

Abstract
Metastatic colonization is an ominous feature of cancer progression. Recent studies have established the importance of pre-mRNA alternative splicing (AS) in cancer biology. However, little is known about the transcriptome-wide landscape of AS associated with metastatic colonization. Both in vitro and in vivo models of metastatic colonization were utilized to study AS regulation associated with cancer metastasis. Transcriptome profiling of prostate cancer cells and derivatives crossing in vitro or in vivo barriers of metastasis revealed splicing factors with significant gene expression changes associated with metastatic colonization. These include splicing factors known to be differentially regulated in epithelial-mesenchymal transition (ESRP1, ESRP2, RBFOX2), a cellular process critical for cancer metastasis, as well as novel findings (NOVA1, MBNL3). Finally, RNA-seq indicated a large network of AS events regulated by multiple splicing factors with altered gene expression or protein activity. These AS events are enriched for pathways important for cell motility and signaling, and affect key regulators of the invasive phenotype such as CD44 and GRHL1.
Keywords: alternative splicing, splicing factor, cancer, metastasis, RNA-seq
Introduction
A central goal of cancer research is to elucidate molecular events that contribute to the life-threatening metastasis of cancer cells. Hematogenous dissemination and metastatic colonization of distant organs is a salient feature of malignant cancers (1). An emerging theme in our understanding of this critical process of cancer progression is that epithelial-to-mesenchymal transition (EMT) may facilitate metastasis (2). EMT is a normal process in development whereby cells phenotypically switch from an epithelial state to a mesenchymal state enabling, among other things, invasive and migratory cell behavior (2). It is also considered as a key process by which epithelial tumors acquire the capability to metastasize (3). This phenotypically plastic EMT-like state is driven by global changes of the gene regulatory program at both the transcriptional and post-transcriptional levels (4–7). Transcriptome profiling studies have identified the core transcriptional regulatory network of EMT involving key transcription factors and their upstream regulators and downstream targets (4). Recent studies have also begun to reveal widespread changes in mRNA isoforms during EMT arising from pre-mRNA alternative splicing (AS) (6–8).
AS is an important mechanism of generating regulatory complexity in higher eukaryotes (9). Through alternative choices of exons and splice sites during splicing, a single gene can generate functionally distinct mRNA and protein isoforms. Many genes are differentially spliced during development or among different tissues and cell types (10). Cell-type-specific AS patterns are largely controlled by a small number of master splicing regulators (“splicing factors”) with cell-type-specific gene expression or protein activity (11–13). Pioneering work by Carstens and colleagues identified the epithelial-specific splicing factors ESRP1 and ESRP2 as the master splicing regulators in epithelial cells (14). During EMT, the expression levels of ESRPs are greatly diminished (15). This triggers a transcriptome-wide remodeling of the AS regulatory program, resulting in the loss of epithelial-specific protein isoforms that promote cell-cell adhesion and the acquisition of mesenchymal-specific protein isoforms that promote cell migration and motility (6, 16–18). Ectopic expression or knockdown of ESRPs leads to changes in cell morphology and phenotypes that resemble the transitions between epithelial and mesenchymal cells (6, 7). Using cell culture models of EMT, a large number of EMT-associated AS changes have been identified (7). Many of these AS events are regulated by ESRPs, while additional splicing factors such as those from the RBFOX, MBNL, CELF, and hnRNP families may also play roles in AS regulation during EMT (7). However, it should be noted that these AS events are identified using in vitro models of EMT or by perturbing the cellular concentrations of EMT-relevant splicing factors. The transcriptome-wide landscape of AS regulation in cancer cells undergoing metastatic colonization remains to be elucidated.
To study the metastatic colonization of cancer cells, we previously derived a series of cell lines from the commonly used PC-3 prostate cancer cell line by selecting for subpopulations of PC-3 cells that crossed in vitro or in vivo barriers for metastasis (19). The PC-3 cell line was used in classic studies by Fidler and colleagues to demonstrate that passage of these cells through mice selects for variants of greater metastatic colonization potential (20), and thus makes this an experimentally advantageous model with which to probe the determinants of metastatic colonization. Specifically, we used an in vitro transendothelial migration model to isolate variants of PC-3 cells on the basis of their ability to cross an endothelial monolayer, which is related to the process of extravasation during cancer metastasis (19). We also isolated cells from an in vivo murine metastatic colonization model, in which we injected PC-3 cells (or derivative cells) into mouse tail vein and subsequently harvested cells from metastatic tumors in distal organs. Compared to the parental PC-3 cell line, these derivative cell lines exhibited molecular and phenotypic hallmarks of EMT including the loss of E-cadherin and the expression of mesenchymal transcription factors such as ZEB1, which is required for their enhanced invasive phenotype (19). These in vitro and in vivo models thus represent unbiased phenotypic selection experiments and allow us to comprehensively characterize transcriptome profiles including AS events and splicing factor expression associated with metastatic colonization.
In this study, we performed comprehensive transcriptome analyses of the parental PC-3 cell line and a series of derivative cell lines to elucidate AS regulation associated with metastatic colonization. Using microarray and RNA-seq, we discovered splicing factors with significant changes in gene expression between the PC-3 and its derivative cell lines. The ESRP splicing factors had the most dramatic change in expression levels through serial passages across the in vitro and in vivo barriers of metastasis, while several additional splicing factors were also significantly up- or down-regulated. From the RNA-seq data, we identified nearly a thousand splice variants with significant differential AS between the parental and derivative cells, providing novel splicing signatures for cancer metastasis. Our data demonstrate that transcriptome-wide remodeling of the AS network is an integral regulatory process underlying metastatic colonization, and reveal an extensive resource of AS events that may impact the invasiveness and metastatic behavior of cancer cells.
Materials and Methods
Gene expression microarray analysis
The PC-3 cell line and its derivative cell lines were profiled for gene expression using the Affymetrix HG-U133_plus_2 GeneChip. Microarray data were deposited into the NCBI Gene Expression Omnibus (GEO) with the accession ID GSE14405 and GSE56410. RNA samples were processed by the University of Iowa DNA Core Facility. Probe intensity values from the Affymetrix CEL files, which summarize the pixel intensities, were used for further analyses. Reading and processing of the probe intensities were done using the affy package in Bioconductor (21). Gene expression estimates (log2 scale) were obtained using the default parameters of the GCRMA algorithm from the gcrma package (22). All samples were quantile normalized as a group in GCRMA. Ensembl Gene IDs corresponding to Affymetrix probe set IDs were obtained using the biomaRt package (23, 24). The gene expression matrices were then refined by excluding control probe sets and probe sets with ambiguous gene assignments (i.e. annotated to multiple Ensembl gene IDs). For genes with multiple probe sets, we selected the probe set with the highest overall probe signal to represent its Ensembl gene. In total, we compiled 20,595 probe sets representing 20,595 unique Ensembl human genes. Principal component analysis (PCA) based on all 20,595 genes’ expression levels was performed using the pcaMethods package (25). All data processing steps were carried out in the R statistical computing environment (http://www.r-project.org).
Prostate cancer cell lines and real-time qPCR analysis of splicing factors
The PC-3 cell line and its derivatives were cultured in DMEM/F12 with 10% FBS, 1% NEAA and 400 μg/ml G-418. All cell lines were grown at 37°C and 5% CO2. Total RNAs were extracted using the RNeasy® Mini kit (Qiagen Science, Germantown, MD) and then subjected to reverse transcription by the High-Capacity cDNA Kit (Applied Biosystems, Foster city, CA). Quantitative real-time PCR was carried out using SYBR Green reagents. GAPDH was used as the reference gene. Relative gene expression level was measured by the comparative Ct (2−ΔΔCt) method (26). Realtime qPCR primer sequences were provided in Supplementary Table S1.
RNA sequencing and data analysis
The PC-3E and GS689.Li cell lines were used for Illumina RNA sequencing, using RNAs from three independent cell cultures per cell line. Total RNA samples with RIN (RNA integrity number) >9.5 (measured by the Agilent 2100 BioAnalyzer) were used for RNA-seq library preparation using the TruSeq™ RNA Sample Preparation Kit (Illumina). 101x2 bp paired-end RNA-seq reads were generated on the HiSeq-2000 sequencer. The RNA-seq data were deposited into the NCBI Sequence Read Archive (accession ID: SRS354082).
We mapped RNA-seq reads to the human genome (hg19) and transcriptome (Ensembl, release 65) using the software TopHat (v1.4.1) (27) allowing up to 3 bp mismatches per read and up to 2 bp mismatches per 25 bp seed. We used Cufflinks (v2.1.1) (28) to calculate RNA-seq based gene expression levels using the FPKM metric (fragments per kilobase of exon per million fragments mapped). We used edgeR (29) to identify differential gene expression between the two cell types under FDR<1% and fold change >2. To identify differential AS events between the two cell types, we used rMATS (http://rnaseq-mats.sourceforge.net, version 3.0.7) to identify differential AS events corresponding to all five basic types of AS patterns (see the entire list in Supplementary Table S2). Briefly, rMATS uses a modified version of the generalized linear mixed model to detect differential AS from RNA-seq data with replicates. It accounts for exon-specific sequencing coverage in individual samples as well as variation in exon splicing levels among replicates. For each AS event, we used both the reads mapped to the splice junctions and the reads mapped to the exon body as the input for rMATS.
Gene Ontology enrichment analysis
We performed GO enrichment analysis using DAVID (30). Genes with differential AS or differential gene expression were submitted to DAVID as the gene list of interest. We used all expressed genes with average FPKM>1 in at least one of the two cell types as the background gene list. Only GO terms with at least 10 counts (genes) in the gene list of interest were considered. Significant GO terms were defined as those with Benjamini corrected FDR of < 0.05.
Motif enrichment analysis
We sought to identify binding sites of splicing factors and other RNA binding proteins that were significantly enriched in differential exon skipping events between the PC-3E and GS689.Li cell lines as compared to control (non-regulated) alternative exons. We collected 112 known binding sites of human RNA binding proteins including many well-characterized splicing factors from the literature (16, 31–33) (Supplementary Table S3). For each motif, we scanned for motif occurrences separately in exons or their 250 bp upstream or downstream intronic sequences. For intronic sequences, we excluded the 20 bp sequence within the 3′ splice site and the 6 bp sequence within the 5′ splice site. 5,671 alternative exons without splicing changes (rMATS FDR>50%) in highly expressed genes (FPKM>5.0 in at least one cell type) were treated as control exons. For each motif, after we counted the number of occurrences in the differentially spliced exons and the control exons, we calculated p-value for motif enrichment via Fisher’s exact test (one-sided) and used Benjamini FDR correction to adjust for multiple testing for exons, upstream intronic sequences, and downstream intronic sequences separately.
Compilation of known target exons of ESRP1, RBFOX2, and NOVA1
276 ESRP1 target exons were collected from our exon microarray and RNA-seq analysis of ESRP1-regulated alternative splicing events (6, 16). 121 RBFOX2 target exons were collected from the work of Venables and colleagues which compiled a list of RBFOX2-regulated alternative splicing events based on RT-PCR and CLIP-seq studies (34). 287 NOVA1 target exons were compiled by converting the coordinates of NOVA1-regulated mouse exons (35) to the human genome.
Fluorescently labeled RT-PCR
Fluorescently labeled RT-PCR was performed to quantify exon splicing levels as described previously (36). RT-PCR primers were shown in Supplementary Table S1.
Ectopic expression of ESRP1, RBFOX2, and NOVA1 in prostate cancer cells
Murine Esrp1 coding sequence (amplified from mouse kidney cDNA) and human NOVA1 coding sequence (obtained from Open Biosystems; clone ID: 30915356) were subcloned into the pQCXIP vector. Final plasmids were confirmed by DNA Sanger sequencing (University of Iowa DNA facility, Iowa city, IA, USA). pcDNA-RBFOX2 was a gift from Dr. Douglas Black (UCLA). Based on the expression levels of these three splicing factors, we ectopically expressed Esrp1 in GS689.Li and RBFOX2 and NOVA1 in PC-3E. Retroviruses containing Esrp1 and NOVA1 were generated by transient transfection of retroviral vector together with a packaging plasmid pVSV-G into the packaging cell line GP2-293. The supernatant containing retroviruses was collected, filtered through 0.45μM filter and used to infect recipient cells. After infection, cells were selected in puromycin (0.5 μg/ml) and further passaged in culture. Transient transfection of pcDNA-RBFOX2 was carried out using Lipofectamine 2000 following the manufacturer’s protocol. RNA extraction was done ~ 48 hours after transfection. RT-PCR primer sequences for the 30 randomly selected exons were provided in Supplementary Table S1.
Ectopic expression of EMT-relevant transcription factors in prostate cancer cells
For ectopic expression in PC-3E cells, coding sequences of ZEB1 (purchased from Origene) and TWIST2 (amplified from cDNA of PC-3 cells) were subcloned into the pQCXIP or pLEX vector. All plasmids constructed using PCR were confirmed by DNA sequencing. pWZL-Blast-SNAI1-ER were purchased from Addgene. Retroviruses containing TWIST2 and SNAI1-ER were generated by transient transfection in packaging cell line GP2-293. Lentiviruses containing ZEB1 were produced in packaging cell line 293FT. The supernatant containing retroviruses or lentiviruses was collected, filtered through 0.45-μm filter and used to infect PC-3E cells. Infected cells were selected in puromycin (1 μg/ml) or blasicidin (20 μg/ml) and further passaged in culture.
Western blot analysis
Protein lysates were scraped and harvested in RIPA buffer. Primary antibodies reported in this study are: anti-β-actin (Sigma A1978), anti-NOVA1 (a gift from Dr. Douglas Black), anti-FLAG (Sigma F3165), anti-E-cadherin (R&D System), anti-TWIST2 (Sigma, clone:3C8), anti-SNAI1 (Cell signaling, clone:C15D3), anti-ZEB1 (a gift from Dr. Douglas Darling). Secondary antibodies are: goat anti-mouse IgG FITC (Millipore), Odyssey goat anti-mouse IRDye 800CW and Odyssey goat anti-rabbit IRDye 680 (Li-Cor Biosciences), and IgG Isotype (Sigma). Western blot was performed following the standard protocol. Signals were detected by the Odyssey Infrared Imager (LI-COR Biosciences).
Survival analysis of The Cancer Genome Atlas (TCGA) data
The RNA-seq data of 13 cancer types were downloaded from The Cancer Genome Atlas (TCGA) data portal (http://tcga-data.nci.hih.gov/tcga/). We obtained the RNA-seq data of 877 breast cancer patients (BRCA), 152 glioblastoma patients (GBM), 380 lung adenocarcinoma patients (LUAD), 353 lung squamous cell carcinoma patients (LUSC), 469 clear cell renal cell carcinoma patients (ccRCC), 260 ovarian cancer patients (OV), 149 rectum adenocarcinoma patients (READ), 156 prostate cancer patients (PRAD), 390 colon adenocarcinoma patients (COAD), 26 cutaneous melanoma patients (SKCM), 466 thyroid cancer patients (THCA), 303 head and neck squamous cell carcinoma patients (HNSC), and 441 uterine corpus endometrial carcinoma patients (UCEC). The RNA-seq data were mapped to genes and gene expression levels were calculated by the TCGA consortium. We obtained the expression levels of ESRP1 from the processed gene expression levels provided by TCGA. To test the association of ESRP1 expression level with patient survival, for each cancer type unsupervised 2-means clustering based on ESRP1 expression levels was performed to segregate patients into two distinct groups, one group with high expression of ESRP1 and the other group with low expression of ESRP1. The Kaplan-Meier survival estimator was plotted for the two patient groups in ccRCC and breast cancer separately. Cox regression was used to calculate the p-value of association between gene expression and survival.
Results
Altered expression of pre-mRNA splicing factors in models of metastatic colonization
Tissue invasion and metastasis are a hallmark of cancer (1). To study transcriptome changes associated with metastasis, we previously used in vitro and in vivo models of metastatic colonization to derive a series of cell lines from the PC-3 prostate cancer cell line (Figure 1A). Specifically, starting from the parental PC-3 cell line, we utilized an in vitro transendothelial migration (TEM) model to derive cell lines after single passage (TEM2-5, TEM3-8) or double passage (TEM4-18) of PC-3 cells through an endothelial monolayer (19). We also injected the PC-3 or TEM4-18 cells into the tail vein of mice and harvested cells through single or double rounds of selection in vivo from metastatic tumors in distal organs, such as left axillary lymph node (denoted as LALN), lung (Lu), urogenital (Ug), kidney (Ki), LAd (left adrenal), liver (Li). Of note, these derived cell lines have enhanced ability to migrate through the endothelial monolayer and are more aggressive in the murine metastatic colonization model in vivo, with cell lines derived from in vivo passage having a somewhat greater metastatic colonization potential than those derived in vitro by selection for transendothelial migration (19). A detailed schematic diagram describing the derivation and relationships of these cell lines is shown in Figure 1A.
Figure 1.
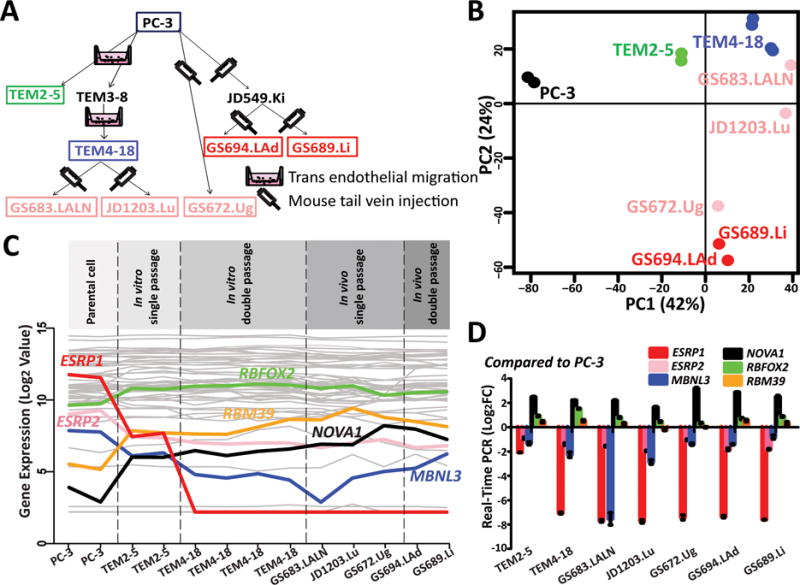
Microarray analysis of global gene expression profiles of PC-3 and derivative cell lines.
(A) A schematic diagram describing the generation of the PC-3 derivative cell lines using the in vitro transendothelial migration model or the in vivo murine metastatic colonization model. The relationship between the parental PC-3 cell line and all derivative cell lines is depicted. Derivative cell lines selected for gene expression microarray analysis are indicated by boxes. (B) Principal component analysis (PCA) of global gene expression profiles of PC-3 and derivative cell lines. (C) Gene expression levels of 60 well-known splicing factors. The six splicing factors with significant expression differences between PC-3 and derivative cell lines are highlighted. (D) Realtime qPCR results of six splicing factors. GAPDH was used as the reference gene. Relative gene expression level was normalized to PC-3 cells. Error bar indicates the standard error of the mean of technical triplicates.
To characterize the transcriptome profiles of these cell lines, we carried out global gene expression profiling of the parental PC-3 cell line and its derivative cell lines (boxed in Figure 1A) using the Affymetrix HG-U133_plus_2 gene expression microarray, with multiple replicates for some of the cell lines. We used principal component analysis (PCA) to compare and contrast the global gene expression profiles among these cell lines. As shown in Figure 1B, the PCA analysis revealed a clear separation between the parental PC-3 cell line and other derivative cell lines. This pattern was consistent with our previous report that all derivative cell lines showed molecular and phenotypic features of EMT as compared to PC-3 (19). Additionally, we note that these derivative cell lines could be further classified on the basis of their gene expression profiles in a way that matched their number of passages and relationships in the in vitro or in vivo model of metastatic colonization. The two cell lines derived in vitro (TEM2-5, TEM4-18) were clustered, along with two additional cell lines (GS683.LALN, JD1203.Lu) derived from the TEM4-18 cell line after single round of selection in vivo. Similarly, the two cell lines from two rounds of selection in vivo (GS694.LAd and GS689.Li) that shared the same immediate parental cell line JD549.Ki were clustered together, despite the fact that they were isolated from metastatic tumors in different organs (left adrenal and liver, respectively). Taken together, this PCA analysis suggests that the global expression profiles of these cell lines reflect an intrinsic transcriptome signature of metastatic colonization rather than the specific sites of the secondary metastatic tumors.
To investigate the possibility of a transcriptome-wide remodeling of splicing regulation during metastatic colonization, we next sought to identify splicing factors with significant differences in gene expression levels between the PC-3 and its derivative cell lines. We compiled a list of 60 splicing factors with well-established roles in AS regulation (37). Requiring at least two-fold change in gene expression levels between the parental PC-3 cell line and the average of all derivative cell lines, we identified six splicing factors (ESRP1, ESRP2, MBNL3, NOVA1, RBFOX2, RBM39) with significant increase or decrease in gene expression associated with metastatic colonization (Figure 1C). Realtime qPCR analysis using RNAs from independent cell cultures confirmed the significant expression change of 5 of these 6 splicing factors, with the only exception being RBM39 which had a fairly modest expression difference between the PC-3 and derivative cells according to qPCR (Figure 1D). The most striking pattern was observed for the epithelial-specific splicing factor ESRP1. Its expression level was high in the parental PC-3 cell line, dropped by 17-fold in the TEM2-5 cell line after one round of selection in vitro, and was almost completely non-expressed (i.e. with its microarray signal at the background level) after two rounds of selection in vitro and in cell lines selected in vivo (Figure 1C). Its close homolog ESRP2 showed a similar (although less dramatic) trend. These observations are consistent with the key role of ESRP1 and ESRP2 in maintaining epithelial-specific AS patterns, and suggest that the down-regulation of the ESRP splicing network is a key feature of cancer cells with greater metastatic colonization potential. We also observed the up-regulation or down-regulation of three other splicing factors (RBFOX2, NOVA1, and MBNL3). These include RBFOX2 which has been implicated in regulating mesenchymal AS patterns during EMT (7, 8, 38), as well as novel findings (NOVA1, MBNL3). As each splicing factor may regulate AS of hundreds or even thousands of genes, the substantial change in gene expression levels of these splicing factors suggests the global remodeling of the AS regulatory network associated with metastatic colonization.
Transcriptome-wide changes in pre-mRNA alternative splicing associated with metastatic colonization
To discover and characterize AS changes associated with metastatic colonization, we selected one of the in vivo selected cell lines GS689.Li for comparison to the E-cadherin-positive fraction of PC-3 (referred to as PC-3E in this manuscript). PC-3E cells were derived from the parental PC-3 population by fluorescence-activated cell sorting for E-cadherin-positive cells (19). PC-3E constitutes approximately 75% of the PC-3 cells and contains cells that lack the ability to cross the metastatic barrier. We collected RNAs from three independent cell cultures of both PC-3E and GS689.Li. As a pilot analysis, we took 10 ESRP-regulated EMT signature exons (6) and used RT-PCR to quantify their inclusion levels in these two cell types. Except for one exon of the gene RALGPS2 with poor RT-PCR amplification, we observed significant changes of 9 exons between these two cell lines (Figure 2A). These splicing switches are consistent with the mesenchymal features of GS689.Li (19) and indicate that the global AS regulatory program is significantly different in GS689.Li as compared to the PC-3E cells.
Figure 2.
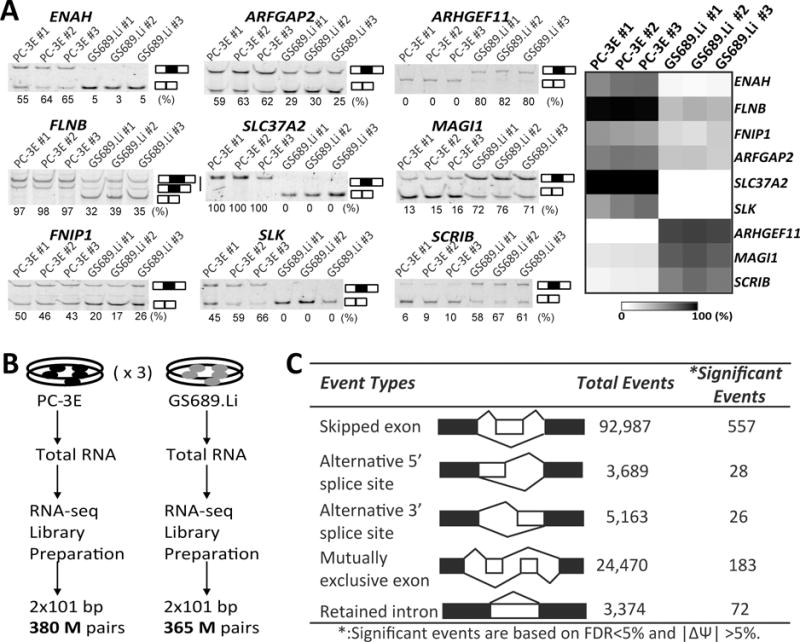
Comparison of AS profiles between the PC-3E and GS689.Li cell lines.
(A) RT-PCR based exon inclusion levels of nine EMT signature exons in PC-3E and GS689.Li. Left panel: fluorescently labeled RT-PCR gel images; the estimated exon inclusion levels are indicated below each gel picture. Right panel: heatmap of nine exons’ exon inclusion levels. (B) Flowchart of RNA-seq analysis of PC-3E and GS689.Li. (C) Summary of differential AS events detected between PC-3E and GS689.Li.
To globally compare AS patterns between the GS689.Li and PC-3E cell lines, we performed deep RNA sequencing (RNA-seq) with three biological replicates for each cell line (Figure 2B). In total, 114~132 million 101bp x 2 paired-end reads were generated per RNA sample, with ~80% of reads mapped uniquely to the human genome and transcriptome (hg19). From the RNA-seq data, we used our rMATS algorithm (see Materials and Methods) to detect differential AS events between these two cell lines corresponding to five basic types of AS patterns (SE: skipped exon, A5SS: alternative 5′ splice site, A3SS: alternative 3′ splice site, MXE: mutually exclusive exons, and RI: retained intron). In total, 129,683 AS events were analyzed and rMATS was used to calculate the p-value and false discovery rate (FDR) that the average splicing level (denoted as ψ, or percent-spliced-in (39)) differed by more than 5% between these two cell types. Under FDR<0.05, we identified 866 differential AS events in 494 genes (Supplementary Table S2). Of these events, the largest category of AS patterns was skipped exon (557), followed by mutually exclusive exons (183), retained intron (72), alternative 5′ splice site (28), and alternative 3′ splice site (26) (Figure 2C and Figure 3A). The RNA-seq data and RT-PCR validation of selected AS events are shown in Figure 3B–C. Of note, among these 866 events, 353 (41%) events showed a change of percent-spliced-in (ψ) of more than 25% (|Δψ|>25%), including 82 (9.5%) events that had “switch-like” AS patterns (40, 41) with the average ψ <34% in one cell line and >66% in the other cell line. These data indicate that numerous genes have a significant shift in isoform usage between these two phenotypically distinct cell types.
Figure 3.
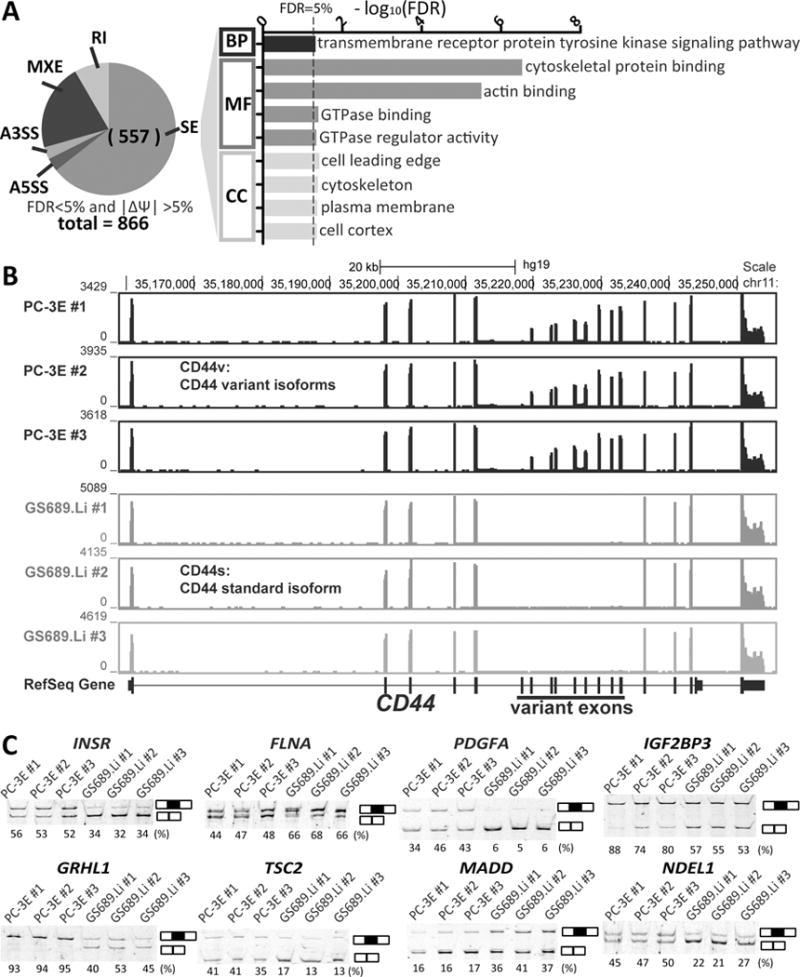
Differential AS between PC-3E and GS689.Li preferentially targets pathways important for cell motility and signaling.
(A) Gene Ontology enrichment analysis of differential exon skipping events. All significant GO terms (Benjamini-corrected FDR < 5%) in the GO BP (“biological process”), MF (“molecular function”) and CC (“cellular component”) categories are listed in the bar graph. SE: skipped exon; A3SS and A5SS: alternative 3′ or 5′ splice sites; MXE: mutually exclusive exons; RI: retained intron. (B) RNA-seq read density plot indicates a metastasis-relevant isoform switch of CD44 between the two cell types. (C) RT-PCR gel images of selected differential AS events. The RT-PCR estimated exon inclusion levels are indicated below each gel picture.
Metastasis-associated alternative splicing events are enriched in pathways important for cell motility and signaling
To explore gene functional groups preferentially influenced by AS events associated with metastatic colonization, we performed Gene Ontology (GO) enrichment analysis of genes with differential AS between PC-3E and GS689.Li. We identified 9 significantly enriched GO terms at FDR<5% (Figure 3A). Genes with these enriched GO terms are listed in Supplementary Table S4. We found several enriched GO terms related to the regulation of cell motility and migration, such as cytoskeletal protein binding, actin binding, GTPase binding, and cell leading edge, suggesting the role of these AS events in regulating cellular processes related to cell invasion and migration. For example, our RNA-seq data revealed significant isoform switch of CD44 from the CD44v (variant) isoforms in PC-3E to the CD44s (standard) isoforms in GS689.Li (Figure 3B). This isoform switch is known to occur during EMT and is associated with enhanced invasiveness and aggressiveness of cancer cells (42–44).
We also found a significant enrichment (FDR=0.048) of genes involved in the transmembrane receptor tyrosine kinases (RTKs) pathway (Figure 3A), which plays a key role in cancer development and progression (45). Of 19 differential AS events in the RTK pathway (Table 1), 18 were in protein-coding regions and one (in AKT1) was in the 5′-UTR. These include several AS events previously demonstrated to have functional impact. For example, the insulin receptor (IR) gene INSR has two isoforms (IR-A and IR-B) resulting from the alternative splicing of exon 11. IR-B is the full length mRNA isoform with exon 11 included. It is highly expressed in liver and is reported to be the major INSR isoform involved in insulin metabolism, while IR-A which lacks exon 11 plays a role in promoting cell proliferation (46–48). Here, the average exon inclusion level of INSR exon 11 was 54% in PC-3E but dropped significantly to 33% in GS689.Li (Figure 3C). In other words, the ratio of the proliferative isoform (IR-A) over the metabolic isoform (IR-B) increased significantly in cells with greater metastatic colonization potential. Interestingly, an increased ratio of IR-A over IR-B was independently observed in clinical tissue specimens of breast cancer, prostate cancer, and liver cancer (49–51), suggesting the functional and clinical relevance of this AS event in cancer. FLNA encodes an actin-binding protein that regulates cell shape and migration through cytoskeleton remodeling (52–55). An in-frame alternatively spliced exon (exon 30) of FLNA encodes eight amino acids within one filamin repeat domain with a crucial role in remodeling the cytoskeleton. We found that GS689.Li had a significantly higher inclusion level of this exon as compared to PC-3E (67% versus 46%, see Figure 3C), indicating a higher percentage of the full-length protein isoform with intact filamin repeat domain in GS689.Li. Interestingly, we also observed a higher gene expression level of FLNA in GS689.Li (fold change over PC-3E = 1.43, FDR=0.0005), suggesting that cells underwent concurrent changes in gene expression and AS to produce more full-length FLNA in metastatic cancer cells. Another example was the exon 6 of Platelet-derived growth factor A (PDGFA). PDGFA is a mitogen which affects tumor growth and drives pathological mesenchymal response in various diseases (56–58). Previous studies showed that the inclusion of PDGFA exon 6 primarily occurred in epithelial tissues (57). Consistent with this previous observation, we found a significantly higher exon inclusion level of PDGFA exon 6 in PC-3E as compared to GS689.Li (41% versus 6%, Figure 3C). Vascular endothelial growth factor A (VEGFA) is another member of the PDGF/VEGF growth factor family. VEGFA induces angiogenesis and promotes cell migration (59–61). The skipping of VEGFA exon 7 was increased in GS689.Li (Table 1), and it has been reported that the skipping of VEGFA exon 7 promotes tumor angiogenesis of human prostate cancer (62). Given the importance of the RTK pathway in cancer cell signaling, it is possible that coordinated AS changes in the RTK pathway affect key aspects of gene regulation and signaling pathways of metastatic cancer cells. Importantly, of the 18 differential AS events in the protein-coding regions of these RTK-related genes, 15 (83%) involve an in-frame alternative exon with an exact multiple of 3 nt in length. This percentage is much higher than the transcriptome-wide average (40, 63), suggesting that these metastasis-associated AS events produce functional protein isoforms in distinct cell types.
Table 1.
Differential AS events involved in the transmembrane receptor protein tyrosine kinase signaling pathway.
| Gene Symbol | Gene Name | Exon Coordinates (hg19) | Exon Size | Region | Δψ(GS689.Li – PC-3E) by RNA-seq |
|---|---|---|---|---|---|
| PDGFA | platelet-derived growth factor alpha polypeptide | −chr7:540067-540136 | 69 | CDS + 3′UTR | −49% |
| INSR | insulin receptor | −chr19:7150507-7150543 | 36 | CDS | −48% |
| BAIAP2 | BAI1-associated protein 2 | +chr17:79084713-79084759 | 46 | CDS + 3′UTR | −48% |
| SORBS1 | sorbin and SH3 domain containing 1 | −chr10:97174250-97174619 | 369 | CDS | −48% |
| TSC2 | tuberous sclerosis 2 | +chr16:2127598-2127727 | 129 | CDS | −44% |
| ERBB2IP | erbb2 interacting protein | +chr5:65364704-65364848 | 144 | CDS | 39% |
| ERBB2IP | erbb2 interacting protein | +chr5:65367996-65368119 | 123 | CDS | −35% |
| GHR | growth hormone receptor | +chr5:42629139-42629205 | 66 | CDS | −30% |
| ABI1 | abl-interactor 1 | −chr10:27044583-27044670 | 87 | CDS | 29% |
| FLNA | filamin A, alpha (actin binding protein 280) | −chrX:153585618-153585642 | 24 | CDS | 25% |
| PTK2 | PTK2 protein tyrosine kinase 2 | −chr8:141679825-141679834 | 9 | CDS | 24% |
| VEGFA | vascular endothelial growth factor A | +chr6:43749692-43749824 | 132 | CDS | −24% |
| PXN | paxillin | −chr12:120653362-120653464 | 102 | CDS | −21% |
| AKT1 | v-akt murine thymoma viral oncogene homolog 1 | −chr14:105259463-105259547 | 84 | 5′UTR | −20% |
| SOCS7 | suppressor of cytokine signaling 7 | +chr17:36520634-36520739 | 105 | CDS | 19% |
| MPZL1 | myelin protein zero-like 1 | +chr1:167745300-167745403 | 103 | CDS | −17% |
| ARF4 | ADP-ribosylation factor 4 | −chr3:57569624-57569734 | 110 | CDS | −15% |
| RAPGEF1 | Rap guanine nucleotide exchange factor (GEF) 1 | −chr9:134479347-134479440 | 93 | CDS | −12% |
| PXN | paxillin | −chr12:120657009-120657894 | 885 | CDS | 11% |
Splicing factors responsible for alternative splicing regulation associated with metastatic colonization
The widespread differences in AS between PC-3E and GS689.Li raised a question about the mechanisms for AS switches associated with metastatic colonization. To address this question, we investigated which splicing factors may be responsible for these AS differences, by identifying enriched binding sites of splicing factors and other RNA binding proteins around the differentially spliced exons. We focused this motif analysis on alternatively spliced exons involved in exon skipping events. From the rMATS results, after removing redundancies we obtained a list of 424 unique alternative exons with significant differential exon skipping between the two cell types. Of them, 221 and 203 exons had significantly lower or higher exon inclusion in GS689.Li, respectively. We also compiled a control list of 5,671 alternatively spliced exons without splicing differences between the two cell types (see details in Materials and Methods). Using a comprehensive list of 112 binding sites of human RNA binding proteins including many well-characterized splicing factors (Supplementary Table S3), we scanned for motif occurrences separately in exons or their 250nt upstream or downstream intronic sequences and identified significantly enriched motifs in GS689.Li down-regulated or up-regulated exons as compared to the control (non-regulated) alternative exons.
Of all 112 motifs analyzed, six motifs of well-known splicing factors (PTBP1, SRSF3, RBFOXs, QKI, ESRPs and MBNLs) had a Benjamini-adjusted p-value of < 0.05 within or around exons down-regulated in GS689.Li, while four motifs (corresponding to MBNLs, PTBP1, U2AF2 and RBFOXs) were enriched in upstream or downstream intronic sequences of exons up-regulated in GS689.Li (Figure 4A). Of note, the binding site of ESRPs (UGGUGG) was significantly enriched in the downstream intronic regions of GS689.Li down-regulated exons. Given that ESRP expression was largely diminished in the GS689.Li cell line, this motif pattern is consistent with the literature that exons with downstream intronic binding sites of ESRPs generally are positively regulated by ESRPs (16). Similarly, the binding motif of the RBFOX family of splicing factors (WGCAUGM) was significantly enriched within GS689.Li down-regulated exons and in the downstream intronic regions of GS689.Li up-regulated exons. This effect was most likely mediated by RBFOX2, which had a higher expression level in the GS689.Li cell line (Figure 1 and 4B). Moreover, the positions of the motif enrichment matched the well-known position-dependent effects of RBFOX2 on its target alternative exons (64).
Figure 4.
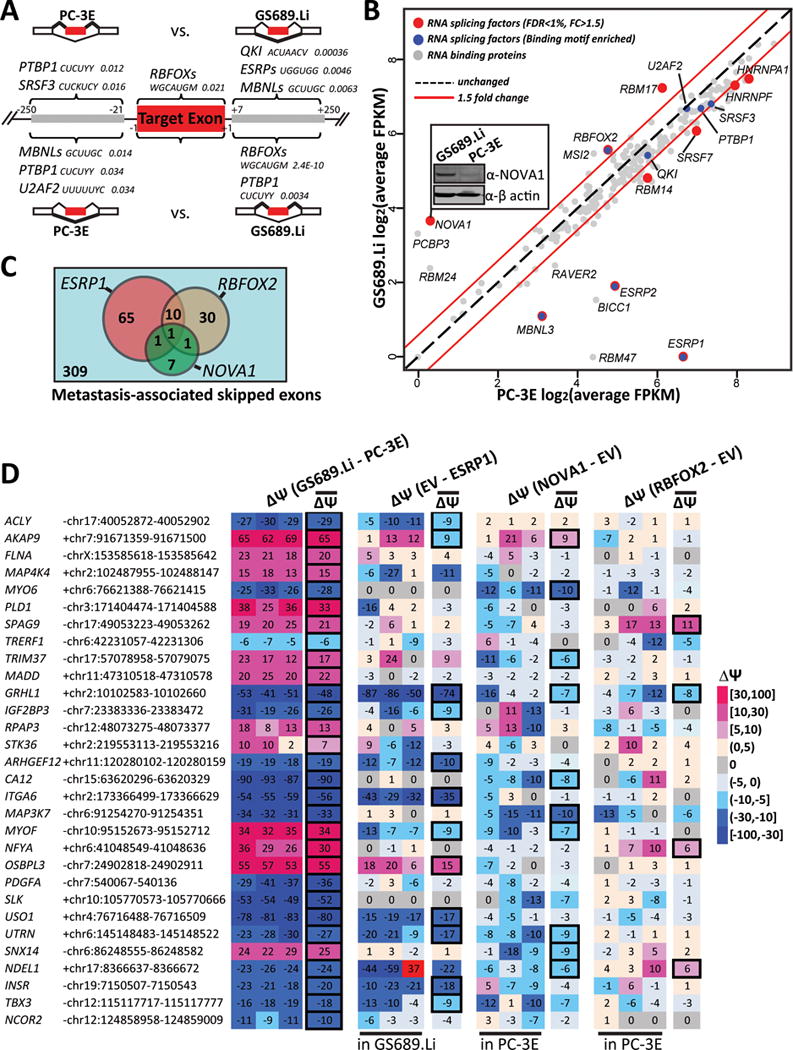
Splicing factors responsible for alternative splicing regulation associated with metastatic colonization.
(A) Significantly enriched binding sites of splicing factors and other RNA binding proteins in differential exon skipping events between PC-3E and GS689.Li. Gene symbols of splicing factors are followed by the consensus binding motifs and their Benjamini-adjusted p-values. Y: C/U; K: G/U; W: A/U; H: U/C/A; V: A/C/G; M: A/C. (B) Scatter plot of 221 RNA binding proteins’ average gene expression levels (log2 FPKM) in PC-3E and GS689.Li. Splicing factors with significant gene expression change are colored in red, while splicing factors whose binding motifs are significantly enriched are colored in blue. Some splicing factors belong to both categories. The inset image shows the western blot confirmation of NOVA1 protein expression levels. (C) The number of known target exons of ESRP1, RBFOX2, or NOVA1 among differential exon skipping events between PC-3E and GS689.Li. (D) Heatmap showing the effects on 30 randomly selected metastasis-associated skipped exons by ectopic expression of ESRP1, NOVA1, and RBFOX2. The Δψ values between PC-3E and GS689.Li or upon ectopic expression of a specific splicing factor are color coded for three individual replicates and the average of all three replicates. Significant splicing changes are highlighted by the bold box. EV: empty vector (control).
We correlated the enriched motifs with the gene expression profiles of their corresponding splicing factors (Figure 4B). As expected, several enriched motifs corresponded to splicing factors with significant differential gene expression between these two cell types, such as ESRP1/2, MBNL3, and RBFOX2. We also identified splicing factors whose motifs were significantly enriched but the splicing factors themselves were not differentially expressed. Examples include PTBP1, SRSF3, and QKI, which have been previously linked to AS regulation in cancer (65–70). It is possible that the functional activities of these splicing factors are altered at the post-transcriptional or post-translational levels, without significant changes in their steady-state gene expression levels. Lastly, our RNA-seq data also revealed significant differential expression of additional splicing factors, despite the fact that their binding sites did not show up among the enriched motifs. An intriguing example is NOVA1, whose gene expression level was significantly higher in GS689.Li (fold change=13, FDR=8.24e-121, see Figure 4B; also see Figure 1 for the microarray and qPCR data of NOVA1). We further confirmed the up-regulation of NOVA1 protein expression in GS689.Li using western blot (Figure 4B, inset). NOVA1 has traditionally been studied as an important neuronal-specific splicing factor (33, 71), and little is known about NOVA1 in non-neural tissues. In future studies it would be interesting to elucidate the endogenous targets and functional relevance of NOVA1 in metastatic cancer cells. Collectively, our motif enrichment analysis in conjunction with the expression analysis of splicing factors suggests a complex network of multiple splicing factors that drives AS changes associated with metastatic colonization.
We performed additional analyses and experiments to investigate the functional association between the AS events and specific splicing factors. As ESRP1, RBFOX2, and NOVA1 were among the most differentially expressed splicing factors between PC-3E and GS689.Li and the transcriptome-wide targets of these splicing factors were characterized in a series of published work, we asked which exons identified in our study were known targets of ESRP1, RBFOX2, and NOVA1. To do this, we collected from the literature 276 ESRP1 target exons, 121 RBFOX2 target exons, and 287 NOVA1 target exons (see details in Materials and Methods). Of the 424 unique skipped exons identified in our study, 77, 42, and 10 exons were known targets of ESRP1, RBFOX2, and NOVA1 (Figure 4C), representing a statistically significant overlap with the targets of all three splicing factors (Fisher’s exact test p-value=7.5e-53 for ESRP1, 8.7e-35 for RBFOX2, and 0.0001 for NOVA1 respectively). The overlap with known NOVA1 targets was the lowest among the three splicing factors. This was not surprising, as previous studies of NOVA1 exon targets were conducted exclusively in neuronal tissues (35). Furthermore, to experimentally test the functional associations between the AS events and the variation in the levels of specific splicing factors in our prostate cancer cell lines, we ectopically expressed ESRP1, RBFOX2, and NOVA1 in PC-3E or GS689.Li cells (Materials and Methods), and tested by RT-PCR if the endogenous splicing levels of specific AS events were affected. We randomly tested 30 metastasis-associated AS events including several from the receptor tyrosine kinase pathway (Figure 3C), without using any prior information about the known targets or binding sites of these splicing factors. This allowed us to assess in an unbiased manner what fraction of metastasis-associated AS events were influenced by the levels of these three splicing factors. We performed the experiments in triplicates and defined an exon as being regulated by a splicing factor if all three replicates showed the same direction of splicing change and an average change of exon inclusion levels of greater than 5%. Of these 30 exons, 12 (40%), 4 (13%), and 10 (33%) were affected by ectopic expression of ESRP1, RBFOX2, and NOVA1 respectively (Figure 4D). Four exons were affected by two of the three splicing factors and another one was affected by all three. This suggests that multiple splicing factors may coordinately regulate the splicing of certain metastasis-associated AS events. For example, the splicing level of the GRHL1 exon was strongly enhanced by ESRP1, while both RBFOX2 and NOVA1 had measurable repressive effects on this exon. In total, up to two thirds (20 of 30) of tested exons were regulated by at least one of these three splicing factors. As expected, ESRP1 had the broadest and most potent effects on these AS events. By contrast, RBFOX2 had the weakest effects on these randomly selected exons, consistent with the fact that it had the least dramatic expression change among these three splicing factors between PC-3E and GS689.Li. Of note, a third of these randomly selected exons were influenced by variation in NOVA1 levels. This underscores the significant differential expression of NOVA1 between PC-3E and GS689.Li and the likely functional relevance of NOVA1 to the metastasis-associated AS program.
ESRP1 as a prognostic marker of patient survival for certain cancer types
To explore the potential clinical significance of our findings, we asked whether the expression level of ESRP1 could serve as a molecular or prognostic marker of human cancers. Specifically, we tested the association of ESRP1 expression with patient survival using The Cancer Genome Atlas (TCGA) RNA-seq data across 13 types of cancers (see details in Materials and Methods). In two cancer types we found a significant positive association between ESRP1 expression and patient survival (Cox regression model, p=0.018 for clear cell renal cell carcinoma (ccRCC), and p=0.009 for breast cancer). For example, in ccRCC, while the vast majority of patients had low expression of ESRP1, we found a subgroup (approximately 5%) of ccRCC patients with high expression levels of ESRP1, and these patients had significantly better prognosis (Figure 5A). In both cancer types, the expression level of ESRP1 may mark specific subtypes of cancer with epithelial characteristics and less aggressive clinical behaviors (6, 72).
Figure 5.
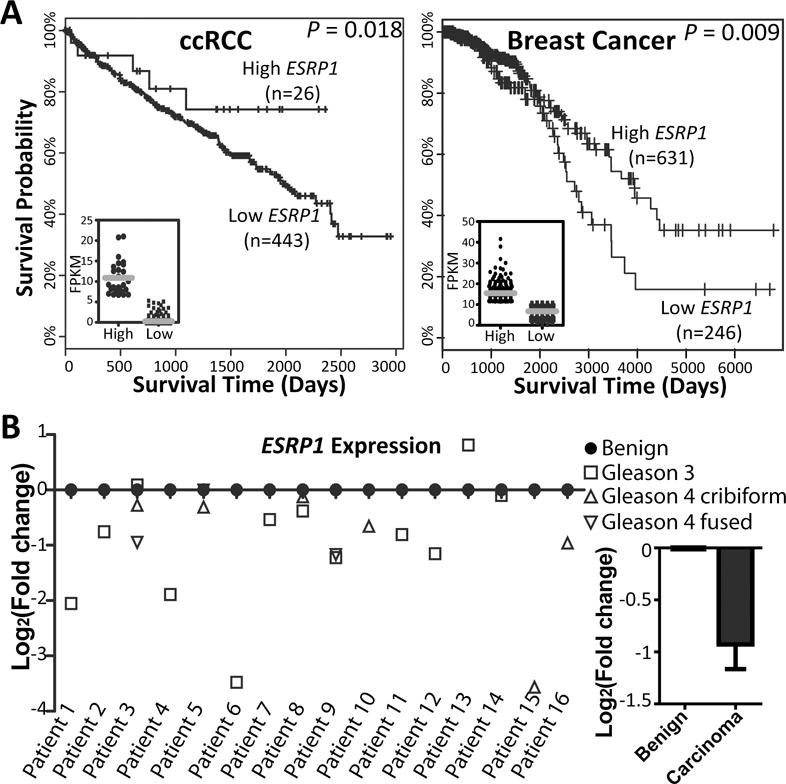
ESRP1 gene expression level as molecular markers of cancer specimens.
(A) Kaplan-Meier survival curves of two patient sub-groups with low or high ESRP1 expression levels in ccRCC (clear cell renal cell carcinoma) and breast cancer. Unsupervised 2-means clustering based on ESRP1 expression levels was performed to segregate patients into two sub-groups. Cox regression was used to calculate the p-value of association between ESRP1 expression level and patient survival. The inset image shows the dot plot of ESRP1 expression levels in individual patients from the two sub-groups, with the grey line indicating the average gene expression level in each sub-group. (B) Realtime qPCR analysis of ESRP1 expression levels in laser-capture microdissected tumor/benign samples from 16 prostate cancer patients. GAPDH was used as the reference gene. All tumor samples were normalized to their patient-matched benign tissues. The bar plot on the right was generated by pooling all prostate cancer samples with different Gleason grades and all patient-matched benign samples. Error bar indicates standard error of the mean.
It should be noted that the ability to detect association with patient survival depends on a variety of factors, such as sample size, follow-up time, and mortality rate. For example, in the TCGA cohort of prostate cancer, the mortality rate is very low (0.6%) within the follow-up time, making it almost impossible to identify survival signals. To assess ESRP1 expression levels in prostate cancer, we analyzed laser-capture microdissected (LCM) samples from 16 prostate cancer patients. Realtime qPCR was performed to compare ESRP1 expression levels in tumor specimens and patient-matched benign tissues. As shown in Figure 5B, except for one patient (patient 13), ESRP1 was consistently down regulated in prostate cancer specimens. Of note, recent studies have reported the functional roles of ESRP1 in breast cancer and prostate cancer cells (6, 7). In pancreatic ductal adenocarcinoma, ESRP1 inhibits cancer cell migration and metastasis in vitro and in vivo, and high ESRP1 expression level is associated with favorable patient survival (73). Here our results corroborate the role of ESRP1 in cancer progression and indicate the value of ESRP1 as a prognostic marker for multiple types of cancers.
Discussion
In this work, we performed transcriptome analyses of cancer cell lines derived from in vitro and in vivo models of metastatic colonization to study AS events and splicing factors associated with metastatic cancer cells. Although previous studies have surveyed cell-type-specific AS during EMT (6, 7), no study to date has investigated transcriptome-wide changes in AS in cancer cells under phenotypic selection for metastatic colonization. The panel of PC-3 and derivative cell lines crossing in vitro or in vivo barriers of metastasis thus provides an excellent and unbiased system for us to comprehensively characterize AS events and identify key splicing factors that impact splicing regulation underlying metastasis. Our data confirm the important role of several known EMT-relevant splicing factors (ESRPs, RBFOX2) in determining the splicing profiles of metastatic cancer cells. The most striking pattern was observed for ESRP1, which had dramatic stepwise reduction in gene expression levels as cells went through multiple rounds of selection across the in vitro endothelial migration barrier. Meanwhile, our data also indicate that ESRPs alone only account for a minor fraction of AS switches observed in the PC-3 derivative cell lines, as only 18% of differential exon skipping events were known ESRP targets and 32% of differential exon skipping events contained putative ESRP binding sites within the exons or flanking intronic sequences. Indeed, our motif analysis revealed significant enrichment of binding sites of other splicing factors such as those from the MBNL and PTB families of splicing factors. Some of these splicing factors (such as MBNL3) were differentially expressed between the two cell types. Others (such as PTBP1, SRSF3, and QKI) may be regulated at the post-transcriptional or post-translational levels to produce differential splicing factor activities between the two cell types. Our RNA-seq data also revealed significant differential expression of several less-characterized RNA binding proteins, such as BICC1 and RBM47 (Figure 4B). BICC1 and RBM47 had dramatically decreased expression levels in GS689.Li (BICC1: fold change=7, FDR=3.24e-84; RBM47: fold change=67, FDR=3.42e-261; see Figure 4B). Interestingly, prior work revealed that loss of mouse Bicc1 disrupts E-cadherin-based cell-cell adhesion and epithelial cell polarity (74). RBM47 was recently found to inhibit breast cancer re-initiation and growth and act as a suppressor of breast cancer metastasis (75). Taken together, our data suggest that multiple RNA binding proteins collectively contribute to AS switches (and likely other post-transcriptional changes) associated with metastatic colonization.
Using deep RNA-seq, we identified 866 significant differential AS events between PC-3E and one of its derivative cell lines (GS689.Li). GO enrichment analyses suggest that these AS events preferentially target genes involved in pathways associated with cancer development and metastasis. For example, genes in the receptor tyrosine kinase signaling pathway are significantly enriched (Figure 3A). Besides these enriched functional categories, we also identified AS events in master regulators of cancer cell gene expression and signaling with intriguing functional implications. For example, GRHL1 is a member of the Grainyhead family of transcription factors. These transcription factors are highly expressed in epithelial cells over mesenchymal cells and have been recently implicated in transcriptome regulation during EMT (76). We found an alternative exon of GRHL1 (exon 5) with differential AS between the PC-3E and GS689.Li cell lines. This exon had close to 100% exon inclusion in PC-3E. By contrast, its exon inclusion level was much lower (~50%) in GS689.Li (Figure 3C). The skipping of this exon resulted in a transcript isoform containing a premature termination codon, which would be targeted for degradation by the mRNA nonsense-mediated decay (NMD) pathway. Consistent with this finding, the expression level of GRHL1 was 5.4-fold lower in GS689.Li cells, suggesting that AS of this exon contributed to the molecular processes that turned off GRHL1 expression in metastatic cancer cells. Of all the identified AS events, cell-type-specific isoforms arising from a small number of events have been functionally characterized previously (described in Results). However, for the vast majority of events the consequences of AS are unknown. This represents an extensive resource for detailed functional investigations.
Shapiro et al. previously carried out AS profiling of an in vitro EMT model triggered by ectopic expression of the EMT transcription factor Twist, and generated 27–30 million 39bp single-end RNA-seq reads on the epithelial or mesenchymal cell state respectively (7). To compare the similarities and differences in AS profiles between the in vitro EMT model and our in vivo model of metastatic colonization, we re-analyzed the RNA-seq data of Shapiro et al. using the same bioinformatics pipeline as applied to our RNA-seq data. Under the same FDR cutoff, we identified 166 unique exon skipping events with significant differential splicing between the epithelial and mesenchymal cell states (RNA-seq data of Shapiro et al.), as compared to 424 events in our data (PC-3E versus GS689.Li). Of these, 40 events were in common, representing a highly statistically significant overlap (Fisher’s exact test p-value=9.3e-23). On the other hand, the majority of the identified differential exon skipping events were unique to one of the two datasets. Such differences in AS profiles may be attributed to multiple biological or technical factors. First, Shapiro et al. analyzed a human mammary epithelial cell line (HMLE) while we analyzed AS profiles of two prostate cancer cell lines. AS events in cell-type-specific genes may not be shared between these two datasets. Second, our cell lines (PC-3E and GS689.Li) were derived from an in vivo model of metastatic colonization (19), while Shapiro et al. specifically overexpressed one EMT transcription factor Twist to trigger EMT in vitro (7). Third, our RNA-seq coverage was substantially deeper than that of the Shapiro et al. data. Specifically, we analyzed three biological replicates for each cell line, with 114~132 million 101bp x 2 paired-end reads per replicate. By contrast, Shapiro et al. had no replicate for RNA-seq and 27–30 million 39bp single-end RNA-seq reads per RNA sample. Our increased RNA-seq coverage would lead to improved statistical power, as evidenced by the fact that the number of significant exon skipping events identified from the Shapiro et al. dataset was less than half of the events identified from our data. Consistent with the trend observed at the exon level, we found a similar pattern when we compared the gene expression profiles of RNA binding proteins between these two datasets. While certain RNA binding proteins had strong differential gene expression in both systems (ESRP1, ESRP2, RBM47), a few others were differentially expressed in only one of the two systems. For example, NOVA1, MBNL3, and RBM24 were among the most differentially expressed splicing factors between PC-3E and GS689.Li cells (Figure 4B), but there was no evidence of differential expression in the in vitro EMT model. Another example is BICC1, which had opposite directions of gene expression change in the two datasets. Specifically, BICC1 had 7-fold reduction in expression levels in our in vivo model of metastatic colonization (Figure 4B), but ~3-fold increase in expression levels in the in vitro EMT model. These expression differences in RNA binding proteins are expected to contribute to downstream differences in AS. In summary, our comparison to the Shapiro et al. dataset demonstrated a conserved core AS program and splicing regulators associated with the invasiveness and metastatic properties of cancer cells, but also revealed many events unique to specific cell types and biological systems.
It is noteworthy that we did not find significant overlap between differentially spliced genes (DSGs) and differentially expressed genes (DEGs) between PC-3E and GS689.Li. The vast majority of DSGs had similar steady-state gene expression levels between the two cell types (Figure 6A). Additionally, DSGs and DEGs had distinct sets of enriched GO terms (see Figure 3A for enriched GO terms of DSGs and Supplementary Table S5 for enriched GO terms of DEGs). This pattern is similar to observations made in other developmental or evolutionary systems (77, 78), suggesting that differential gene transcription and splicing act in an orthogonal manner to regulate and fine-tune the global gene expression programs and cellular phenotypes (Figure 6B).
Figure 6.
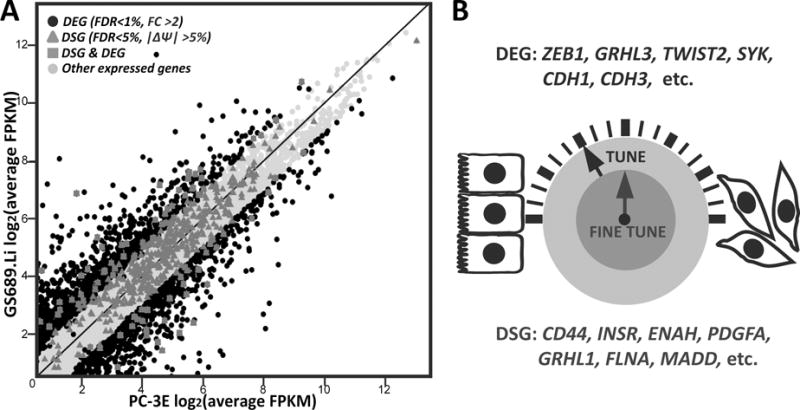
Differential alternative splicing and differential gene expression act in an orthogonal manner to promote metastatic colonization.
(A) Scatter plot of gene expression levels indicate a low overlap between differentially spliced genes (DSGs) and differentially expressed genes (DEGs). (B) Differential AS and differential gene expression influence distinct sets of genes with important roles in regulating cellular phenotypes associated with cancer metastasis.
The extent to which EMT is involved in prostate cancer progression has been somewhat controversial (79). However, support for its involvement in prostate cancer progression includes evidence of down-regulation of E-cadherin, up-regulation of N-cadherin and expression of EMT transcription factors such as ZEB1 in primary prostate cancers (80–82). Previously, we determined that ZEB1 was partially responsible for the aggressive metastatic behavior evident in the PC-3 variants used in this study (19). Here we found significant down-regulation of ESRP1, which is repressed by ZEB1 (83), in laser-capture microdissected glands from prostate cancer patients. This is also supportive of the notion that EMT results in progressive loss of ESRP1 expression in clinical specimens. An interesting direction for further research would be to determine whether increased ZEB1 and decreased ESRP1 are observed in the same cells from prostate cancer tissue, which might mark a subpopulation of cells undergoing EMT. It should also be noted that EMT may be reversed during productive metastatic colonization, consistent with the finding that E-cadherin is often detected in bone metastases in prostate cancer patients (79, 84). The PC-3 model employed here may not adequately reflect this situation since in mouse xenografts cells in an EMT-like state exhibit productive metastatic colonization (19). In this regard, it would be of interest to determine the AS patterns and splicing factor expression in metastatic lesions of prostate cancer.
One surprising observation in this study was the significant up-regulation of the splicing factor NOVA1 in all derivative cell lines. This up-regulation was observed in both in vitro and in vivo models (Figure 1C), and was confirmed at both the mRNA and protein levels (Figure 1D and 4B). NOVA1 has been extensively studied as a neuronal-specific splicing regulator (33, 71). Little is known about the function of NOVA1 outside the neuronal system. However, we note that NOVA1 was originally identified in a lung cancer cell line (85). Moreover, increased expression of NOVA1 mRNA has been observed in docetaxel resistant PC-3 prostate cancer cells (86) and in lymph node metastasis (87), although neither study commented on the functional significance of this observation. It was also reported recently that higher NOVA1 expression was associated with worse survival outcomes in hepatocellular carcinoma (HCC) patients, and the overexpression of NOVA1 in HCC cells promoted cell proliferation (88). Collectively, these data imply a yet unappreciated role of NOVA1 in metastatic cancer cells. Using cells with ectopic expression of several EMT-relevant transcriptional regulators such as SNAI1, TWIST2, and ZEB1, we obtained the preliminary evidence that the protein expression of NOVA1 can be induced in a time-dependent manner by SNAI1, but not by TWIST2 or ZEB1 (see Supplementary Figure S1), suggesting that NOVA1 may be a downstream target of the SNAI1-regulated gene network. We should note that the regulatory and functional significance of NOVA1 up-regulation in the PC-3 derivative cell lines is currently unclear. Motif analysis did not find significant enrichment of the NOVA1 motif around the differential AS events, but our RT-PCR analysis of 30 randomly selected exons did indicate that a considerable portion of metastasis-associated AS events were influenced by the variation in NOVA1 levels. It is also possible that NOVA1 may play a role in other aspects of RNA regulation in addition to splicing control in these cancer cells. Alternatively, given that GO terms such as “neuron differentiation” and “cell morphogenesis involved in neuron differentiation” are significantly enriched among the differentially expressed genes (see enriched GO terms among DEGs in Supplementary Table S5), the up-regulation of NOVA1 in the PC-3 derivative cells may reflect a neuronal-like gene expression program associated with neuroendocrine differentiation occurring in aggressive prostate cancers (89, 90).
Supplementary Material
Implications.
Transcriptome-wide remodeling of AS is an integral regulatory process underlying metastatic colonization and AS events impact the metastatic behavior of cancer cells.
Acknowledgments
We thank Hong Wu (UCLA), Douglas Black (UCLA), and Peter Stoilov (WVU) for discussions on this work, Douglas Black (UCLA) for providing the NOVA1 antibody, and the Biospecimen Core of the Pacific Northwest Prostate Cancer SPORE (CA097186) for providing prostate cancer cDNAs.
Grant Support
The work was supported by the National Institutes of Health [CA130916 to MDH and GM105431 to YX]. YX is supported by an Alfred Sloan Research Fellowship.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 4.Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010;107:15449–54. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–96. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, et al. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. Embo J. 2010;29:3286–300. doi: 10.1038/emboj.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, et al. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011;7:e1002218. doi: 10.1371/journal.pgen.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venables JP, Brosseau JP, Gadea G, Klinck R, Prinos P, Beaulieu JF, et al. RBFOX2 is an important regulator of mesenchymal tissue-specific splicing in both normal and cancer tissues. Mol Cell Biol. 2013;33:396–405. doi: 10.1128/MCB.01174-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–63. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalsotra A, Cooper TA. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet. 2011;12:715–29. doi: 10.1038/nrg3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–54. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de la Grange P, Gratadou L, Delord M, Dutertre M, Auboeuf D. Splicing factor and exon profiling across human tissues. Nucleic Acids Res. 2010;38:2825–38. doi: 10.1093/nar/gkq008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–13. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell. 2009;33:591–601. doi: 10.1016/j.molcel.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tavanez JP, Valcarcel J. A splicing mastermind for EMT. Embo J. 2010;29:3217–8. doi: 10.1038/emboj.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dittmar KA, Jiang P, Park JW, Amirikian K, Wan J, Shen S, et al. Genome-wide determination of a broad ESRP-regulated posttranscriptional network by high-throughput sequencing. Mol Cell Biol. 2012;32:1468–82. doi: 10.1128/MCB.06536-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warzecha CC, Carstens RP. Complex changes in alternative pre-mRNA splicing play a central role in the epithelial-to-mesenchymal transition (EMT) Semin Cancer Biol. 2012;22:417–27. doi: 10.1016/j.semcancer.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warzecha CC, Shen S, Xing Y, Carstens RP. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol. 2009;6:546–62. doi: 10.4161/rna.6.5.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drake JM, Strohbehn G, Bair TB, Moreland JG, Henry MD. ZEB1 enhances transendothelial migration and represses the epithelial phenotype of prostate cancer cells. Mol Biol Cell. 2009;20:2207–17. doi: 10.1091/mbc.E08-10-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pettaway CA, Pathak S, Greene G, Ramirez E, Wilson MR, Killion JJ, et al. Selection of highly metastatic variants of different human prostatic carcinomas using orthotopic implantation in nude mice. Clin Cancer Res. 1996;2:1627–36. [PubMed] [Google Scholar]
- 21.Gautier L, Cope L, Bolstad BM, Irizarry RA. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–15. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 22.Wu Z, Irizarry RA. Preprocessing of oligonucleotide array data. Nat Biotechnol. 2004;22:656–8. doi: 10.1038/nbt0604-656b. author reply 8. [DOI] [PubMed] [Google Scholar]
- 23.Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–40. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- 24.Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4:1184–91. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stacklies W, Redestig H, Scholz M, Walther D, Selbig J. pcaMethods–a bioconductor package providing PCA methods for incomplete data. Bioinformatics. 2007;23:1164–7. doi: 10.1093/bioinformatics/btm069. [DOI] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 31.Anderson ES, Lin CH, Xiao X, Stoilov P, Burge CB, Black DL. The cardiotonic steroid digitoxin regulates alternative splicing through depletion of the splicing factors SRSF3 and TRA2B. RNA. 2012;18:1041–9. doi: 10.1261/rna.032912.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–7. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, et al. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–6. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- 34.Venables JP, Klinck R, Koh C, Gervais-Bird J, Bramard A, Inkel L, et al. Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol. 2009;16:670–6. doi: 10.1038/nsmb.1608. [DOI] [PubMed] [Google Scholar]
- 35.Zhang C, Frias MA, Mele A, Ruggiu M, Eom T, Marney CB, et al. Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science. 2010;329:439–43. doi: 10.1126/science.1191150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao K, Lu Z-x, Park JW, Zhou Q, Xing Y. GLiMMPS: Robust statistical model for regulatory variation of alternative splicing using RNA-Seq data. Genome Biol. 2013;14:R74. doi: 10.1186/gb-2013-14-7-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J, Zhao K, Jiang P, Lu ZX, Wang J, Murray JC, et al. Transcriptome landscape of the human placenta. BMC Genomics. 2012;13:115. doi: 10.1186/1471-2164-13-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braeutigam C, Rago L, Rolke A, Waldmeier L, Christofori G, Winter J. The RNA-binding protein Rbfox2: an essential regulator of EMT-driven alternative splicing and a mediator of cellular invasion. Oncogene. 2014;33:1082–92. doi: 10.1038/onc.2013.50. [DOI] [PubMed] [Google Scholar]
- 39.Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7:1009–15. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xing Y, Lee CJ. Protein modularity of alternatively spliced exons is associated with tissue-specific regulation of alternative splicing. PLoS Genet. 2005;1:e34. doi: 10.1371/journal.pgen.0010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–6. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, et al. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest. 2011;121:1064–74. doi: 10.1172/JCI44540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho SH, Park YS, Kim HJ, Kim CH, Lim SW, Huh JW, et al. CD44 enhances the epithelial-mesenchymal transition in association with colon cancer invasion. Int J Oncol. 2012;41:211–8. doi: 10.3892/ijo.2012.1453. [DOI] [PubMed] [Google Scholar]
- 44.Zoller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11:254–67. doi: 10.1038/nrc3023. [DOI] [PubMed] [Google Scholar]
- 45.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giudice J, Leskow FC, Arndt-Jovin DJ, Jovin TM, Jares-Erijman EA. Differential endocytosis and signaling dynamics of insulin receptor variants IR-A and IR-B. J Cell Sci. 2011;124:801–11. doi: 10.1242/jcs.076869. [DOI] [PubMed] [Google Scholar]
- 47.Kellerer M, Lammers R, Ermel B, Tippmer S, Vogt B, Obermaier-Kusser B, et al. Distinct alpha-subunit structures of human insulin receptor A and B variants determine differences in tyrosine kinase activities. Biochemistry. 1992;31:4588–96. doi: 10.1021/bi00134a008. [DOI] [PubMed] [Google Scholar]
- 48.Kosaki A, Pillay TS, Xu L, Webster NJ. The B isoform of the insulin receptor signals more efficiently than the A isoform in HepG2 cells. J Biol Chem. 1995;270:20816–23. doi: 10.1074/jbc.270.35.20816. [DOI] [PubMed] [Google Scholar]
- 49.Chettouh H, Fartoux L, Aoudjehane L, Wendum D, Claperon A, Chretien Y, et al. Mitogenic insulin receptor-A is overexpressed in human hepatocellular carcinoma due to EGFR-mediated dysregulation of RNA splicing factors. Cancer Res. 2013;73:3974–86. doi: 10.1158/0008-5472.CAN-12-3824. [DOI] [PubMed] [Google Scholar]
- 50.Heni M, Hennenlotter J, Scharpf M, Lutz SZ, Schwentner C, Todenhofer T, et al. Insulin receptor isoforms A and B as well as insulin receptor substrates-1 and -2 are differentially expressed in prostate cancer. PLoS One. 2012;7:e50953. doi: 10.1371/journal.pone.0050953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang J, Morehouse C, Streicher K, Higgs BW, Gao J, Czapiga M, et al. Altered expression of insulin receptor isoforms in breast cancer. PLoS One. 2011;6:e26177. doi: 10.1371/journal.pone.0026177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baldassarre M, Razinia Z, Burande CF, Lamsoul I, Lutz PG, Calderwood DA. Filamins regulate cell spreading and initiation of cell migration. PLoS One. 2009;4:e7830. doi: 10.1371/journal.pone.0007830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guiet R, Verollet C, Lamsoul I, Cougoule C, Poincloux R, Labrousse A, et al. Macrophage mesenchymal migration requires podosome stabilization by filamin A. J Biol Chem. 2012;287:13051–62. doi: 10.1074/jbc.M111.307124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leung R, Wang Y, Cuddy K, Sun C, Magalhaes J, Grynpas M, et al. Filamin A regulates monocyte migration through Rho small GTPases during osteoclastogenesis. J Bone Miner Res. 2010;25:1077–91. doi: 10.1359/jbmr.091114. [DOI] [PubMed] [Google Scholar]
- 55.Xu Y, Bismar TA, Su J, Xu B, Kristiansen G, Varga Z, et al. Filamin A regulates focal adhesion disassembly and suppresses breast cancer cell migration and invasion. J Exp Med. 2010;207:2421–37. doi: 10.1084/jem.20100433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li SH, Lee RK, Chen PW, Lu CH, Wang SH, Hwu YM. Differential expression and distribution of alternatively spliced transcripts of PDGF-A and of PDGF receptor-alpha in mouse reproductive tissues. Life Sci. 2005;77:2412–24. doi: 10.1016/j.lfs.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 58.Ostman A. PDGF receptors-mediators of autocrine tumor growth and regulators of tumor vasculature and stroma. Cytokine Growth Factor Rev. 2004;15:275–86. doi: 10.1016/j.cytogfr.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 59.Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004;56:549–80. doi: 10.1124/pr.56.4.3. [DOI] [PubMed] [Google Scholar]
- 60.Morales-Ruiz M, Fulton D, Sowa G, Languino LR, Fujio Y, Walsh K, et al. Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ Res. 2000;86:892–6. doi: 10.1161/01.res.86.8.892. [DOI] [PubMed] [Google Scholar]
- 61.Wang S, Li X, Parra M, Verdin E, Bassel-Duby R, Olson EN. Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc Natl Acad Sci U S A. 2008;105:7738–43. doi: 10.1073/pnas.0802857105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Catena R, Muniz-Medina V, Moralejo B, Javierre B, Best CJ, Emmert-Buck MR, et al. Increased expression of VEGF121/VEGF165–189 ratio results in a significant enhancement of human prostate tumor angiogenesis. Int J Cancer. 2007;120:2096–109. doi: 10.1002/ijc.22461. [DOI] [PubMed] [Google Scholar]
- 63.Resch A, Xing Y, Alekseyenko A, Modrek B, Lee C. Evidence for a subpopulation of conserved alternative splicing events under selection pressure for protein reading frame preservation. Nucleic Acids Res. 2004;32:1261–9. doi: 10.1093/nar/gkh284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol. 2009;16:130–7. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cheung HC, Hai T, Zhu W, Baggerly KA, Tsavachidis S, Krahe R, et al. Splicing factors PTBP1 and PTBP2 promote proliferation and migration of glioma cell lines. Brain. 2009;132:2277–88. doi: 10.1093/brain/awp153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He X, Arslan AD, Ho TT, Yuan C, Stampfer MR, Beck WT. Involvement of polypyrimidine tract-binding protein (PTBP1) in maintaining breast cancer cell growth and malignant properties. Oncogenesis. 2014;3:e84. doi: 10.1038/oncsis.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He X, Arslan AD, Pool MD, Ho TT, Darcy KM, Coon JS, et al. Knockdown of splicing factor SRp20 causes apoptosis in ovarian cancer cells and its expression is associated with malignancy of epithelial ovarian cancer. Oncogene. 2011;30:356–65. doi: 10.1038/onc.2010.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jia R, Li C, McCoy JP, Deng CX, Zheng ZM. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int J Biol Sci. 2010;6:806–26. doi: 10.7150/ijbs.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tang Y, Horikawa I, Ajiro M, Robles AI, Fujita K, Mondal AM, et al. Downregulation of splicing factor SRSF3 induces p53beta, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene. 2013;32:2792–8. doi: 10.1038/onc.2012.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zong FY, Fu X, Wei WJ, Luo YG, Heiner M, Cao LJ, et al. The RNA-Binding Protein QKI Suppresses Cancer-Associated Aberrant Splicing. PLoS Genet. 2014;10:e1004289. doi: 10.1371/journal.pgen.1004289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, et al. Nova regulates brain-specific splicing to shape the synapse. Nat Genet. 2005;37:844–52. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- 72.Zhao Q, Caballero OL, Davis ID, Jonasch E, Tamboli P, Yung WK, et al. Tumor-specific isoform switch of the fibroblast growth factor receptor 2 underlies the mesenchymal and malignant phenotypes of clear cell renal cell carcinomas. Clin Cancer Res. 2013;19:2460–72. doi: 10.1158/1078-0432.CCR-12-3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ueda J, Matsuda Y, Yamahatsu K, Uchida E, Naito Z, Korc M, et al. Epithelial splicing regulatory protein 1 is a favorable prognostic factor in pancreatic cancer that attenuates pancreatic metastases. Oncogene. 2013 doi: 10.1038/onc.2013.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fu Y, Kim I, Lian P, Li A, Zhou L, Li C, et al. Loss of Bicc1 impairs tubulomorphogenesis of cultured IMCD cells by disrupting E-cadherin-based cell-cell adhesion. Eur J Cell Biol. 2010;89:428–36. doi: 10.1016/j.ejcb.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vanharanta S, Marney CB, Shu W, Valiente M, Zou Y, Mele A, et al. Loss of the multifunctional RNA-binding protein RBM47 as a source of selectable metastatic traits in breast cancer. Elife. 2014:e02734. doi: 10.7554/eLife.02734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cieply B, Riley Pt, Pifer PM, Widmeyer J, Addison JB, Ivanov AV, et al. Suppression of the epithelial-mesenchymal transition by Grainyhead-like-2. Cancer Res. 2012;72:2440–53. doi: 10.1158/0008-5472.CAN-11-4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Calarco JA, Xing Y, Caceres M, Calarco JP, Xiao X, Pan Q, et al. Global analysis of alternative splicing differences between humans and chimpanzees. Genes Dev. 2007;21:2963–75. doi: 10.1101/gad.1606907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Giudice J, Xia Z, Wang ET, Scavuzzo MA, Ward AJ, Kalsotra A, et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat Commun. 2014;5:3603. doi: 10.1038/ncomms4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nauseef JT, Henry MD. Epithelial-to-mesenchymal transition in prostate cancer: paradigm or puzzle? Nat Rev Urol. 2011;8:428–39. doi: 10.1038/nrurol.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.De Marzo AM, Knudsen B, Chan-Tack K, Epstein JI. E-cadherin expression as a marker of tumor aggressiveness in routinely processed radical prostatectomy specimens. Urology. 1999;53:707–13. doi: 10.1016/s0090-4295(98)00577-9. [DOI] [PubMed] [Google Scholar]
- 81.Graham TR, Zhau HE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, et al. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008;68:2479–88. doi: 10.1158/0008-5472.CAN-07-2559. [DOI] [PubMed] [Google Scholar]
- 82.Tomita K, van Bokhoven A, van Leenders GJ, Ruijter ET, Jansen CF, Bussemakers MJ, et al. Cadherin switching in human prostate cancer progression. Cancer Res. 2000;60:3650–4. [PubMed] [Google Scholar]
- 83.Horiguchi K, Sakamoto K, Koinuma D, Semba K, Inoue A, Inoue S, et al. TGF-beta drives epithelial-mesenchymal transition through deltaEF1-mediated downregulation of ESRP. Oncogene. 2012;31:3190–201. doi: 10.1038/onc.2011.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Putzke AP, Ventura AP, Bailey AM, Akture C, Opoku-Ansah J, Celiktas M, et al. Metastatic progression of prostate cancer and e-cadherin regulation by zeb1 and SRC family kinases. Am J Pathol. 2011;179:400–10. doi: 10.1016/j.ajpath.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Buckanovich RJ, Posner JB, Darnell RB. Nova, the paraneoplastic Ri antigen, is homologous to an RNA-binding protein and is specifically expressed in the developing motor system. Neuron. 1993;11:657–72. doi: 10.1016/0896-6273(93)90077-5. [DOI] [PubMed] [Google Scholar]
- 86.Marin-Aguilera M, Codony-Servat J, Kalko SG, Fernandez PL, Bermudo R, Buxo E, et al. Identification of docetaxel resistance genes in castration-resistant prostate cancer. Mol Cancer Ther. 2012;11:329–39. doi: 10.1158/1535-7163.MCT-11-0289. [DOI] [PubMed] [Google Scholar]
- 87.Farnsworth RH, Karnezis T, Shayan R, Matsumoto M, Nowell CJ, Achen MG, et al. A role for bone morphogenetic protein-4 in lymph node vascular remodeling and primary tumor growth. Cancer Res. 2011;71:6547–57. doi: 10.1158/0008-5472.CAN-11-0200. [DOI] [PubMed] [Google Scholar]
- 88.Zhang YA, Zhu JM, Yin J, Tang WQ, Guo YM, Shen XZ, et al. High expression of neuro-oncological ventral antigen 1 correlates with poor prognosis in hepatocellular carcinoma. PLoS One. 2014;9:e90955. doi: 10.1371/journal.pone.0090955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beltran H, Tomlins SA, Aparicio AM, Arora VK, Rickman DS, Ayala GE, et al. Aggressive Variants of Castration Resistant Prostate Cancer. Clin Cancer Res. 2014 doi: 10.1158/1078-0432.CCR-13-3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tai S, Sun Y, Squires JM, Zhang H, Oh WK, Liang CZ, et al. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate. 2011;71:1668–79. doi: 10.1002/pros.21383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.