and calculate data.
Acknowledgments
This work was supported by NIH (DK 52398, DK 080448, and P30 DK 046200) to SKF and Evans Biomedical Research Foundation pilot grant to MJLMJL and SKF take full responsibility for this chapter.
References
- Bieback K, Hecker A, Schlechter T, Hofmann I, Brousos N, Redmer T, et al. Replicative aging and differentiation potential of human adipose tissue-derived mesenchymal stromal cells expanded in pooled human or fetal bovine serum. Cytotherapy. 2012;14:570–583. doi: 10.3109/14653249.2011.652809. [DOI] [PubMed] [Google Scholar]
- Bordicchia M, Liu D, Amri EZ, Ailhaud G, Dessi-Fulgheri P, Zhang C, et al. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. The Journal of Clinical Investigation. 2012;122:1022–1036. doi: 10.1172/JCI59701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnell BA, Flaat M, Gagliardi C, Patel B, Ripoll C. Adipose-derived stem cells: Isolation, expansion and differentiation. Methods. 2008;45:115–120. doi: 10.1016/j.ymeth.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthorn WP, Scheller EL, Macdougald OA. Adipose tissue stem cells meet preadipocyte commitment: Going back to the future. Journal of Lipid Research. 2012a;53:227–246. doi: 10.1194/jlr.R021089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthorn WP, Scheller EL, Macdougald OA. Adipose tissue stem cells: The great WAT hope. Trends in Endocrinology and Metabolism. 2012b;23:270–277. doi: 10.1016/j.tem.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieregato K, Castegnaro S, Madeo D, Astori G, Pegoraro M, Rodeghiero F. Epidermal growth factor, basic fibroblast growth factor and platelet-derived growth factor-bb can substitute for fetal bovine serum and compete with human platelet-rich plasma in the ex vivo expansion of mesenchymal stromal cells derived from adipose tissue. Cytotherapy. 2011;13:933–943. doi: 10.3109/14653249.2011.583232. [DOI] [PubMed] [Google Scholar]
- Deslex S, Negrel R, Vannier C, Etienne J, Ailhaud G. Differentiation of human adipocyte precursors in a chemically defined serum-free medium. International Journal of Obesity. 1987;11:19–27. [PubMed] [Google Scholar]
- Dicker A, Ryden M, Naslund E, Muehlen IE, Wiren M, Lafontan M, et al. Effect of testosterone on lipolysis in human pre-adipocytes from different fat depots. Diabetologia. 2004;47:420–428. doi: 10.1007/s00125-003-1324-0. [DOI] [PubMed] [Google Scholar]
- Entenmann G, Hauner H. Relationship between replication and differentiation in cultured human adipocyte precursor cells. The American Journal of Physiology. 1996;270:C1011–C1016. doi: 10.1152/ajpcell.1996.270.4.C1011. [DOI] [PubMed] [Google Scholar]
- Fischer-Posovszky P, Newell FS, Wabitsch M, Tornqvist HE. Human SGBS cells—A unique tool for studies of human fat cell biology. Obesity Facts. 2008;1:184–189. doi: 10.1159/000145784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried SK, Tittelbach T, Blumenthal J, Sreenivasan U, Robey L, Yi J, et al. Resistance to the antilipolytic effect of insulin in adipocytes of African-American compared to Caucasian postmenopausal women. Journal of Lipid Research. 2010;51:1193–1200. doi: 10.1194/jlr.P000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson B, Smith U. The WNT inhibitor Dickkopf 1 and bone morphogenetic protein 4 rescue adipogenesis in hypertrophic obesity in humans. Diabetes. 2012;61:1217–1224. doi: 10.2337/db11-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauner H, Entenmann G. Regional variation of adipose differentiation in cultured stromal-vascular cells from the abdominal and femoral adipose tissue of obese women. International Journal of Obesity. 1991;15:121–126. [PubMed] [Google Scholar]
- Hauner H, Schmid P, Pfeiffer EF. Glucocorticoids and insulin promote the differentiation of human adipocyte precursor cells into fat cells. The Journal of Clinical Endocrinology and Metabolism. 1987;64:832–835. doi: 10.1210/jcem-64-4-832. [DOI] [PubMed] [Google Scholar]
- Hauner H, Skurk T, Wabitsch M. Cultures of human adipose precursor cells. Methods in Molecular Biology. 2001;155:239–247. doi: 10.1385/1-59259-231-7:239. [DOI] [PubMed] [Google Scholar]
- Hauner H, Wabitsch M, Pfeiffer EF. Differentiation of adipocyte precursor cells from obese and nonobese adult women and from different adipose tissue sites. Hormone and Metabolic Research Supplement. 1988;19:35–39. [PubMed] [Google Scholar]
- Honnor RC, Dhillon GS, Londos C. cAMP-dependent protein kinase and lipolysis in rat adipocytes. I. Cell preparation, manipulation, and predictability in behavior. The Journal of Biological Chemistry. 1985;260:15122–15129. [PubMed] [Google Scholar]
- Im W, Chung JY, Kim SH, Kim M. Efficacy of autologous serum in human adipose-derived stem cells; cell markers, growth factors and differentiation. Cell Mol Biol (Noisy-le-grand) 2011;57(Suppl):OL1470–OL1475. [PubMed] [Google Scholar]
- Isakson P, Hammarstedt A, Gustafson B, Smith U. Impaired preadipocyte differentiation in human abdominal obesity: Role of Wnt, tumor necrosis factor-alpha, and inflammation. Diabetes. 2009;58:1550–1557. doi: 10.2337/db08-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Fried SK. Glucocorticoids antagonize tumor necrosis factor-alpha-stimulated lipolysis and resistance to the antilipolytic effect of insulin in human adipocytes. American Journal of Physiology. Endocrinology and Metabolism. 2012;303:E1126–E1133. doi: 10.1152/ajpendo.00228.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Wu Y, Fried SK. A modified protocol to maximize differentiation of human preadipocytes and improve metabolic phenotypes. Obesity (Silver Spring) 2012;20:2334–2340. doi: 10.1038/oby.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lystedt E, Westergren H, Brynhildsen J, Lindh-Astrand L, Gustavsson J, Nystrom FH, et al. Subcutaneous adipocytes from obese hyperinsulinemic women with polycystic ovary syndrome exhibit normal insulin sensitivity but reduced maximal insulin responsiveness. European Journal of Endocrinology. 2005;153:831–835. doi: 10.1530/eje.1.02027. [DOI] [PubMed] [Google Scholar]
- Nimitphong H, Holick MF, Fried SK, Lee MJ. 25-hydroxyvitamin d(3) and 1,25-dihydroxyvitamin d(3) promote the differentiation of human subcutaneous preadipocytes. PLoS One. 2012;7:e52171. doi: 10.1371/journal.pone.0052171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez LM, Bernal A, San MN, Lorenzo M, Fernandez-Veledo S, Galvez BG. Metabolic rescue of obese adipose-derived stems cells by Lin28/Let7 pathway. Diabetes. 2013;62:2368–2379. doi: 10.2337/db12-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepe A, Tchkonia T, Thomou T, Zamboni M, Kirkland JL. Aging and regional differences in fat cell progenitors—A mini-review. Gerontology. 2011;57:66–75. doi: 10.1159/000279755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahparaki A, Grunder L, Sorisky A. Comparison of human abdominal subcutaneous versus omental preadipocyte differentiation in primary culture. Metabolism. 2002;51:1211–1215. doi: 10.1053/meta.2002.34037. [DOI] [PubMed] [Google Scholar]
- Skurk T, Ecklebe S, Hauner H. A novel technique to propagate primary human preadipocytes without loss of differentiation capacity. Obesity (Silver Spring) 2007;15:2925–2931. doi: 10.1038/oby.2007.349. [DOI] [PubMed] [Google Scholar]
- Skurk T, Hauner H. Primary culture of human adipocyte precursor cells: Expansion and differentiation. Methods in Molecular Biology. 2012;806:215–226. doi: 10.1007/978-1-61779-367-7_15. [DOI] [PubMed] [Google Scholar]
- Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453:783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- Strawford A, Antelo F, Christiansen M, Hellerstein MK. Adipose tissue triglyceride turnover, de novo lipogenesis, and cell proliferation in humans measured with 2H2O. American Journal of Physiology. Endocrinology and Metabolism. 2004;286:E577–E588. doi: 10.1152/ajpendo.00093.2003. [DOI] [PubMed] [Google Scholar]
- Tchkonia T, Giorgadze N, Pirtskhalava T, Tchoukalova Y, Karagiannides I, Forse RA, et al. Fat depot origin affects adipogenesis in primary cultured and cloned human preadipocytes. American Journal of Physiology Regulatory, Integrative and Comparative Physiology. 2002;282:R1286–R1296. doi: 10.1152/ajpregu.00653.2001. [DOI] [PubMed] [Google Scholar]
- Tchkonia T, Giorgadze N, Pirtskhalava T, Thomou T, DePonte M, Koo A, et al. Fat depot-specific characteristics are retained in strains derived from single human preadipocytes. Diabetes. 2006;55:2571–2578. doi: 10.2337/db06-0540. [DOI] [PubMed] [Google Scholar]
- Tchkonia T, Tchoukalova YD, Giorgadze N, Pirtskhalava T, Karagiannides I, Forse RA, et al. Abundance of two human preadipocyte subtypes with distinct capacities for replication, adipogenesis, and apoptosis varies among fat depots. American Journal of Physiology. Endocrinology and Metabolism. 2005;288:E267–E277. doi: 10.1152/ajpendo.00265.2004. [DOI] [PubMed] [Google Scholar]
- Tchoukalova YD, Koutsari C, Votruba SB, Tchkonia T, Giorgadze N, Thomou T, et al. Sex- and depot-dependent differences in adipogenesis in normal-weight humans. Obesity (Silver Spring) 2010;18:1875–1880. doi: 10.1038/oby.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunaitis V, Borutinskaite V, Navakauskiene R, Treigyte G, Unguryte A, Aldonyte R, et al. Effects of different sera on adipose tissue-derived mesenchymal stromal cells. Journal of Tissue Engineering and Regenerative Medicine. 2011;5:733–746. doi: 10.1002/term.374. [DOI] [PubMed] [Google Scholar]
- Yu G, Floyd ZE, Wu X, Hebert T, Halvorsen YD, Buehrer BM, et al. Adipogenic differentiation of adipose-derived stem cells. Methods in Molecular Biology. 2011;702:193–200. doi: 10.1007/978-1-61737-960-4_14. [DOI] [PubMed] [Google Scholar]
- Zierath JR, Livingston JN, Thorne A, Bolinder J, Reynisdottir S, Lonnqvist F, et al. Regional difference in insulin inhibition of non-esterified fatty acid release from human adipocytes: Relation to insulin receptor phosphorylation and intra-cellular signalling through the insulin receptor substrate-1 pathway. Diabetologia. 1998;41:1343–1354. doi: 10.1007/s001250051075. [DOI] [PubMed] [Google Scholar]