

Abstract
A number of endogenous and exogenous agents, and cellular processes create abasic (AP) sites in DNA. If unrepaired, AP sites cause mutations, strand breaks and cell death. Aldehyde-reactive agent methoxyamine reacts with AP sites and blocks their repair. Another alkoxyamine, ARP, tags AP sites with a biotin and is used to quantify these sites. We have combined both these abilities into one alkoxyamine, AA3, which reacts with AP sites with a better pH profile and reactivity than ARP. Additionally, AA3 contains an alkyne functionality for bioorthogonal click chemistry that can be used to link a wide variety of biochemical tags to AP sites. We used click chemistry to tag AP sites with biotin and a fluorescent molecule without the use of proteins or enzymes. AA3 has a better reactivity profile than ARP and gives much higher product yields at physiological pH than ARP. It is simpler to use than ARP and its use results in lower background and greater sensitivity for AP site detection. We also show that AA3 inhibits the first enzyme in the repair of abasic sites, APE-1, to about the same extent as methoxyamine. Furthermore, AA3 enhances the ability of an alkylating agent, methylmethane sulfonate, to kill human cells and is more effective in such combination chemotherapy than methoxyamine.
Introduction
Abasic sites are created in cellular DNA through water-mediated depurination or depyrimidination (1). About 10,000 abasic sites are generated in this way in a human cell every day and these apurinic/apyrimidinic (AP) sites are considered to be the most commonly generated lesions in DNA (2). AP sites are also created through the action of agents that react with DNA. For example, alkylation of 7-nitrogen of guanine destabilizes the glycosidic linkage and increases the rate of depurination (3). Furthermore, many damaged bases are repaired via the base excision-repair (BER) pathway, which starts with the excision of the damaged base by a glycosylase creating an AP site (4). Although the glycosylase action is normally coupled with other enzymes that process the AP sites, an imbalance in the repair enzymes may cause the AP sites to persist.
Replicative DNA polymerases cannot copy AP sites and the progress of the replication fork is blocked at AP sites causing single- and double-strand breaks. Alternately, AP sites may be copied by error-prone translesion-synthesis polymerases that cause base substitution mutations, but allow replication to continue (5). The strand breaks resulting from unrepaired AP sites may be repaired error-free using homologous recombination, or may be repaired by a non-homologous end-joining process that creates small addition/deletion mutations. If unrepaired, the strand breaks lead to gross chromosome alterations such as translocations and cause cell death (6). Thus creation of AP sites in the genome and their processing by cellular machinery has profound implications to genome integrity.
Many different techniques have been used to label, identify and quantify AP sites (7–14). It is difficult to use some of the techniques with a large number of samples because they either use equipment such as HPLC or use radioisotopes that are incompatible with a clinical setting. Consequently, the most commonly used method for the detection and quantification of AP sites is based on the reaction of an alkoxyamine called aldehyde-reactive probe (ARP) which reacts with the open form of deoxyribose sugar in AP sites forming an oxime and tagging the site with a biotin [Fig. 1A; (8,10)]. An advantage of the use of ARP in labeling AP sites is that multiple samples can be processed in parallel and the reaction products can be spotted on a membrane to create an ELISA-like assay. The biotin is subsequently bound with streptavidin that is linked with horseradish peroxidase (10) and incubated with chemiluminescent substrate, or directly bound to fluorescently tagged streptavidin (15) to obtain an optical readout. ARP has been used to determine AP sites in different mammalian tissues (16), to monitor changes in AP sites during aging (17) and AP sites generated as a result of treatment of cells with carcinogens (18). It has also been adapted to quantify genomic uracils by excising uracils by uracil-DNA glycosylase to create AP sites followed by ARP treatment (19–21). It has been used to determine uracil levels in normal and repair-deficient Escherichia coli cells (20–22), in normal mammalian tissue (15,19) and in cancer cells (15).
Figure 1.
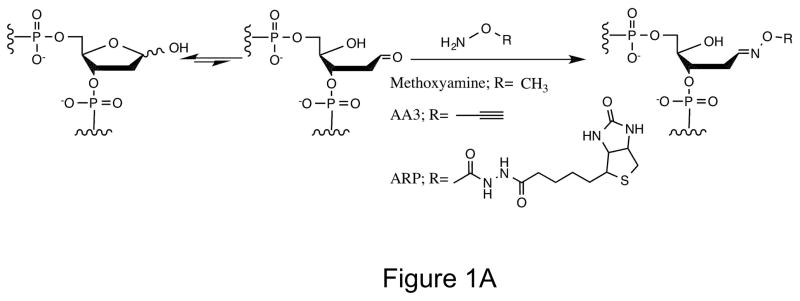

Labeling AP sites with alkoxyamines. (A) The open-chain aldehyde of an AP site in DNA reacts with alkoxyamines (Methoxyamine, AA3 or ARP). (B) Use of click chemistry to label AA3 adducted DNA (★ can be biotin, a fluorophore or any other molecule).
However, ARP-based assays for AP sites suffer from several drawbacks. ARP contains biotin which is also present in cells, and hence fluorescent labeling of AP sites in living or fixed tissues using ARP results in considerable background (unpublished results). ARP is bulky (MW 331.4) and its reaction with AP sites is likely to be significantly hindered. The presence of biotin within ARP also necessitates use of a protein like streptavidin making the labeling scheme cumbersome and somewhat expensive. Finally, a detailed study of ARP reactivity with AP sites has shown that the reaction creates side products in addition to ARP linked to full-length DNA (23).
Another use of alkoxyamines is in combination chemotherapy. Alkylating agents such as Temozolomide (TMZ) kill cancer cells by methylating DNA bases and backbone. However, cells can excise products of TMZ treatment such as 7-methylguanine and 3-methyladenine using DNA glycosylases, repair the resulting AP sites and suppress the effects of TMZ. The alkoxyamine that has been used in combination chemotherapy is methoxyamine (MX; Fig. 1A). MX potentiates the cytotoxic effects of TMZ by reacting with the AP sites created by DNA glycosylases and inhibiting the cleavage of AP sites by the AP endonuclease, APE-1 (24). Multiple clinical trials using MX as one part of combination chemotherapy are underway or have been completed (Clinical trials.gov identifiers NCT00892385, NCT01658319 and NCT00692159).
To create a more versatile chemical for labeling AP sites, we synthesized a compound, AA3, that couples alkoxyamine chemistry with chemistry of 1,3-dipolar cycloaddition (click chemistry; (25)). The latter chemistry is a well-established bioorthogonal reaction that creates stable triazoles and has been used to label sugars, proteins, DNA and other biomolecules both in vitro and in situ (26–28). We show here that AA3 can be used for labeling AP sites without the use of proteins or enzymes and for AP site quantification. Furthermore, we show that AA3 inhibits APE-1 about as well as MX, and is better than MX in a combination chemotherapy regimen.
Materials and Methods
Synthesis of O-2-Propynylhydroxylamine hydrochloride (AA3)
AA3 was synthesized according to scheme 1
Scheme 1.
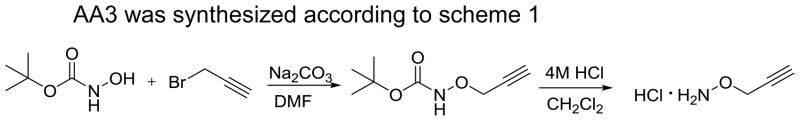
Synthesis of AA3
Propargyl bromide (11.3 mmol, Sigma Aldrich) was added drop wise into a mixture of tert-butyl-N-hydroxycarbonate (3.7 mmol, Sigma Aldrich) and sodium carbonate (7.4 mmol) in N,N-dimethylformamide. The reaction mixture was stirred overnight at 70°C, washed with water and extracted with ethyl acetate three times. The combined organic solution was washed with saturated aqueous sodium chloride (50 mL), dried over sodium sulfate and concentrated. The crude residue was purified by silica gel column chromatography (hexane: ethyl acetate = 10:1) to give the intermediate product tert-butyl N-(2-propynyloxy)-carbamate. HCl solution (4 M in 1,4-dioxane, 2 mL, Sigma Aldrich) was added to a concentrated solution of tert-butyl N-(2-propynyloxy)-carbamate (2.1 mmol) in dichloromethane in an ice bath and stirred for approximately 20 minutes. The white precipitate was filtered and recrystallized with diethyl ether and ethanol to give O-2-propynylhydroxylamine hydrochloride (AA3, 0.08 g). Its structure was confirmed by 1H NMR and 13C NMR. 1H NMR (400 MHz, CD3OD) δ: 4.748 (s, 2H, CH2), 3.376 (t, 1H, CCH, J= 2.4 Hz); 13C NMR (500 MHz, CD3OD), δ: 79.8 (CH2), 74.6 (C), 62.3 (CH).
AP site labeling reaction
A 6-carboxyfluorescein-(6-FAM-) labeled oligonucleotide U-17-mer (5′-6-FAM-ATTATTAUCCATTTATT-3′, Integrated Device Technology) was used for the AP site labeling reaction. The oligomer (4 pmol) was incubated with E. coli uracil DNA glycosylase (UDG, 1 unit, New England Labs) at 37°C for 30 minutes in reaction buffer containing 20 mM Tris-HCl (pH 8.0), 1 mM DTT and 1 mM EDTA to create AP site containing DNA. The alkoxyamine [methoxyamine, MX (Sigma Aldrich); aldehyde reactive probe, ARP (Dojindo Laboratories) or AA3] was added into the oligomer solution at indicated concentrations, and incubated at 37°C for another 30 minutes. In some experiments, the pH of the solution was adjusted with HCl or NaOH solution following removal of uracil and the pH was confirmed using pH paper. Following the reaction of alkoxyamine, unlabeled AP sites were reduced by the addition of NaBH4 (Sigma-Aldrich) to 100 mM and further incubation for 5 minutes. If this step was omitted, we observed significant degradation of the DNA (Supplementary Fig. S1 and data not shown). The reactions were stopped by the addition of formamide, loading dye and heating to 95°C for 5 minutes. The DNAs were electrophoresed in 20% polyacrylamide gels containing 7 M urea and scanned using a Typhoon 9210 phosphorimager (GE Healthcare). Product bands in images were quantified using ImageQuant software.
In experiments involving competition between alkoxyamines, AP site containing DNA was reacted with the first alkoxyamine at 37°C for 30 minutes. This was followed by the addition of the second alkoxyamine and incubation for an additional 30 minutes. The reactions were stopped and the products were analyzed as described above.
Click reaction
Biotin azide (29) or Cy5 azide (Lumiprobe) was added to a solution of AA3-linked 17-mer to 0.5 mM followed by the addition of a freshly prepared solution of CuBr/TBTA (1:3 in DMSO/t-BuOH 3:1, 0.5 mM, Sigma-Aldrich). The mixture was shaken at 45°C for 1 Hr. The DNAs were electrophoresed and analyzed as described above.
Genomic DNA isolation
HeLa cells were grown in DMEM with 10% fetal bovine serum (HyClone). The cells were harvested by centrifugation and lysed by incubation for 1 Hr at 37°C in Tris-EDTA buffer (TE) containing 2 μg/ml of RNase A and 0.5% SDS, followed by incubation with Proteinase K (100 μg /ml, Qiagen) at 56°C for 3 hours. The DNA was isolated by phenol/chloroform extraction and ethanol precipitation and dissolved in TE.
Creation of AP sites in genomic DNA by heat and acid treatment
HeLa genomic DNA was digested with HaeIII (New England Biolabs) and endogenous AP sites in DNA were reduced by the addition of NaBH4 to 100 mM. The reducing agent was removed by gel filtration with MicroSpin G-25 column (GE Healthcare) and this DNA was incubated in sodium citrate buffer (10 mM sodium citrate, 10 mM NaH2PO4, 10 mM NaCl, pH 5.0) at 70°C for various lengths of time (0, 15, 30, 45, or 60 minutes). The DNA was rapidly chilled on ice and filtered by precipitation with ethanol.
AP site quantification assay using ARP and AA3
ARP or AA3 was added to solution of genomic DNA to 2 mM and the DNA was incubated at 37°C for 30 minutes. In parallel, a 75 base pair duplex DNA with one uracil (5′-T37UT37-3′/5′-A37GA37-3′) was treated with UDG to create AP sites and was also treated with ARP or AA3. This served as an AP site standard. All DNAs were purified by phenol/chloroform extraction and ethanol precipitation, followed by MicroSpin G-25 column (GE Healthcare).
The ARP tagged DNA was heated at 95°C for 5 minutes, prior to transfer to a positively charged nylon membrane. The DNA was UV cross-linked to the membrane using a Beckman UV Stratalinker 1800. The membrane was then incubated with Starting Block Blocking Buffer (Fisher) at room temperature for 1Hr, followed by the incubation of streptavidin-conjugated horseradish peroxidase (HRP, Thermo Scientific) in blocking buffer at room temperature for 30 minutes. Alternatively, ARP-DNA on the membrane was incubated with 5×10−4 mg/ml of Cy5-streptavidin at room temperature for 1hr. After washing with Tris-buffered Saline containing Tween-20 (TBS-T) for 15 minutes, the membrane was incubated in SuperSignal West Dura Chemiluminescent Substrate (Thermo Scientific) for five minutes (when HRP-Streptavidin was used). The emitted light was captured by FluorChem Imaging System (Alpha Innotech). The resulting images were analyzed using ImageQuant software. When Cy5-Streptavidin was used the fluorescence was quantified using Typhoon 9210 phosphorimager (GE Healthcare).
AA3-tagged DNA was linked with Cy5 azide using the click reaction and purified by ethanol precipitation and filtration through a MicroSpin G-25 column (GE Healthcare). The Cy5-labeled DNA was heated at 95°C for 5 minutes prior to being transferred to a positively charged nylon membrane. The membrane was scanned using a Typhoon 9210 phosphorimager. All the images from chemiluminescence or fluorescence were analyzed using ImageQuant software. Alternately, the fluorescence of AA3/Cy5 tagged samples was directly measured using Synergy H1 Hybrid Reader (BioTEK) and the fluorescence intensities were obtained directly from the instrument.
AP endonuclease activity assay
The cleavage activity of AP endonuclease APE-1 (1 unit, New England Biolabs) was assayed using a 6-FAM labeled oligomer (4 pmol) containing a single uracil. The uracil was excised using UDG to create an AP site and the AP site was labeled with an alkoxyamine as described above. The APE-1 reaction was performed in the reaction buffer (50 mM potassium acetate, 20 mM Tris-acetate, 10 mM Magnesium acetate, 1 mM DTT, pH 7.9) at 37°C for 1 hour. DNA products were stabilized by incubation with NaBH4 and analyzed by gel electrophoresis as described above.
Killing of HeLa cells by a combination of MMS and MX or AA3
Methyl methanesulfonate (MMS, Sigma Aldrich) was diluted in phosphate buffered saline. MX or AA3 was dissolved in sterile water, and the pH was adjusted to 7 using NaOH solution. The solutions of all the chemicals were freshly prepared for each cytotoxicity experiment. HeLa cells were seeded in 48-well tissue culture plates at 3×104 to 6×104 cell/mL and grown overnight in DMEM with 10% fetal bovine serum (HyClone). Cells were treated with MMS or as a combination of MMS with MX or AA3 at indicated concentrations and harvested after 24 hours. The viability of the cells was assessed via trypan blue (HyClone) exclusion assay performed using TC20 Automated Cell Counter (Bio-Rad Laboratories).
Results and Discussion
Design and Synthesis of AA3
Methoxyamine (MX) and aldehyde-reactive probe (ARP) are well-known chemicals that react with AP sites (Fig. 1A). While MX does not allow tagging of AP sites, ARP is bulky, contains biotin as the only tag and requires proteins and enzymes for its use (8,10). To create a more versatile agent for labeling AP sites with good reactivity, we synthesized a small alkoxyamine with alkyne functionality (see Materials and Methods). This chemical, AA3 (Fig. 1A) should react with AP sites in the same manner as MX, but should allow facile labeling of the sites with different biochemical tags using copper-catalyzed azide-alkyne cycloaddition reaction (click chemistry; Figure 1B). AA3 should be useful for intracellular labeling of AP sites in addition to in vitro DNA labeling, because click chemistry is considered bioorthogonal (30,31).
Labeling of AP sites using AA3
To demonstrate that AA3 reacts with AP sites we showed that it inhibits the ability of ARP to label AP sites. AP sites were created by the excision of uracils by uracil-DNA glycosylase (UDG) in a synthetic oligomer. They were reacted with ARP and the products separated on a denaturing gel. The linking of ARP to DNA caused a shift in mobility of oligomer [Fig. 2A; lane 3; (23)]. This shift was eliminated completely if the AP sites were pretreated with AA3 (Fig. 2A; lane 6). This shows that AA3 forms a stable adduct at AP sites and blocks the reaction of ARP. Treatment of the oligomer with AA3 alone did not create an observable shift in oligomer mobility (Fig. 2A; lane 5) probably because of the small size of AA3 compared to ARP (MW 71.0 vs. 331.4). However, treating the oligomer with the two chemicals in the reverse order still created the mobility shift (Fig. 2A; lane 4) suggesting that ARP-DNA adducts are stable and cannot be replaced with AA3.
Figure 2.



Reactivity of AA3 towards AP sites in DNA. A scheme for each experiment is shown at the top of each part of the figure. (A) Inhibition of ARP reaction by AA3. Briefly, a 5′-6-FAM labeled DNA was incubated with UDG to create AP sites and reacted with AA3 and ARP in the indicated order. For the reaction in lane 7, DNA was incubated with UDG and AA3, followed by reaction with biotin azide using click chemistry. Asterisks (*) mark both the 17-mer+ARP and 17-mer+AA3+Biotin bands. (B) Comparison of reactions of AA3 with AP sites in double-stranded DNA (lane 3 and 4) and single-stranded DNA (lane 7 and 8). Reactions of ARP with double- and single- stranded DNA are shown in lanes 2 and 6, respectively. (C) Labeling of AP sites with Cy5 using AA3. AP sites were reacted with AA3 followed by click reaction with biotin azide (lane 3) or Cy5 azide (lane 4).
AA3 is both versatile and more efficient than ARP in labeling AP sites. AP sites reacted with AA3 can be labeled with an appropriate azide using click chemistry. Following a reaction with AA3, biotin azide was used to routinely convert greater than 70% of the AP sites to biotinylated form (Fig. 2A, lane 7). It may be possible to increase the reaction yield through further optimization of the click reaction (32–34). Prior treatment of the AP sites with a reducing agent, sodium borohydride, eliminated labeling showing that AA3 reacts with only the unreduced form of the AP site (Supplementary Fig. S2). AA3 reacted equally well with AP sites in ssDNA and dsDNA converting overwhelming majority of substrate to product (Fig. 2B). In contrast, ARP converted less than 20% of the substrate to biotinylated product (Fig. 2A, lanes 3, Fig. 2B, lanes 2 and 6).
To show that AA3 can be also used to label AP sites with different tags, we replaced biotin azide with Cy5 azide in the click chemistry. Using this strategy 67% of AP sites in the oligomer were labeled with the fluorescent label (Fig. 2C, lane 4). As a large number of fluorescent dyes and other molecules are commercially available in azide form, it should be possible to choose appropriate labels for AP sites based on the intended application.
AA3 has better reactivity profile than ARP
We were surprised that ARP labeled <20% AP sites under the standard conditions (Tris buffer, pH 8.0). To investigate this, the pH of the reaction buffer was changed after uracil excision, but prior to addition of ARP. When the products were quantified by gel electrophoresis the gel showed that ARP reacts well with AP sites only at acidic conditions (Fig. 3A). We consistently found that with 0.2 μM oligomer and 5 mM ARP, <20% of the substrate was converted to product at pH 7 or 8 (Figs. 2 and 3A). In contrast, AA3 was much more reactive with AP sites over the pH range of 4 to 8, and converted 57% or greater AP sites to AP-AA3 adduct (Figs. 2 and 3B). Therefore, AA3 is more suitable for in situ labeling of AP sites in cells than ARP.
Figure 3.


pH dependence of reaction of ARP and AA3 with AP-sites. A scheme for each experiment is shown at the top of each part of the figure. (A) Reactions of ARP (5 mM) with AP sites at pH 4, 5, 6 and 7 (lanes 3 through 6). (B) Reactions of AA3 (5 mM) with AP sites at pH 4, 5, 6 and 7 (lanes 3 through 6) followed by the addition of biotin azide.
Under physiological conditions, AA3 was more reactive towards AP sites than ARP even at much lower concentrations. Increasing the ARP concentration from 5 to 10 mM resulted in an increase in product formation from 13% to 20% (Fig. 4A), while AA3 converted greater than that amount of substrate to product even at 1 mM (Fig. 4B). The results from three independent experiments are presented in Fig. 4C. They show that AA3 was much more reactive at pH 7 towards AP sites than ARP over a range of concentrations and AA3 was about as reactive towards AP sites at 1 mM as ARP was at 10 mM (Fig. 4C).
Figure 4.

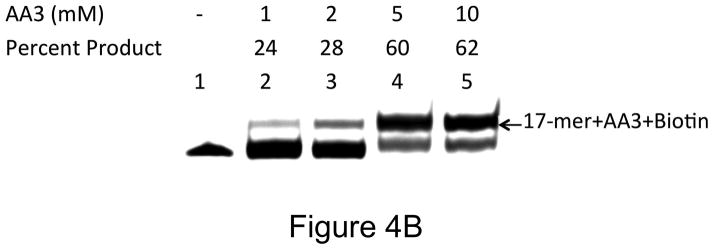

Concentration dependence of reactivity of ARP and AA3 with AP sites at pH 7. (A) DNA was incubated with UDG to create AP sites and reacted with 2, 1, 5, or 10 mM ARP (lanes 2 through 5). (B) DNA was incubated with UDG to create AP sites and reacted with 1, 2, 5, or 10 mM AA3 (lanes 2 through 5) followed by click reaction with biotin azide. (C) Quantification of the reaction products of different concentrations of ARP and AA3 with AP sites at pH 7. Mean and standard deviation of triplicate samples are shown.
AA3 can be used to quantify genomic aldehydic lesions and AP sites
As ARP has been used to quantify AP sites in genomic DNA of normal cells (11,16,17) and cancer cell lines (16,35), we wished to compare the use of AA3 in a similar setting. The comparison was done in three different ways. First, genomic DNA was extracted from HeLa cells and the AP sites were quantified using ARP or AA3. When ARP was used, the DNA adducts were quantified using streptavidin-conjugated horseradish peroxidase (16), and when AA3 was used the adducts were tagged with Cy5 for quantification. In both cases, the samples were spotted on a nylon membrane using a dot-blot apparatus and the light or fluorescence from each dot was quantified. It should be noted that ARP and AA3 would react with all aldehydic lesions in DNA including intact unoxidized AP sites, cleaved AP sites and formamidopyrimidines which result from alkylation or oxidation of purines in DNA (16,36). Such lesions occur routinely in cellular DNA, and hence the sites labeled by ARP or AA3 in HeLa DNA are referred simply as aldehydic lesions in DNA.
The results from these experiments are shown in Figure 5. The ARP-based method gave about twice as many aldehydic lesions in HeLa as AA3 (Fig. 5A). A previous study of HeLa DNA using ARP reported (35) somewhat higher number of aldehydic sites (~20 per 106 bp). The differences in those numbers and the numbers obtained in our study may be due to methods of DNA preparation, age of DNA used and use of different AP site standards. The Mendez et al study used depurinated pBR322 as the standard (35), while we used a synthetic oligomer containing uracil that was treated with UDG as the AP site standard (Supplementary figure S3).
Figure 5.


Quantification of aldehydic lesions and AP sites using ARP and AA3. (A) HeLa DNA was labeled with ARP and vacuum-spotted onto a nylon membrane. Aldehydic lesions in this DNA were quantified using streptavidin-conjugated horseradish peroxidase (HRP) and detected using a chemiluminescent substrate (open bar). In parallel, aldehydic lesions in HeLa DNA were also labeled with AA3 followed by reaction with Cy5 azide and quantification of fluorescence on a nylon membrane (black bar). AA3-labeled aldehydic lesions reacted with Cy5 azide were also quantified in solution using a microplate reader (gray bars). Mean and standard deviation of triplicate samples is shown. (B) HeLa DNA was pre-treated with NaBH4 to reduce endogenous aldehydic lesions and AP sites were generated by heat and acid treatment for different lengths of time. The AP sites were labeled using ARP or AA3 for labeling and the DNA was spotted onto a nylon membrane and the membranes scanned to quantify AP sites (Left). AP sites in the same DNAs were also quantified by reacting them successively with AA3 and Cy5 azide, and measuring fluorescence intensity directly using a microplate reader (right). Mean and standard deviation of triplicate samples is shown for each time point.
In a second set of experiments, HeLa DNA was first treated with sodium borohydride to reduce preexisting aldehydic lesions and make them resistant to ARP or AA3. The DNA was then heated under acidic conditions to create new AP sites through depurination. The depurination reaction was terminated at various times and the AP sites were quantified using the two chemicals as described above. The data showed there was a linear time-dependent increase in the number of AP sites and the two methods gave comparable numbers for AP sites at all time points (Fig. 5B, left panel).
Sensitivity and ease of use of AA3
To determine whether AA3-based quantification of AP sites was as sensitive as ARP-based quantification, we treated different amounts of a DNA oligomer containing AP sites with ARP or AA3 and quantified the products. The membrane images from the two parallel experiments are shown in Figure 6A. The sample containing 109 AP sites was visible using ARP-chemiluminescence assay, but the sample with 108 sites could not be detected above background. In contrast, 108 AP site sample could be detected in the image from AA3-fluorescence assay. Adjusting the image brightness did not change the relative sensitivities of AA3 and ARP. The membrane containing ARP-labeled samples has much higher background than the membrane with AA3-labeled samples (Supplementary figure S4). Thus under these conditions, AA3-based detection of AP sites has lower background and greater sensitivity than ARP.
Figure 6.

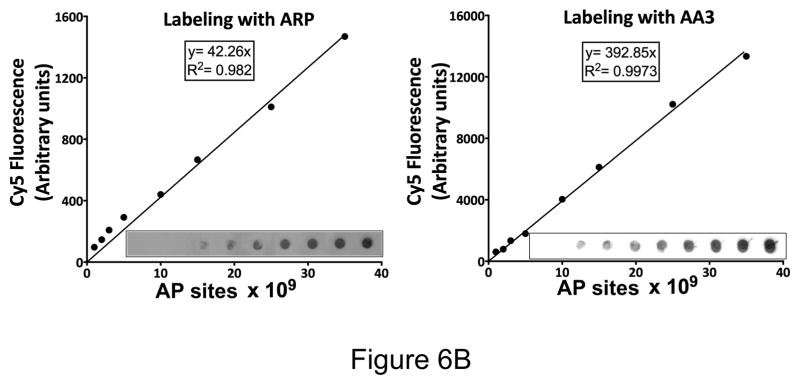
Sensitivity of detection of AP sites using ARP and AA3. (A) Image of the nylon membranes where different amounts of DNA containing a synthetic oligomer with one AP site were labeled using either ARP-HRP (top) or AA3-Cy5 (bottom). (B) Synthetic duplex containing one AP site was labeled with either ARP or AA3. Different dilutions of ARP-labeled DNA was spotted on a membrane and bound with Cy5-streptavidin. AA3-labeled DNA was reacted with Cy5 azide and different dilutions were spotted on a membrane. Cy5 fluorescence was quantified in each case and is plotted against the number of AP sites in each spot. (Inset) Image of the membrane in each case.
However, the readout for the comparison of two techniques used to quantify AP sites in Fig. 6A were different, and we wanted to eliminate this variable in the comparison. To accomplish this we labeled different amounts of the AP site-containing DNA duplex with ARP and then bound it to Cy5-streptavidin. In parallel reactions the DNA was reacted with AA3 followed by reaction with Cy5 azide. Both sets of samples were spotted on nylon membranes and Cy5 fluorescence was quantified. The results show that both methods result in a linear relationship between the number of AP sites and Cy5 fluorescence (Fig. 6B), but the use of AA3 results in lower background and hence greater signal-to-noise ratio (Fig. 6B, inset).
One problem of using ARP to quantify AP sites is the difficulty of separating unbound protein (HRP or streptavidin) from protein that is bound to DNA. This is the likely source of the high background seen in membranes with ARP-DNA (Fig. 6). In contrast, AA3-based method does not use a protein and hence it is possible to eliminate unreacted Cy5 azide from the much larger Cy5-AA3-DNA using a G-25 mini-column. The Cy5 fluorescence can then be directly measured using a microplate fluorometer (Supplementary Fig. S5). We performed this simplified procedure on endogenous aldehydic lesions in HeLa DNA and on AP sites created by heat and acid treatment, and the results were comparable to those obtained by the other two methods (Fig. 5A, and 5B, right panel). Thus use of AA3 simplifies AP site quantification (Supplementary Fig. S5).
AA3 inhibits APE-1
The reaction of MX with AP sites is known to inhibit its repair by AP endonuclease (37) and this is the basis of its proposed use as part of anti-cancer combination chemotherapy (24). To find out whether ARP and AA3 similarly inhibit AP endonuclease APE-1, we reacted an oligomer containing an AP site with MX, ARP or AA3 and then challenged the DNA with APE-1. The results showed that while MX and AA3 were very effective in blocking action of APE-1, ARP was a poor inhibitor of the enzyme under physiological condition.
When AP sites were reacted with ARP at pH 7 and the DNA was then treated with APE-1, about 90% of the DNA was cleaved by the enzyme showing poor protection of AP sites by ARP (Fig. 7A, lane 8). This is probably because of the poor reactivity of ARP at pH 7 (Fig. 4A). In contrast, both MX and AA3 protected an overwhelming majority of AP sites at pH 7 (Fig. 7A, respectively lanes 4 and 6). When the pH of ARP reaction was lowered to 5, protection of AP sites against cleavage by APE-1 increased to 74% (Fig. 7B, lane 8). Under the same conditions, the protection by MX and AA3 was ~100% (Fig. 7B, lanes 4 and 6). Thus, MX and AA3 protect AP sites against APE-1 cleavage equally well at both pH conditions.
Figure 7.


Inhibition of APE-1 activity by MX, ARP or AA3. A scheme for each experiment is shown at the top of each part of the figure. (A) DNA containing an AP site was reacted with MX, AA3 or ARP at pH 7. This was followed by incubation of DNA with APE-1. (B) DNA containing an AP site was reacted with MX, AA3 or ARP at pH 5. This was followed by incubation of DNA with APE-1.
AA3 kills Cells containing DNA base damage
Inhibition of base-excision repair has been proposed as a strategy for anti-cancer chemotherapy (38,39). In particular, it has been shown that coupling treatment of cancers with alkylating agents such as MMS or temozolomide with BER inhibitor MX increases killing of tumor cells (24,40). To determine whether AA3 is also able to enhance killing cells treated with an alkylating agent, we combined MMS treatment of HeLa cells with AA3 treatment. The results are shown in Fig. 8.
Figure 8.


Killing of HeLa cells by combining MMS with MX or AA3. (A) HeLa cells were treated with 50 μM MMS and different concentrations of AA3 and cell killing was determined by Trypan Blue exclusion assay. Each circle represents the result from one independent culture and the broken line connects median values at each AA3 concentration. (B) HeLa cells were treated with only AA3 (10 mM) or MMS (50 μM); or treated together with MMS and MX (10 mM) or AA3 (10 mM), and cell killing was quantified. The results are from six independent cultures and the mean and standard deviation is shown in each case.
When the cells were treated with a low concentration of MMS (50 μM, Fig. 8A) or 10 mM AA3 (Fig. 8B), very little loss of viability was observed after one day. In fact, even 20 mM AA3 killed only ~10% of HeLa cells (Supplementary Fig. S6). However, when HeLa cells were treated with 50 μM MMS and different concentrations of AA3, cell viability decreased with increasing concentration of AA3 dropping to about 50% survival at 10 mM AA3 (Fig. 8A). We then directly compared the ability of MX and AA3 to enhance killing by MMS at this concentration using six independent cultures for each chemical. The results showed that while MX did enhance killing by MMS, AA3 had a stronger lethal effect (Fig. 8B). The difference between the killing enhancement caused by MX and AA3 was statistically significant and suggests that AA3 would be better than MX as a component in anti-cancer combination chemotherapy regimen. It is unclear why AA3 kills MMS treated cells better than MX, despite the fact that both chemicals appear to be equally effective at inhibiting APE-1 and we are investing this phenomenon further.
In summary, we have designed a small molecule, AA3, that can be used to quantify aldehydic lesions and AP sites in DNA in multi-titer plate format without the use of proteins. AA3 has much higher reactivity towards AP sites at physiological pH than ARP and this opens up the possibility that AA3 may be more effective at labeling AP sites in live cells. The use of click chemistry allows introducing a variety of fluorescent tags at AP sites and the biorthogonal nature of click chemistry should allow the fluorescent labeling reaction to be performed in permeabilized or fixed cells. AA3, like MX, is an efficient inhibitor of mammalian APE-1 and works better than the latter in killing cells treated with the alkylating agent MMS. Therefore, AA3 is more sensitive and versatile than ARP for labeling and quantifying AP sites, and is more effective than MX in combination chemotherapy against HeLa cells. Experiments are underway to determine whether AA3 is more effective in killing other types of cancer cells in combination with known anticancer agents.
Supplementary Material
Highlight.
A new alkoxyamine that reacts with abasic sites in DNA
Bioorthogonal click chemistry
Labeling of abasic sites with biotin or fluorophores
Higher sensitivity and better biocompatibility than aldehyde-reactive probe
Inhibition of AP endonuclease 1
Better killing in combination with methyl methanesulfonate than methoxyamine
Acknowledgments
The work reported here was supported by NIH grant GM 57200 (to A.S.B.), a Wayne State University graduate fellowship (to S.S.) and funds from Wayne State University (to both A.S.B. and Y-H Ahn).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 2.Kunkel TA. The high cost of living. Trends in genetics : TIG; American Association for Cancer Research Special Conference: endogenous sources of mutations; Fort Myers, Florida, USA. 11–15 November 1998; 1999. pp. 93–94. [DOI] [PubMed] [Google Scholar]
- 3.Lawley PD, Brookes P. Further Studies on the Alkylation of Nucleic Acids and Their Constituent Nucleotides. The Biochemical journal. 1963;89:127–138. doi: 10.1042/bj0890127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim YJ, Wilson DM., 3rd Overview of base excision repair biochemistry. Current molecular pharmacology. 2012;5:3–13. doi: 10.2174/1874467211205010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodman MF, Woodgate R. Translesion DNA polymerases. Cold Spring Harbor perspectives in biology. 2013;5:a010363. doi: 10.1101/cshperspect.a010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boiteux S, Guillet M. Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA repair. 2004;3:1–12. doi: 10.1016/j.dnarep.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Fundador E, Rusling J. Detection of labeled abasic sites in damaged DNA by capillary electrophoresis with laser-induced fluorescence. Analytical and bioanalytical chemistry. 2007;387:1883–1890. doi: 10.1007/s00216-006-1041-x. [DOI] [PubMed] [Google Scholar]
- 8.Ide H, Akamatsu K, Kimura Y, Michiue K, Makino K, Asaeda A, Takamori Y, Kubo K. Synthesis and damage specificity of a novel probe for the detection of abasic sites in DNA. Biochemistry. 1993;32:8276–8283. doi: 10.1021/bi00083a031. [DOI] [PubMed] [Google Scholar]
- 9.Kow YW, Dare A. Detection of abasic sites and oxidative DNA base damage using an ELISA-like assay. Methods. 2000;22:164–169. doi: 10.1006/meth.2000.1057. [DOI] [PubMed] [Google Scholar]
- 10.Kubo K, Ide H, Wallace SS, Kow YW. A novel, sensitive, and specific assay for abasic sites, the most commonly produced DNA lesion. Biochemistry. 1992;31:3703–3708. doi: 10.1021/bi00129a020. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura J, Walker VE, Upton PB, Chiang SY, Kow YW, Swenberg JA. Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer research. 1998;58:222–225. [PubMed] [Google Scholar]
- 12.Roberts KP, Sobrino JA, Payton J, Mason LB, Turesky RJ. Determination of apurinic/apyrimidinic lesions in DNA with high-performance liquid chromatography and tandem mass spectrometry. Chemical research in toxicology. 2006;19:300–309. doi: 10.1021/tx0502589. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Liu L, Wu C, Bulgar A, Somoza E, Zhu W, Gerson SL. Direct detection and quantification of abasic sites for in vivo studies of DNA damage and repair. Nuclear medicine and biology. 2009;36:975–983. doi: 10.1016/j.nucmedbio.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou X, Liberman RG, Skipper PL, Margolin Y, Tannenbaum SR, Dedon PC. Quantification of DNA strand breaks and abasic sites by oxime derivatization and accelerator mass spectrometry: application to gamma-radiation and peroxynitrite. Analytical biochemistry. 2005;343:84–92. doi: 10.1016/j.ab.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Shalhout S, Haddad D, Sosin A, Holland TC, Al-Katib A, Martin A, Bhagwat AS. Genomic Uracil Homeostasis during Normal B Cell Maturation and Loss of this Balance During B Cell Cancer Development. Molecular and cellular biology. 2014 doi: 10.1128/MCB.00589-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura J, Swenberg JA. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer research. 1999;59:2522–2526. [PubMed] [Google Scholar]
- 17.Atamna H, Cheung I, Ames BN. A method for detecting abasic sites in living cells: age-dependent changes in base excision repair. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:686–691. doi: 10.1073/pnas.97.2.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melendez-Colon VJ, Luch A, Seidel A, Baird WM. Cancer initiation by polycyclic aromatic hydrocarbons results from formation of stable DNA adducts rather than apurinic sites. Carcinogenesis. 1999;20:1885–1891. doi: 10.1093/carcin/20.10.1885. [DOI] [PubMed] [Google Scholar]
- 19.Cabelof DC, Nakamura J, Heydari AR. A sensitive biochemical assay for the detection of uracil. Environmental and molecular mutagenesis. 2006;47:31–37. doi: 10.1002/em.20165. [DOI] [PubMed] [Google Scholar]
- 20.Lari SU, Chen CY, Vertessy BG, Morre J, Bennett SE. Quantitative determination of uracil residues in Escherichia coli DNA: Contribution of ung, dug, and dut genes to uracil avoidance. DNA repair. 2006;5:1407–1420. doi: 10.1016/j.dnarep.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parisien R, Bhagwat AS. In: DNA and RNA Modification Enzymes: Comparative Structure, Mechanism, Functions, Cellular Interactions and Evolution. Grosjean H, editor. Landes Bioscience; Austin, TX: 2009. pp. 127–143. [Google Scholar]
- 22.Wijesinghe P, Bhagwat AS. Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic acids research. 2012;40:9206–9217. doi: 10.1093/nar/gks685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennett SE, Kitner J. Characterization of the aldehyde reactive probe reaction with AP-sites in DNA: influence of AP-lyase on adduct stability. Nucleosides, nucleotides & nucleic acids. 2006;25:823–842. doi: 10.1080/15257770600726133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L, Taverna P, Whitacre CM, Chatterjee S, Gerson SL. Pharmacologic disruption of base excision repair sensitizes mismatch repair-deficient and -proficient colon cancer cells to methylating agents. Clinical cancer research : an official journal of the American Association for Cancer Research. 1999;5:2908–2917. [PubMed] [Google Scholar]
- 25.Huisgen R. 1.3-Dipolare Cycloadditionen - Ruckschau Und Ausblick. Angew Chem Int Edit. 1963;75:604. [Google Scholar]
- 26.Cavanagh BL, Walker T, Norazit A, Meedeniya AC. Thymidine analogues for tracking DNA synthesis. Molecules. 2011;16:7980–7993. doi: 10.3390/molecules16097980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mamidyala SK, Finn MG. In situ click chemistry: probing the binding landscapes of biological molecules. Chemical Society reviews. 2010;39:1252–1261. doi: 10.1039/b901969n. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Zhang Y. Applications of azide-based bioorthogonal click chemistry in glycobiology. Molecules. 2013;18:7145–7159. doi: 10.3390/molecules18067145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahn YH, Hwang Y, Liu H, Wang XJ, Zhang Y, Stephenson KK, Boronina TN, Cole RN, Dinkova-Kostova AT, Talalay P, et al. Electrophilic tuning of the chemoprotective natural product sulforaphane. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9590–9595. doi: 10.1073/pnas.1004104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Best MD. Click chemistry and bioorthogonal reactions: unprecedented selectivity in the labeling of biological molecules. Biochemistry. 2009;48:6571–6584. doi: 10.1021/bi9007726. [DOI] [PubMed] [Google Scholar]
- 31.Prescher JA, Bertozzi CR. Chemistry in living systems. Nature chemical biology. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 32.Hong V, Presolski SI, Ma C, Finn MG. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angewandte Chemie. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paredes E, Das SR. Optimization of acetonitrile co-solvent and copper stoichiometry for pseudo-ligandless click chemistry with nucleic acids. Bioorganic & medicinal chemistry letters. 2012;22:5313–5316. doi: 10.1016/j.bmcl.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 34.Winz ML, Samanta A, Benzinger D, Jaschke A. Site-specific terminal and internal labeling of RNA by poly(A) polymerase tailing and copper-catalyzed or copper-free strain-promoted click chemistry. Nucleic acids research. 2012;40:e78. doi: 10.1093/nar/gks062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mendez F, Goldman JD, Bases RE. Abasic sites in DNA of HeLa cells induced by lucanthone. Cancer investigation. 2002;20:983–991. doi: 10.1081/cnv-120005914. [DOI] [PubMed] [Google Scholar]
- 36.Jena NR, Mishra PC. Formation of ring-opened and rearranged products of guanine: mechanisms and biological significance. Free radical biology & medicine. 2012;53:81–94. doi: 10.1016/j.freeradbiomed.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 37.Liuzzi M, Talpaert-Borle M. A new approach to the study of the base-excision repair pathway using methoxyamine. The Journal of biological chemistry. 1985;260:5252–5258. [PubMed] [Google Scholar]
- 38.Fishel ML, Kelley MR. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Molecular aspects of medicine. 2007;28:375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 39.Liu L, Nakatsuru Y, Gerson SL. Base excision repair as a therapeutic target in colon cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8:2985–2991. [PubMed] [Google Scholar]
- 40.Yan L, Bulgar A, Miao Y, Mahajan V, Donze JR, Gerson SL, Liu L. Combined treatment with temozolomide and methoxyamine: blocking apurininc/pyrimidinic site repair coupled with targeting topoisomerase IIalpha. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:1532–1539. doi: 10.1158/1078-0432.CCR-06-1595. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.