

Abstract
Nephritis as a result of autoimmunity is a common morbidity associated with Systemic Lupus Erythematosus (SLE). There is substantial clinical and industry interest in medicinal intervention in the SLE nephritic process; however, clinical trials to specifically treat lupus nephritis have not resulted in complete and sustained remission in all patients. Multiple mouse models have been used to investigate the pathologic interactions between autoimmune reactivity and SLE pathology. While several models bear a remarkable similarity to SLE-driven nephritis, there are limitations for each that can make the task of choosing the appropriate model for a particular aspect of SLE pathology challenging. This is not surprising given the variable and diverse nature of human disease. In many respects, features among murine strains mimic some (but never all) of the autoimmune and pathologic features of lupus patients. Although the diversity often limits universal conclusions relevant to pathogenesis, they provide insights into the complex process that result in phenotypic manifestations of nephritis. Thus nephritis represents a microcosm of systemic disease, with variable lesions and clinical features. In this review, we discuss some of the most commonly used models of lupus nephritis (LN) and immune-mediated glomerular damage examining their relative strengths and weaknesses, which may provide insight in the human condition.
Introduction
SLE is a prototypic systemic autoimmune disease characterized by loss of tolerance to nuclear autoantigens including DNA, histones, and ribonucleoproteins (1). Hallmark features of SLE are significant immune system activation, lymphoproliferation, and widespread inflammatory damage as the result of massive immune complex (IC) formation in tissues (1, 2). In particular, vascular beds of certain organs (e.g. kidney, liver) appear to be exceptionally susceptible to IC mediated pathology. SLE is a genetic disease driven by multiple susceptibility alleles in a heritable fashion, however inheritance of any particular genetic lesion does not significantly impact overall risk of SLE development. Genome-wide association studies (GWAS) indicate a large number of genes involved in inflammatory processes may contribute to disease susceptibility (3). As is the case with most autoimmune diseases, environmental influence of disease is significant, and numerous exogenous stimuli (e.g. sunburn, viral infection, exposure to heavy metals) exacerbate disease in susceptible individuals or can cause temporary disease in otherwise healthy people.
Nephritis is a common (and one of the most serious) complication of SLE. Given the genetic heterogeneity of SLE it is not surprising that LN can take a variety of manifestations including proliferative lesions with cellular crescent formation, sclerosis, necrosis, and podocyte foot process effacement as common features. Although LN is genetically influenced, GWAS studies indicate genes driving autoimmunity and nephritis are distinct (4). This implies LN is the result of combined inheritance of both susceptibility to development of systemic autoimmune reactivity and increased end organ sensitivity to inflammatory pathology.
Nevertheless, immune complex formation is a hallmark of nephritis, especially glomerulonephritis. Immune complex formation is commonly initiated by autoantibody binding to glomerular antigens, with differences in autoantigenic specificity for glomerular (and tubular) antigens accounting for differences in the location of immune deposits (5, 6). Circulating immune complexes engage IgG Fc receptors (FcγRs) on glomerular and infiltrating cells (e.g. PMN, monocytes) to further incite the inflammatory process. The IgG sublclass is relevant to both engagement of FcγR and complement activation.
Thereafter the inflammatory response, which is at least in part genetically determined, is very influential in the course of disease. Cytokine and chemokine responses at the systemic and renal level contribute to the severity of inflammation, the extent of fibrosis, response to therapy and progression to end stage renal disease. These processes are complex, and they likely contribute to the variable response to immunosuppression therapies, since the agents may not always affect the dominant immunologic/inflammatory response in a given individual. Thus the challenge of modeling of SLE and LN is a daunting one. Over the years multiple animal models of spontaneous and induced SLE and LN have been described that reproduce one or multiple aspects of human disease. Below we discuss some of the most commonly used mouse models and characteristics of autoimmunity and renal pathology that develop in murine SLE relevant to human LN.
Spontaneous mouse models of SLE and LN
The MRLlpr and gld models
Spontaneous SLE in mice represents an excellent model of human disease as a large body of data suggests that the defects fundamental to systemic autoimmunity in humans operate in a fairly analogous fashion in mice. In many murine SLE models the kidney is a primary target organ (Fig. 1), and in most murine models SLE is more common and severe in female mice (with the exception of BXSB mice, discussed below).
Figure 1. Various manifestations of glomerulonephritis in SLE-prone mice.
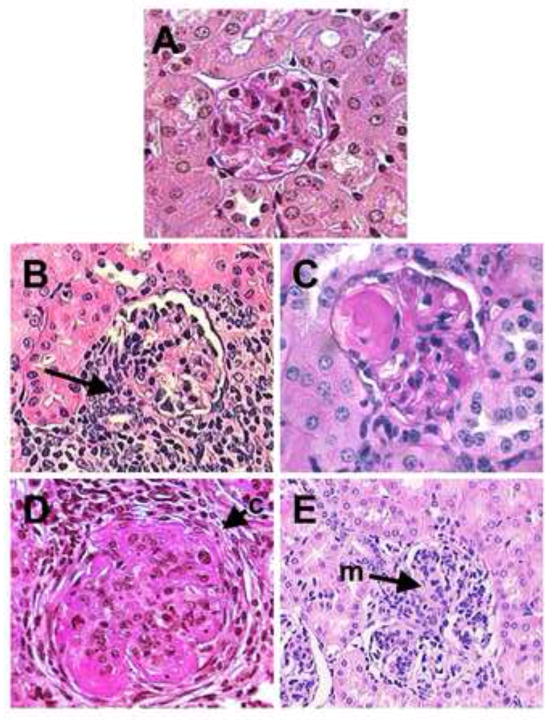
A) Normal glomerulus from C57BL/6 mouse. B) Glomerulus from MRLlpr mouse with significant peri-glomerular and interstitial infiltrate (arrow). C) NZM2410 glomerulus with fibrinoid necrosis. D) Glomerulus from male BXSB mouse with significant sclerosis and cellular crescent formation (c). E) Glomerulus from Fcgr2b−/− mouse with marked proliferative glomerulonephritis and mesangial expansion (m).
The MRL strain was developed in 1976 by Murphy and Roths in an effort to transfer a mutation for anchrondroplasia from the leukemic strain AKR (7). The MRL genome is primarily derived from the LG/J strain with contributions from AKR, C3H and C57BL/6 (7, 8). In the 12th generation of inbreeding a MRL substrain was identified that developed lymphadenopathy characterized by massive accumulation of lymphocytes including an unusual population of CD4negCD8neg B220+ T cells. Backcross experiments revealed the defect to be the result of a recessive mutation termed lymphoproliferation (lpr). In the early 1990s Shigekazu Nagata’s group identified lpr as insertion of an early transposable element in intron 2 of the Fas gene resulting in a loss of protein expression as a result of truncated translation (9). MRLlpr mice are unusual in that they show a full spectrum of SLE features including arthritis, inflammatory skin lesions and glomerulonephritis. Both male and female mice are affected by the lpr mutation, although disease is more severe in females (10). In comparison to other SLE models MRLlpr mice show accelerated mortality with 50% death at approximately 24 weeks due to renal failure (10).
In MRLlpr mice glomerulonephritis involves proliferation of both endothelial and mesangial cells as well as thickening of the basement membrane with wire-loop capillaries reminiscent of proliferative LN. However, in contrast to other spontaneous SLE strains of mice, MRLlpr mice show a relatively low incidence of glomerular crescent formation (10). There is significant infiltration of inflamed glomeruli, consisting chiefly of macrophages with occasional T cells and neutrophils. IgG deposition is granular and generally mesangial and sub-endothelial, with some subepithelial deposits. Glomerular Ig deposits can be observed usually beginning around 2 months of age accompanied with significant complement C3 deposition. Complement driven inflammation is a critical factor in the renal pathogenesis as MRLlpr mice with a deficiency in the regulatory factor H exhibit markedly accelerated kidney damage and become azotemic (elevated blood urea nitrogen) with 70% of the mice dead by 14 weeks (11). Likewise deficiency in factor B (rendering the alternative pathway defective) protects MRLlpr mice from renal disease (12) and mice deficient in the complement pathway attenuator decay acceleration factor (DAF) show increased renal pathology (13). In general, the nephritis parallels hypocomplementemia and autoreactivity suggesting a direct link between systemic autoimmunity, complement activation and progressive kidney disease. However, unlike other lupus models (e.g. NZB/NZW F1 discussed below), nephritis in MRLlpr mice is independent of FcγRs so the models relevance to human lupus may not be totally appropriate (14). Nevertheless, inhibition of the monocyte chemokine CCL2/MCP-1 reduces LN pathology indicating macrophage recruitment is a key mechanism of autoimmune renal injury (15).
The gld mutation, was first reported by Roths, Murphy and Eicher in C3H/HeJ mice with lymphoproliferation similar to that observed in MRLlpr mice (16). The gld mutation was later identified as a point mutation in the cognate receptor for Fas, Fas ligand (FasL), that abrogated functional Fas/FasL interactions (17). Lymphocyte expansion and humoral autoimmunity is similar in gld compared to the MRLlpr strain, however gld mice have a much longer lifespan. Gld mouse glomeruli show significant renal IgG deposition by 22 weeks, although a majority of mice do not show signs of glomerular pathology (16). Moreover, gld mice exhibit reduced renal pathology with anti-glomerular antibody-driven injury suggesting a dichotomy in the contribution of Fas and FasL in LN (18). Thus the use of gld mice can provide valuable insight of the relative contribution of Fas and FasL to mechanisms of autoimmune pathology in the kidney.
The BXSB model
The BXSB strain was developed in the 1970s by crossing of C57BL/6 and SB/Le mice (hence the BXSB nomenclature) (19). This strain is unique in that male mice show significantly enhanced disease compared to females due to the presence of the y-linked autoimmune accelerator (Yaa) driving autoimmunity (19, 20). Elegant chromosomal hybridization studies showed the Yaa mutation is a translocation of a telomeric region of the X chromosome to the Y chromosome resulting in duplication of several genes including toll like receptor 7 (TLR7) increasing both expression and function (21). As TLR7 binds RNA, this may explain the unique serum autoantibody profile BXSB mice exhibit. Serum reactivity to DNA and histones is comparatively low, however BXSB mice develop reactivity to nucleolar antigens including ribonucleoproteins (RNP) giving rise to a “speckled” pattern when anti-nuclear antibody (ANA) assays are performed (22). Male mice show significant lymphoproliferation, but in contrast to MRLlpr mice the cellular composition is primarily B cells. Male BXSB mice show prominent deposition of IgG and C3 in the mesangium and capillary wall and develop fulminant nephritis with 50% mortality at 5 months of age and 90% mortality at 8 months (10, 22). Death is due to renal failure as male BXSB mice develop exudative, proliferative nephritis with a significant neutrophil infiltration and prominent proteinuria by 3 months (8, 10). Female mice show a weaker disease phenotype with 50% mortality around 15 months; however both male and female mice die due to glomerulonephritis and renal failure.
The NZB/NZW F1 model
The NZB/NZW F1 hybrid (B/W) model of SLE and LN is the oldest of the “classical” models of spontaneous disease (23). B/W mice are regarded by many to be the closest approximation of human SLE due to the characteristics of disease development and the underlying genetics driving autoimmunity. B/W mice are derived by crossing two strains with mild autoimmune characteristics (New Zealand black [NZB] and New Zealand White [NZW]). The resultant F1 generation animals show significant autoimmunity with high levels of anti-DNA and anti-chromatin antibodies (23). Female mice are more severely affected and serum IgG reactivity to DNA is detectible by 3 months (24). Unlike BXSB or MRLlpr mice there is little lymphoid hyperplasia, although mice show develop splenomegaly and signs of chronic polyclonal B cell activation. Female B/W mice have 50% mortality around 8 months and >90% mortality after 1 year of age (10). Death is due to LN and B/W mice develop progressive proteinuria beginning around 5 months and azotemia from approximately 7 months onward (8, 10). The nephritis is sclerotic with heavy proteinaceous deposits in the mesangium, tubular cast formation, diffuse proliferation of glomerular cells, prominent crescent formation, and a significant periglomerular and interstitial monocytic infiltrate. Female B/W mice exhibit heavy mesangial and occasional capillary IgG and C3 deposition at 5 months and as B/W mice age there is increased extra-glomerular, peri-tubular, and arteriolar deposition (10).
An accidental backcross between B/W and NZW mice lead to the establishment of the inbred New Zealand Mixed (NZM) mouse strains, several of which show highly penetrant SLE and LN resembling disease in parental B/W mice (25, 26). This has facilitated the study of genetics driving autoimmunity and pathology in murine SLE. In particular, the NZM2410 stain has been used extensively to elucidate the genetics underlying SLE and its pathologic features. In summary, LN in NZM2410 mice is driven primarily by 3 susceptibility loci (Sle1, Sle2, and Sle3 on chromosome 1, 4, and 7 respectively) that act in a threshold manner to drive disease. In other words, each loci drives certain characteristics of immune dysfunction (e.g. Sle2 causes B cell hyper-activation) but is insufficient to cause outright SLE pathology and LN in otherwise healthy congenic mice (26). In particular, Sle1 is located in an area of chromosome 1 syntenic with human chromosome 1, a region with several identified SLE susceptibility loci (27). As congenics for each susceptibility loci are available these strains may provide useful tools to study genetic interactions and the development of LN.
Genetic manipulation: Gene disruption models of LN
A large number of studies have reported that genetic disruption of genes involved in a range of immune functionality leads to the development of murine SLE and occasionally outright LN. A full discussion is beyond the scope of this review, however it should be pointed out that this approach could provide valuable tools to study LN. In particular, since the majority of knock out models are generated by gene disruption in either the C57BL/6 or BALB/c mouse strains, there is a significant number of tools available to the researcher to dissect mechanisms of pathology or therapy.
One model worth mention is the Fcgr2b knock out (Fcgr2b−/−) mouse. Fcgr2b encodes for a single chain FcγR unique among the classic FcγRs in that the cytoplasmic domain contains an inhibitory ITIM motif negatively regulating signal transduction in B cells, macrophages, and dendritic cells. Originally derived from 129/SvJ cells, the Fcgr2b−/− mouse was on a mixed genetic background and showed no signs autoimmunity. However, when backcrossed 12 generations to the C57BL/6 (BL/6) and BALB/c strains a striking dichotomy was observed. BALB/c Fcgr2b−/− mice showed no overt autoimmunity, but female BL/6 Fcgr2b−/− mice developed SLE with high-titer anti-DNA and anti-chromatin IgG by 5 to 6 months paralleling polyclonal B cell activation and splenomegaly (28). Likewise, female BL/6 Fcgr2b−/− mice exhibited severe LN (manifesting between 5 and 7 months) with proliferative lesions, crescent formation, peri-glomerular and interstitial infiltration of macrophages and B cells, and significant glomerular IgG and C3 deposition (22, 28, 29). The immune deposits are primarily IgG and all subclasses are represented, although IgG2b and IgG2c predominate. IgG deposition is mainly mesangial but subendothelial deposits are frequent (TLM, unpublished observations). Mortality is 50% at 9 months and 80% at 1 year due to LN in female mice while males show mild autoimmunity but a normal lifespan (22, 28). Genetic studies have suggested the presence of susceptibility loci on the BL/6 background that drive autoimmunity and proteinuria in Fcgr2b−/− mice (30). However, several reports have called into question the relative role of FcγRIIb in the disease phenotype, and it is likely the combination of Fcgr2b gene disruption and flanking 129/SvJ genomic material inherited via linkage disequilibrium drives SLE and LN in this model (31). Nevertheless, FcγRIIB is implicated as an SLE risk factor in a number of genetic studies (32, 33), and Fcgr2b promoter polymorphisms affecting expression and function are a common feature of the autoimmunity-prone strains NZB, BXSB, and MRLlpr (34, 35). Moreover, overexpression of FcγRIIB by retroviral transduction was sufficient to prevent SLE and LN in BXSB and NZM2410 mice indicating FcγRIIB is an important regulator or inflammatory autoimmunity (22). Thus, the subtleties of the underlying genetics of disease coupled with the utility of the BL/6 genetic background make this a versatile model to study LN.
Inducible models of nephritis: Pristane and other approaches
Intraperitoneal injection of pristane (2,6,10,14 tetramethylpentadecane) is a standard approach to create ascitic fluid for monoclonal antibody production. However, Satoh and Reeves reported within 2 months of pristane administration, otherwise healthy BALB/c mice developed anti-RNP (and later anti-DNA and anti-histone) antibodies (36, 37). Interestingly, anti-RNP antibodies class switched to IgG while anti-DNA antibodies were almost entirely IgM, in stark contrast to most mouse SLE models (37). Pristane injected BALB/c mice developed some features of nephritis 6 months after injection with focal to diffuse proliferative glomerulonephritis and moderate proteinuria (37). Moreover, pristane injected mice exhibited a renal monocytic infiltrate and mesangial and sub-endothelial deposition of IgM and IgG with mesangial C3 deposits. Survival studies with this model are confounded by other pathologies induced by pristane injection including plasmacytoma (38). Nevertheless, it is unlikely the mice die due to nephritis, and there is little mortality up to 6 months post-pristane injection. Most mouse strains are susceptible to pristane-induced autoimmunity although the development of LN is variable (39). Sex is also a factor in pristane susceptibility with female mice showing heightened susceptibility to pristane-driven SLE in the SJL/J strain (40). It is worth noting that pristane-treated BALB/c mice develop other autoimmune phenotypes such as capillary hemorrhagic pathology in the lung and TNFα-driven arthritis that may resemble arthralgia and capillaritis occurring in SLE patients (41–43). Thus this model may useful in examining environmental triggers of LN in the context of more broad-spectrum pathology.
In addition to pristane, several other models of inducible nephritis have been developed that can resemble LN pathology and may be useful in examining mechanisms of renal tissue damage. These include the nephrotoxic serum nephritis model of crescentic glomerulonephritis (44) and the serum sickness model of IC nephritis (45). However, while the reader should be aware of these approaches, these models are most useful investigating downstream effector responses common to many forms of nephritis including LN and are beyond the scope of this review.
Conclusion
Overall, breakdown in immunologic self-tolerance leads to autoimmunity that may manifest as organ/antigen specific or systemic autoimmunity (i.e. lupus). Spontaneous forms of murine lupus have provided important insights. Multiple pathways lead to autoimmunity in general and lupus nephritis in particular, and these may differ among individual lupus strains and individual patients. Nevertheless, it is the specificity of the autoimmune response, coupled with both the systemic and the local organ inflammatory responses that determine the type and severity of the lesions and the accompanying clinical parameters. Understanding the interplay among the contributors and which dominates during the course of disease should provide therapeutic insights, which translate into more timely and effective therapy.
Table 1.
Comparision of mouse models of SLE and lupus nephritis
| Clinical | Histologic | autoantibodies | |
|---|---|---|---|
| MRLlpr |
|
|
|
|
NZB/NZW F1 NZM2410 |
|
|
|
| BXSB |
|
|
|
| B6.Fcgr2b−/− |
|
|
|
| Pristane |
|
|
|
Acknowledgments
The authors would like to thank Dr. Andrew Mellor for critical comments on the manuscript. This work is is supported by grants from the Lupus Research Institute and NIH grants AI092213 and AI105500 (TLM) and DK081140 (MPM).
Footnotes
Conflict of interests
The authors have no conflict of interests to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu Z, Davidson A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nature medicine. 2012;18:871–882. doi: 10.1038/nm.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsokos GC. Systemic lupus erythematosus. The New England journal of medicine. 2011;365:2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 3.Rullo OJ, Tsao BP. Recent insights into the genetic basis of systemic lupus erythematosus. Annals of the rheumatic diseases. 2013;72(Suppl 2):ii56–61. doi: 10.1136/annrheumdis-2012-202351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung SA, Brown EE, Williams AH, Ramos PS, Berthier CC, Bhangale T, Alarcon-Riquelme ME, Behrens TW, Criswell LA, Graham DC, Demirci FY, Edberg JC, Gaffney PM, Harley JB, Jacob CO, Kamboh MI, Kelly JA, Manzi S, Moser-Sivils KL, Russell LP, Petri M, Tsao BP, Vyse TJ, Zidovetzki R, Kretzler M, Kimberly RP, Freedman BI, Graham RR, Langefeld CD for the International Consortium for Systemic Lupus Erythematosus, Lupus Nephritis Susceptibility Loci in Women with Systemic Lupus Erythematosus. . Journal of the American Society of Nephrology: JASN. 2014 doi: 10.1681/ASN.2013050446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Madaio MP. The relevance of antigen binding to the pathogenicity of lupus autoantibodies. Kidney international. 2012;82:125–127. doi: 10.1038/ki.2012.159. [DOI] [PubMed] [Google Scholar]
- 6.Krishnan MR, Wang C, Marion TN. Anti-DNA autoantibodies initiate experimental lupus nephritis by binding directly to the glomerular basement membrane in mice. Kidney international. 2012;82:184–192. doi: 10.1038/ki.2011.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy ED, Roths JB. A single gene model for massive lymphoproliferation with immune complex disease in a new mouse strain MRL. Proceedings of the 16th International Congress of Hematology; 1976. [Google Scholar]
- 8.Theofilopoulos AN, Dixon FJ. Etiopathogenesis of murine SLE. Immunological reviews. 1981;55:179–216. doi: 10.1111/j.1600-065x.1981.tb00343.x. [DOI] [PubMed] [Google Scholar]
- 9.Adachi M, Watanabe-Fukunaga R, Nagata S. Aberrant transcription caused by the insertion of an early transposable element in an intron of the Fas antigen gene of lpr mice. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:1756–1760. doi: 10.1073/pnas.90.5.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, McConahey PJ, Murphy ED, Roths JB, Dixon FJ. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. The Journal of experimental medicine. 1978;148:1198–1215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bao L, Haas M, Quigg RJ. Complement factor H deficiency accelerates development of lupus nephritis. Journal of the American Society of Nephrology: JASN. 2011;22:285–295. doi: 10.1681/ASN.2010060647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe H, Garnier G, Circolo A, Wetsel RA, Ruiz P, Holers VM, Boackle SA, Colten HR, Gilkeson GS. Modulation of renal disease in MRL/lpr mice genetically deficient in the alternative complement pathway factor B. Journal of immunology. 2000;164:786–794. doi: 10.4049/jimmunol.164.2.786. [DOI] [PubMed] [Google Scholar]
- 13.Miwa T, Maldonado MA, Zhou L, Sun X, Luo HY, Cai D, Werth VP, Madaio MP, Eisenberg RA, Song WC. Deletion of decay-accelerating factor (CD55) exacerbates autoimmune disease development in MRL/lpr mice. The American journal of pathology. 2002;161:1077–1086. doi: 10.1016/S0002-9440(10)64268-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsumoto K, Watanabe N, Akikusa B, Kurasawa K, Matsumura R, Saito Y, Iwamoto I, Saito T. Fc receptor-independent development of autoimmune glomerulonephritis in lupus-prone MRL/lpr mice. Arthritis and rheumatism. 2003;48:486–494. doi: 10.1002/art.10813. [DOI] [PubMed] [Google Scholar]
- 15.Kulkarni O, Pawar RD, Purschke W, Eulberg D, Selve N, Buchner K, Ninichuk V, Segerer S, Vielhauer V, Klussmann S, Anders HJ. Spiegelmer inhibition of CCL2/MCP-1 ameliorates lupus nephritis in MRL-(Fas)lpr mice. Journal of the American Society of Nephrology: JASN. 2007;18:2350–2358. doi: 10.1681/ASN.2006121348. [DOI] [PubMed] [Google Scholar]
- 16.Roths JB, Murphy ED, Eicher EM. A new mutation, gld, that produces lymphoproliferation and autoimmunity in C3H/HeJ mice. The Journal of experimental medicine. 1984;159:1–20. doi: 10.1084/jem.159.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76:969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 18.Tarzi RM, Sharp PE, McDaid JP, Fossati-Jimack L, Herbert PE, Pusey CD, Cook HT, Warrens AN. Mice with defective Fas ligand are protected from crescentic glomerulonephritis. Kidney international. 2012;81:170–178. doi: 10.1038/ki.2011.319. [DOI] [PubMed] [Google Scholar]
- 19.Murphy ED, Roths JB. A Y chromosome associated factor in strain BXSB producing accelerated autoimmunity and lymphoproliferation. Arthritis and rheumatism. 1979;22:1188–1194. doi: 10.1002/art.1780221105. [DOI] [PubMed] [Google Scholar]
- 20.Hudgins CC, Steinberg RT, Klinman DM, Reeves MJ, Steinberg AD. Studies of consomic mice bearing the Y chromosome of the BXSB mouse. Journal of immunology. 1985;134:3849–3854. [PubMed] [Google Scholar]
- 21.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 22.McGaha TL, Sorrentino B, Ravetch JV. Restoration of tolerance in lupus by targeted inhibitory receptor expression. Science. 2005;307:590–593. doi: 10.1126/science.1105160. [DOI] [PubMed] [Google Scholar]
- 23.Helyer BJ, Howie JB. Renal disease associated with positive lupus erythematosus tests in a cross-bred strain of mice. Nature. 1963;197:197. doi: 10.1038/197197a0. [DOI] [PubMed] [Google Scholar]
- 24.Papoian R, Pillarisetty R, Talal N. Immunological regulation of spontaneous antibodies to DNA and RNA. II. Sequential switch from IgM to IgG in NZB/NZW F1 mice. Immunology. 1977;32:75–79. [PMC free article] [PubMed] [Google Scholar]
- 25.Rudofsky UH, Evans BD, Balaban SL, Mottironi VD, Gabrielsen AE. Differences in expression of lupus nephritis in New Zealand mixed H-2z homozygous inbred strains of mice derived from New Zealand black and New Zealand white mice. Origins and initial characterization. Laboratory investigation; a journal of technical methods and pathology. 1993;68:419–426. [PubMed] [Google Scholar]
- 26.Morel L, Rudofsky UH, Longmate JA, Schiffenbauer J, Wakeland EK. Polygenic control of susceptibility to murine systemic lupus erythematosus. Immunity. 1994;1:219–229. doi: 10.1016/1074-7613(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 27.Tsao BP. Lupus susceptibility genes on human chromosome 1. International reviews of immunology. 2000;19:319–334. doi: 10.3109/08830180009055502. [DOI] [PubMed] [Google Scholar]
- 28.Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity. 2000;13:277–285. doi: 10.1016/s1074-7613(00)00027-3. [DOI] [PubMed] [Google Scholar]
- 29.Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. The Journal of experimental medicine. 2006;203:553–561. doi: 10.1084/jem.20052438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolland S, Yim YS, Tus K, Wakeland EK, Ravetch JV. Genetic modifiers of systemic lupus erythematosus in FcgammaRIIB(−/−) mice. The Journal of experimental medicine. 2002;195:1167–1174. doi: 10.1084/jem.20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boross P, Arandhara VL, Martin-Ramirez J, Santiago-Raber ML, Carlucci F, Flierman R, van der Kaa J, Breukel C, Claassens JW, Camps M, Lubberts E, Salvatori D, Rastaldi MP, Ossendorp F, Daha MR, Cook HT, Izui S, Botto M, Verbeek JS. The inhibiting Fc receptor for IgG, FcgammaRIIB, is a modifier of autoimmune susceptibility. Journal of immunology. 2011;187:1304–1313. doi: 10.4049/jimmunol.1101194. [DOI] [PubMed] [Google Scholar]
- 32.McGaha TL, Karlsson MC, Ravetch JV. FcgammaRIIB deficiency leads to autoimmunity and a defective response to apoptosis in Mrl-MpJ mice. Journal of immunology. 2008;180:5670–5679. doi: 10.4049/jimmunol.180.8.5670. [DOI] [PubMed] [Google Scholar]
- 33.Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nature reviews Immunology. 2010;10:328–343. doi: 10.1038/nri2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiu Y, Nakamura K, Abe M, Li N, Wen XS, Jiang Y, Zhang D, Tsurui H, Matsuoka S, Hamano Y, Fujii H, Ono M, Takai T, Shimokawa T, Ra C, Shirai T, Hirose S. Transcriptional regulation of Fcgr2b gene by polymorphic promoter region and its contribution to humoral immune responses. Journal of immunology. 2002;169:4340–4346. doi: 10.4049/jimmunol.169.8.4340. [DOI] [PubMed] [Google Scholar]
- 35.Rahman ZS, Niu H, Perry D, Wakeland E, Manser T, Morel L. Expression of the autoimmune Fcgr2b NZW allele fails to be upregulated in germinal center B cells and is associated with increased IgG production. Genes and immunity. 2007;8:604–612. doi: 10.1038/sj.gene.6364423. [DOI] [PubMed] [Google Scholar]
- 36.Satoh M, Reeves WH. Induction of lupus-associated autoantibodies in BALB/c mice by intraperitoneal injection of pristane. The Journal of experimental medicine. 1994;180:2341–2346. doi: 10.1084/jem.180.6.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Satoh M, Kumar A, Kanwar YS, Reeves WH. Anti-nuclear antibody production and immune-complex glomerulonephritis in BALB/c mice treated with pristane. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:10934–10938. doi: 10.1073/pnas.92.24.10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Potter M, Wax JS, Anderson AO, Nordan RP. Inhibition of plasmacytoma development in BALB/c mice by indomethacin. The Journal of experimental medicine. 1985;161:996–1012. doi: 10.1084/jem.161.5.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Satoh M, Richards HB, Shaheen VM, Yoshida H, Shaw M, Naim JO, Wooley PH, Reeves WH. Widespread susceptibility among inbred mouse strains to the induction of lupus autoantibodies by pristane. Clinical and experimental immunology. 2000;121:399–405. doi: 10.1046/j.1365-2249.2000.01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith DL, Dong X, Du S, Oh M, Singh RR, Voskuhl RR. A female preponderance for chemically induced lupus in SJL/J mice. Clinical immunology. 2007;122:101–107. doi: 10.1016/j.clim.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chowdhary VR, Grande JP, Luthra HS, David CS. Characterization of haemorrhagic pulmonary capillaritis: another manifestation of Pristane-induced lupus. Rheumatology. 2007;46:1405–1410. doi: 10.1093/rheumatology/kem117. [DOI] [PubMed] [Google Scholar]
- 42.Nguyen VA, Gotwald T, Prior C, Oberrnoser G, Sepp N. Acute pulmonary edema, capillaritis and alveolar hemorrhage: pulmonary manifestations coexistent in antiphospholipid syndrome and systemic lupus erythematosus. Lupus. 2005;14:557–560. doi: 10.1191/0961203305lu2107cr. [DOI] [PubMed] [Google Scholar]
- 43.Wooley PH, Seibold JR, Whalen JD, Chapdelaine JM. Pristane-induced arthritis. The immunologic and genetic features of an experimental murine model of autoimmune disease. Arthritis and rheumatism. 1989;32:1022–1030. doi: 10.1002/anr.1780320812. [DOI] [PubMed] [Google Scholar]
- 44.Du Y, Fu Y, Mohan C. Experimental anti-GBM nephritis as an analytical tool for studying spontaneous lupus nephritis. Archivum immunologiae et therapiae experimentalis. 2008;56:31–40. doi: 10.1007/s00005-008-0007-4. [DOI] [PubMed] [Google Scholar]
- 45.Iskandar SS, Jennette JC, Wilkman AS, Becker RL. Interstrain variations in nephritogenicity of heterologous protein in mice. Laboratory investigation; a journal of technical methods and pathology. 1982;46:344–351. [PubMed] [Google Scholar]