

Abstract
The tumor suppressor protein p53, encoded by TP53, inhibits tumorigenesis by inducing cell cycle arrest, senescence and apoptosis. Several genetic polymorphisms exist in TP53, including a proline to arginine variant at amino acid 72 (P72 and R72, respectively); this polymorphism alters p53 function. In general, the P72 variant shows increased ability to induce cell cycle arrest, while the R72 variant possesses increased ability to induce apoptosis, relative to P72. At present, the underlying mechanisms for these functional differences are not fully understood. Toward elucidating the molecular basis for these differences a gene expression microarray analysis was conducted on normal human fibroblast cells that are homozygous for P72 and R72 variants, along with subclones of these lines that express a p53 short hairpin (shp53). Approximately three dozen genes were identified whose transactivation is affected by the codon 72 polymorphism. One of these is the tripartite motif family-like 2 (TRIML2) gene, which is preferentially induced by the R72 variant. Importantly, the accumulated data indicate that TRIML2 interacts with p53, and facilitates the modification of p53 with SUMO2. TRIML2 also enhances the ability of p53 to transactivate a subset of pro-apoptotic target genes associated with prolonged oxidative stress, including PIDD, PIG3 (TP53I3) and PIG6 (PRODH). These data indicate that TRIML2 is part of a feed-forward loop that activates p53 in cells expressing the R72 variant, particularly after prolonged stress.
Keywords: p53, TRIML2, apoptosis, DNA damage, Pidd, Pig3, Pig6
INTRODUCTION
The p53 tumor suppressor gene (TP53) is inactivated by mutation in more than half of human cancers (1). Following stresses such as DNA damage or oncogene activation, p53 is activated to trigger a variety of biological functions to suppress tumor development, including cell cycle arrest, senescence and apoptosis. This versatility in p53 outcomes lies primarily in its role as a transcription factor, and its ability to transcriptionally activate different classes of p53 target genes that play roles in different outcomes (2). For example, the p53 target genes CDKN1A (p21) and Gadd45 play key roles in p53-induced cell cycle arrest, while the BH3-only-encoding target genes BBC3 (Puma) and PMAIP1 (Noxa) are critical players in p53-mediated apoptotic cell death (3). One of the challenges in p53 research has been to identify how this protein selectively induces different classes of target genes, and different cell fates, in response to different stresses. To date, protein-protein interactions and post-translational modification have been linked to the ability of p53 to selectively transactivate cell cycle arrest or pro-apoptotic target genes (4). In addition, the cell-of-origin, and the level and type of stress, are also implicated in the p53 decision between growth arrest and apoptosis (5). Finally, data from our group and others indicate that this decision can also be affected by a common polymorphism in the p53 gene at codon 72, encoding either proline (P72) or arginine (R72).
The codon 72 polymorphism in p53 is the most common coding region polymorphism in the TP53 gene (6). There is a distinct latitudinal bias in the frequencies of P72 and R72 alleles, with the P72 allele more common in populations near the equator (7). This latitudinal bias in codon 72 allele frequency has been suggested to be associated with either the level of UV exposure or winter temperature (8). The change from a proline to an arginine at amino acid 72 is predicted to result in a significant structural change of p53 (9), and several functional differences between these polymorphic variants have been described. Specifically, under the same DNA damage signals, the P72 variant preferentially promotes cell cycle arrest, while the R72 variant shows superior ability to induce apoptosis (9, 10). At present, the underlying basis for the differences in growth arrest and apoptosis between these variants is incompletely understood. In this study we undertook an unbiased approach toward this question, and identified a p53 target gene that is transactivated to a significantly greater extent by the R72 variant of p53, in multiple different cell lines containing endogenous or inducible p53. We show that this gene, TRIML2, encodes a protein that feeds back on p53 to bind to it and target it for SUMO-2 modification. We further show that cells with higher levels of TRIML2 show superior ability to transactivate a subset of p53 target genes that are associated with prolonged DNA damage and apoptosis, including PIDD (PIDD1), PIG3 (TP53I3) and PIG6 (PRODH). We posit that the R72 variant possesses enhanced apoptotic potential, at least in part, because of its superior ability to transactivate TRIML2; this leads to increased transactivation of a subset of p53 target genes that are associated with prolonged DNA damage and apoptosis.
MATERIALS AND METHODS
Cell culture, reagents and plasmids
Unless otherwise mentioned, all cell lines were obtained from the American Type Culture Collection, and cells were used during early passages after being thawed from frozen stocks. Normal Human Fibroblast (NHF) cells were purchased from the Coriell Institute for Medical Research (Camden, NJ, U.S.A.) and cultured in 15% fetal bovine serum (Gembio), Penicillin (100 I.U./ml)-Streptomycin (100μg/ml) (Pen/Strep, Cellgro, 30-002-CI), Non-Essential Amino Acids (NEA, Cellgro, 25-030-CI), L-glutamine (Cellgro, 25-005-CI) in Dulbecco’s Modification of Eagle’s Medium (DMEM, Cellgro). Temperature-sensitive Saos2 cells containing P72 or R72 variant of p53 were described previously (11). These cells were maintained at 39°C in DMEM supplemented with 10% FBS and Pen/Strep. H1299 cells containing Tet-inducible variants of p53 P72 and R72 were provided by Steven McMahon (Thomas Jefferson University) (12) and cultured in DMEM supplemented with 10% Tet approved system FBS (Clontech, 631106) and Pen/Strep. Hct116 p53 WT and Hct116 p53−/− cells were provided by Bert Vogelstein from Johns Hopkins School of Medicine (13). These cells were cultured in McCoy’s 5A medium (Cellgro, 10-050-CV) supplemented with 10% FBS and Pen/Strep. U2OS-Trex cells were provided by Pradip Raychaudhuri (University of Illinois, Chicago) and maintained in DMEM supplemented with 10% Tet system approved FBS and Pen/Strep. Etoposide (Sigma, E1383) was used at a concentration of 20-100 uM. 5-fluoruracil (5-FU, Sigma, 858471) was used at a concentration of 5 μM. A concentration of 0.75 μg/ml doxycycline (BD Biosciences, 631311) was used to induce gene expression from tetracycline-inducible constructs. Ginkgolic acid (Sigma, 75741) was used as sumoylation inhibitor at a concentration of 100μM. TRIML2 cDNA was purchased in pCR4-TOPO vector from Open Biosystems (8327427) and subcloned into pCR2.1-TOPO vector through TOPO TA cloning according to manufacturer’s protocols (Invitrogen, K4575-01SC). TRIML2 overexpressing construct was created by subcloning TRIML2 to pcDNA3.1D/V5-His-TOPO vector through restriction enzyme digestions (Hind III and Xho I) and ligation. TRIML2 was subsequently subcloned into pcDNA4/TO vector through Hind III/Xho I digestions and ligation to generate tetracycline-inducible construct. Stable cells overexpressing pcDNA3.1-TRIML2 or pcDNA4/TO-TRIML2 were maintained under the selection using 400μg/ml G418 and 100μg/ml Zeocin, respectively. Expression constructs (all in pRK5 vector) of TRIM27 (Flag-tagged), PML (isoform IV, Flag-tagged), Ubiquitin (HA-tagged), SUMO1 (His-tagged), and SUMO2 (His-tagged) were obtained from Xiaolu Yang (University of Pennsylvania) (14). Fugene 6 transfection reagent (Promega) was used for all transfection experiments.
Human p53 knock-in (Hupki) mice
Hupki P72 and R72 mice were described previously (12). All studies with mice complied with all federal and institutional guidelines as per IACUC protocols. Mice were housed in plastic cages with ad libitum diet and maintained at 22°C with a 12-hour dark/12- hour light cycle. Primary murine embryonic fibroblasts (MEFs) obtained from 13.5-day-old Hupki mouse containing either homozygous P72 or R72 p53 were grown in DMEM supplemented with 10% FBS and 1% Pen/Strep. For irradiation experiments, mice were exposed to a cesium-137 gamma source (The Wistar Institute) and tissues harvested were subjected to RNA extraction using RNeasy Mini kit (Qiagen, 74104).
Gene expression microarray
Normal Human Fibroblast (NHF) cells expressing homozygous P72 or R72 forms of p53 as well as cells expressing a short hairpin RNA against p53 (shp53) were treated with 5 Gy of gamma radiation. RNA was isolated from the cells using TRIzol (Invitrogen, 15596-026) before being amplified and labeled using the Agilent Quick Amp labeling kit. Amplified cDNAs were hybridized onto human gene expression 4×44K v2 arrays (Agilent, G4845A) according to the Agilent protocol. Hybridized slides were scanned at a 5-μm resolution on an Agilent scanner, and fluorescence intensities of hybridization signals were extracted using Agilent Feature Extraction software. Raw expression data obtained from Agilent microarrays were background corrected and quantile normalized across the experimental conditions (15). The LIMMA (Linear Models for Microarray Data) methodology was applied to the log2-transformed expression data to identify differentially expressed genes in each comparison. The LIMMA module in the Open Source R/Bioconductor package was utilized in the computations (16). Differentially expressed genes were identified based on statistical significance (p<0.01) as well as biological significance using fold change cutoff. Genes identified through microarray were analyzed through the use of IPA (Ingenuity® Systems,www.ingenuity.com) for their associated functions and diseases. Gene expression data were deposited into the GEO database with accession number GSE61124.
Lentiviral transduction of shRNA
Stable cell lines for shRNA knockdowns were generated by infection with the lentiviral vector pLKO.1-puro carrying a shRNA sequence against TRIML2: shA(TCCAATGTTAAATGTCTCTGG) TRCN0000150366, shB(TTTAGCTGCTTCAAGTTTCTC) TRCN0000150766, and shC(AAATCCAATCTTTCTGGGTTG ) TRCN0000150389 (Open Biosystems). VSVG-pseudotyped lentivirus was generated by cotransfection of 293-FT cells with shRNA constructs and packaging vectors according to the manufacturer’s protocols (Invitrogen, K4960-00). Lentivirus was added to cells with Polybrene (6μg/ml) for maximum viral transduction. Stable cells were selected using puromycin (1μg/ml), and gene knockdown was confirmed by qRT-PCR and western blot analysis.
Western blot analysis and Immunoprecipitation
For western blot analysis, 25-50 μg of protein was resolved over sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using pre-cast NuPAGE® Bis-Tris gels (Life Technologies) and transferred onto PVDF membranes (Bio-Rad). Primary antibodies used in this study include p53 (Ab6, Calbiochem, OP43), p53 Ser-15-P (Cell Signaling, 9284), p53 Ser-46-P (Cell Signaling, 2521), TRIML2 (Sigma, HPA043838), TRIML2 (Abcam, ab87292), FLAG-Tag (M2, Sigma, F3165), GAPDH (14C10, Cell Signaling, 2118), Actin (AC-15, Sigma, A5441), p21 (Ab6, Calbiochem, OP79), MDM2 (Ab1-OP46, Ab2-OP115, Calbiochem), Cleaved lamin A (Cell Signaling, 2035), Cleaved caspase 3 (Cell Signaling, 9061), PIDD (Anto-1, Novus Biologicals, NBP1-97595), Caspase 2 (Cell Signaling, 2224), PIG3 (Ab1, Oncogene, PC268), PML (Santa Cruz, sc-5621), PARP (46D11, Cell Signaling, 9532), SUMO2/3 (Cell Signaling, 4971), SUMO1 (Cell Signaling, 4930), Ubiquitin (Cell Signaling, 3933). Secondary antibodies conjugated to Horseradish peroxidase were used at a dilution of 1:10,000 (Jackson Immunochemicals). ECL (Amersham, RPN2232) was then applied to blots and protein levels were detected using autoradiography. Densitometry quantification of protein signals was performed using ImageJ software (NIH). For immunoprecipitation, a total of 500-2000 μg of whole cell lysate was pre-cleared by protein G-agarose beads (Millipore, 16266) and incubated with 1 μg of antibody overnight at 4°C. Protein G-agarose beads were then added for 1 h, followed by washes using lysis buffer. Pulled-down proteins were eluted using 2X Laemmli sample buffer (62.5 mM Tris-HCl, pH 6.8, 25 % glycerol, 2 % SDS, 0.01 % Bromophenol Blue) and subjected to western blot analysis as described earlier. Horseradish peroxidase-conjugated light-chain-specific secondary antibody was used (Jackson Immunochemicals).
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
Treated cells were lysed using the QIAshredder columns (Qiagen, 79656) and total RNA was isolated using RNeasy Mini kit (Qiagen, 74104) including on-column DNase digestion (Qiagen, 79254) following the manufacturer’s protocol. Equal amounts of RNA from samples were used to create cDNA with the High Capacity cDNA Reverse Transcription Kit (Applied Biosciences, 4368814). Quantitative PCR (qPCR) was performed using Brilliant III Ultra Fast SYBR® Green QPCR Mix kit (Agilent Technologies, 600882) on the Stratagene Mx3005P device (Agilent Technologies), according to the manufacturer’s protocol. Data analysis was done using the MxPro program (Stratagene). Messenger RNA expression levels were normalized to cyclophilin A. Primers used in qRT-PCR analyses were designed based on published sequence information (Ensembl genome browser) and to prevent amplication of genomic DNA of target genes (Table S2).
Chromatin immunoprecipitation
Chromatin immunoprecipitation (16) analysis was performed according to the manufacturer’s protocol (EZ-ChIP; Millipore) with slight modifications. Briefly, cells were cultivated in 100-mm plates to 90% confluence and scrape harvested. Cells were then fixed for 10 min in freshly made fixation solution (1% formaldehyde, 10mM NaCl, 0.1mM EDTA, 5mM HEPES, pH7.9). Fixation was stopped by 0.125M Glycine, followed by PBS wash, and cells were lysed in SDS lysis buffer (1% SDS, 10mM EDTA, 50mM Tris pH8.1) containing protease inhibitors. Chromatin was sheared by sonication to an average size of ~200 to 500 bp, clarified, and precleared for 1 h at 4°C with salmon sperm DNA-saturated protein A-Sepharose beads (Millipore, 16-157). The supernatant was incubated with normal mouse immunoglobulin G or anti-p53 (Ab6, Calbiochem, OP43) and rotated overnight at 4°C. Lysates were immunoprecipitated with salmon sperm DNA-saturated protein A-Sepharose beads for 1 h at 4°C and washed extensively according to the manufacturer's instructions. Input and immunoprecipitated protein/DNA complexes were eluted at room temperature, and the cross-linking was reversed overnight at 65°C in the presence of 200 mM NaCl. After RNase A (37°C for 30 min) and proteinase K (45°C for 2 h) treatment, sample DNAs were purified through DNA columns for further analysis. Conventional PCR of ChIP products was performed with GoTaq® Hot Start Polymerase (Promega, M5001). Quantitative PCR (qPCR) was performed as described above. p53 responsive elements (REs) on TRIML2 were predicted through MatInspector software of Genomatix using consensus the p53 binding sequence RRRCWWGYYY (R=A or G; W =A or T; Y = C or T), allowing no more than 2 mismatches (17). Primer pairs used for different ChIP target sequences are listed in Table S2.
Immunofluorescence; Annexin V and Cell viability assays; Statistical analysis
p53-null H1299 cells seeded in 6-well plates were co-transfected with pcDNA3-TRIML2 and p53 (in pRc/CMV vector) expression constructs for 48 h, followed by 4% paraformaldehyde fixation and 0.2% Triton X-100 permeabilization. Transfected cells were stained with antibodies to p53 (Ab6, Calbiochem, OP43) and TRIML2 (Sigma, HPA043838) and then incubated with fluorescence-conjugated secondary antibodies (Jackson Immunoresearch Laboratories, Rhodamine Red-X RRX anti-rabbit, 111-295-144; Alexa Fluor 488 anti-mouse, 115-545-146). Counterstaining with DAPI was applied to stain the cell nucleus. The samples were imaged using a Leica TCS SP5 II scanning laser confocal system (Wistar Institute Imaging Facility). Annexin V staining to detect apoptotic cells and cell viability assay were performed using Guava Nexin Reagent (Millipore, 4500-0450) and ViaCount Reagent (Millipore, 4000-0041) on the Guava easyCyte HT system according to the manufacturer’s protocols, respectively. For statistical analysis, data were analyzed by two-sided unpaired Student's t test using a GraphPad software package. Data were considered significant if p<0.05.
RESULTS
Identification of genes that are differentially regulated by the P72 and R72 variants of p53
In order to identify p53 target genes that are differentially regulated by the P72 and R72 variants of p53, we purchased and analyzed over two dozen normal human fibroblast (NHF) cell lines from the Coriell Institute by genotyping them for the codon 72 polymorphism. This led to the identification of four NHF cell lines, two of which were homozygous for P72, and two of which were homozygous for R72; these were all early passage and showed similar doubling times of approximately twenty-four hours (not shown). Two of these lines were selected for further analysis. We first set out to show that these cell lines, which are from non-related individuals, respond similarly to DNA damage. In response to etoposide, these P72 and R72 NHF lines showed nearly identical p53 induction, phosphorylation, and upregulation of MDM2 and CDKN1A (p21) (Figure 1A).
Figure 1. TRIML2 is differentially regulated between the P72 and R72 variants of p53.

(A) Normal human fibroblasts (NHFs) homozygous for P72 or R72 variants of p53 were treated with etoposide (20μM and 100μM) for 24 hr. Whole cell lysates were extracted and western blot analysis was performed for p53 (p53, Ser15-P-p53 and Ser46-P-p53) and target genes (MDM2 and p21). Actin serves as the loading control.
(B) Western blot analysis for TRIML2 protein in P72 and R72 NHF lines four hours following 5 Gy gamma radiation. GAPDH serves as the loading control.
(C) Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) analysis in two independent sets of P72 and R72 normal human fibroblasts (NHFs) following treatment with 4 hr 5-Gy gamma-irradiation for the expression level of CDKN1A (p21) and TRIML2, normalized to control (cyclophilin A). The data depicted are the averaged results from three independent experiments; error bars mark standard error. The asterisk denotes a p-value <0.05.
(D) Top panel: the non small-cell lung carcinoma cell line H1299 containing doxycycline-inducible versions of P72 and R72 were analyzed by qRT-PCR for the induction of TRIML2 mRNA following treatment with doxycycline (0.75μg/ml) and etoposide (100μM) for 24 hr, relative to control (cyclophilin A). Bottom panel: the level of p53 and TRIML2 protein induced is depicted. GAPDH serves as a loading control. The data depicted are the averaged results from three independent experiments; error bars mark standard error. The asterisk denotes a p value <0.05. P=P72, R=R72. Doxy = doxycycline. Eto = etoposide.
(E) Mouse embryo fibroblasts generated from Humanized p53 knock-in (Hupki) mice were treated with etoposide (100 μM) for twenty-four hours and RNA was isolated and used for qRT-PCR analysis; the fold induction of TRIML2 is normalized to control (cyclophilin A). In the upper panel, the level of p53 protein is depicted. GAPDH serves as a loading control. The data depicted are the averaged results from three independent experiments; error bars mark standard error. The asterisk denotes a p value <0.05.
(F) Humanized p53 knock-in (Hupki) mice were treated with 5-Gy gamma-irradiation for four hours and RNA was isolated from small intestine and used for qRT-PCR analysis; the fold induction of TRIML2 is normalized to control (cyclophilin A). The data depicted are the averaged results from three independent experiments; error bars mark standard error. The asterisk denotes a p value <0.05.
Two of the P72 and R72 NHF lines, which we denote P72-1 and R72-1, were used to create sub-clones expressing a p53 short hairpin (shp53), and were confirmed to express significantly reduced p53 levels (data not shown). Subsequently all six cell lines (P72-1, P72-1shp53, P72-2, R72-1, R72-1shp53, and R72-2) were subjected to 5 Gy gamma radiation and RNA was isolated after 0, 2 and 4 hours for micro-array analysis on Agilent arrays (Figure S1A); gamma irradiation was chosen as the stressor so that these findings could be compared with a previous analysis from our group of P72- and R72-induced genes in the mouse (12). This analysis was performed in triplicate on independent samples; for data analysis we focused on genes whose expression level was changed more than 50% post-irradiation in all four P72 and R72 cell lines, and whose upregulation was absent or significantly reduced in NHFs expressing the short hairpin to p53 (Figure S1B). This analysis revealed over two hundred genes regulated in a p53-dependent manner; the overwhelming majority of these genes were regulated similarly in P72 and R72 NHFs. A significant number of these “commonly-regulated” genes were previously identified p53 target genes whose protein products are associated with known p53-mediated functions, including cell death, cell cycle, and DNA repair (Figure S1C). This analysis also revealed approximately three dozen genes that were differentially regulated between P72 and R72 NHFs; this subset included genes that were induced only in P72, or only in R72 (Table S1 and Figure S1D). Interestingly, the majority of the “differentially-regulated” genes could not be linked to classic p53-associated functions (Figure S1C, bottom panel).
Several of the genes showing greater up-regulation by the P72 variant were identical to those previously identified by our group as showing preferential up-regulation by this variant in mouse cells (12). Conversely, there was only one gene identified as being specifically upregulated in R72 NHFs; this was the gene encoding TRIML2 (identified as FLJ25801 in the microarray gene list), which was up-regulated in the R72 NHFs, but not P72, and whose upregulation was lost in shp53-expressing cells (Fig S2A). Western blot analysis confirmed the increased expression of TRIML2 protein following gamma radiation in R72 but not P72 NHFs (Figure 1B). Importantly, qRT-PCR validated that this gene was significantly upregulated in both sets of R72 NHFs, but was not induced in either P72 NHF cell line following gamma radiation (p<0.05 comparing P72 versus R72, Figure 1C).
To validate the above findings, we next tested three other cell line model systems for the preferential induction of TRIML2 by the R72 variant of p53. One of these model systems consisted of H1299 (p53 null non-small cell lung carcinoma) cells containing a doxycycline-inducible version of p53 encoding either the P72 or R72 variant (11). In these cells, the P72 variant showed some ability to upregulate TRIML2 following etoposide treatment, but the R72 variant reproducibly showed significantly increased ability to induce TRIML2 (p<0.05 comparing P72 versus R72, Figure 1D). This difference was evident at the protein level as well (Figure 1D, bottom panel). Similar findings were made in Saos2 (human osteosarcoma) cells containing temperature-sensitive versions of the P72 and R72 alleles (Figure S2B). Moreover, in mouse embryonic fibroblasts (MEFs) from the P72 and R72 Humanized p53 knock-in (Hupki) mouse, the up-regulation of Triml2 in response to etoposide was significantly greater in R72 MEFs compared to P72 (p<0.05, Figure 1E). Finally, increased induction of Triml2 by the R72 variant was also seen in the small intestine from Hupki mice treated with gamma radiation (p<0.05, Figure 1F); in this tissue we have previously seen increased apoptosis in R72 mice, compared to P72 (18). The combined data indicate that in five different cell line systems, three with endogenous p53 and two with inducible versions, the TRIML2 gene is consistently preferentially transactivated by the R72 variant.
TRIML2 is a p53 target gene
To determine whether TRIML2 is a direct p53 target gene, Hct116 human colorectal cancer cells, and the somatic cell knockout counterpart Hct116 p53 −/− were treated with etoposide for twenty-four hours, and the RNA and protein level of TRIML2 was assessed. This analysis revealed that TRIML2 is induced by etoposide at both the RNA and protein levels, and that this induction is significantly (but not completely) reduced in p53 −/− cells (Figure 2A). We also analyzed TRIML2 induction in response to other stresses. p53-dependent induction of TRIML2 is evident in Hct116 cells in response to 5-fluoruracil (5-FU) treatment and following ultraviolet (UV) irradiation (Figure S2C). Analysis of the TRIML2 promoter and intronic regions revealed the presence of three consensus p53 binding sites (Figure 2B). To test the possibility that p53 could bind to these, we performed chromatin immunoprecipitation analysis using p53 antibody in Hct116 cells treated with 5-FU. As positive controls for p53-binding, we analyzed the known p53 consensus elements in the CDKN1A (p21) and MDM2 target genes. ChIP analysis of the three p53 consensus elements in the TRIML2 gene revealed that two of these sites, both located in the TRIML2 promoter (−1439 and −2855, relative to the start site of transcription, +1), were consistently immunoprecipitated with p53 antibody, while the third site was not (Figure 2B). We next used ChIP in order to test the ability of P72 and R72 proteins to bind to the TRIML2 promoter. For this experiment we used H1299 cells expressing doxycycline-inducible versions of P72 and R72, as these express equivalent levels of both p53 variants (Figure 2C). As controls, we analyzed the interaction of these variants with the CDKN1A (p21) promoter, which we and others showed demonstrates enhanced binding by P72 protein (19), and the p53 consensus element in the BTG2 gene, which according to our microarray was induced to identical levels in P72 and R72 cells (Figure S1B and data not shown). As expected, these analyses revealed that the P72 and R72 protein bound identically to the BTG2 target gene, and that the P72 variant bound to greater extent to the p21 consensus site (Figure 2C). Interestingly, however, the R72 protein immunoprecipitated both RE1 and RE2 of the TRIML2 promoter significantly more efficiently than P72 (Figure 2C). These data suggest that the R72 variant preferentially transactivates the TRIML2 gene, at least in part, due to enhanced binding of the R72 protein to the promoter elements in this gene.
Figure 2. TRIML2 is a p53 target gene.

(A) The human colorectal cancer cell line Hct116 containing wild-type p53 (WT) or the somatic cell knock-out for p53 (p53−/−) were treated with etoposide (100μM) and monitored after twenty-four hours. Whole cell lysates and extracted RNA were subjected to western blot analysis (top panel) and qRT-PCR (bottom panel), respectively. Densitometry quantification of TRIML2 levels, normalized to GAPDH, is depicted. Expression levels of mRNA were normalized to cyclophilin A. The data depicted are the averaged results from three independent experiments; error bars mark standard error. The asterisk denotes a p-value <0.05.
(B) Left panel: Chromatin immunoprecipitation of Hct116 cells treated with DMSO or 5-FU (5μM) for 72 hr. Immunoprecipitated DNA was eluted and analyzed by q-PCR and regular PCR using primers flanking p53 response elements (RE) on the known p53 target genes MDM2 and CDKN1A (p21) as positive controls. Upper right panel: diagram of the TRIML2 gene, along with three potential consensus p53 response elements (RE). The start site of transcription (TSS) is denoted +1, and the location of p53 REs is shown relative to this. Ex= exon. Bottom right panel: Immunoprecipitated DNA was eluted and analyzed by PCR using primers flanking the p53 REs (Table S2). In each case the percent of DNA bound, normalized to input, is depicted. The data depicted are the averaged results from three independent experiments; error bars mark standard error. The asterisk denotes a p-value <0.05.
(C) Left panel: western blot analysis for p53, p21, TRIML2 and loading control (GAPDH) in H1299 cells containing doxycycline-inducible P72 and R72 forms of p53 following doxycycline treatment (0.75μg/ml) for 24 hr. Right panels: Chromatin immunoprecipitation analyses in H1299 cells expressing doxycycline-inducible P72 or R72 forms of p53 using primers that span the p53 binding site in CDKN1A (p21), BTG2, or RE1 and RE2 of TRIML2 following doxycycline treatment (0.75μg/ml) for 24 hr. The data depicted are the averaged results from three independent experiments; error bars mark standard error. The asterisk denotes a p-value <0.05.
TRIML2 positively regulates p53-mediated apoptosis
In order to assess the impact of TRIML2 on downstream p53 functions, we next sought to silence this gene in cells with WT p53, and assess the impact of this silencing on p53-mediated programmed cell death and transactivation. Toward this goal, we first tested three different short hairpins for their ability to silence TRIML2. We identified one (sh-A, see Materials and Methods) that routinely led to the most efficient silencing (Figure 3A). Lentiviral transduction and selection for this short hairpin into Hct116 cells led to the isolation of several clones with silenced TRIML2, and this silencing did not appear to markedly impact cell behavior (data not shown). As expected, treatment of these clones with etoposide led to decreased induction of TRIML2 protein, compared to vector-transduced controls; interestingly however, this also led to decreased steady state levels of p53 (Figure 3A, bottom panel). These data suggest that TRIML2 positively regulates p53 protein levels.
Figure 3. TRIML2 is a positive regulator of p53 activation and apoptosis.

(A) Upper panel: lentiviral transduction of control vector and three different short hairpins for TRIML2 (sh-A, -B and –C) in Hct116 cells, followed by western blot analysis for TRIML2 and loading control (Actin). Lower panel: western blot analysis for TRIML2, p53, CDKN1A (p21) and GAPDH in Hct116 cells infected with parental vector or sh-A for TRIML2. Stably-infected cells were treated with etoposide (100μM) for 24 hr. The data depicted are representative of three independent experiments. Densitometry quantification of TRIML2 levels, normalized to Actin or GAPDH, is depicted.
(B) Left panel: western blot analysis of TRIML2, p53, cleaved caspase 3 and cleaved lamin A (CC3 and CLA, both markers of apoptosis) in Hct116 cells infected with control vector or sh-A to TRIML2, treated with 5-FU (5μM) for 48 or 72 hr. Densitometry quantification of gel images was done with ImageJ software and normalized to GAPDH. The data depicted are representative of three independent experiments. Upper right panel: flow cytometric analysis of Annexin V staining in Hct116 cells stably-infected with control vector or sh-A of TRIML2, following treatment with 5-FU (5μM) for 72 hr. Bottom right panel: flow cytometric analysis of cell viability in Hct116 cells stably infected with control vector of sh-A of TRIML2, following treatment with 5-FU (5 μM) for 24, 48, or 72 hr. The averaged results from three independent experiments are shown, and error bars mark standard error. Asterisk denotes p<0.05.
(C) Left panel: western blot analysis of U2OS cells containing doxycycline-inducible vector alone (vector) or doxycycline-inducible TRIML2, following treatment with 0.75 μg/mL doxycycline and 100 μM etoposide for twenty-four hours. Cleaved lamin A (CLA) and cleaved caspase 3 (CC3) were used as markers for apoptosis. The data depicted are representative of three independent experiments. Right panel: flow cytometric analysis of Annexin V staining in U2OS cells containing doxycycline-inducible vector alone (vector) or doxycycline-inducible TRIML2, following treatment with 0.75 μg/mL doxycycline, and 100 μM etoposide for twenty-four hours. The averaged results from three independent experiments are shown, and error bars mark standard error. The triple asterisk (***) denotes p<0.0005.
We next extended these analyses to include an assessment of programmed cell death in TRIML2 silenced cells. In Hct116 cells treated with 5-FU, knockdown of TRIML2 led to decreased steady state level of p53, as well as decreased levels of cleaved lamin A and cleaved caspase 3 at late timepoints (72 hours), indicating that apoptosis was impaired (Figure 3B). Similar findings were made when a different short hairpin (sh-B) to TRIML2 was used, indicating this was not an off-target effect (Figure S2D). A similar decrease in apoptosis was evident in U2OS cells with TRIML2 silenced (Figure S2E). Annexin V staining and cell viability assays using flow cytometry confirmed that TRIML2 knockdown results in significant inhibition of 5-FU-induced apoptosis and increased viability of Hct116 cells after 72 hours (Figure 3B, upper and lower right panels). We next sought to test the impact of overexpression of TRIML2 on p53 stabilization and apoptosis. Toward this end, we created a doxycycline-inducible TRIML2 cell line, in the background of U2OS cells, which contain WT p53. Doxycycline treatment of these but not parental cells followed by etoposide revealed that overexpression of TRIML2 protein led to increased levels of p53, along with increased induction of the apoptosis markers cleaved lamin A and cleaved caspase-3 (Figure 3C). This increase in apoptosis was particularly evident when apoptosis was assayed by Annexin V staining (Figure 3C, right panel). Similar findings were made in Hct116 cells stably-transfected with TRIML2 (Figure S2F). The combined data indicate that TRIML2 positively regulates p53 levels, and can also enhance p53-mediated apoptosis.
We next sought to test the hypothesis that the increased induction of TRIML2 in cells containing the R72 variant of p53 might contribute to the higher apoptotic potential of this variant, relative to P72. In order to test this hypothesis, we generated sub-clones of tetracycline-inducible P72 and R72 cells, each stably-transfected with parental vector or a CMV promoter-driven construct of TRIML2. Analyses of these cells revealed that overexpression of TRIML2 in tetracycline-inducible P72 cells resulted in increased apoptosis (indicated by cleaved PARP and cleaved caspase-3), to levels seen in R72 control cells (Figure S3A). Similarly, we found that silencing TRIML2 in R72-inducible cells led to decreased apoptosis (indicated by cleaved PARP and cleaved lamin A), to levels nearly identical to that in P72 cells infected with the short hairpin control (Figure S3B). These findings support the premise that the increased up-regulation of TRIML2 in R72 cells contributes significantly to the increased apoptotic potential of this variant, relative to P72.
TRIML2 affects the up-regulation of a select set of p53 target genes
We next analyzed the impact of TRIML2 on p53-mediated transactivation of target gene expression. Toward this goal we used a qRT-PCR approach to analyze the expression levels of p53 target genes associated with growth arrest and apoptosis in Hct116 cells expressing vector alone or the short hairpin for TRIML2, in the presence and absence of 5-FU. Analysis of over two dozen p53 target genes revealed that, despite causing a decrease in steady state p53 levels, silencing of TRIML2 did not affect the transactivation of the majority of p53 target genes analyzed, and actually led to increased ability of p53 to transactivate the growth arrest target genes CDKN1A (p21), GADD45, and CYCLIN G1 (Figure 4A). Conversely, at the timepoint analyzed (5-FU treatment for 72 hr), there was no impact of TRIML2 silencing on the transactivation of the pro-apoptotic genes BBC3 (PUMA), Bax, DRAM1 or EI24, with the exception of PMAIP1 (NOXA) (Figure 4A). Interestingly, however, TRIML2 silencing significantly impaired the transactivation of a small subset of p53 pro-apoptotic target genes, consisting of the pro-death genes PIDD (PIDD1), PIG3 (TP53I3) and PIG6 (PRODH). Taken together, these findings implicate TRIML2 in the growth arrest versus cell death decision by p53, as TRIML2 silencing resulted in up-regulation of growth arrest genes (CDKN1A, GADD45 and CYCLIN G1), and decreased expression of three p53 pro-apoptotic genes (PIDD, PIG3 and PIG6).
Figure 4. TRIML2 influences the transcriptional potential of p53.

(A) Upper left panel: western blot analysis of TRIML2 and p53 in Hct116 cells stably-infected with vector alone or sh-A of TRIML2, and treated with 5-FU (5μM) for 72 hours. Upper right panel: qRT-PCR analysis of the samples in (A) for p53 pro-apoptotic target genes that are not downregulated by TRIML2 silencing. Bottom panel: selective p53 target genes whose expression was affected by TRIML2 knockdown. The data depicted are the averaged results from three independent experiments, normalized to control (cyclophilin A); error bars mark standard error. The asterisk denotes a p-value <0.05.
(B) qRT-PCR analysis of the levels of TRIML2-regulated p53 target genes in U2OS cells containing doxycycline-inducible vector alone (vector) or doxycycline-inducible TRIML2, following treatment with 0.75 μg/mL doxycycline, and 100 μM etoposide for twenty-four hours. The level of each gene in untreated, TRIML2-overexpressing cells was set to 1-fold and the data are normalized to cyclophilin A. The data depicted are the averaged results from three independent experiments; error bars mark standard error. The asterisk (*) denotes p<0.05, and the double asterisk (**) denotes p<0.005.
(C) Western blot analysis for TRIML2 and cleaved caspase-2 in Hct116 cells stably-infected with vector alone or sh-A of TRIML2, and treated with 5-FU (5μM) for 48 hours. The data depicted are representative of three independent experiments.
(D) Western blot analysis for TRIML2, PIDD and cleaved caspase-2 in U2OS cells containing doxycycline-inducible vector alone (vector) or doxycycline-inducible TRIML2, following treatment with 0.75 μg/mL doxycycline and 100 μM etoposide for twenty-four hours. Actin serves as the loading control; the data depicted are representative of three independent experiments.
(E) qRT-PCR analysis of the levels of BAX, PIG3, BBC3 (Puma) and PIDD in H1299 cells with tetracycline-inducible P72 or R72, following treatment with 0.75 μg/mL doxycycline plus 100 μM etoposide for twenty-four hours. The level of each gene in DMSO-treated P72 cells was set to 1 and the data are normalized to cyclophilin A. The data depicted are the averaged results from three independent experiments; error bars mark standard error. The double asterisk (**) denotes p<0.005. P=P72, R=R72. Doxy = doxycycline. Eto = etoposide.
To extend our findings, we expanded our qRT-PCR analysis to include several dozen p53 target genes (Figure S4A-B). This analysis was performed using RNA isolated from Hct116 cells with TRIML2 silenced compared to vector control, in the absence or presence of 5-FU. This analysis confirmed that TRIML2 silencing led to reduced expression of not only pro-apoptotic p53 target genes PIDD, PIG3 and PIG6, but also several p53 metabolism target genes, such as GLS2, GLUT1 (SLC2A1), and TIGAR (C12orf5), as well as two genes that require the p53 transactivation domain II (TA II) for transactivation, CRIP2 and KANK3 (Figure S4C-E) (20). We next sought to confirm these findings by analyzing the expression of these p53 target genes in the U2OS cell line containing a tetracycline-inducible version of TRIML2. qRT-PCR analysis confirmed that the p53 pro-apoptotic genes PIDD, PIG3 and PIG6 showed increased expression in etoposide-treated cells when TRIML2 was induced with doxycycline (Figure 4B). Similar increases in gene expression were also seen in Hct116 cells stably transfected with TRIML2 following 5-FU treatment (Figure S5A). In sum, in two different cell types (U2OS and Hct116), using TRIML2 over-expression and silencing, we were able to detect the same set of genes affected by TRIML2.
The protein product of the p53 target gene PIDD regulates p53-mediated apoptosis through the formation of the PIDDosome. The PIDDosome consists of PIDD, RAIDD, and Caspase 2; this complex controls the cleavage and activation of caspase 2 en route to apoptotic cell death (21). We reasoned that altered transactivation of PIDD following TRIML2 overexpression or knockdown might be accompanied by differences in the level of cleaved caspase-2. To test this premise, we analyzed Hct116 cells infected with short hairpin control or TRIML2 short hairpin for the presence of activated (cleaved) caspase 2 following p53 induction. We also analyzed cleaved caspase-2 levels in U2OS cells that overexpress TRIML2. We found that silencing of TRIML2 in Hct116 cells led to significantly decreased level of cleaved caspase-2 following 5-FU treatment (Figure 4C). Further, overexpression of TRIML2 in doxycycline-inducible U2OS cells led to significantly increased PIDD protein, along with increased cleaved caspase-2 upon etoposide treatment (Figure 4D). These data support the premise that the alterations of PIDD level following silencing or overexpression of TRIML2 could lead to the observed effects on apoptosis, as manifested by cleavage and activation of caspase-2.
The impact of TRIML2 on the expression of target genes like PIDD suggests that some of these target genes might show increased expression in cells expressing the R72 variant of p53. To test this premise, we performed qRT-PCR on PIDD and PIG3, compared to BAX and BBC3 (PUMA), in H1299 cells that express tetracycline-inducible versions of P72 and R72 forms of p53. These analyses revealed similar transactivation of BAX and BBC3 (PUMA) in P72 and R72 tet-inducible cells, but significantly increased transactivation of both PIG3 and PIDD in R72 cells (Figure 4E; PIG6 was not expressed in this cell line). Increased PIDD level was also evident in temperature sensitive Saos2-R72 cells, compared to P72 (Figure S5B). The combined data suggest that the R72 variant of p53 possesses increased apoptotic potential due in part to increased ability to transactivate TRIML2, along with enhanced transactivation of genes like PIG3 and PIDD.
TRIML2 regulates apoptosis at late stages of p53 induction
Our data indicate that TRIML2 knockdown reduces apoptosis following p53 induction, but that this was evident only following prolonged periods of p53 induction (72 hr, Figure 3B). To further explore this finding, we performed a time course analysis of apoptosis and p53 target gene expression in Hct116 cells infected with control vector or TRIML2 short hairpin; in this case we used treatment with 5-FU as the genotoxic stress, as this agent has a longer half-life in cell culture medium compared to other cytotoxic agents (22). qRT-PCR analysis revealed that cells with TRIML2 silenced showed a significant decrease in PIG3, PIG6 and PIDD, compared to cells infected with vector alone, as early as twenty four hours following 5-FU treatment (Figure 5A and B). However, the changes in PIG3, PIG6 and PIDD following TRIML2 silencing were most evident at later timepoints (72 and 96 hr, Figure 5B). Similarly, cells expressing TRIML2 short hairpin showed decreased apoptosis at 24 and 72 hours, but this difference was most pronounced at 96 hours following DNA damage (Figure 5C). In contrast, the cell cycle arrest genes CDKN1A (p21) and CYCLIN G1 are not affected at 48 hours and become inversely correlated with TRIML2 expression only at later timepoints (72 and 96 hr, Figure 5A). These combined data all support the premise that TRIML2 has the most pronounced effects on p53 target expression at late timepoints of p53 induction.
Figure 5. TRIML2 shows the most significant effects on late stages of apoptosis.

(A and B) qRT-PCR analysis of Hct116 cells stably-infected with vector control or TRIML2 sh-A following treatment with DMSO or 5-FU (5μM) for 8, 24, 48, 72, or 96 hr. The level of each gene in Vector-infected, DMSO-treated cells at the 0 timepoint was set to 1. The data depicted are the averaged results from three independent experiments, normalized to control (cyclophilin A); error bars mark standard error. The asterisk denotes a p-value <0.05. In (B), genes negatively regulated by TRIML2-silencing are presented.
(C) Flow cytometric analysis of Annexin V positive cells in Hct116 cells stably infected with vector or sh-A of TRIML2, following 24, 72 or 96 hours (36) of treatment with 5-FU (5μM). The percent of Annexin V positive cells are depicted; the data depicted are the averaged results from four independent experiments; error bars mark standard error. The asterisk (*) denotes p<0.05, and the triple asterisk (***) denotes p<0.0005.
TRIML2 interacts with p53, and modifies p53 with SUMO-2/3
TRIML2 is a member of the tripartite-motif family of proteins, the majority of which are reported to possess ubiquitin and/or SUMO-ligase activities (23). As p53 can be modified by ubiquitin, SUMO-1, and SUMO-2/3, we next sought to test the hypothesis that TRIML2 might control p53 function by specifically interacting with, and/or post-translationally modifying p53. Because of the lack of availability of a TRIML2 antibody that works in immunoprecipitation, we performed these assays in Hct116 cells engineered to stably express a V5-tagged form of TRIML2, and we monitored a TRIML2-p53 interaction in these cells following exposure to etoposide. p53 was readily immunoprecipitated with TRIML2 by V5 antibody following DNA damage in stably-transfected cells with endogenous p53 (Figure 6A). We found no evidence for the ubiquitin ligase MDM2 in this complex. We next monitored the sub-cellular localization of p53 and TRIML2. Toward this end we performed immunofluorescence of p53 and TRIML2 in transfected cells, and found that TRIML2 appears to localize to both the cytoplasm and nucleus of cells, and that the nuclear-localized TRIML2 showed significant colocalization with p53 (Figure S6A). These combined findings indicate that p53 and TRIML2 can be associated in a complex together, and that TRIML2 might post-translationally modify p53.
Figure 6. TRIML2 interacts with p53, and regulates p53 modification with SUMO-2/3.

(A) Immunoprecipitation-western blot analysis of TRIML2 in Hct116 cells transfected with V5-tagged TRIML2, following incubation with etoposide (100 uM) for twenty-four hours. Five-hundred μg of whole cell lysate was immunoprecipitated with 1 μg of V5 antibody, and immunoprecipitates were subjected to western blot analysis for p53 and MDM2. WCL: whole cell lysate. IP: immunoprecipitation. The doublet detected by the V5-TRIML2 N-terminally-tagged construct is believed to represent an internal translation species from the first ATG of TRIML2 in this construct, which is detectable using TRIML2 antisera by western blot.
(B) Western blot analysis for p53 and indicated proteins in H1299 cells transfected with TRIML2, SUMO-2, and p53, along with the positive control proteins indicated (TRIM27, PML). The right brace denotes the higher-molecular weight species of p53 that are induced when TRIML2 and SUMO-2 are co-transfected.
(C) Western blot analysis in H1299 cells transfected with p53, TRIML2 and SUMO-2. Ginkgolic acid was used to inhibit sumoylation and verify TRIML2-mediated sumoylation of p53. The right brace denotes the higher-molecular weight species of p53 that are induced. Note that the SUMO-2 tagged version of p53 is increased when TRIML2 is transfected and decreased with the addition of ginkgolic acid.
(D) Immunoprecipitation analysis in H1299 cells transfected with p53, TRIML2 and SUMO-2. p53 was immunoprecipitated with polyclonal antisera against p53, followed by western blot analysis with SUMO-2 antibody. The right brace denotes the higher-molecular weight species of p53 that are induced. IP: immunoprecipitation. IgG HC: Immunoglobulin heavy chain.
We next chose to determine whether TRIML2 might enhance the ubiquitylation or SUMOylation of p53. To test this possibility, we transfected TRIML2 along with WT p53 and either tagged-ubiquitin or tagged SUMO-1 into H1299 cells. We then analyzed the ubiquitylation and SUMOylation patterns of p53 in these cells; as positive controls, we analyzed p53 ubiquitylation and SUMOylation following transfection with TRIM27 and PML, which are known to regulate these processes (14). In these experiments, we failed to detect evidence for increased ubiquitylation or SUMOylation of p53 in TRIML2-transfected cells (Figure S6B-C). As p53 can also be modified with SUMO-2/3 (24, 25), and because some TRIM proteins are reported to be SUMO-2/3 ligases (14), we next tested whether TRIML2 might modify p53 with SUMO-2/3. Toward this end we transfected H1299 cells with p53, TRIML2, and SUMO-2, and analyzed p53 by western blot, looking for increases in the higher molecular weight species that would be indicative of modification with SUMO-2. In these experiments western blot analysis for p53 revealed a readily apparent upward shift in p53 mobility following transfection with TRIML2, similar to what was seen following transfection with the positive controls TRIM27 and PML; these findings were consistent with modification of p53 with SUMO-2 (Figure 6B). To further confirm TRIML2-mediated SUMO-2 sumoylation of p53 in H1299 cells, we used an inhibitor of sumoylation, ginkgolic acid, to specifically inhibit this process (26). In this experiment, the higher molecular weight species of p53 that were induced by TRIML2 expression were decreased by addition of the inhibitor ginkgolic acid, supporting that they are likely SUMO-2-modified versions of p53 (Figure 6C). We next sought to confirm this by immunoprecipitating p53 in TRIML2-transfected cells, followed by western analysis using SUMO2 antibody. This experiment confirmed that the ~70 kDa and 130 kDa species of p53 induced by TRIML2 transfection were in fact p53 modified by SUMO-2 (Figure 6D). These data also suggest that TRIML2 may induce poly-SUMOylation of p53 with SUMO-2; this feature is distinct from SUMO-1, which can be used only to mono-SUMOylate p53 (27, 28).
DISCUSSION
This report marks the first attempt to identify codon 72 allele-specific target genes in normal, non-transformed cells. We identified nearly a hundred genes whose expression was altered significantly in a p53-dependent manner after gamma-irradiation treatment of P72 or R72 NHFs (Figure S1). A small subset of these showed differential regulation by the P72 and R72 variants. The majority of these differentially-regulated genes were preferentially transactivated by the P72 variant, and several of these genes were previously reported by our group (12), including CSF-1, which we showed participates in a feed forward loop to enhance p53-mediated growth arrest in P72 cells (29). The only gene identified in this study as being preferentially induced by R72 is TRIML2. We show that this gene is preferentially induced in cells containing the R72 variant in five different cell line systems, of both mouse and human origin. Notably, in a recent study of hemizygous p53 knockout RKO colon carcinoma cells containing either R72 or P72 alleles, several codon 72 allele-specific transcripts were identified following etoposide treatment; this group also reported TRIML2 as an R72 preferentially-induced p53 target gene (30). We show here-in that this preferential transactivation by R72 is the result of increased ability of this variant to bind to the TRIML2 promoter. How the codon 72 polymorphism, which is located proximal but not within the DNA binding domain of p53, affects the ability of p53 to bind to different response elements in promoters remains to be determined.
TRIML2 is a member of a large tri-partite motif (TRIM) protein family that has more than 70 members in humans to date (23). TRIM protein family members are involved in diverse biological functions, including interferon-induced antiviral and antimicrobial activities (31); recent evidence also implicates TRIM proteins in transcriptional regulation, apoptosis, and cancer (23). There are no published studies on the TRIM family member described here, TRIML2. However, a recent study showed that many TRIM proteins function as ligases for SUMO-1, -2 or -3 (the latter two are 95% identical, so are often referred to as SUMO-2/3) (14). Additionally, a recent study identified TRIML2 as part of the SUMO-2 containing complex in the nucleus of HeLa cells (32). TRIML2 is not the first p53-induced TRIM, nor is it the first TRIM that impacts p53 modification and function. Four other TRIM genes are known to be transactivated by p53, including TRIM8, TRIM19 (PML), TRIM22 and TRIM24 (33-36). qRT-PCR data in our cell lines did not indicate that any of these other TRIMs show clear preferential transactivation by codon 72 variants of p53 (Figure S6D). Interestingly, whereas we found that TRIML2 enhances p53’s pro-apoptotic gene expression and apoptosis and diminishes transactivation of growth arrest genes, TRIM8 does the opposite: it facilitates p53-mediated cell cycle arrest by selectively up-regulating expression of CDKN1A(p21) and GADD45, and decreases transactivation of apoptosis-associated p53 targets (34). These data suggest that TRIMs may be used by the cell to fine-tune the growth arrest versus cell death decision by p53, and indeed that different TRIMs, some of which can hetero-oligomerize, may be able to antagonize each other in order to achieve this function.
Other TRIMs also have been shown to post-translationally modify p53. TRIM8, TRIM19 (PML) and TRIM13 can increase p53 stability and function by inhibiting MDM2-mediated p53 degradation (34, 37, 38). TRIM24, TRIM28, TRIM29 (also known as ataxia-telangiectasia group D complementing, or ATDC) and TRIM39 all inhibit p53 activity by increasing p53 degradation and/or nuclear export, largely by controlling p53 ubiquitylation (39-42). Finally, a recent study showed that TRIM21 inhibits p53 functions indirectly by blocking the formation of the deubiquitylation complex GMPS-USP7 (43). In our hands TRIML2 did not appear to alter ubiquitylation of p53, but rather caused increases in p53 modification with SUMO-2/3. Little is known about modification of p53 with SUMO-2/3. In general, modification of p53 with SUMO-1 is believed to inhibit the DNA binding activity and transcriptional function of p53 (44), but the available data suggest that modification with SUMO-2/3 is different. Specifically, conjugation of p53 with SUMO-2/3 has been seen on lysine residue (K386), and shown to selectively inhibit the transactivation of a subset of p53 transcriptional targets, and possibly to activate other p53 target genes (24, 25). Our findings support the hypothesis that SUMO-2 modification may differentially affect different p53 target genes: we show that TRIML2 increases the transactivation of a subset of pro-apoptotic target genes (PIG3, PIG6 and PIDD) and at the same time decreases the transactivation of growth arrest genes (CDKN1A, GADD45, CYCLIN G1). How modification with SUMO-2/3 affects p53 target gene transactivation remains to be determined. The combined data support a model wherein the R72 variant, through increased transactivation of TRIML2, then shows enriched modification with SUMO-2/3 and increased ability to transactivate a subset of pro-apoptotic p53 target genes, thus leading to increased ability to induce apoptosis (Figure 7).
Figure 7.
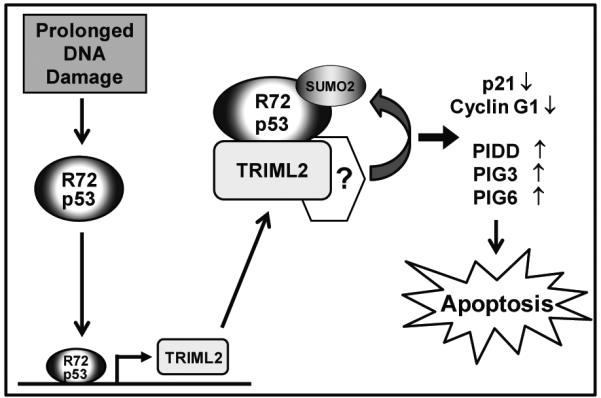
Working model of the positive feed-forward loop between the R72 variant of p53 and TRIML2.
Our data indicate that TRIML2 should be added to the growing list of proteins that control the growth arrest versus cell death decision by p53. Our data also support the hypothesis that increased ability of the R72 variant to transactivate TRIML2 explains part of the increased ability of this variant to induce programmed cell death, relative to P72. Interestingly, the collected cancer microarray data from the ONCOMINE database reveals that TRIML2 is frequently downregulated in a variety of cancers, including gastric, colorectal, lung and liver cancers (data not shown), suggesting that TRIML2 may have anti-cancer or anti-proliferative function. As the P72 variant of p53 has been identified as a risk factor for these same cancers (45-48), these findings provide a potential link between the failure of the P72 variant to induce TRIML2 and its reduced ability to suppress tumor development in these tissues.
Supplementary Material
Implications.
The defined actions of TRIML2, in part, explain the underlying molecular basis for increased apoptotic potential of the R72 variant of p53.
ACKNOWLEDGMENTS
We thank members of the Murphy laboratory for helpful discussions and review of the manuscript. We thank Frederick Keeney and James Hayden of the Imaging Facility (The Wistar Institute) and Yue-Sheng Li of the Microarray Facility at Fox Chase Cancer Center.
Grant support: R01 CA102184 (M.E.M)
Footnotes
The authors declare there are no conflicts of interest.
REFERENCES
- 1.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–58. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–12. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 3.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 4.Carvajal LA, Manfredi JJ. Another fork in the road--life or death decisions by the tumour suppressor p53. EMBO Rep. 2013;14:414–21. doi: 10.1038/embor.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson JG, Post SM, Lozano G. Regulation of tissue- and stimulus-specific cell fate decisions by p53 in vivo. J Pathol. 2011;223:127–36. doi: 10.1002/path.2783. [DOI] [PubMed] [Google Scholar]
- 6.Murphy ME. Polymorphic variants in the p53 pathway. Cell Death Differ. 2006;13:916–20. doi: 10.1038/sj.cdd.4401907. [DOI] [PubMed] [Google Scholar]
- 7.Beckman G, Birgander R, Sjalander A, Saha N, Holmberg PA, Kivela A, et al. Is p53 polymorphism maintained by natural selection? Hum Hered. 1994;44:266–70. doi: 10.1159/000154228. [DOI] [PubMed] [Google Scholar]
- 8.Shi H, Tan SJ, Zhong H, Hu W, Levine A, Xiao CJ, et al. Winter temperature and UV are tightly linked to genetic changes in the p53 tumor suppressor pathway in Eastern Asia. Am J Hum Genet. 2009;84:534–41. doi: 10.1016/j.ajhg.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas M, Kalita A, Labrecque S, Pim D, Banks L, Matlashewski G. Two polymorphic variants of wild-type p53 differ biochemically and biologically. Mol Cell Biol. 1999;19:1092–100. doi: 10.1128/mcb.19.2.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pim D, Banks L. p53 polymorphic variants at codon 72 exert different effects on cell cycle progression. Int J Cancer. 2004;108:196–9. doi: 10.1002/ijc.11548. [DOI] [PubMed] [Google Scholar]
- 11.Dumont P, Leu JI, Della Pietra AC, 3rd, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet. 2003;33:357–65. doi: 10.1038/ng1093. [DOI] [PubMed] [Google Scholar]
- 12.Frank AK, Leu JI, Zhou Y, Devarajan K, Nedelko T, Klein-Szanto A, et al. The codon 72 polymorphism of p53 regulates interaction with NF-{kappa}B and transactivation of genes involved in immunity and inflammation. Mol Cell Biol. 2011;31:1201–13. doi: 10.1128/MCB.01136-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 14.Chu Y, Yang X. SUMO E3 ligase activity of TRIM proteins. Oncogene. 2011;30:1108–16. doi: 10.1038/onc.2010.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–93. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 16.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–4. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 17.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, et al. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–42. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 18.Azzam GA, Frank AK, Hollstein M, Murphy ME. Tissue-specific apoptotic effects of the p53 codon 72 polymorphism in a mouse model. Cell Cycle. 2011;10:1352–5. doi: 10.4161/cc.10.9.15344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salvioli S, Bonafe M, Barbi C, Storci G, Trapassi C, Tocco F, et al. p53 codon 72 alleles influence the response to anticancer drugs in cells from aged people by regulating the cell cycle inhibitor p21WAF1. Cell Cycle. 2005;4:1264–71. doi: 10.4161/cc.4.9.1978. [DOI] [PubMed] [Google Scholar]
- 20.Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–83. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–6. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 22.Park JG, Kramer BS, Steinberg SM, Carmichael J, Collins JM, Minna JD, et al. Chemosensitivity testing of human colorectal carcinoma cell lines using a tetrazolium-based colorimetric assay. Cancer Res. 1987;47:5875–9. [PubMed] [Google Scholar]
- 23.Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11:792–804. doi: 10.1038/nrc3139. [DOI] [PubMed] [Google Scholar]
- 24.Stindt MH, Carter S, Vigneron AM, Ryan KM, Vousden KH. MDM2 promotes SUMO-2/3 modification of p53 to modulate transcriptional activity. Cell Cycle. 2011;10:3176–88. doi: 10.4161/cc.10.18.17436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li T, Santockyte R, Shen RF, Tekle E, Wang G, Yang DC, et al. Expression of SUMO-2/3 induced senescence through p53- and pRB-mediated pathways. J Biol Chem. 2006;281:36221–7. doi: 10.1074/jbc.M608236200. [DOI] [PubMed] [Google Scholar]
- 26.Fukuda I, Ito A, Hirai G, Nishimura S, Kawasaki H, Saitoh H, et al. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem Biol. 2009;16:133–40. doi: 10.1016/j.chembiol.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 27.Tatham MH, Jaffray E, Vaughan OA, Desterro JM, Botting CH, Naismith JH, et al. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J Biol Chem. 2001;276:35368–74. doi: 10.1074/jbc.M104214200. [DOI] [PubMed] [Google Scholar]
- 28.Tammsalu T, Matic I, Jaffray EG, Ibrahim AF, Tatham MH, Hay RT. Proteome-wide identification of SUMO2 modification sites. Sci Signal. 2014;7:rs2. doi: 10.1126/scisignal.2005146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azzam G, Wang X, Bell D, Murphy ME. CSF1 is a novel p53 target gene whose protein product functions in a feed-forward manner to suppress apoptosis and enhance p53-mediated growth arrest. PLoS One. 2013;8:e74297. doi: 10.1371/journal.pone.0074297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weige CC, Birtwistle MR, Mallick H, Yi N, Berrong Z, Cloessner E, et al. Transcriptomes and shRNA Suppressors in a TP53 Allele-specific Model of Early-onset Colon Cancer in African Americans. Mol Cancer Res. 2014 doi: 10.1158/1541-7786.MCR-13-0286-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ozato K, Shin DM, Chang TH, Morse HC., 3rd TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol. 2008;8:849–60. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ouyang J, Shi Y, Valin A, Xuan Y, Gill G. Direct binding of CoREST1 to SUMO-2/3 contributes to gene-specific repression by the LSD1/CoREST1/HDAC complex. Mol Cell. 2009;34:145–54. doi: 10.1016/j.molcel.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jain AK, Allton K, Duncan AD, Barton MC. TRIM24 is a p53-Induced E3-Ubiquitin Ligase that undergoes ATM-Mediated Phosphorylation and Autodegradation during DNA Damage. Mol Cell Biol. 2014 doi: 10.1128/MCB.01705-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caratozzolo MF, Micale L, Turturo MG, Cornacchia S, Fusco C, Marzano F, et al. TRIM8 modulates p53 activity to dictate cell cycle arrest. Cell Cycle. 2012;11:511–23. doi: 10.4161/cc.11.3.19008. [DOI] [PubMed] [Google Scholar]
- 35.de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G, et al. PML is a direct p53 target that modulates p53 effector functions. Mol Cell. 2004;13:523–35. doi: 10.1016/s1097-2765(04)00062-0. [DOI] [PubMed] [Google Scholar]
- 36.Obad S, Brunnstrom H, Vallon-Christersson J, Borg A, Drott K, Gullberg U. Staf50 is a novel p53 target gene conferring reduced clonogenic growth of leukemic U-937 cells. Oncogene. 2004;23:4050–9. doi: 10.1038/sj.onc.1207524. [DOI] [PubMed] [Google Scholar]
- 37.Bernardi R, Scaglioni PP, Bergmann S, Horn HF, Vousden KH, Pandolfi PP. PML regulates p53 stability by sequestering Mdm2 to the nucleolus. Nat Cell Biol. 2004;6:665–72. doi: 10.1038/ncb1147. [DOI] [PubMed] [Google Scholar]
- 38.Joo HM, Kim JY, Jeong JB, Seong KM, Nam SY, Yang KH, et al. Ret finger protein 2 enhances ionizing radiation-induced apoptosis via degradation of AKT and MDM2. Eur J Cell Biol. 2011;90:420–31. doi: 10.1016/j.ejcb.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 39.Allton K, Jain AK, Herz HM, Tsai WW, Jung SY, Qin J, et al. Trim24 targets endogenous p53 for degradation. Proc Natl Acad Sci U S A. 2009;106:11612–6. doi: 10.1073/pnas.0813177106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang C, Ivanov A, Chen L, Fredericks WJ, Seto E, Rauscher FJ, 3rd, et al. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 2005;24:3279–90. doi: 10.1038/sj.emboj.7600791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan Z, Villagra A, Peng L, Coppola D, Glozak M, Sotomayor EM, et al. The ATDC (TRIM29) protein binds p53 and antagonizes p53-mediated functions. Mol Cell Biol. 2010;30:3004–15. doi: 10.1128/MCB.01023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L, Huang NJ, Chen C, Tang W, Kornbluth S. Ubiquitylation of p53 by the APC/C inhibitor Trim39. Proc Natl Acad Sci U S A. 2012;109:20931–6. doi: 10.1073/pnas.1212047110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reddy BA, van der Knaap JA, Bot AG, Mohd-Sarip A, Dekkers DH, Timmermans MA, et al. Nucleotide Biosynthetic Enzyme GMP Synthase Is a TRIM21-Controlled Relay of p53 Stabilization. Mol Cell. 2014;53:458–70. doi: 10.1016/j.molcel.2013.12.017. [DOI] [PubMed] [Google Scholar]
- 44.Wu SY, Chiang CM. Crosstalk between sumoylation and acetylation regulates p53-dependent chromatin transcription and DNA binding. EMBO J. 2009;28:1246–59. doi: 10.1038/emboj.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou Y, Li N, Zhuang W, Liu GJ, Wu TX, Yao X, et al. P53 codon 72 polymorphism and gastric cancer: a meta-analysis of the literature. Int J Cancer. 2007;121:1481–6. doi: 10.1002/ijc.22833. [DOI] [PubMed] [Google Scholar]
- 46.Jones JS, Chi X, Gu X, Lynch PM, Amos CI, Frazier ML. p53 polymorphism and age of onset of hereditary nonpolyposis colorectal cancer in a Caucasian population. Clin Cancer Res. 2004;10:5845–9. doi: 10.1158/1078-0432.CCR-03-0590. [DOI] [PubMed] [Google Scholar]
- 47.Wu X, Zhao H, Amos CI, Shete S, Makan N, Hong WK, et al. p53 Genotypes and Haplotypes Associated With Lung Cancer Susceptibility and Ethnicity. J Natl Cancer Inst. 2002;94:681–90. doi: 10.1093/jnci/94.9.681. [DOI] [PubMed] [Google Scholar]
- 48.Zhu ZZ, Cong WM, Liu SF, Xian ZH, Wu WQ, Wu MC, et al. A p53 polymorphism modifies the risk of hepatocellular carcinoma among non-carriers but not carriers of chronic hepatitis B virus infection. Cancer Lett. 2005;229:77–83. doi: 10.1016/j.canlet.2005.04.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.