

Abstract
In response to oncogene activation and oncogene-induced aberrant proliferation, mammalian cells activate apoptosis and senescence, usually via the p53-ARF tumor suppressor pathway. Apoptosis is a known barrier to cancer and is usually down-regulated prior to full malignancy, but senescence as an anticancer barrier is controversial due to its presence in the tumor environment. In addition, senescence may aid cancer progression via releasing senescence-associated factors that instigate neighboring tumor cells. Here, it is demonstrated that apoptosis unexpectedly remains robust in ErbB2 (ERBB2/HER2)-initiated mammary early lesions arising in adult mice null for either p53 or ARF. These early lesions, however, down-regulate senescence significantly. This diminished senescence response is associated with accelerated progression to cancer in ARF-null mice compared to ARF-wild-type mice. Thus, the ARF-p53 pathway is dispensable for the apoptosis anticancer barrier in the initiation of ErbB2 breast cancer, the apoptosis barrier alone cannot halt mammary tumorigenesis, and that senescence is a key barrier against carcinogenesis.
Implications
Findings in this relevant mouse model of HER2-driven breast cancer suggest that effective prevention relies upon preserving both ARF/p53-independent apoptosis and ARF/p53-dependent senescence.
Keywords: Breast cancer, p53, ARF, senescence, RCAS, TVA
Introduction
Oncogene activation in otherwise normal cells can trigger the induction of apoptosis and senescence (1). Apoptosis is widely accepted to be one of the most critical safety mechanisms employed by cells to protect against unbridled proliferation and malignant transformation(2). In accordance with it being a barrier to cancer, apoptosis usually subsides as precancerous lesions progress to full malignancy(3,4). Furthermore, forced downregulation of genes that activate apoptosis accelerates the progression to cancer(reviewed in 2).
Senescence is also detected in precancerous lesions in humans and animal models (5), and has also been reported to function as a physiologic barrier to the development of tumors of the hematopoietic system, lung, prostate, and skin (6-10). However, senescence has also been detected in some tumors, and senescent cells within a cancer have been found to aid tumor progression viareleasing senescence-associated factors that instigate neighboring tumor cells(11-14). Therefore, it remains controversial whether senescence actually imposes a significant barrier to tumorigenesis.
We have reported mouse models of sporadic breast cancer by using intraductal injection of retrovirus to deliver oncogene into a small subset of mammary epithelial cells with an intact mammary gland(15-17). These models more closely mimic human breast cancer initiation than conventional transgenic and knockout models(18). Using retrovirus to introduce the gene encoding a activated version of ErbB2, a member of the epidermal growth factor receptor family of tyrosine kinases that is commonly over-activated in human breast cancers, we detected robust apoptosis and senescence in the resulting early lesions(19,20). As these lesions progress to frank tumors, apoptosis diminishes, but senescence remains resilient(19). Here, we use this mouse model to examine the role of senescence as a barrier to mammary tumorigenesis and show that loss of p53-ARF-dependent senescence is associated with rapid tumor induction by ErbB2 despite the erection of a robust apoptotic response, suggesting that senescence is, indeed, a critical barrier to tumor formation in the mammary gland.
Materials and Methods
Mice
The animal protocol used in this study was approved by the IACUC of Baylor College of Medicine, Houston, TX. ARF-null mice (21) on B6.129 background were acquired from the NCI Mouse Repository (strain number 01XG7), then back-crossed 2-3 generations to FVB wildtype mice before interbreeding for ARF-wildtype, -heterozygous, and -null females for experiments. Mouse genotype was determined using primers and PCR settings published by the NCI (http://mouse.ncifcrf.gov/available_details.asp?ID=01XG7). p16-null mice (22) on FVB.129 background were acquired from the NCI Mouse Repository (strain number 01XE4), then mated once to FVB wildtype mice before generating p16-wildtype, -heterozygous, and -null females for experiments. Mouse genotype was determined using primers and PCR settings published by the NCI (http://mouse.ncifcrf.gov/available_details.asp?ID=01XE4). Tyrosinase-tagged p53-null mice (23)were graciously provided by Dr. Lawrence A. Donehower, Baylor College of Medicine, Houston, TX. Mice were crossed 3 generations to FVB wildtype females to generate p53-wildtype and -null females for experiments. Mouse genotype was determined by color and confirmed with PCR, as described (23). All animals were euthanized according to NIH guidelines.
Viral preparation and delivery
Constitutively activated ErbB2 (caErbB2) oncogene used to induce premalignant lesions and tumors was carried by either the lentiviral vector FUCGW (24)or the avian leucosis virus-derived RCAS(Replication-Competent ASLV long terminal repeat with a Splice acceptor)(25). Virus was prepared as described (15,24). Viral particles were concentrated by ultracentrifugation at 27000 rpm for 90 minutes, and then stored at -80°C until titration or intraductal injection. Viral titer was determined via limiting dilution transduction of either 293T (for lentivirus) or DF1 (for RCAS virus) cells. To preserve titer, intraductal injection was performed within 3 hours of thawing virus.
Generation of premalignant lesions
Virgin female mice aged 12-14 weeks old (for ARF WT/KO and p16 WT/KO) or 6-10 weeks old (for p53 WT/KO) were intraductally injected with 106 IU of virus harboring the caErbB2 oncogene. Mammary glands were collected two-to-three weeks after injection, embedded in paraffin or frozen tissue matrix (OCT), and sectioned for analysis. Uninjected mammary glands were used as controls.
Immunofluorescence (IF) and immunohistochemical (IHC) staining and quantification
Staining and image capture were performed on 3μm formalin-fixed and paraffin-embedded sections as described(15,20). Primary antibodies used included mouse monoclonal antibodies against hemagglutinin tag (HA, MMS-101P, Covance, 1:500), p21 (cat#sc-6246 Santa Cruz, 1:200), gamma-H2AX (cat#05-636 Millipore, 1:500), and pATM (cat#200-301-500 Rockland, 1:200), as well as rabbit polyclonal antibodies against cleaved caspase 3 (CC3, cat#9661S Cell Signaling Technology, 1:200), Ki67 (cat#NCL-Ki67P Novocastra, 1:200), phospho-histone 3 (pH3, cat#06-570 Millipore, 1:200), macroH2A (cat#ab37264 Abcam, 1:150), p16 (cat#sc-1207 Santa Cruz, 1:200), and p53 (cat#NCL-p53-CM5p Novacastra, 1:1000). Cell counting was achieved using Image J software as well as Adobe Photoshop.
Terminal Deoxynucleotidyl Transferase dUTP Nick-end Labeling (TUNEL) Assay
Formalin-fixed and paraffin-embedded 3μm sections were prepared and stained using the ApopTag® Red In Situ Apoptosis Detection Kit (cat#S7165, Chemicon) according to manufacturer’s instructions.
Senescence-associated beta-galactosidase staining
Mammaryglands bearing premalignant lesions embedded in OCT were sectioned at 10 μm and stained for senescence-associated β-galactosidase activity as described (26-28). Briefly, frozen sections were fixed with glutaraldehyde, treated with X-gal, incubated for 12-16 hours until color developed, counterstained with hematoxylin, and then mounted.
Infection rate determination
Virgin female mice aged 12-14 weeks old were intraductally injected into left and right #4 mammary glands with106 IU of RCAS virus harboring GFP. Mammary epithelial cells were isolated from injected glands four days post-injection and analyzed for GFP by flow cytometry to determine the percentage of infected mammary epithelial cells. Uninjected mammary glands were used as GFP-negative controls.
Wholemount preparation and quantification
Mice that were WT or KO for ARF were intraductally injected with 106 IU of virus harboring the caErbB2 oncogene. Mammary glands were collected two weeks later, neutral-red stained (29), and wholemounted. Images of the wholemounted gland were captured using the Leica MZ16 F stereomicroscope (Leica, Houston TX) and Leica DFC300 FX Digital Color Camera (Leica, Houston TX). The number of lesions of diameters 200-299um, 300-399um, 400-499um, 500-599um, and ࣙ600um were quantified using Image J.
Tumor latency and growth rate determination
Virgin female mice aged 12-14 weeks old were intraductally injected in one #4 mammary gland with106 IU of RCAS virus harboring the caErbB2 oncogene and monitored for tumor incidence by palpation of the mammary gland at least twice a week. Tumor latency was determined by recording the number of days post injection at which tumor was first palpable. Tumor growth was monitored by taking caliper and/or palpation measurements of up to three dimensions. Tumor volume was calculated using the formula 4/3*Pi*(x/2)(y/2)(z/2), where x, y, and z are the three measured dimensions. The uninjected contralateral #4 mammary gland was used as control.
Results
Early lesions arisingin p53-null mammary glands exhibit diminishedsenescence but intact apoptosis
We have reported that intraductal injection of retrovirus carrying the gene encoding constitutively activated ErbB2 (caErbB2) led to early lesions with elevated levels of pATM, γH2AX, and other markers of an active DNA damage response (DDR) pathway(19). Genetic ablation of ATM, with an accompanying decrease in p53 levels, diminished both apoptosis and senescence in these premalignant lesions(19), suggesting that an intact DDR (perhaps ATM-p53, specifically) is necessary for robust induction of both apoptosis and senescence.
To determine the effect of p53loss on apoptosis and senescence, we generated premalignant lesions in wildtype and p53-null mice(23)(Fig1A) by intraductally injecting female virgin mammary glands with a lentiviralvector (FUCGW) harboring caErbB2(FUCGW-caErbB2)(17). Injected mammary glands were collected two-to-three weeks post-injection. The resultant premalignant lesions were evaluated for senescence by measuring levels of macroH2A, a histone variant that is enriched in senescence-associated heterochromatic foci(30), as well as expression of senescence-mediator p16 (31). We observed that loss of p53 led to a severe decrease in senescence response compared to wildtype (Fig 1B, C), suggesting that p53 is required for senescence following ErbB2 activation in the mammary gland.
Figure 1. ErbB2-initiatedearly lesions in p53-null mammary glands exhibit decreased senescence but intact apoptosis.
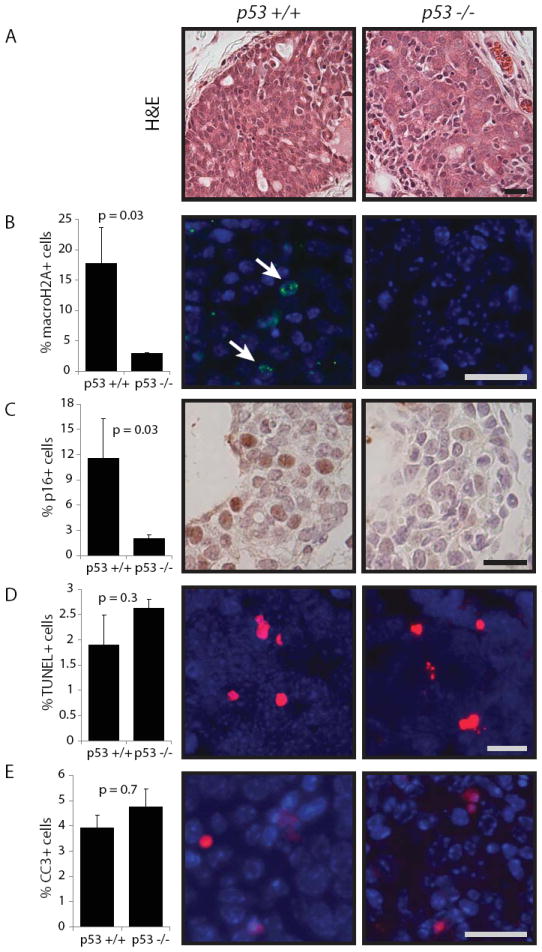
Lentivirus was used to carry caErbB2 into the mammary glands. Early lesions were analyzed two weeks following viral injection. Scale bar = 20μm. For bar graphs, columns represent the mean, and error bars represent the SEM.
A. H&E of premalignant lesions from wildtype and p53-null mammary glands.
B. Quantification of macroH2A-positive cells in premalignant lesions from widltype and p53-null mice (n=3, 4). Representative images shown.
C. Quantification of p16-positive cells in premalignant lesions from widltype and p53-null mice (n=4, 5). Representative images shown.
D. Quantification of TUNEL-positive cells (red) in premalignant lesions from widltype and p53-null mice (n=4). Representative images shown.
E. Quantification of CC3-positive cells (red) in premalignant lesions from widltype and p53-null mice (n=3,4). Representative images shown.
We next determined effect of p53 on the apoptotic response via TUNEL detection of DNA fragmentation characteristic of dying cells and via presence of cleaved caspase 3 (CC3), a critical player in programmed cell death. Surprisingly, loss of p53 did not impair the apoptotic response in premalignant lesions (Fig 1D, E), which is contrary to the expected role of p53 in mediating apoptosis in early stages of tumorigenesis.
Loss of ARF recapitulates p53ablation in failing to activate senescence but maintaining intact apoptosis
Like ATM, ARF is a key upstream regulator of p53 (reviewed in 23). While ARF has been reported to regulate senescence as well as apoptosis via p53(32), its role in regulating apoptosis and senescence as well tumorigenesis in the mammary gland has not been rigorously tested(10,33). We have reported that ARF is activated in mammary early lesions induced by caErbB2(19). To determine whether the ARF tumor suppressor can mediate apoptosis and senescence following oncogene activation, we again generated premalignant lesions in wildtypeand ARF-null mice(21)using FUCGW-caErbB2(Fig 2A), confirmed diminished p53 activity by assessing p21 levels (Supplemental fig 1A), and then evaluated lesions for senescence and apoptosis. As in p53-null mice, we observed a significant impairment of the senescence response in premalignant lesions arising inthe ARF-null mammary epithelium (Fig 2B-D) based on staining for macroH2A, SA-β-gal, and p16. Interestingly, there was no impairment of the induction of apoptosis(Fig 2E, F). This impairment of senescence with preservation of the apoptosis response was also observed when caErbB2 was delivered via RCAS retrovirus (RCAS-caErB2) (15)(Supplementary Fig 1A-E). Therefore, these data suggest that ARF, like p53, is required for full induction of senescence in early lesions but is dispensable for the apoptosis response. This phenocopying of p53 by ARF, given the existing knowledge of ARF as a p53 regulator(34), suggests that any role of ARF in mediating a barrier to mammary tumorigenesis is likely and primarily via p53 signaling.
Figure 2. ErbB2-initiatedearly lesions in ARF-null mammary glands exhibit decreased senescence but intact apoptosis.
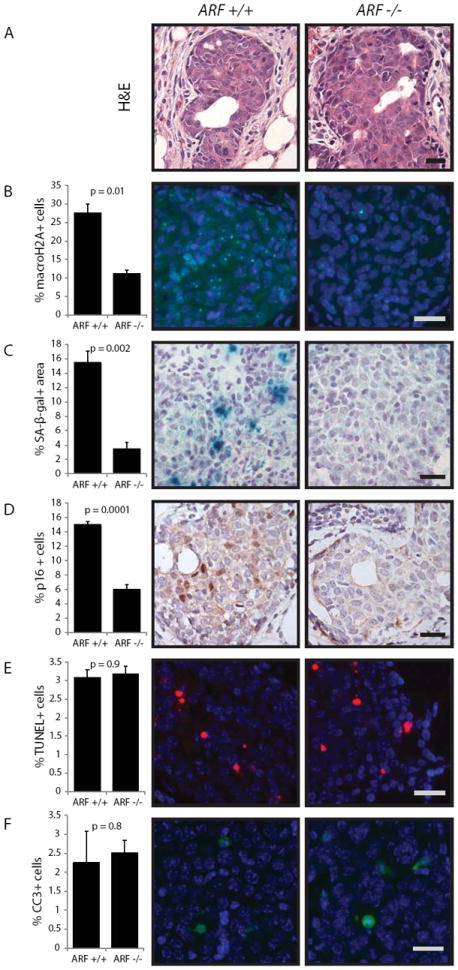
Lentivirus was used to carry caErbb2 into the mammary glands. Early lesions were analyzed two weeks following viral injection.Scale bar = 20μm. For bar graphs, columns represent the mean, and error bars represent the SEM.
A. H&E of premalignant lesions from wildtype and ARF-null mammary glands.
B. Quantification of cells positive for macroH2A-positive foci (green) in premalignant lesions from wildtype and ARF-null mice (n=3). Representative images shown.
C. Quantification of positivity of early lesions for senescence-associated β-galactosidase from wildtype and ARF-null mice (n=4, 6). Positivity was scored based on area of staining as a percentage of area of lesion. Representative images shown.
D. Quantification of p16-positive cells in premalignant lesions from wildtype and ARF-null mice (n= 4). Representative images shown.
E. Quantification of TUNEL-positive cells (red) in premalignant lesions from wildtype and ARF-null mice (n=4). Representative images shown.
F. Quantification of CC3-positive cells (green) in premalignant lesions from wildtype and ARF-null mice (n=4). Representative images shown.
Of note, although p16 has also been reported to be an alternative mediator of the senescence response(9,22,35,36), we found that caErbB2-initiated premalignant lesions in p16-null mice (22)did not exhibit a diminished senescence response following oncogene-activation (Supplementary Fig 2), suggesting that pathways converging upon p53 (such as the ATM-p53 and ARF-p53 axes), rather than those regulating p16, play more critical roles in mediating the senescence response in the mammary cells that have suffered an oncogenic mutation.
Loss of the senescence response in ARF-null mice is associated with a heavier premalignant lesion load
Next, we determined whether senescence is truly a barrier to tumorigenesis by examining the effect of senescence-loss on premalignant lesion advancement and tumor latency. Historically, this study has been difficult to conduct since loss of senescence and loss of apoptosis often occur concurrently, confounding interpretation of the results. However, we have presented above two mouse models in which, at least in the mammary gland, senescence is severely diminished but apoptosis is completely preserved following oncogene activation. The early lethality experienced by p53-null mice (due to the early and frequent development of lymphoma and other non-mammary cancers) (37)precluded the use of these mice in long term studies. Therefore, we elected to use the ARF-null model, which recapitulates the effect of p53-loss on apoptosis and senescence, but has a longer lifespan amenable to long term tumor latency studies.
To determine whether loss of senescence is associated with failure of the mammary epithelium to eliminate premalignant lesions, we first quantified the premalignant lesion load following oncogene activation. After confirming that both mammary gland development and RCAS viral infection rates were comparable between wildtype and ARF-null mammary glands (Supplementary Fig 3), we quantified lesion load of the entire wholemounted (Fig 3A) mammary gland following RCAS-mediated caErbB2 delivery, and observed a significant increase in number of both total (>200 μm; Fig 3B) and advanced (>600 μm; Fig 3C) premalignant lesions in ARF-null mammary glands compared to wildtype. To ensure that the observed increase in premalignant lesion load was due to primarily to impairment of senescence, as opposed to loss of other functionsof ARF, we examined the effect of ARF-loss on proliferation rates and induction of autophagy, both of which have been reported to be at least partially regulated by ARF(38,39). We found that both proliferation rates and levels of autophagy were comparable between wildtype and ARF-null early lesions (Supplementary Fig 4). Taken together, these data suggest that ARF regulates a senescence-mediated barrier that functions to (1) prevent oncogene-activated mammary epithelial cells from forming premalignant lesions and (2) subsequently impair the progression of these early lesions to advanced lesions.
Figure 3. Loss of senescence in ARF-null mice is associated with a heavier premalignant lesion load.
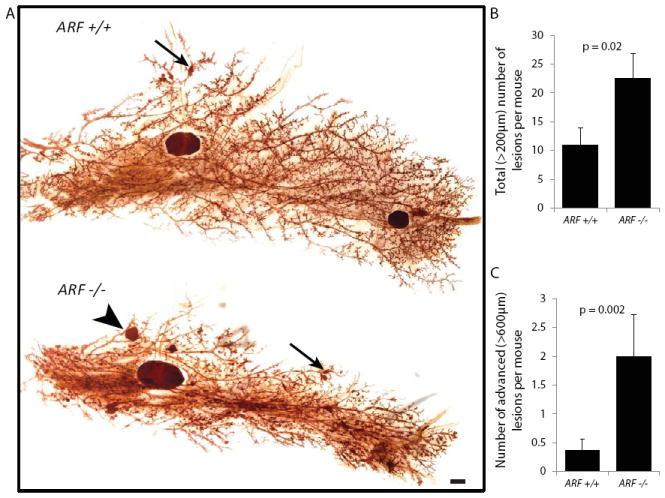
RCAS retrovirus was used to carry caErbb2 into the mammary glands. Early lesions were analyzed two weeks following viral injection.
A. Wholemounted mammary glands from ARF-null and wildtype stained with neutral red. Arrows indicate examples of structures considered to be lesions <600μm, arrowhead indicates lesion >600 μm. (Scale bar = 1mm)
B &C. Quantification of the number of lesions in total (B) and the number of larger lesions (C) based on images of wholemounted glands (n =11,7). Columns represent the mean, and error bars represent the SEM.
Loss of senescence is associated with more rapid tumor induction
To determine whether loss of senescence equates to the loss of a critical tumor barrier in the mammary gland, we carried out a tumor study in which mammary epithelia of ARF-null and wildtype mice were infected with RCAS-caErbB2 and then palpated for tumors. We found that loss of ARF ledto significantly decreased tumor latency (Fig 4A), strongly suggesting that oncogene-induced senescence truly inhibits tumorigenesis independently of apoptosis. ARF-null tumors also grew more rapidly (Fig 4B).Together, these findings suggest that senescence playsa critical role as a barrier to tumor initiation and growth.
Figure 4. Loss of senescencein ARF-null miceis associated with more rapid tumor induction.
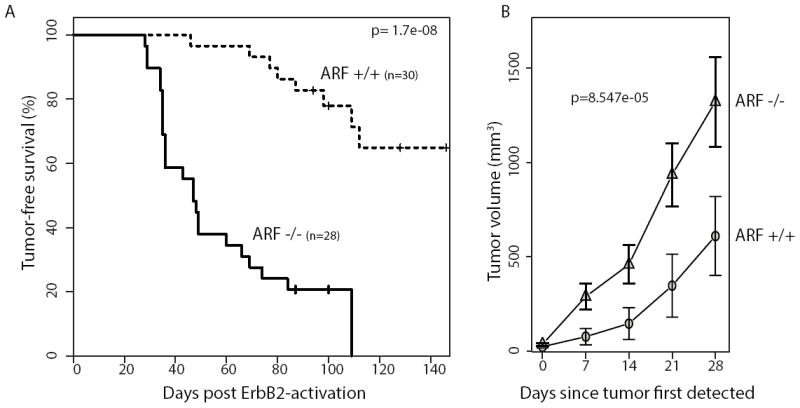
A. Kaplan-Meier survival curve comparing mammary tumor-free survival between wildtype and ARF-null mice (n=30, 28) injected with RCAS retrovirus carrying caErbB2.
B. Tumor growth curves of the above mice (n=17, 27).
Discussion
Previous studies have identified the DNA-damage response pathways as critical for mediating apoptosis and senescence following oncogene activation(19,40-45). A large volume of data points to the p53 tumor suppressor as an important mediator of apoptosis, cell cycle arrest, and senescence under a variety of cellular circumstances(reveiwed in 38-41).However, it has been recently reported (44), and we have confirmed (Supplementary Fig 5A), that complete loss of p53 does not perturb the oncogene-induced DNA damage complex formation and upstream signaling. In this report, we show that p53 is critical for at least the senescence response to oncogene-activation, and that the ARF-p53 axis likely works in concert with the ATM-p53 axis to execute a robust senescence response and inhibit tumorigenesis. In support of this, we find that loss of ARF leads to a compensatory increase in the DDR (Supplementary figure 4D), which is diminishedin frank tumors (Supplementary figure 6B). These findings are in agreement with those of Evangelou et al.(45)and Gupta et al.(44), both of which suggest that the DNA damage response and the ARF pathway interact cooperatively to erect barriers to tumorigenesis. However, in contrast with the suggestion by Gupta et al. (44)that the ARF pathway is required as a tumor barrier only in the context of impaired DDR, our studies indicate that ARF is a necessary tumor suppressor even in the context of an intact (and even elevated) DDR. Based on the observation that ARF is induced at a higher threshold of oncogenic stress than is the DDR(45), we hypothesize that impaired DDR leads to a more rapid accumulation of DNA damage and replicative stress, thereby triggering an early robust induction of ARF. It would be interesting to determine whether oncogene-activated DDR-wild type cells are able to induce ARF to a similar degree as DDR-impaired cells, albeit in a delayed manner.
Our observation thatp53-null lesions exhibit robust apoptosisis heavily contrary to conventional wisdom that places p53 as a central mediator of apoptosis, oncogene-induced or otherwise(46-49). It is possible that oncoprotein-induced cellular stresses (such as replicative and metabolic stress) can access multiple p53-independent pathways that culminate in apoptosis. Based on our findings, it seems that the senescence barrier, more than the apoptotic barrier, relies primarily on the ARF-p53 axis for full induction. Because of the abundance of evidence identifying p53 as a pivotal and multifaceted tumor suppressor, it would certainly be of keen interest to clearly delineate additional processes by which p53 plays its most decisive tumor suppressive roles.
Our data revealed that loss of ARF led to an increased premalignant lesion load and more rapid tumor induction. These findings are in agreement with the documented role of ARF as tumor suppressor since the early 1990s(21,50,51). Previous attempts to determine the role of ARF in the mammary gland in vivo have yielded limited conclusions, presumably because ARF-null mice succumb to lymphoma prior to the incidence of mammary tumors using transgenic models (50,52). Intraductally delivering an activated oncogene to the mammary epithelium allows us to generate premalignant lesions and tumors before the development of lymphoma in ARF-null mice. Incidentally, though it is not the primary aim of study to focus on ARF, the set of data we have presented is the first to prove, using a completely in vivo model, that ARF functions as a bona fide mammary tumor suppressor.
Although ARF has been reported to regulate other processes in the cell, including autophagy and proliferation via ribosome biogenesis (34), the only difference we have been able to detect between wildtype and ARF-null mice is in the induction of senescence following oncogene-activation, leading us to propose that it truly is this senescence difference that is responsible for the ultimate ARF-null phenotype of increased lesion load and shortened tumor latency. Further, we found that senescence in wildtype tumors are reduced to levels comparable to that in ARF-null tumors (Supplementary Fig 6A), suggesting that senescence in wildtype mice must have been disabled over the course of tumor progression, in line with the role of senescence as a barrier to tumorigenesis.
Mammary glands from ARF-null virgin mice have been reported to be similar to those of wild type mice with the exception of possibly increased dilation of primary ducts and enhanced tertiary branches (53). Our whole mount staining confirmed similar ductal trees between ARF-null versus wild type mice (Supplementary fig 3). It has also been reported that ductal proliferation in virgin mice was unaltered by AFR loss (53). Nevertheless, we cannot exclude the possibility that ARF loss skewed the mammary gland cell fate, resulting into a more susceptible state to transformation by ErbB2. ARF loss-induced reduction in cellular senescence (Fig 2) and increase in transplantation potential of mammary epithelial cells (53)suggest an expanded stem cell population in ARF-null mammary glands. However, the mammary cell subtype that is most susceptible to tumor induction by ErbB2 seems to be committed luminal cells expressing the alveolar cell marker whey acidic protein(20,54)or luminal progenitor cells (55)but not the less differentiated cells expressing keratin 6 (56)or cells with active Wnt signaling (57).
Our in vivo evidence that the induction of senescence at the premalignant lesion stage can greatly inhibit progression of premalignant mammary lesions has significant implications for cancer chemoprevention; our observations suggest that chemoprevention administered to patients bearing premalignant lesions may be more effective in halting or delaying tumor formation if both apoptosis and senescence are efficiently induced. However, because of increasing reports that within an established cancer senescence instigate tumor progressionvia a pro-tumor secretory mechanism(12), it is essential to elucidate why senescent cells within an established cancer are tumor-supportive while senescent cells within a premalignant lesion are tumor-suppressive.
In summary, p53 and ARF are required for a robust senescence response in breast cells that have suffered an oncogenic mutation while they are unexpectedly dispensable to an apoptosis response. Furthermore, the ARF gene function providesa critical barrier to mammary tumorigenesis, most likely via induction of a robust senescence response following oncogene activation. In addition, our data strongly suggest that weakening of the senescence barrier at the premalignant stage promote tumor formation.
Supplementary Material
Acknowledgments
The authors thank Dr. Lawrence Donehower for advice in experimental design and data interpretation and for the p53 knockout mouse line, and Drs. Jeffrey Rosen, Hua Dai, Jie Dong, and Svasti Haricharan for advice in experimental design and data interpretation. This work was supported in part by funds from the Cancer Prevention Research Institute of Texas RP101499 (to V.S); NIH CA124820 (to Y.L) and U54CA149196 (to Y. L; PI: Stephan Wong); from CDMRP BC085050 (to Y. L) and BC073703 (to Y. L); and from the Nancy Owens Memorial Foundation (to Y. L); as well as by the resources from the Dan L. Duncan Cancer Center (P30CA125123) and the Sue & Lester Breast Center (P50-CA058183).
Footnotes
Conflict of interest: The authors declare no conflict of interest
Supplementary information accompanies the paper on the Cancer Research website (http://cancerres.aacrjournals.org/)
References
- 1.Reddy JP, Li Y. Oncogene-induced senescence and its role in tumor suppression. J Mammary Gland Biol Neoplasia. 2011;16:247–56. doi: 10.1007/s10911-011-9221-5. [DOI] [PubMed] [Google Scholar]
- 2.Cotter TG. Nat Rev Cancer. Vol. 9. Nature Publishing Group; 2009. Apoptosis and cancer: the genesis of a research field; pp. 501–7. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg R. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg R. Cell. Vol. 144. Elsevier Inc.; 2011. Hallmarks of cancer: the next generation; pp. 646–74. [DOI] [PubMed] [Google Scholar]
- 5.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–7. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AHFM, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–5. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 7.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z, Trotman LC, Shaffer D, Lin H-K, Dotan Za, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, van der Horst CMaM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 10.Sarkisian CJ, Keister Ba, Stairs DB, Boxer RB, Moody SE, Chodosh La. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 11.Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. Dec;2008:2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davalos AR, Coppe J-P, Campisi J, Desprez P-Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29:273–83. doi: 10.1007/s10555-010-9220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–90. doi: 10.1038/ncb2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang T-W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–51. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 15.Du Z, Podsypanina K, Huang S, McGrath A, Toneff MJ, Bogoslovskaia E, et al. Introduction of oncogenes into mammary glands in vivo with an avian retroviral vector initiates and promotes carcinogenesis in mouse models. Proc Natl Acad Sci U S A. 2006;103:17396–401. doi: 10.1073/pnas.0608607103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bu W, Chen J, Morrison GD, Huang S, Creighton CJ, Huang J, et al. Keratin 6a marks mammary bipotential progenitor cells that can give rise to a unique tumor model resembling human normal-like breast cancer. Oncogene. 2011;30:4399–409. doi: 10.1038/onc.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haricharan S, Hein SM, Dong J, Toneff MJ, Aina OH, Rao PH, et al. Contribution of an alveolar cell of origin to the high-grade malignant phenotype of pregnancy-associated breast cancer. Oncogene. 2013 doi: 10.1038/onc.2013.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reddy JP, Li Y. The RCAS-TVA system for introduction of oncogenes into selected somatic mammary epithelial cells in vivo. J Mammary Gland Biol Neoplasia. 2009;14:405–9. doi: 10.1007/s10911-009-9157-1. [DOI] [PubMed] [Google Scholar]
- 19.Reddy JP, Peddibhotla S, Bu W, Zhao J, Haricharan S, Du Y-CN, et al. Defining the ATM-mediated barrier to tumorigenesis in somatic mammary cells following ErbB2 activation. Proc Natl Acad Sci U S A. 2010;107:3728–33. doi: 10.1073/pnas.0910665107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haricharan S, Dong J, Hein S, Reddy JP, Du Z, Toneff M, et al. Elife. Vol. 2. eLife Sciences Publications Limited; 2013. Mechanism and preclinical prevention of increased breast cancer risk caused by pregnancy; p. e00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun Ra, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–59. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 22.Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- 23.Zheng B, Vogel H, Donehower LA, Bradley A. Visual genotyping of a coat color tagged p53 mutant mouse line. Cancer Biol Ther. 2002;1:433–5. doi: 10.4161/cbt.1.4.24. [DOI] [PubMed] [Google Scholar]
- 24.Bu W, Xin L, Toneff M, Li L, Li Y. Lentivirus vectors for stably introducing genes into mammary epithelial cells in vivo. J Mammary Gland Biol Neoplasia. 2009;14:401–4. doi: 10.1007/s10911-009-9154-4. [DOI] [PubMed] [Google Scholar]
- 25.Pao W, Klimstra DS, Fisher GH, Varmus HE. Use of avian retroviral vectors to introduce transcriptional regulators into mammalian cells for analyses of tumor maintenance. Proc Natl Acad Sci U S A. 2003;100:8764–9. doi: 10.1073/pnas.1133333100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bandyopadhyay D, Gatza C, Donehower LA, Medrano EE. Analysis of cellular senescence in culture in vivo: the senescence-associated beta-galactosidase assay. Curr Protoc Cell Biol. 2005;Chapter 18(Unit 18.9) doi: 10.1002/0471143030.cb1809s27. [DOI] [PubMed] [Google Scholar]
- 28.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4:1798–806. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- 29.Landua JD, Visbal AP, Lewis MT. Methods for preparing fluorescent and neutral red-stained whole mounts of mouse mammary glands. J Mammary Gland Biol Neoplasia. 2009;14:411–5. doi: 10.1007/s10911-009-9155-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005;8:19–30. doi: 10.1016/j.devcel.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Ibrahim JG, et al. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell. 2009;8:439–48. doi: 10.1111/j.1474-9726.2009.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer Nature Publishing Group. 2006;6:663–73. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- 33.Swarbrick A, Roy E, Allen T, Bishop JM. Id1 cooperates with oncogenic Ras to induce metastatic mammary carcinoma by subversion of the cellular senescence response. Proc Natl Acad Sci U S A. 2008;105:5402–7. doi: 10.1073/pnas.0801505105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ozenne P, Eymin B, Brambilla E, Gazzeri S. The ARF tumor suppressor: structure, functions and status in cancer. Int J Cancer. 2010;127:2239–47. doi: 10.1002/ijc.25511. [DOI] [PubMed] [Google Scholar]
- 35.Serrano M, Lin A, McCurrach M, Beach D, Lowe S. Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16 INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 36.Brenner AJ, Stampfer MR, Aldaz CM. Increased p16 expression with first senescence arrest in human mammary epithelial cells and extended growth capacity with p16 inactivation. Oncogene. 1998;17:199–205. doi: 10.1038/sj.onc.1201919. [DOI] [PubMed] [Google Scholar]
- 37.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 38.Sugimoto M, Kuo M-L, Roussel MF, Sherr CJ. Mol Cell. Vol. 11. Elsevier; 2003. Nucleolar Arf Tumor Suppressor Inhibits Ribosomal RNA Processing; pp. 415–24. [DOI] [PubMed] [Google Scholar]
- 39.Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, et al. A Short Mitochondrial Form of p19ARF Induces Autophagy and Caspase-Independent Cell Death. Mol Cell. 2006;22:463–75. doi: 10.1016/j.molcel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 40.Te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA Damage Is Able to Induce Senescence in Tumor Cells in Vitro and in Vivo. Cancer Res. 2002;62:1876–83. [PubMed] [Google Scholar]
- 41.Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, et al. Nature. Vol. 434. Macmillian Magazines Ltd; 2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis; pp. 864–70. [DOI] [PubMed] [Google Scholar]
- 42.Gorgoulis VG, Vassiliou L-VF, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Nature. Vol. 434. Macmillian Magazines Ltd; 2005. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions; pp. 907–13. [DOI] [PubMed] [Google Scholar]
- 43.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 44.Gupta GP, Vanness K, Barlas A, Manova-Todorova KO, Wen YH, Petrini JHJ. The Mre11 Complex Suppresses Oncogene-Driven Breast Tumorigenesis and Metastasis. Mol Cell. 2013 doi: 10.1016/j.molcel.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evangelou K, Bartkova J, Kotsinas A, Pateras IS, Liontos M, Velimezi G, et al. The DNA damage checkpoint precedes activation of ARF in response to escalating oncogenic stress during tumorigenesis. Cell Death Differ. 2013;20:1485–97. doi: 10.1038/cdd.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donehower LA, Lozano G. Nat Rev Cancer. Vol. 9. Nature Publishing Group; 2009. 20 years studying p53 functions in genetically engineered mice; pp. 831–41. [DOI] [PubMed] [Google Scholar]
- 47.Levine AJ, Oren M. Nat Rev Cancer. Vol. 9. Nature Publishing Group; 2009. The first 30 years of p53: growing ever more complex; pp. 749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meek DW. Nat Rev Cancer. Vol. 9. Nature Publishing Group; 2009. Tumour suppression by p53: a role for the DNA damage response? pp. 714–23. [DOI] [PubMed] [Google Scholar]
- 49.Brady CA, Attardi LD. p53 at a glance. J Cell Sci. 2010;123:2527–32. doi: 10.1242/jcs.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59:2217–22. [PubMed] [Google Scholar]
- 51.Weber JD. p53-independent functions of the p19ARF tumor suppressor. Genes Dev. 2000;14:2358–65. doi: 10.1101/gad.827300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.D’Amico M, Wu K, Di Vizio D, Reutens AT, Stahl M, Fu M, et al. The Role of Ink4a/Arf in ErbB2 Mammary Gland Tumorigenesis. Cancer Res. 2003;63:3395–402. [PubMed] [Google Scholar]
- 53.Yi Y, Shepard A, Kittrell F, Mulac-Jericevic B, Medina D, Said TK. p19ARF determines the balance between normal cell proliferation rate and apoptosis during mammary gland development. Mol Biol Cell. 2004;15:2302–11. doi: 10.1091/mbc.E03-11-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Henry MD, Triplett AA, Oh KB, Smith GH, Wagner K-U. Parity-induced mammary epithelial cells facilitate tumorigenesis in MMTV-neu transgenic mice. Oncogene. 2004;23:6980–5. doi: 10.1038/sj.onc.1207827. [DOI] [PubMed] [Google Scholar]
- 55.Jeselsohn R, Brown NE, Arendt L, Klebba I, Hu MG, Kuperwasser C, et al. Cyclin D1 kinase activity is required for the self-renewal of mammary stem and progenitor cells that are targets of MMTV-ErbB2 tumorigenesis. Cancer Cell. 2010;17:65–76. doi: 10.1016/j.ccr.2009.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Y, Welm B, Podsypanina K, Huang S, Chamorro M, Zhang X, et al. Evidence that transgenes encoding components of the Wnt signaling pathway preferentially induce mammary cancers from progenitor cells. Proc Natl Acad Sci U S A. 2003;100:15853–8. doi: 10.1073/pnas.2136825100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bu W, Zhang X, Dai H, Huang S, Li Y. PLoS One. Vol. 8. Public Library of Science; 2013. Mammary cells with active Wnt signaling resist ErbB2-induced tumorigenesis; p. e78720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.