

Abstract
One X chromosome, selected at random, is silenced in each female mammalian cell. Xist encodes a noncoding RNA that influences the probability that the cis-linked X chromosome will be silenced. We found that the A-repeat, a highly conserved element within Xist, is required for the accumulation of spliced Xist RNA. In addition, the A-repeat is necessary for X-inactivation to occur randomly. In combination, our data suggest that normal Xist RNA processing is important in the regulation of random X-inactivation. We propose that modulation of Xist RNA processing may be part of the stochastic process that determines which X chromosome will be inactivated.
To balance X-linked gene dosage between males and females, female mammals silence one X chromosome in each cell. In eutherian embryos, X-inactivation is random such that each X has an equal probability of being inactivated1. Female mouse embryonic stem (ES) cells also have two active Xs and undergo random X-inactivation when differentiated2, providing a model system in which to study this process.
The choice of which X will be silenced is regulated in cis by the X-inactivation center (Xic). The Xic encodes an antisense pair of genes, Xist and Tsix, each of which produces a noncoding RNA3. Heterozygous loss-of-function mutations in either Xist or Tsix cause nonrandom X-inactivation4–7, indicating that these RNAs influence the probability of silencing. Xist also has a role in silencing the inactive X (Xi)8. When X-inactivation is triggered, Xist RNA coats the X that will become the Xi and recruits chromatin-modifying factors9,10. A conserved repeat element at the 5′ end of Xist, the A-repeat, is necessary for silencing when Xist is ectopically expressed from an inducible cDNA transgene11. This element is also necessary for silencing during imprinted X-inactivation in mouse extraembryonic tissues, where the paternally inherited X is silenced in every cell12.
Female mice heterozygous for a deletion of the A-repeat (ΔA) undergo nonrandom X-inactivation, with the ΔA X always remaining active12. This nonrandom X-inactivation may occur because the cells that attempt to silence the mutant X die owing to the Xist mutation (secondary nonrandom X-chromosome inactivation, or secondary XCI) or because the mutant X is never selected for silencing in the first place (primary nonrandom X-chromosome inactivation, or primary XCI). To distinguish between these possibilities, we deleted the A-repeat from one X in female mouse ES cells and assayed the effects on random X inactivation. Our results show that female ΔA cells undergo primary XCI, demonstrating that the A-repeat is necessary for random choice. In addition, we identify two new functions of the A-repeat that may explain why X-inactivation is nonrandom in ΔA cells. First, the A-repeat is important for Xist RNA processing, and second, the A-repeat binds alternative splicing factor, or splicing factor-2 (ASF/SF2). In combination, our data suggest a model in which Xist RNA splicing is a regulatory step used to ensure that X-inactivation occurs randomly.
RESULTS
Deletion of the A-repeat causes primary XCI
To investigate the role of the A-repeat, we generated a female ES cell line bearing an A-repeat deletion (XΔAX). We targeted the 129/Sv (129) X in female ES cells carrying one X of 129 origin and one of Mus castaneous (cas) origin (Supplementary Fig. 1). To determine the effect of the A-repeat deletion on X-inactivation, we differentiated XΔAX and parental XX cells and used allele-specific strand-specific reverse transcriptase (RT)-PCR to determine the ratio of 129 to cas Xist RNA. In wild-type 129/cas cell lines, X-inactivation is skewed away from a 1:1 ratio because the 129 and cas X chromosomes contain different alleles of the X controlling element13. The differentiated parental XX cells showed a skewed ratio of 129 transcripts to cas transcripts, whereas differentiated XΔAX cells expressed only cas Xist transcripts (Fig. 1a). This result indicates that the ΔA chromosome never becomes the Xi.
Figure 1.
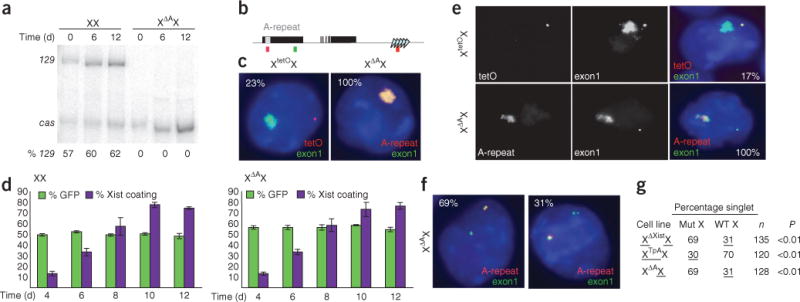
XΔAX cells undergo primary nonrandom X-inactivation. (a) Allele-specific RT-PCR for spliced Xist RNA (exon 1–exon 3) in wild-type and XΔAX cells at 0, 6 and 12 d of differentiation. % 129, percentage of total Xist RNA expressed from 129 X. (b) Genomic structure of Xist (black boxes indicate exons), showing positions of the A-repeat (grey box) and the tetO arrays (triangles) in XtetOX. Positions of FISH probes for A-repeat (red), tetO (red) and Xist exon 1 (green) are indicated below. (c) Allele-specific FISH in differentiated XtetOX (left) and XΔAX (right) ES cells. TetO DNA FISH probes and A-repeat RNA FISH probes were used to identify the tetO and wild-type alleles, respectively, whereas exon1 probes identified all Xist transcripts. DAPI-stained nuclei are blue, the A-repeat or tetO array is in red, and exon 1 is in green. Inset numbers indicate the percentage of cells with the pattern shown for each cell type. Scale bar indicates 2 μm. (d) Survival assay measuring the competitiveness of wild-type (left) and XΔAX (right) cells when codifferentiated with GFP-expressing male ES cells. Green bars indicate the percentage of GFP-negative cells. Purple bars indicate the percentage of female cells, as determined by female-specific patterns of Xist and Tsix expression, with Xist RNA coating. At each time point, the cells were trypsinized and samples divided in half for analysis by RNA FISH or for GFP fluorescence. At least 100 cells were counted for each time point in each replicate. Bars indicate 1 s.d. (e) Allele-specific FISH in XtetOX (top) and XΔAX (bottom) cells, showing both Xist RNA coating and Xist-Tsix pinpoint expression. In the merged image, nuclei are blue, exon 1 is in green, and tetO or the A-repeat is in red. Numbers inset in the right panels indicate the percentage of cells with the pattern shown. Scale bar indicates 1 μm. (f) Allele-specific FISH for Xist and Tsix RNA in XΔAX cells, using A-repeat (red) and exon 1 (green) probes. Both cells show singlet-doublet FISH signals, in which one locus shows a singlet signal and the other a doublet. Inset numbers indicate the percentages of singlet-doublet cells with the patterns shown: the wild-type allele gave the doublet FISH signal in 69% of cells (left) and the singlet signal in 31% of cells (right). Scale bars indicate 1 μm. (g) Summary of results of allele-specific FISH in XΔXistX, XTpAX and XΔAX ES cells. The proportion of singlet-doublet cells in which each allele gives the singlet signal is indicated (Percentage singlet). Mut X, mutant X; WT X, wild-type X. For XΔXistX and XTpAX cells, underline indicates the percentages for the future Xi. n indicates number of nuclei scored. P values indicate that the frequency with which the mutant X in XΔAX ES cells shows a singlet FISH signal is comparable to the frequency with which the future Xa shows a singlet signal in Xist and Tsix heterozygous mutant ES cells.
Next, we used allele-specific fluorescence in situ hybridization (FISH) as an independent assay to determine the frequency with which Xist RNA coats each X in differentiated XΔAX and control cells. A control line, XtetOX5, which is derived from the same parental female ES cell line as XΔAX, carries an insertion of a tet operator (tetO) array that marks the 129 X (Fig. 1b). We used DNA FISH to detect the tetO sequences and RNA FISH to detect Xist transcripts in differentiated XtetOX cells (Fig. 1c, left). Xist RNA coated the unmarked cas X in ~25% of XtetOX cells (9 of 40), consistent with the expected frequency of cas silencing in a 129/cas cross (P = 0.72)5,13. Two RNA FISH probes, one within the A-repeat and a second downstream of the A-repeat in exon 1, were used to identify the wild-type and ΔA alleles in XΔAX cells (Fig. 1b). In 100% of differentiated XΔAX cells (55 of 55), wild-type cas Xist RNA coated the Xi (Fig. 1c, right). This result is significantly different from the 25% of cells expected to silence the cas X (P < 0.0001). Both wild-type and A-repeat–mutant cells showed silencing of X-linked genes on the Xist RNA–coated chromosome (Supplementary Fig. 1). In combination, these allele-specific RT-PCR and FISH data indicate that a ΔA mutation changes the frequency of 129 X silencing from 75% to 0%.
To test whether the ΔA mutation causes primary or secondary XCI, we compared the viability of differentiating wild-type and XΔAX cells. When XX or XΔAX ES cells were codifferentiated with green fluorescent protein (GFP)–expressing wild-type male ES cells, the percentage of XΔAX cells showing Xist RNA coating at each time point was similar to that observed in XX cells (Fig. 1d), indicating normal X-inactivation kinetics in XΔAX cells. In addition, there was no change in the ratio of GFP+ to GFP− cells over time (Fig. 1d). Therefore, differentiating XΔAX cells were not at a proliferative disadvantage relative to XX cells, consistent with primary XCI. Differentiating XΔAX cells do not undergo more cell death than XX cells (data not shown), also consistent with the ΔA mutation affecting choice.
To further distinguish between primary XCI and secondary XCI, we examined Xist and Tsix expression on each X in cells during the early stages of X-inactivation. Shortly after ES cells are induced to differentiate, Xist RNA coats the X that will become the Xi while Tsix and Xist expression persists transiently on the active X (Xa), appearing as a pinpoint FISH signal6,10,14. We used allele-specific RNA FISH to determine which X was silenced in this early stage of X-inactivation in XΔAX and XtetOX cells. In differentiating XtetOX cells, the pinpoint Tsix-Xist RNA signal was associated with the tetO-marked 129 X in 4 of 24 cells (Fig. 1e), consistent with the fact that the 129 becomes the Xa in ~25% of differentiated wild-type cells (P = 0.35)5. In XΔAX cells, the pinpoint Tsix-Xist signal arose from the ΔA X in 18 of 18 cells, which is significantly different from the proportion seen in XtetOX cells (P < 0.0001; Fig. 1e). These results further support the conclusion that XΔAX cells undergo primary XCI.
The results of a third assay were also consistent with primary XCI in XΔAX cells. Before female ES cells are differentiated, the X that will remain active (future Xa) and the X that will be silenced (future Xi) show distinctive FISH signatures15. In heterozygous Xist- and Tsix-mutant ES cells, the future Xa shows predominantly doublet FISH signals at Xist, whereas the future Xi shows predominantly singlet FISH signals15. We compared the Xist loci in XΔAX ES cells to those in heterozygous Xist-mutant (XΔXistX5) and Tsix-mutant (XTpAX7) ES cells. In XΔAX cells, the A-repeat–mutant X showed a high frequency of singlet FISH signals at Xist, a pattern similar to that seen on the future Xa in the Xist- and Tsix-mutant ES cells (Fig. 1f,g and Supplementary Fig. 1). In other words, XΔAX cells show the FISH signature characteristic of cells that will undergo primary XCI. In combination, our results indicate that deletion of the A-repeat from one X in female ES cells results in primary XCI upon differentiation. Thus, the system that determines the choice of which X will become the Xa and which will become the Xi is influenced by the A-repeat.
Histone modifications do not correlate with fixed chromosomal fate
In an ES cell line heterozygous for a deletion that removes the major Tsix promoter, primary XCI occurs and the mutant X always becomes the Xi16. In addition, histone modifications that are associated with silent chromatin, such as enrichment of histone H3 trimethylated on lysine 27 (H3K27me3), and depletion of histone H3 dimethylated on lysine 4 (H3K4me2) and acetylated histone H4 (H4ac), have been reported to mark the mutant X in these cells17. These observations led to the suggestion that chromatin marks preemptively designate the X that will become the Xi. We performed chromatin immunoprecipitation for H3K27me3, H3K4me2 and H4ac in wild-type, XΔAX and XTpAX ES cells to determine whether primary XCI caused by an Xist mutation also correlates with these altered chromatin-modification patterns.
We used restriction enzyme polymorphisms to determine the relative abundance of H3K27me3, H3K4me2 and H4ac in the Xist-Tsix region (Fig. 2a) on the 129 and cas Xs in undifferentiated XX, XΔAX and XTpAX ES cells. In wild-type cells, the distribution of all three modifications was roughly equal between the 129 and cas Xs at the Xist promoter region, in the Xist gene body and at the Tsix promoter region (Fig. 2b,c). Insertion of a polyadenylation signal downstream of the Tsix promoter (XTpAX) caused a skewed distribution of H3K27me3 at all three sites, with a greater proportion of this modification occurring on the mutant X. The distribution of H3K4me2 and H4ac did not differ markedly from that in wild-type cells at any of the three sites.
Figure 2.
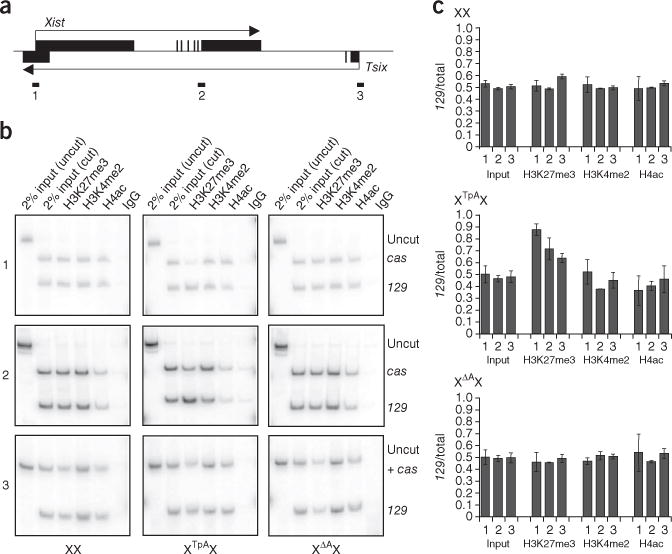
XΔAX cells do not show allelic enrichment for histone modifications. (a) Genomic structure of Xist (above line) and Tsix (below line), with location of PCR amplicons at Xist promoter (1), Xist-Tsix gene body (2) and Tsix promoter (3) indicated. (b) Allele-specific chromatin immunoprecipitation showing distribution of H3K27me3, H3K4me2 and H4ac between the 129 and cas chromosomes in wild-type, XTpAX and XΔAX cells. Both TpA and ΔA mutations are on the 129 chromosome. (c) Data from b plotted as the fraction of products arising from the 129 allele. Fractions represent the average of at least three experiments, and error bars indicate 1 s.d.
In contrast to Tsix-mutant ES cells, XΔAX ES cells showed roughly equal distributions of H3K27me3, H3K4me2 and H4ac between the 129 and cas alleles at all three sites analyzed. Thus, the primary XCI that occurs in differentiated XΔAX ES cells does not correlate with markedly altered distribution of these modifications in undifferentiated ES cells. These data indicate that the unequal distribution of these chromatin marks at Xist and Tsix is not necessary to preemptively mark Xs for primary XCI.
Properly processed ΔA Xist transcripts do not accumulate
We next examined the effect of an A-repeat deletion on Xist RNA processing. An independently generated male XΔAY ES cell line in which the A-repeat and approximately 400 bp of flanking sequence are deleted from the sole Xist locus (Supplementary Fig. 2) was used for this analysis because it enabled us to exclusively measure transcripts derived from ES cells. Populations of ES cells contain a low percentage of differentiated cells even when grown under conditions that promote self-renewal. The amount of Xist RNA in differentiated female cells is several orders of magnitude greater than in undifferentiated ES cells18. As a result, a substantial proportion of the Xist RNA in a population of female ES cells arises from the low percentage of differentiated cells within that population (Supplementary Fig. 2). Because the wild-type cas X always becomes the Xi in XΔAX ES cells, cas Xist RNA from differentiated cells may mask any 129 Xist RNA from the undifferentiated cells in the population. In contrast, Xist is silenced in differentiated male cells19,20, so only ES cell–specific Xist transcripts are assayed.
We compared the levels of spliced Xist RNA in parental XY versus XΔAY ES cells using primers spanning exon 1 to exon 3 (ex1–3) or exon 3 to exon 6 (ex3–6) (Fig. 3a). At 40 cycles of PCR, when detection is in the linear range, correctly spliced ex1–3 and ex3–6 products were observed in XY cells but not in XΔAY ES cells. When mutant cDNA was amplified by 50 cycles of PCR, low levels of Xist cDNA from ex1–3 were detected in a small fraction of reactions (Fig. 3a). A normal ex3–6 product was never detected after 50 cycles of PCR in ΔA cells. However, a smaller-than-expected product, corresponding to aberrantly spliced Xist RNA, was observed at low frequency (Fig. 3a,b and Supplementary Fig. 3). Fifty cycles of PCR amplification using primers in exon 1 and exon 4 or exon 1 and exon 5 also amplified only incorrectly sized products in the mutant cell line (Supplementary Fig. 3). These data indicate that deletion of the A-repeat causes an Xist RNA processing defect.
Figure 3.
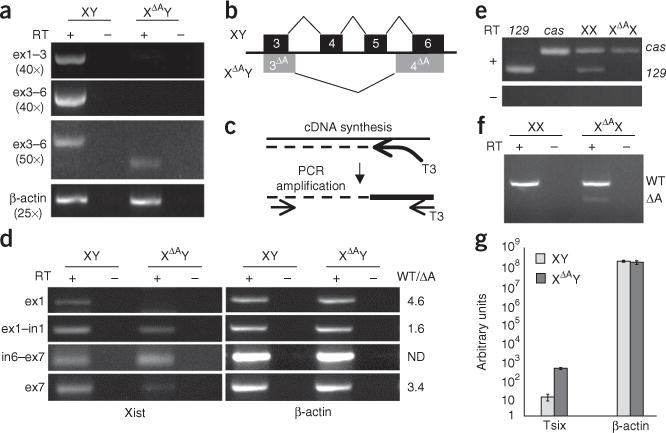
Reduced levels of correctly spliced Xist transcripts from the ΔA mutant chromosome. (a) RT-PCR for spliced Xist exons 1–3 (ex1–3) or exons 3–6 (ex3–6) and β-actin in wild-type (XY) and ΔA (XΔAY) cells. Numbers of PCR cycles used are indicated in parentheses. (b) Diagram showing splicing patterns from Xist exons 3–6 in XY (black) and XΔAY (gray) ES cells. 3ΔA and 4ΔA indicate the aberrant splicing in XΔAY cells. (c) Diagram outlining stand-specific RT-PCR procedure. First-strand synthesis is carried out using a specific primer with a T3 anchor, and PCR is carried out using a second specific primer and a T3 primer. (d) Strand-specific RT-PCR for Xist RNA regions indicated in XY and XΔAY ES cells. The ratio of each transcript in wild-type and mutant cells (WT/ΔA), as determined by RT-qPCR, is indicated at the right. The decrease in transcript abundance within exons 1 and 7 probably reflects the absence of correctly processed Xist RNA in the ΔA mutant. ND, not determined. (e) Allele-specific RT-PCR for spliced Xist RNA (ex3–6) in undifferentiated XX and XΔAX ES cells and control 129 or cas cells. Sizes of 129 and cas bands are indicated on the right. (f) Allele-specific RT-PCR for unspliced Xist RNA in undifferentiated XX and XΔAX ES cells. Primers flank the A-repeat, and as a result the ΔA chromosome produces a shorter transcript. Sizes of wild-type (WT) and ΔA RT-PCR products are indicated on the right. (g) RT-qPCR for Tsix RNA in XY and XΔAY cells. Error bars indicate 1 s.d.
We next examined whether the absence of correctly spliced Xist transcripts in XΔAY ES cells could be attributed to a lack of primary transcripts. Strand-specific RT reactions were performed to ensure that Xist, not Tsix, transcript levels were measured. We found that control RT reactions carried out without primers could yield RT-PCR products (Supplementary Fig. 3), suggesting that Xist and Tsix RNAs can prime each other, as has been reported for other sense-antisense RNA pairs21. As a result, it is unclear whether PCR products from standard strand-specific RT-PCR reactions arise from the Xist or Tsix strand. Therefore, we used a primer containing an anchor sequence for the RT reaction and a primer within the anchor sequence to amplify the cDNA (Fig. 3c). This method ensured that only cDNAs produced from the RT primer were amplified. Across both the first (exon 1–intron 1) and last (intron 6–exon 7) junctions, similar amounts of correctly sized products were detected in wild-type and XΔAY cells (Fig. 3d), as has been previously reported12. Therefore, the absence of correctly spliced Xist RNA cannot be attributed to an absence of primary transcripts from the ΔA allele.
The absence of spliced Xist RNA in XΔAY cells led us to ask whether the ΔA mutation affected accumulation of spliced Xist RNA in female XΔAX cells. We used allele-specific RT-PCR to assess whether spliced Xist RNA was produced from the mutant 129 X. Although both 129 and cas ex1-3 cDNA was detected from wild-type female ES cells, no 129 product was detected in XΔAX cells (Fig. 3e). Thus, spliced Xist RNA is also absent from the mutant X in XΔAX ES cells. Within exon 1 and across the exon 1–intron 1 splice junction, transcripts were detected from the mutant 129 X (Fig. 3f), suggesting that the lack of spliced Xist RNA from the ΔA X is not due to a transcriptional defect. Together, the analyses of male and female ΔA cells indicate that deletion of the A-repeat impairs production or accumulation of spliced Xist RNA.
During imprinted X-inactivation, deletion of the A-repeat results in a loss of Xist RNA and an increase in Tsix RNA levels from the mutant X (ref. 12). Reverse transcriptase quantitative PCR (RT-qPCR) for spliced Tsix RNA (exon 3–exon 4) revealed approximately six-fold more spliced Tsix RNA in XΔAY cells relative to XY control cells (Fig. 3g and data not shown). Therefore, as is the case in imprinted X-inactivation, deletion of the A-repeat causes an increase Tsix RNA levels.
The A-repeat binds ASF/SF2
To gain insight into the molecular role of the A-repeat, we identified proteins that interact with this sequence. Radiolabeled RNA corresponding to a consensus repeat unit (Fig. 4a)11 was incubated with HeLa or ES cell nuclear extracts, and bound complexes were separated by nondenaturing PAGE (Fig. 4b) or by UV cross-linking followed by SDS-PAGE (Supplementary Fig. 4). Two prominent band-shifted complexes were observed with both methods. Both complexes could be competed away with unlabeled A-repeat RNA, but not with a nonspecific competitor, indicating that both complexes were specific to the A-repeat (Fig. 4b). A three-step purification enriched the larger complex (complex II) approximately 50-fold (Supplementary Fig. 4). To determine the protein composition of this complex, this partially purified HeLa nuclear extract was separated by SDS-PAGE. Mass spectrometry identified the most prominent bands that migrated near the molecular weight of the larger complex (Supplementary Fig. 4) as aldolase A and ASF/SF2.
Figure 4.
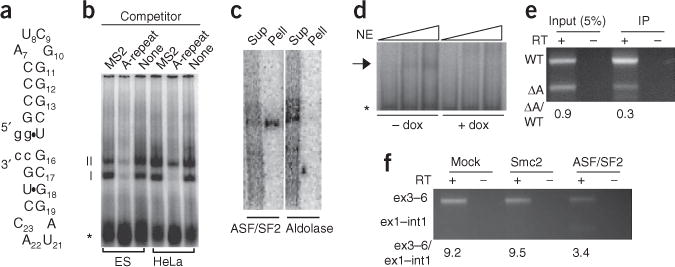
The A-repeat binds ASF/SF2. (a) Predicted hairpin-loop structure of the A-repeat unit consensus. (b) Gel-shift assay using nuclear extracts from ES cells or HeLa cells identified two A-repeat–interacting complexes, I and II. Both complexes were competed away by excess unlabeled A-repeat RNA, but not by control MS2 hairpin RNA. Asterisk indicates unbound radiolabeled A-repeat RNA. (c) Immunoprecipitation of material cross-linked with radiolabeled A-repeat RNA using ASF/SF2 or aldolase A antibodies. Sup, supernatant; Pell, pellet. (d) Gel-shift assay using nuclear extract (NE) prepared from MEFs in which ASF/SF2 is expressed (− dox) or silenced (+ dox) (Supplementary Fig. 4). Triangles above indicate increasing amount of nuclear extract. Arrow indicates the complex that is absent when ASF/SF2 is not expressed. (e) RT-PCR for wild-type (WT) and ΔA XIST RNA. Input shows relative amounts of wild-type and ΔA XIST RNA expressed, and IP shows relative amounts coimmunoprecipitated with HA-ASF/SF2. The numbers below the gel indicate the ratio of ΔA to WT XIST RNA. (f) Simultaneous RT-PCR for spliced (ex3–6) and unspliced (int1–ex1) Xist RNA in male Tsix-mutant ES cells that have been mock depleted or depleted of Smc2 or ASF/SF2 (Supplementary Fig. 4). Numbers beneath the gel indicate the ratio of spliced to unspliced products.
To determine whether either ASF/SF2 or aldolase A bound the A-repeat, radiolabeled A-repeat RNA was incubated with HeLa cell nuclear extract and UV cross-linked. ASF/SF2 antibodies, but not aldolase antibodies, immunoprecipitated the cross-linked, radiolabeled A-repeat RNA–protein complex (Fig. 4c). We next examined whether ASF/SF2 was necessary to band-shift the A-repeat, using a MEF line expressing ASF/SF2 under tet-off control22. Whereas the A-repeat produced a band-shift product when incubated with nuclear extract made from MEFs expressing ASF/SF2, this complex was not present when nuclear extracts were prepared from cells depleted of ASF/SF2 (Fig. 4d). Together these results indicate that ASF/SF2 binds the A-repeat in vitro.
To assess whether ASF/SF2 interacts with XIST RNA in vivo, we simultaneously expressed wild-type human XIST RNA and ΔA XIST RNA in MEFs that express hemagglutinin (HA)–tagged ASF/SF2 (ref. 22). Whole-cell extracts from these cells were immunoprecipitated using an HA antibody. RT-PCR, with primers specific for wild-type XIST RNA and the ΔA XIST RNA, was used to assess the relative amounts of each RNA in the input and the immunopurified samples. Deletion of the A-repeat resulted in a nearly 60% decrease in the amount of XIST RNA that coprecipitates with HA-ASF/SF2 (Fig. 4e). These data suggest that binding of the ~500-bp A-repeat comprises a substantial proportion of the ASF/SF2 binding activity in the ~17-kb XIST transcript.
To determine whether ASF/SF2 is necessary for normal Xist RNA processing in vivo, we compared the effect of ASF/SF2 depletion to the effects of control depletions on the ratio of spliced to unspliced Xist RNA. RNA interference that knocks down expression of ASF/SF2 causes considerable cell death in ES cells. Therefore, we compared ASF/SF2 knockdown to Smc2 knockdown, a control knockdown that causes a similar amount of cell death23. Knockdowns were performed in male Tsix-mutant ES cells7 to eliminate Tsix RNA and simplify analysis. PCR reactions for spliced (exon 3–exon 6) and unspliced (intron 1–exon 1) Xist RNA were carried out simultaneously from the same randomly primed RT reactions. For both mock- and Smc2-depleted cells, this ratio was approximately 10:1. In ASF/SF2-depleted cells, it dropped to 3.5:1 (Fig. 4f). The change in the ratio of unspliced to spliced transcripts was due to both an increase in the abundance of unspliced Xist RNA and a decrease in the amount of spliced RNA. Such an increase in the unspliced precursor and decrease in the spliced product is consistent with inefficient Xist RNA splicing. Collectively, the ASF/SF2 RNA immunoprecipitation and knockdown studies suggest that ASF/SF2 interacts with and regulates Xist RNA processing in vivo.
We next performed mutational analysis on an A-repeat unit to identify sequences necessary for ASF/SF2 binding. Each A-repeat unit is predicted to form a pair of stem-loop structures (Fig. 4a) by the free energy–minimization program MFOLD24,25. In addition, the A-repeat consensus unit contains two sites, identified by ESEfinder26, that conform to the consensus motif for ASF/SF2 (Fig. 5a,b). Complex II did not form with RNAs in which either site was mutated such that it was no longer predicted to bind ASF/SF2 (Fig. 5c and Supplementary Fig. 4, mutations C, E, F, G and H). Mutations designed to disrupt predicted base pairing within either or both stems, but that did not perturb predicted ASF/SF2 consensus sequences, did not affect complex II formation (Fig. 5c and Supplementary Fig. 4, mutations B and D). In combination, these data suggest that the primary sequence, and not the predicted secondary structure, is important for ASF/SF2 to interact with the A-repeat. Because band-shifted complexes required the presence of both predicted ASF/SF2 binding sites within the A-repeat unit, each A-repeat unit may form a bipartite binding site for ASF/SF2, or ASF/SF2 may cooperatively bind within the A-repeat unit.
Figure 5.
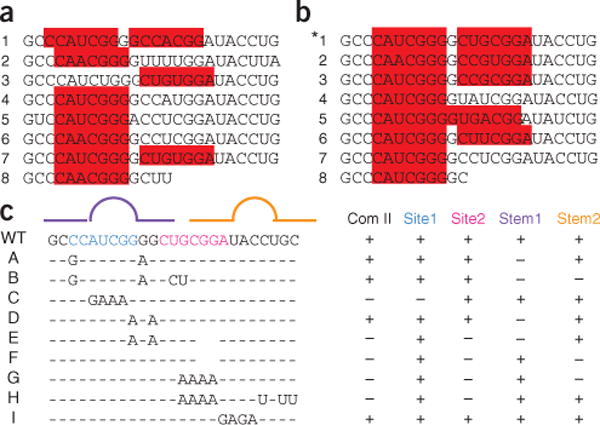
Binding of ASF/SF2 to the A-repeat depends on ASF/SF2 consensus sequences and not predicted secondary structure. (a,b) The sequences of the 7.5 repeat units from mouse (a) and human (b) A-repeat are shown. The consensus repeat unit is indicated by the asterisk. The sequences highlighted in red were identified by ESEfinder as high-score motifs corresponding to an ASF/SF2 consensus26. (c) Summary of A-repeat mutations assayed by gel mobility shift (Supplementary Fig. 4). The lines on top indicate predicted stems (straight lines) and loops (arcs). Stem-loop 1 is purple and stem-loop 2 is orange. The wild-type (WT) A-repeat consensus sequence is written in full. Blue (site 1) and pink (site 2) bases delineate the regions with similarity to the ASF/SF2 consensus sequence. Mutated sequences (A–H) are shown. All sequences are relative to the WT consensus sequence. Dashes represent unchanged bases, letters indicate substitutions and spaces represent deletions. Columns on the right indicate whether each A-repeat mutation maintains (+) or disrupts (−) complex II formation (Com II), base pairing within the predicted stems (Stem1, Stem2), or predicted ASF/SF2 binding at either site (Site1, Site2)24,25.
DISCUSSION
The A-repeat is necessary for Xist RNA to establish silencing in ES cells11, is required for imprinted X-inactivation12 and is sufficient to bind Ezh2 (refs. 27,28). Our studies identify three additional activities for this highly conserved repeat sequence. The A-repeat is necessary to ensure that X-inactivation is random, binds the splicing factor ASF/SF2 and is required for accumulation of spliced Xist RNA. These observations suggest that regulation at the level of Xist RNA processing has a role in the random choice of which X is silenced.
The A-repeat constitutes one of the most conserved regions of Xist11. Mutational analysis within this element indicates that its predicted secondary structure, and not its primary sequence, is important for silencing11. The secondary structure formed by the A-repeat is also recognized by the A-repeat–binding protein Ezh2 (refs. 27,28). In contrast, ASF/SF2’s interaction with the A-repeat is sequence dependent and appears to be independent of predicted secondary structure. Perhaps the A-repeat is so highly conserved because the silencing, Ezh2 binding and ASF/SF2 binding activities need to be maintained simultaneously.
XΔAX cells undergo primary XCI, similar to what is observed in other heterozygous Xist-mutant ES cells29–31. Whereas other mutations abolish or prematurely terminate Xist transcription, deletion of the A-repeat skews choice without eliminating Xist transcription. Therefore, events downstream of Xist transcription must be important for random choice.
In heterozygous Tsix-mutant ES cells, the Xist-Tsix locus on the mutant chromosome has been reported to be enriched for H3K27me3 and depleted for H3K4me2 and H4ac17. Because the Tsix-mutant X is always silenced upon differentiation, it was proposed that the acquisition of these modifications provides a preemptive mark that directs primary XCI. Deletion of the A-repeat also causes primary XCI, but the X that will be silenced in these cells does not acquire H3K27me3 enrichment, depletion of H3K4me2 or histone H4 hypoacetylation. Thus, these epigenetic alterations on the future Xi are not required to predetermine the fates of the Xs. Although differential modification of the Xist-Tsix region is not necessary for primary XCI in XΔAX cells, modifications at other sites may correlate with fate in both Xist- and Tsix-mutant cells. One candidate for such a site is the H3K27me3 hotspot upstream of the Xist promoter32,33. However, analysis of this site did not reveal any allelic bias in H3K27me3, H3K4me2 or H4ac in XΔAX or XTpAX mutant ES cells (Supplementary Fig. 5).
The A-repeat appears to be an unusual regulatory element. In other instances of splicing regulation by exonic sequences, which in many instances contain ASF/SF2 binding sites, mutation of the exonic splicing regulator affects splice-site selection at the exon containing the regulatory sequence, and processing of other exons is unaffected34. In contrast, deletion of the A-repeat disrupts processing of the entire Xist primary transcript, suggesting a novel cis-regulatory role for this highly conserved element. Xist RNA has a very large first exon and an antisense transcript; the position of the A-repeat at the 5′ end of the first exon may aid proper processing of this unusual RNA.
The lack of spliced Xist RNA and elevated levels of spliced Tsix RNA from ΔA chromosomes suggest that the A-repeat is necessary for normal metabolism of both noncoding RNAs. Because Xist, including the A-repeat, is encompassed within Tsix, any mutation of Xist also alters Tsix. As a result, it is difficult to definitively establish whether the altered Xist and Tsix RNA levels that occur in A-repeat–mutant cells are a consequence of the absence of this sequence in Xist, Tsix or both. The possibility that Xist RNA processing is directly regulated by the A-repeat is supported by the fact that only aberrantly spliced Xist transcripts can be detected in ΔA cells, that the A-repeat binds ASF/SF2 and that ASF/SF2 is necessary for normal spliced Xist RNA accumulation. However, it is also possible that the A-repeat does not directly participate in Xist RNA processing, but rather acts indirectly through Tsix. Perhaps the increase in Tsix transcripts in ΔA cells blocks accumulation of spliced Xist RNA in cis. Elucidating the mechanisms by which Xist and Tsix RNA regulate each other and how deletion of the A-repeat affects the production of both transcripts will be important for understanding how these noncoding transcripts ensure that X-inactivation is random.
Any model accurately describing how Xa and Xi fates are assigned in wild-type female cells must meet two criteria. It should account for how the two Xs adopt mutually exclusive fates, and it should explain why each X has an equal probability of becoming the Xi. Most models for stochasticity and mutual exclusivity posit that trans-acting factors asymmetrically assemble on the two Xs in each female cell. As a result of this asymmetric distribution, one X is marked to become the Xa and the other to become the Xi. Transgenic experiments have identified regions of the Xic that, when introduced onto an autosome, trick male ES cells into behaving as if they have two Xs. The minimal transgenes that function in this manner contain Xist and flanking sequences35,36, suggesting that trans-acting factors asymmetrically assemble at a site within or closely linked to Xist. Deletion of the sequences contained in these transgenes does not perturb mutual exclusivity in female cells, indicating either redundancy or an alternative mechanism37. Another model suggests that each X has an independent probability of turning on Xist and being silenced. In this model, secondary selection that eliminates cells that have silenced too many or too few Xs accounts for the mutual exclusivity37.
Here we propose an alternative mechanism in which Xist RNA, and not a DNA element, is central to establishing randomness and mutual exclusivity (Fig. 6). In our model, stochastic differences in Xist RNA levels between the two Xs in each female ES cell underlie stochastic choice. Xist RNA is an attractive candidate as a factor that underlies randomness for several reasons. First, the Xist gene and its promoter are contained in all transgenes that are capable of tricking male cells into ectopically inactivating their sole X35,36. Second, mutations that upregulate Xist expression in ES cells increase the probability that the mutant X will be the Xi6,7,38–40, whereas mutations that decrease Xist expression have the opposite effect31,37,29. In addition, it is estimated that there are approximately ten copies of spliced Xist RNA per female ES cell41. Thus, it is highly likely that the two Xs in wild-type cells will produce different amounts of spliced Xist RNA because of the noise inherent in the production of such a small number of molecules.
Figure 6.
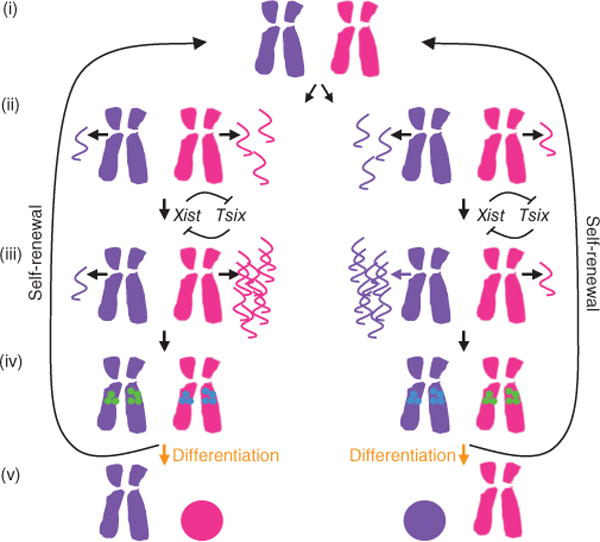
An RNA-based model for randomness and mutual exclusivity. (i) In mitosis, when all transcription ceases, no Xist RNA is produced from either X (purple and magenta). (ii) Once cells enter interphase, the two Xs produce different amounts of mature Xist RNA (purple and magenta wavy lines) by virtue of the noise inherent in production of small numbers of molecules. This noise could be introduced at the level of transcription, splicing or production of a mature ribonucleoprotein complex. (iii) The mutual negative feedback between Xist and Tsix amplifies the initial stochastic difference between the amount of Xist RNA produced from the two Xs. (iv) The X producing less Xist RNA is marked to become the Xa (green circles), and/or the X producing the most Xist RNA is marked to become the Xi (blue circles). (v) Cells that commit to differentiation read out these marks and silence the Xi, whereas cells that self-renew erase these RNA-dependent marks and reset them after the next mitosis.
Once established, any stochastic difference could be amplified by a feedback loop that depends on the mutual regulation of Xist and Tsix. The feedback between Xist and Tsix would ensure that the two Xs robustly adopt mutually exclusive fates by amplifying any small difference in the amount of Xist RNA produced from each chromosome. In our model, the X that produces less mature Xist RNA becomes the Xa upon differentiation. This regulatory network might function in parallel with a mechanism that restricts X-inactivation to cells with more than one Xic42. In such an RNA-based model, stochasticity and feedback ensure that the Xs in XX cells randomly and reliably adopt mutually exclusive fates.
An RNA-based model can also explain why heterozygous Xist and Tsix mutations cause primary XCI without perturbing a cell’s ability to designate one Xa and one Xi. In cells heterozygous for an Xist deletion, the mutant X never produces mature Xist RNA and is always designated as the Xa. In cells heterozygous for an Xist mutation that increases expression, the mutant X always produces more Xist RNA than the wild-type X and always becomes the Xi. When Tsix is mutated, negative regulation of Xist is lost, and as a result the mutant X always produces more Xist RNA than the wild-type X and is designated as the Xi. Our model may also explain why homozygous Tsix-mutant ES cells silence both Xs at high frequency43. Without Tsix, the feedback loops on both chromosomes are broken and small differences in Xist RNA levels are not amplified, impairing the mechanism that ensures mutual exclusivity.
Although the roles of Xist and Tsix in randomness are well established, the molecular framework in which they function remains elusive. We propose that the noise inherent in the production of the small amount of mature Xist RNA expressed before X-inactivation is triggered, combined with the mutual feedback between Xist and Tsix, provide a means to ensure randomness and mutual exclusivity.
METHODS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/nsmb/.
ONLINE METHODS
Cell culture
The wild-type female ES cell line ES 2–1 (ref. 14) and XtetOX (ref. 5), XTpAX (Fa2L; ref. 7), XTpAY (Ma2L; ref. 7), XΔXistX (X1loxX; ref. 31), XΔAY (ref 44), parental XY (ref. 45) and XΔAX ES cells were maintained on male feeder cells. XY-GFP ES cells45 were grown without feeders. For differentiation, ES cells were grown in suspension culture for 4 d to generate embryoid bodies. Embryoid bodies were then plated on gelatinized dishes and grown until the day indicated, with day 0 being the day cells were initially aggregated. For experiments involving undifferentiated ES cells, feeders and differentiated cells were first removed by successive adsorptions on gelatinized dishes. Transformed MEFs expressing HA-ASF/SF2 under control of a tet-off promoter were cultured as described previously22.
Genomic targeting
To produce the ΔA-targeting construct, a puromycin cassette flanked by loxP sites was cloned between the 5′ and 3′ homology arms that directly flank the A-repeats. The 5′ and 3′ homology arms were generated by PCR using primers indicated in Supplementary Table 1.
Fluorescence in situ hybridization
FISH was performed as previously described15. Primers, plasmids or bacterial artificial chromosomes used to generate probes are indicated in Supplementary Table 1. Xist exon 1 or A-repeat concatamer PCR products were used as templates for Xist and A-repeat probes, respectively. The tetO sequences were detected as described previously5.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was carried out using standard protocols46, with the following antibodies: H3K27me3 (Abcam 6002), H3K4me2 (Upstate Biotechnology 07-030), H4ac (Upstate Biotechnology 06-598) or rabbit IgG (Jackson ImmunoResearch 011-000-C03).
Allele-specific PCR
PCR reactions from RT-PCR or ChIP were diluted 1:2 into a new PCR mixture containing a singly radiolabeled primer (indicated by asterisk in Supplementary Table 1) and one final cycle of PCR was performed before digestion with the appropriate restriction enzyme (indicated in Supplementary Table 1). Digested products were separated on an 8% polyacrylamide gel. Products were visualized on a PhosphoImager (Molecular Dynamics), and ImageQuant (Molecular Dynamics) was used to calculate the relative proportion of 129 and cas PCR products in each reaction.
Reverse transcriptase PCR and quantitative PCR
RT was performed on total RNA isolated with Trizol reagent (Invitrogen) and treated with RNase-free DNase (Promega). For RT-PCR of spliced transcripts, first-strand cDNA was synthesized from 0.5 μg of total RNA using random primers, and 40-cycle PCR were performed. To detect unspliced Xist transcripts, cDNA was synthesized from 5 μg of total RNA using a strand-specific primer containing a 5′ T3 anchor sequence. Reactions with and without RT were performed for all samples, and controls without primers were included for all strand-specific RT reactions (data not shown). One-twentieth of the first-strand cDNA was amplified by 50-cycle PCR. Strand-specific RT-qPCR was internally controlled using a T7-tagged β-actin primer in the RT reaction, followed by qPCR with T7- and β-actin–specific primers. All primers are indicated in Supplementary Table 1.
Gel mobility shift and UV cross-linking assays
A-repeat RNA (5′-GGGCCCAUCGGGGCUGCGGAUACCUGCC-3′; Dharmacon) was labeled at the 3′ end with 5′- [32P]pCp and RNA ligase. End-labeled RNA (10 nM) was incubated with 10–20 μg of ES, MEF or HeLa (Chemicon) nuclear extract and 10 μg tRNA in 20 mM HEPES (pH 7.9), 50 mM KCl, 10% (v/v) glycerol and 1 mM DTT in a volume of 20 μl at 4 °C for 30 min. A 100-fold molar excess of wild-type A-repeat or nonspecific competitor RNA (MS2 hairpin, 5′-AGCUGAGGAUCACCCAGCA-3′; Dharmacon) was included where indicated. The mixture was separated by nondenaturing 6% PAGE at 4 °C or UV cross-linked (800 mJ, on ice, 10–15 min). The cross-linked reaction was added to two volumes of sample buffer (20% (v/v) glycerol, 5% (w/v) SDS, 2 M Tris HCl (pH 6.7), 0.2 mg ml−1 bromophenol blue, 5% (w/v) β-mercaptoethanol), boiled for 10 min, placed on ice 5 min and separated by 12% SDS-PAGE. Gels were fixed in methanol and acetic acid, dried and visualized using a PhosphoImager (Molecular Dynamics). For protein purification, unlabeled A-repeat RNA was incubated with HeLa nuclear extract and partially purified as described in Supplementary Figure 4. The mixture was separated by nondenaturing 10% PAGE with a 4% stack and stained with silver.
RNA immunoprecipitation
Transformed MEFs that express HA-tagged ASF/SF2 were transfected using Lipofectamine 2000 (Invitrogen). Cells were simultaneously transfected with vectors that express wild-type and ΔA XIST46,47, and immunoprecipitation was performed as described48, except that phosphatase inhibitors were omitted and Dynal magnetic beads were used, with mouse monoclonal HA-specific antibody (Sigma). Total RNA and randomly primed first-strand cDNA were prepared as described for RT-PCR. PCR was performed using primers specific to wild-type or ΔA XIST (Supplementary Table 1).
RNA interference
ASF/SF2 and Smc2 depletions were carried out using in vitro-diced short interfering RNAs46. Primer sequences for generation of in vitro transcription templates for short interfering RNAs are shown in Supplementary Table 1.
Immunostaining
Immunostaining for ASF/SF2 using a mouse monoclonal antibody (Covance) was carried out using standard procedures49.
Imaging
All microscopy was performed at room temperature, on slides mounted with Vectashield (Vector Laboratories). FISH results were analyzed using an Olympus BX60 fluorescent microscope with a ×100 oil immersion objective, numerical aperture 1.30. Images were captured with a Hamamatsu ORCA-ER digital camera and Openlab 4.0.1 software. Grayscale images were combined into an RGB image using Photoshop (Adobe), with Cy3, FITC and DAPI images pasted into the red, green and blue channels, respectively. The Photoshop Levels tool was used to adjust the upper and lower input levels for each channel to match the upper and lower boundaries of the image histogram. All figures were assembled in PowerPoint (Microsoft).
Statistics
The P values for allele-specific RNA FISH were determined by comparing the observed distribution of signal patterns at each allele to the expected 25–75 distribution in a χ2 distribution test with one degree of freedom. For P-value calculations for singlet-doublet cells, the expected frequency of singlet-doublet cells in which the future Xa showed a singlet FISH signal was 69%15.
Supplementary Material
Acknowledgments
We thank K. Worringer, J. Huff, T. Fazzio, M.K. Alexander, L. Spector, P. O’Farrell and I. Listerman for critical reading of the manuscript, X.-D. Fu and S. Lin (University of California, San Diego) for tet-repressible ASF/SF2 cells, N. Krogan for mass spectrometry and A. Krainer (Cold Spring Harbor) for ASF/SF2 antibodies. This work was funded in part by US National Institutes of Health grant R01 GM088506.
Footnotes
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
AUTHOR CONTRIBUTIONS
M.E.R.-T., A.A.A., H.R.K., D.J.T. and B.P. designed and performed experiments and wrote the manuscript, and A.W., I.D.T. and G.F.K. provided cell lines.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L) Nature. 1961;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- 2.Martin GR, et al. X-chromosome inactivation during differentiation of female teratocarcinoma stem cells in vitro. Nature. 1978;271:329–333. doi: 10.1038/271329a0. [DOI] [PubMed] [Google Scholar]
- 3.Avner P, Heard E. X-chromosome inactivation: counting, choice and initiation. Nat Rev Genet. 2001;2:59–67. doi: 10.1038/35047580. [DOI] [PubMed] [Google Scholar]
- 4.Marahrens Y, Loring J, Jaenisch R. Role of the Xist gene in X chromosome choosing. Cell. 1998;92:657–664. doi: 10.1016/s0092-8674(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 5.Gribnau J, Luikenhuis S, Hochedlinger K, Monkhorst K, Jaenisch R. X chromosome choice occurs independently of asynchronous replication timing. J Cell Biol. 2005;168:365–373. doi: 10.1083/jcb.200405117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JT, Davidow LS, Warshawsky D. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet. 1999;21:400–404. doi: 10.1038/7734. [DOI] [PubMed] [Google Scholar]
- 7.Luikenhuis S, Wutz A, Jaenisch R. Antisense transcription through the Xist locus mediates Tsix function in embryonic stem cells. Mol Cell Biol. 2001;21:8512–8520. doi: 10.1128/MCB.21.24.8512-8520.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wutz A, Jaenisch R. A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol Cell. 2000;5:695–705. doi: 10.1016/s1097-2765(00)80248-8. [DOI] [PubMed] [Google Scholar]
- 9.Panning B, Jaenisch R. DNA hypomethylation can activate Xist expression and silence X-linked genes. Genes Dev. 1996;10:1991–2002. doi: 10.1101/gad.10.16.1991. [DOI] [PubMed] [Google Scholar]
- 10.Sheardown SA, et al. Stabilization of Xist RNA mediates initiation of X chromosome inactivation. Cell. 1997;91:99–107. doi: 10.1016/s0092-8674(01)80012-x. [DOI] [PubMed] [Google Scholar]
- 11.Wutz A, Rasmussen TP, Jaenisch R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat Genet. 2002;30:167–174. doi: 10.1038/ng820. [DOI] [PubMed] [Google Scholar]
- 12.Hoki Y, et al. A proximal conserved repeat in the Xist gene is essential as a genomic element for X-inactivation in mouse. Development. 2009;136:139–146. doi: 10.1242/dev.026427. [DOI] [PubMed] [Google Scholar]
- 13.Cattanach BM, Rasberry C. Identifcation of the Mus castaneus Xce allele. Mouse Genome. 1994;92:114–115. [Google Scholar]
- 14.Panning B, Dausman J, Jaenisch R. X chromosome inactivation is mediated by Xist RNA stabilization. Cell. 1997;90:907–916. doi: 10.1016/s0092-8674(00)80355-4. [DOI] [PubMed] [Google Scholar]
- 15.Mlynarczyk-Evans S, et al. X chromosomes alternate between two states prior to random X-inactivation. PLoS Biol. 2006;4:e159. doi: 10.1371/journal.pbio.0040159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JT, Lu N. Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell. 1999;99:47–57. doi: 10.1016/s0092-8674(00)80061-6. [DOI] [PubMed] [Google Scholar]
- 17.Sun BK, Deaton AM, Lee JT. A transient heterochromatic state in Xist preempts X inactivation choice without RNA stabilization. Mol Cell. 2006;21:617–628. doi: 10.1016/j.molcel.2006.01.028. [DOI] [PubMed] [Google Scholar]
- 18.Norris DP, et al. Evidence that random and imprinted Xist expression is controlled by preemptive methylation. Cell. 1994;77:41–51. doi: 10.1016/0092-8674(94)90233-x. [DOI] [PubMed] [Google Scholar]
- 19.Brockdorff N, et al. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature. 1991;351:329–331. doi: 10.1038/351329a0. [DOI] [PubMed] [Google Scholar]
- 20.Brown CJ, et al. The human XIST gene: analysis of a 17 kb inactive X-specifc RNA that contains conserved repeats and is highly localized within the nucleus. Cell. 1992;71:527–542. doi: 10.1016/0092-8674(92)90520-m. [DOI] [PubMed] [Google Scholar]
- 21.Perocchi F, Xu Z, Clauder-Munster S, Steinmetz LM. Antisense artifacts in transcriptome microarray experiments are resolved by actinomycin D. Nucleic Acids Res. 2007;35:e128. doi: 10.1093/nar/gkm683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin S, Xiao R, Sun P, Xu X, Fu XD. Dephosphorylation-dependent sorting of SR splicing factors during mRNP maturation. Mol Cell. 2005;20:413–425. doi: 10.1016/j.molcel.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 23.Fazzio T, Panning B. Condensin complexes regulate mitotic progression and interphase chromatin structure in embryonic stem cells. J Cell Biol. 2010;188:491–503. doi: 10.1083/jcb.200908026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 25.Zuker M, Jacobson AB. “Well-determined” regions in RNA secondary structure prediction: analysis of small subunit ribosomal RNA. Nucleic Acids Res. 1995;23:2791–2798. doi: 10.1093/nar/23.14.2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–756. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maenner S, et al. 2-D structure of the A region of Xist RNA and its implication for PRC2 association. PLoS Biol. 2010;8:e1000276. doi: 10.1371/journal.pbio.1000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marahrens Y, Panning B, Dausman J, Strauss W, Jaenisch R. Xist-defcient mice are defective in dosage compensation but not spermatogenesis. Genes Dev. 1997;11:156–166. doi: 10.1101/gad.11.2.156. [DOI] [PubMed] [Google Scholar]
- 30.Sado T, Hoki Y, Sasaki H. Tsix silences Xist through modifcation of chromatin structure. Dev Cell. 2005;9:159–165. doi: 10.1016/j.devcel.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 31.Csankovszki G, Panning B, Bates B, Pehrson JR, Jaenisch R. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat Genet. 1999;22:323–324. doi: 10.1038/11887. [DOI] [PubMed] [Google Scholar]
- 32.Heard E, et al. Methylation of histone H3 at Lys-9 is an early mark on the X chromosome during X inactivation. Cell. 2001;107:727–738. doi: 10.1016/s0092-8674(01)00598-0. [DOI] [PubMed] [Google Scholar]
- 33.Rougeulle C, et al. Differential histone H3 Lys-9 and Lys-27 methylation profiles on the X chromosome. Mol Cell Biol. 2004;24:5475–5484. doi: 10.1128/MCB.24.12.5475-5484.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 35.Herzing LB, Romer JT, Horn JM, Ashworth A. Xist has properties of the X-chromosome inactivation centre. Nature [see comments] 1997;386:272–275. doi: 10.1038/386272a0. [DOI] [PubMed] [Google Scholar]
- 36.Lee JT, Lu N, Han Y. Genetic analysis of the mouse X inactivation center defines an 80-kb multifunction domain. Proc Natl Acad Sci USA. 1999;96:3836–3841. doi: 10.1073/pnas.96.7.3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monkhorst K, Jonkers I, Rentmeester E, Grosveld F, Gribnau J. X inactivation counting and choice is a stochastic process: evidence for involvement of an X-linked activator. Cell. 2008;132:410–421. doi: 10.1016/j.cell.2007.12.036. [DOI] [PubMed] [Google Scholar]
- 38.Nesterova TB, et al. Skewing X chromosome choice by modulating sense transcription across the Xist locus. Genes Dev. 2003;17:2177–2190. doi: 10.1101/gad.271203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nesterova TB, et al. Dicer regulates Xist promoter methylation in ES cells indirectly through transcriptional control of Dnmt3a. Epigenetics Chromatin. 2008;1:2. doi: 10.1186/1756-8935-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sado T, Hoki Y, Sasaki H. Tsix defective in splicing is competent to establish Xist silencing. Development. 2006;133:4925–4931. doi: 10.1242/dev.02670. [DOI] [PubMed] [Google Scholar]
- 41.Shibata S, Lee JT. Characterization and quantitation of differential Tsix transcripts: implications for Tsix function. Hum Mol Genet. 2003;12:125–136. doi: 10.1093/hmg/ddg010. [DOI] [PubMed] [Google Scholar]
- 42.Wutz A, Gribnau J. X inactivation Xplained. Curr Opin Genet Dev. 2007;17:387–393. doi: 10.1016/j.gde.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Lee JT. Homozygous Tsix mutant mice reveal a sex-ratio distortion and revert to random X-inactivation. Nat Genet. 2002;32:195–200. doi: 10.1038/ng939. [DOI] [PubMed] [Google Scholar]
- 44.Blewitt ME, et al. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat Genet. 2008;40:663–669. doi: 10.1038/ng.142. [DOI] [PubMed] [Google Scholar]
- 45.Fazzio TG, Huff JT, Panning B. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell. 2008;134:162–174. doi: 10.1016/j.cell.2008.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen HR, Panning B. XIST RNA exhibits nuclear retention and exhibits reduced association with the export factor TAP/NXF1. Chromosoma. 2007;116:373–383. doi: 10.1007/s00412-007-0100-1. [DOI] [PubMed] [Google Scholar]
- 47.Plath K, et al. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300:131–135. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- 48.Huang Y, Yario TA, Steitz JA. A molecular link between SR protein dephosphorylation and mRNA export. Proc Natl Acad Sci USA. 2004;101:9666–9670. doi: 10.1073/pnas.0403533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nusinow DA, et al. The histone domain of macroH2A1 contains several dispersed elements that are each sufficient to direct enrichment on the inactive X chromosome. J Mol Biol. 2007;371:11–18. doi: 10.1016/j.jmb.2007.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.