

Abstract
IgA nephropathy (IgAN) is the most common form of primary glomerulonephritis in the world. IgAN is characterized by the mesangial accumulation of immune complexes containing IgA1, usually with co-deposits of complement C3 and variable IgG and/or IgM. Although more than 40 years have passed since IgAN was first described, the mechanisms underlying the disease development are not fully understood. Small-animal experimental models of IgAN can be very helpful in studies of IgAN, but development of these models has been hindered by the fact that only humans and hominoid primates have IgA1 subclass. Thus, multiple models have been developed, that may be helpful in studies of some specific aspects of IgAN. These models include a spontaneous animal model of IgAN, the ddY mouse first reported in 1985. These mice show mild proteinuria without hematuria, and glomerular IgA deposits, with a highly variable incidence and degree of glomerular injury, due to the heterogeneous genetic background. To obtain a murine line consistently developing IgAN, we intercrossed an earlyonset group of ddY mice, in which the development of IgAN includes mesangial IgA deposits and glomerular injury. After selective intercrossing for >20 generations, we established a novel 100% early-onset grouped ddY murine model. All grouped ddY mice develop proteinuria within eight weeks of age. The grouped ddY mouse model can be a useful tool for analysis of multiple aspects of the pathogenesis of IgAN and may aid in assessment of some approaches for the treatment of IgAN.
Introduction
IgA nephropathy (IgAN) is one of the most frequent forms of glomerulonephritis worldwide, representing 25%–50% of patients with primary glomerulonephritis. The major histologic characteristics of IgAN are mesangial-cell proliferation and matrix expansion associated with granular mesangial immunodeposits of IgA, consisting of polymeric IgA1 (pIgA1) (1), and complement 3 (C3) with variable IgG and/or IgM codeposits. IgAN was initially thought to be a benign chronic glomerulonephritis, but there is an increasing evidence that 30%–40% of the patients progress to end-stage renal disease within 20 years of diagnosis (2). There are no effective disease-specific treatment strategies.
Although the major diagnostic criterion for IgAN is presence of dominant or co-dominant IgA deposits in the glomerular mesangium (3), clinical and histopathologic features of IgAN patients are heterogeneous. Fundamental pathogenic factors are of extrarenal origin, as evidenced by the fact that about half of IgAN patients develop recurrent disease after renal transplantation (4). Recently, many researchers have advanced the understanding of pathogenesis of IgAN at the biochemical, immunological, and genetic levels. Current data indicate that multiple processes contribute to development of IgAN in genetically susceptible individuals (2, 5, 6). Patients with IgAN often have elevated circulating levels of aberrantly glycosylated IgA1, galactose-deficient in some O glycans (Gd-IgA1) (Hit 1). This glycosylation aberrancy is, however, not sufficient to induce renal injury. Autoantibodies directed against Gd-IgA1 bind the aberrant IgA1, resulting in the formation of high-molecular-mass immune complexes (Hit 2), some of which deposit in the glomerular mesangium (Hits 3). These immune complexes activate mesangial cells, inducing proliferation and secretion of extracellular matrix, cytokines, and chemokines, thus inciting a glomerular injury (Hit 4) (6).
Small-animal models of IgAN can be very helpful in studies of disease pathogenesis, but development of such models for IgAN has been hindered by the fact that only humans and hominoid primates have IgA1 subclass. In spite of these obstacles, several different models have been developed, that may be helpful in studies of various specific aspects of primary IgAN. Although attempts have been made to also develop models of secondary IgAN, such as Akita mouse (mouse with mutation in the insulin 2 gene; Ins2 (Akita)) that shows both mesangial sclerosis and IgA deposition (7), we have not covered these more complex models in this review due to a limited space. In this manuscript, we review several selected animal models of primary IgAN (Table 1) that may contribute to elucidating specific steps in the pathogenesis of IgAN, with special emphasis on grouped ddY model.
Table 1. Selected animal models of IgA nephropathy.
| Mouse strain or model name | Year published Brief description | Advantages and deficiencies of each model in capturing key features of IgA nephropathy | |
|---|---|---|---|
| Spontaneous ddY mouse | 1986 | A spontaneous animal model of IgAN, in which the development of IgAN includes mesangial IgA deposits and glomerular injury | A major disadvantage of the ddY mouse is a high degree of variability in the age of onset and severity of the disease, due to the heterogeneous genetic background; Mouse IgA, not human IgA1 |
| Gluten-immunized mouse | 1989 | A lectin-like fraction of gluten, called gliadin, induces IgA mesangial deposits in BALB/c mice, concurrent with an increased level of serum IgA | Model of food-antigen-containing immune complexes with IgA; mouse IgA, not human IgA1 |
| Vomitoxin (deoxynivalenol)-exposed mouse | 1989 | Dietary exposure to the trichothecene vomitoxin, fungal contaminant of cereal grains, induces elevated serum IgA, elevation of serum IgA, IgA-containing immune complexes, and glomerular deposits | Model of antigen-containing immune complexes with IgA; mouse IgA, not human IgA1 |
| High-IgA strain of ddY mouse (HIGA) | 1997 | Established by interbreeding of ddY strains with high serum levels of IgA | HIGA mice have high serum IgA levels, however, serum IgA levels are not associated with the severity of glomerular injury and incidence of the disease; Mouse IgA, not human IgA1 |
| Uteroglobin antisense transgenic mouse | 2000 | Uteroglobin antisense-Tg mice develop IgAN, characterized by microhematuria, albuminuria, and glomerular deposits of IgA, fibronectin, collagen, and C3, accompanied by increased levels of serum IgA-fibronectin complexes | Follow-up studies did not confirm the role of uteroglobin in reducing circulating IgA-fibronectin complexes; mouse IgA, not human IgA1 |
| CD89-transgenic mouse | 2000 | Complexes of Tg IgA1 heavy chain-containing IgA with Tg human soluble CD89; mesangial deposits of the complexes induce hematuria and proteinuria | Tg mice expressing human CD89 (mice do not have a homologue of human CD89) develop mesangial IgA deposits, glomerular and interstitial macrophage infiltration, mesangial matrix expansion, hematuria, and mild proteinuria; can be combined with Tg human IgA1 heavy chain; follow-up studies raised a question whether CD89 is involved in a similar manner in human IgAN |
| Mucosal immunization with Sendai virus | 2001 | Oral immunization and intranasal challenge with Sendai virus, a rodent parainfluenza virus, | Sendai virus is similar to some human respiratory-tract viruses; this model may be useful to assess some infection-related aspects of IgAN; mouse IgA, not human IgA1 |
| Human Bcl-2 transgenic mice | 2004 | Overexpression of Bcl-2 in B cells selectively enhances systemic IgA immune responses; Serum IgA purified from human Bcl-2 TG mice, has an increased ability to deposit in the glomeruli | Selectively enhanced systemic IgA immune responses; mouse IgA, not human IgA1 |
| Human BAFF-transgenic mouse | 2006 | Over-expression of human BAFF in mice results in elevated serum IgA, and fatal glomerulonephritis associated with mesangial deposits of IgA | IgA deposits co-dependent on microbiota; mouse IgA, not human IgA1; light-microscopic features include glomerular sclerosis |
| β1, 4-galactosyltransferase-I-deficient mou | se 2007 | K/O mice exhibit high serum levels of IgA with elevated representation of polymeric IgA and mesangial deposits | Mouse IgA, not human IgA1; N-glycans affected |
| Grouped-ddY mouse | 2012 | The early-onset group of ddY mice, in which the development of IgAN includes mesangial IgA deposits and glomerular injury, were intercrossed over 20 generations. | 100% early-onset model of IgAN. All grouped ddY mice develop proteinuria within eight weeks of age; Mouse IgA, not human IgA1 |
| Passive mouse model of IgA nephropathy | 2012 | SCID or nude mice injected with pre-formed complexes comsisting of human IgA1 deficient in galactose and bound by anti-glycan human IgG deposit in glomerular mesangium with murine C3; IgA1 deposits with concurrent mesangial proliferation, hematuria and proteinuria | Human IgA1 autoantigen and autoantibodies used; requires several injections of preformed complexes |
Animal models of IgA nephropathy
Exogenous antigen
Mucosal antigen exposure is implicated in the pathogenesis of IgAN. Food antigens, such as casein, ovalubumin, and gluten, may contribute to the pathogenesis of IgAN in some patients (8–10). Coppo et al. reported that the effect of gluten and its lectin-like fraction gliadin in inducing IgA mesangial deposits in BALB/c mice with an increased level of serum IgA (11) (Table 1). Administration of lactalbumin with concurrent blockade of the reticuloendothelial system induced mesangial IgA deposition and increase of serum level of IgA (12). Extended dietary exposure to the trichothecene vomitoxin, a naturally occurring fungal contaminant of cereal grains, induces elevated serum IgA. Oral exposure to the trichothecene vomitoxin in mice induces marked elevation of serum levels of IgA, IgA-containing immune complexes, and mesangial IgA deposition in a similar manner as in human IgAN (13). It is thought that vomitoxin can enhance CD4+ T cell-mediated help and promote terminal differentiation of IgA-secreting cells in the intestine.
Chintalacharuvu et al. reported that oral immunization and intranasal challenge with Sendai virus, a rodent parainfluenza virus similar to some human respiratory-tract viruses, can induce IgAN in mice through the hyper-activation of Th2 cells (14). Notably, these experimental conditions mimic acute exposure to a respiratory-tract virus and, thus, this model may be useful to assess some infection-related aspects of IgAN.
Protein uteroglobin is a multifunctional anti-inflammatory protein that modulates immune responses. A uteroglobin gene-knockout mice and uteroglobin antisense-transgenic mice develop IgAN, characterized by microhematuria, albuminuria, and renal glomerular deposits of IgA, fibronectin, collagen, and C3, accompanied by increased levels of serum IgA-fibronectin complexes (15). However, follow-up study did not confirm the role of uteroglobin in reducing circulating IgA-fibronectin complexes (16).
IgA receptors
A role of two IgA receptors, the FcαR (CD89) expressed by blood myeloid cells and the transferrin receptor (TfR1) on mesangial cells, in the pathogenesis of IgAN has been considered (17, 18). It was postulated that abnormal IgA may enhance release of soluble fragment of CD89 from the cell membrane, resulting in the formation of circulating IgA-CD89 complexes. These complexes can be then bound by TfR1 that is overexpressed by mesangial cells of IgAN patients, and induce expression of transglutaminase 2 (19). Transglutaminase 2 stabilizes IgA deposits at the surface of mesangial cells, the cells are then activated, start to proliferate and produce proinflammatory cytokines, leading to glomerular injury. A transgenic mice expressing human CD89 (mice do not have a homologue of human CD89) develop mesangial IgA deposits, glomerular and interstitial macrophage infiltration, mesangial matrix expansion, hematuria, and mild proteinuria (17). However, follow-up studies raised a question whether CD89 is involved in a similar manner in human IgAN (20).
Cellular factors
The Bcl-2 (B-cell lymphoma 2) regulates cell death. Mutations of Bcl-2 gene have been identified as a cause of a number of cancers and autoimmunity. The overexpression of Bcl-2 in B cells selectively enhances systemic IgA immune responses (21). Serum IgA purified from human Bcl-2 transgenic mice, compared to control mice, shows an increased level of aberrantly glycosylated IgA and an increased ability to deposit in the glomeruli, as observed in human IgAN (21).
B-cell activating factor (BAFF), a member of tumor necrosis factor family, is a peripheral B-cell survival factor and involved in antibody class switching. Over-expression of human BAFF in mice results in B-cell hyperplasia, elevated serum immunoglobulins, and fatal glomerulonephritis. BAFF-Tg mice have mesangial deposits of IgA along with elevated circulating levels of polymeric IgA that is aberrantly glycosylated (22).
Spontaneous model of IgAN
The ddY mouse strain is a model of spontaneous IgAN, which develops glomerulonephritis with a striking deposition of IgA in the mesangium, as well as co-deposits of IgG, IgM, and C3 (23). A major disadvantage of the ddY mouse model is a high degree of variability in the age of onset and severity of the disease, because the strain has been maintained as an outbred stock. The high-IgA (HIGA) mouse strain was established by interbreeding of ddY strains with high serum levels of IgA to assess possible correlation of serum IgA levels with the development of IgAN (24). These models revealed that although HIGA mice have high IgA levels, serum IgA levels are not associated with the severity of glomerular injury and incidence of the disease (25). We previously reported that ddY mice could be classified into the early-onset, late-onset, and quiescent groups, based on analyses of serial renal biopsies (25). Genome-wide association study identified four genetic loci (D1Mit216, D1Mit16, D9Mit252, and D10Mit86) linked with the early-onset phenotype (25). D1Mit16 is located close to the selectin gene: single-nucleotide polymorphisms of this gene are associated with human IgAN (26), and D10Mit86 lies within a region of synteny with human 6q22–23 containing IGAN1, which is implicated in familial IgAN (27). These results suggest that IgAN in ddY mice and in humans may be, at least partly, affected by the same susceptibility genes.
Grouped ddY mouse
To overcome a high degree of variability in the age of onset and severity of the disease in ddY mouse model, we intercrossed early-onset ddY mice. After selective intercrossing for >20 generations, we established a novel 100% early-onset grouped ddY mice model (28). All grouped ddY mice develop proteinuria within 8 weeks of birth. The urinary protein output in grouped ddY mice is elevated compared with HIGA mice at 8 weeks of age and the serum creatinine level is increased in association with renal failure at 24 weeks of age (28). The grouped ddY mice show severe glomerular and tubulointerstitial lesions, characterized by mesangial proliferation, mesangial matrix expansion, and tubulointerstitial infiltrations. Glomerular cell numbers and glomerular sclerosis scores at 8 and 24 weeks of age in female grouped ddY mice are significantly higher than those in HIGA mice (28). Electron microscopy showed electrondense deposits mainly in the paramesangial area similar to those found in human IgAN (Figure 1). Immunofluorescence staining revealed glomerular deposits of IgA with IgG and C3 co-deposits in female grouped ddY mice (28).
Figure 1. Histopathological analysis of grouped ddY mouse.
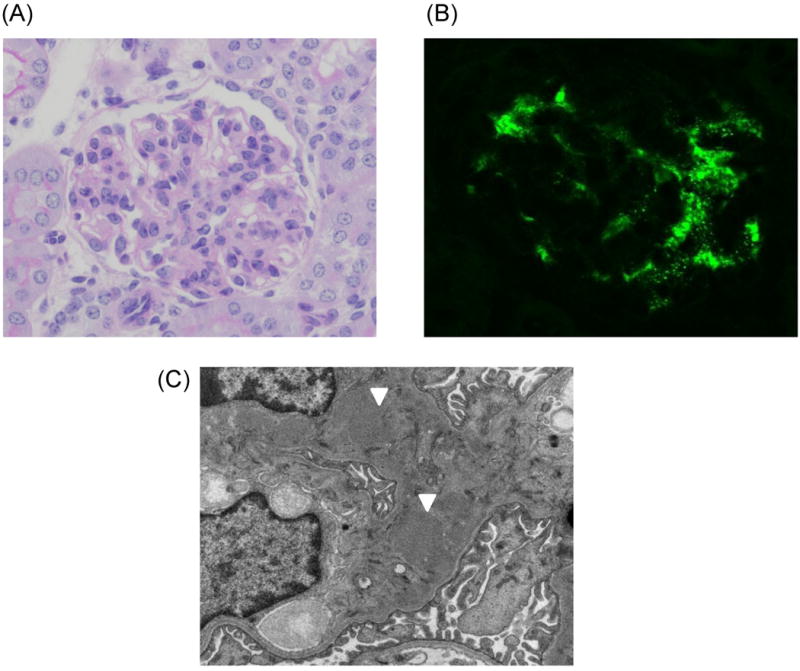
(A) Light-microscopic analysis reveals mesangial-cell proliferation and expansion of mesangial matrix (PAS staining, X400). (B) Immunohistochemical image of granular IgA deposits in the mesangium. (C) Electron-microscopic image with paramesangial immune deposits (white arrows).
Aberrant glycosylation of IgA
The pathogenesis of IgAN in humans involves deposition of immune complexes consisting of polymeric aberrantly O-glycosylated IgA1 (galactose-deficient in some O-glycans; Gd-IgA1) in the mesangium (29, 30). Glycosylation changes of human IgA1 play a key role in the development of the disease, with Gd-IgA1 being recognized by autoantibodies and forming immune complexes (6, 31-33). Importantly, grouped ddY mice showed high serum levels of IgA and IgA-IgG2a immune complexes, compared with those in HIGA mice (34). Notably, rodents do not have IgA isotype with structural features of human IgA1 with respect to O-glycans. In humans, aberrant galactosylation and sialylation of O-glycans in IgA1 hinge region is thought to contribute to the pathogenesis of IgAN (35, 36). Murine IgA has N-glycans, but not O-glycans. p-1,4-galactosyltransferase transfers galactose to the terminal N-acetylglucosamine residues of N-glycans. Notably, β-1,4-galactosyltransferase-deficient mice spontaneously develop IgAN (37). These knock-out mice exhibit high serum levels of IgA with elevated representation of polymeric IgA (37). Of note, a recent study has demonstrated the presence of O-glycans in the hinge region of an IgA rheumatoid factor, and its potential to induce IgAN-like glomerular lesions was associated with increased levels of O-glycosylation (38, 39).
To further analyze potential aberrancy in glycosylation of IgA in grouped ddY and HIGA mice, more extensive biochemical analysis of sugar contents was performed by using monosaccharide compositional analysis with gas-liquid chromatography. The analysis of sugar contents revealed that purified IgA from gddY mice had lower contents of saccharides compared with IgA from HIGA mice (28). This finding, thus, indicates that a glycosylation abnormality of IgA, whether involving O-glycans or N-glycans, can promote formation of macromolecular IgA-containing complexes and that IgG autoantibodies against aberrant IgA glycoform(s) may promote immune-complex formation, with subsequent complement activation in grouped ddY mice, similarly as in human IgAN (Figure 2) (34). Notably, a passive mouse model of IgAN has been recently developed, based on injection of immune complexes pre-formed from human IgA1 with galactose-deficient O-glycans mimicking IgAN bound by anti-glycan autoantibodies from IgAN patients (40). This observation further supports the role of aberrant O-glycosylation of IgA1 and autoantibodies binding to this glycoform in formation of immune deposits in IgAN (Table 1).
Figure 2. Proposed pathways involved in the pathogenesis of lgAN: multi-hit mechanism in human disease vs. grouped ddY mouse model.

Hit 1: Production of aberrantly glycosylated IgA1 (human; Hu) / aberrantly glycosylated IgA (mice; Mo) is regulated by a genetic factor(s) and/or due to a dysregulation of mucosal immune system. Other hits may also be affected by genetic and/or environmental factors. Hit 2: Formation of anti-glycan IgG and/or IgA1 autoantibodies (Hu); production of pathogenic IgG2a that recognize aberrantly glycosylated IgA. Hit 3: Formation of immune complexes from autoantigen (aberrantly glycosylated IgA) and autoantibodies. Hit 4: Deposition of pathogenic immune complexes in the mesangium, activation of mesangial cells, and induction of glomerular injury. Hits 3 and/or 4 may involve complement activation. (Hu: human, Mo: Mouse)
Mucosal Immunity
The impact of mucosal infections in IgAN has been established, as the disease is frequently exacerbated by upper-respiratory-tract or gastrointestinal-tract infections. Some dietary antigens promote B-cell terminal differentiation into IgA-secreting progenitors in the intestine and form IgA-containing immune complexes (10). Production of aberrantly glycosylated IgA1 may be enhanced by mucosal infection (41, 42). A series of studies focused on the role of Toll-like receptors (TLRs), which are evolutionarily conserved regulators of the innate immune response. TLR activation may represent the final common pathway for exogenous antigens, which have a negative effect on the mucosa in patients with IgAN. We have reported that IgAN severity correlates with splenic TLR9 expression in IgAN-prone mice (43). In addition, nasal challenge with CpG DNA (a ligand of TLR9) exacerbated glomerular damage in these mice and was accompanied by an increase in serum IgA levels and mesangial IgA deposition. This observation suggested that mucosal stimulation of TLR may be related, at least in part, to the production of nephritogenic IgA. In patients with IgAN, the expression of tonsillar TLR9 and a TLR9 single-nucleotide polymorphism correlates with the responses to the tonsillectomy combined with steroid-pulse therapy (44)
Conclusion
IgAN is a polygenic disease and the clinical and histopathologic findings of IgAN patients are heterogeneous. Moreover, molecular features of human IgA1, the autoantigen that plays a key role in the pathogenesis of human IgAN, are different from rodents’ IgA. However, it is possible to analyze selected phenotypes and pathological pathways that are common for an animal model and human disease or delineate the differences between the two. Even though murine IgA is different from human IgA1, aberrant glycosylation of IgA tends to enhance production of polymeric IgA and formation immune complexes with autoantibodies in both human and murine IgAN (Figure 2). Furthermore, genetic factors, dysregulation of mucosal immunity, and complement factors are important for initiation and progression of IgAN in animal models and human disease. The grouped ddY mouse model may be useful tool for studies of many of these aspects.
Acknowledgments
HS was supported in part by a research grant from the Study Group on IgA Nephropathy, a Grant-in-Aid for Progressive Renal Disease Research, Research on Intractable Disease from the Ministry of Health, Labour and Welfare of Japan, and by KAKENHI (22790802). JN was supported in part by the NIH grants DK078244, GM098539, and DK082753, and by a gift from the IGA Nephropathy Foundation of America.
Footnotes
Conflict of interest statement: All the authors declared no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tomino Y, Sakai H, Miura M, Endoh M, Nomoto Y. Detection of polymeric IgA in glomeruli from patients with IgA nephropathy. Clin Exp Immunol. 1982;49:419–425. [PMC free article] [PubMed] [Google Scholar]
- 2.Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. 2013;368:2402–14. doi: 10.1056/NEJMra1206793. [DOI] [PubMed] [Google Scholar]
- 3.Berger J, Hinglais N. Intercapillary deposits of IgA-IgG. J Urol Nephrol (Paris) 1968;74:694–695. [PubMed] [Google Scholar]
- 4.Floege J. Recurrent IgA nephropathy after renal transplantation. Semin Nephrol. 2004;24:287–291. doi: 10.1016/j.semnephrol.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 5.Novak J, Julian BA, Mestecky J, Renfrow MB. Glycosylation of IgA1 and pathogenesis of IgA nephropathy. Semin Immunopathol. 2012;34:365–82. doi: 10.1007/s00281-012-0306-z. [DOI] [PubMed] [Google Scholar]
- 6.Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, Wyatt RJ, Scolari F, Mestecky J, Gharavi AG, Julian BA. The pathophysiology of IgA nephropathy. J Am Soc Nephrol. 2011;22:1795–803. doi: 10.1681/ASN.2011050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haseyama T, Fujita T, Hirasawa F, Tsukada M, Wakui H, Komatsuda A, Ohtani H, Miura AB, Imai H, Koizumi A. Complications of IgA nephropathy in a non-insulin-dependent diabetes model, the Akita mouse. Tohoku J Exp Med. 1998;198:233–244. doi: 10.1620/tjem.198.233. [DOI] [PubMed] [Google Scholar]
- 8.Yagame M, Tomino Y, Eguchi K, Miura M, Suga T, Endoh M, Nomoto Y, Sakai H. Levels of circulating IgA immune complexes after gluten-rich diet in patients with IgA nephropathy. Nephron. 1988;49:104–6. doi: 10.1159/000185033. [DOI] [PubMed] [Google Scholar]
- 9.Fornasieri A, Sinico RA, Maldifassi P, Paterna L, Benuzzi S, Colasanti G, D'Amico G. Food antigens, IgA-immune complexes and IgA mesangial nephropathy. Nephrol Dial Transplant. 1988;3:738–43. [PubMed] [Google Scholar]
- 10.Russell MW, Mestecky J, Julian BA, Galla JH. IgA-associated renal diseases: antibodies to environmental antigens in sera and deposition of immunoglobulins and antigens in glomeruli. J Clin Immunol. 1986;6:74–86. doi: 10.1007/BF00915367. [DOI] [PubMed] [Google Scholar]
- 11.Coppo R, Mazzucco G, Martina G, Roccatello D, Amore A, Novara R, Bargoni A, Piccoli G, Sena LM. Gluten-induced experimental IgA glomerulopathy. Lab Invest. 1989;60:499–506. [PubMed] [Google Scholar]
- 12.Sato M, Ideura T, Koshikawa S. Experimental IgA nephropathy in mice. Lab Invest. 1986;54:377–84. [PubMed] [Google Scholar]
- 13.Pestka JJ, Moorman MA, Warner RL. Dysregulation of IgA production and IgA nephropathy induced by the trichothecene vomitoxin. Food Chem Toxicol. 1989;27:361–8. doi: 10.1016/0278-6915(89)90141-5. [DOI] [PubMed] [Google Scholar]
- 14.Chintalacharuvu SR, Nagy NU, Sigmund N, Nedrud JG, Amm ME, Emancipator SN. T cell cytokines determine the severity of experimental IgA nephropathy by regulating IgA glycosylation. Clin Exp Immunol. 2001;126:326–33. doi: 10.1046/j.1365-2249.2001.01678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Z, Kundu GC, Zheng F, Yuan CJ, Lee E, Westphal H, Ward J, DeMayo F, Mukherjee AB. Insight into the physiological function(s) of uteroglobin by gene-knockout and antisense-transgenic approaches. Ann N Y Acad Sci. 2000;923:210–33. doi: 10.1111/j.1749-6632.2000.tb05532.x. [DOI] [PubMed] [Google Scholar]
- 16.Coppo R, Chiesa M, Cirina P, Peruzzi L, Amore A European IgACE Study Group. In human IgA nephropathy uteroglobin does not play the role inferred from transgenic mice. Am J Kidney Dis. 2002;40:495–503. doi: 10.1053/ajkd.2002.34890. [DOI] [PubMed] [Google Scholar]
- 17.Launay P, Grossetête B, Arcos-Fajardo M, Gaudin E, Torres SP, Beaudoin L, Patey-Mariaud de Serre N, Lehuen A, Monteiro RC. Fcα receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger's disease). Evidence for pathogenic soluble receptor-IgA complexes in patients and CD89 transgenic mice. J Exp Med. 2000;191:1999–2009. doi: 10.1084/jem.191.11.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moura IC, Centelles MN, Arcos-Fajardo M, Malheiros DM, Collawn JF, Cooper MD, Monteiro RC. Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J Exp Med. 2001;194:417–25. doi: 10.1084/jem.194.4.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berthelot L, Papista C, Maciel TT, Biarnes-Pelicot M, Tissandie E, Wang PH, Tamouza H, Jamin A, Bex-Coudrat J, Gestin A, Boumediene A, Arcos-Fajardo M, England P, Pillebout E, Walker F, Daugas E, Vrtosvnik F, Flamant M, Benhamou M, Cogné M, Moura IC, Monteiro RC. Transglutaminase is essential for IgA nephropathy development acting through IgA receptors. J Exp Med. 2012;209:793–806. doi: 10.1084/jem.20112005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Boog PJ, De Fijter JW, Van Kooten C, Van Der Holst R, Van Seggelen A, Van Es LA, Daha MR. Complexes of IgA with FcαRI/CD89 are not specific for primary IgA nephropathy. Kidney Int. 2003;63:514–21. doi: 10.1046/j.1523-1755.2003.00756.x. [DOI] [PubMed] [Google Scholar]
- 21.Marquina R, Díez MA, López-Hoyos M, Buelta L, Kuroki A, Kikuchi S, Villegas J, Pihlgren M, Siegrist CA, Arias M, Izui S, Merino J, Merino R. Inhibition of B cell death causes the development of an IgA nephropathy in (New Zealand white x C57BL/6)F(1)-bcl-2 transgenic mice. J Immunol. 2004;172:7177–85. doi: 10.4049/jimmunol.172.11.7177. [DOI] [PubMed] [Google Scholar]
- 22.McCarthy DD, Kujawa J, Wilson C, Papandile A, Poreci U, Porfilio EA, Ward L, Lawson MA, Macpherson AJ, McCoy KD, Pei Y, Novak L, Lee JY, Julian BA, Novak J, Ranger A, Gommerman JL, Browning JL. Mice overexpressing BAFF develop a commensal flora-dependent, IgA-associated nephropathy. J Clin Invest. 2011;121:3991–4002. doi: 10.1172/JCI45563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imai H, Nakamoto Y, Asakura K, Miki K, Yasuda T, Miura AB. Spontaneous glomerular IgA deposition in ddY mice: An animal model of IgA nephritis. Kidney Int. 1985;27:756–761. doi: 10.1038/ki.1985.76. [DOI] [PubMed] [Google Scholar]
- 24.Muso E, Yoshida H, Takeuchi E, Yashiro M, Matsushima H, Oyama A, Suyama K, Kawamura T, Kamata T, Miyawaki S, Izui S, Sasayama S. Enhanced production of glomerular extracellular matrix in a new mouse strain of high serum IgA ddY mice. Kidney Int. 1996;50:1946–1957. doi: 10.1038/ki.1996.517. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki H, Suzuki Y, Yamanaka T, Hirose S, Nishimura H, Toei J, Horikoshi S, Tomino Y. Genome-wide scan in a novel IgA nephropathy model identifies a susceptibility locus on murine chromosome 10, in a region syntenic to human IGAN1 on chromosome 6q22-23. J Am Soc Nephrol. 2005;16:1289–1299. doi: 10.1681/ASN.2004030219. [DOI] [PubMed] [Google Scholar]
- 26.Takei T, Iida A, Nitta K, Tanaka T, Ohnishi Y, Yamada R, Maeda S, Tsunoda T, Takeoka S, Ito K, Honda K, Uchida K, Tsuchiya K, Suzuki Y, Fujioka T, Ujiie T, Nagane Y, Miyano S, Narita I, Gejyo F, Nihei H, Nakamura Y. Association between single-nucleotide polymorphisms in selectin genes immunoglobulin A nephropathy. Am J Hum Genet. 2002;70:781–786. doi: 10.1086/339077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiryluk K, Novak J, Gharavi AG. Pathogenesis of immunoglobulin A nephropathy: recent insight from genetic studies. Annu Rev Med. 2013;64:339–356. doi: 10.1146/annurev-med-041811-142014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okazaki K, Suzuki Y, Otsuji M, Suzuki H, Kihara M, Kajiyama T, Hashimoto A, Nishimura H, Brown R, Hall S, Novak J, Izui S, Hirose S, Tomino Y. Development of a model of early-onset IgA nephropathy. J Am Soc Nephrol. 2012;23:1364–74. doi: 10.1681/ASN.2011121160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allen AC, Bailey EM, Brenchley PE, Buck KS, Barratt J, Feehally J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: observations in three patients. Kidney Int. 2001;60:969–973. doi: 10.1046/j.1523-1755.2001.060003969.x. [DOI] [PubMed] [Google Scholar]
- 30.Hiki Y, Odani H, Takahashi M, Yasuda Y, Nishimoto A, Iwase H, Shinzato T, Kobayashi Y, Maeda K. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int. 2001;59:1077–1085. doi: 10.1046/j.1523-1755.2001.0590031077.x. [DOI] [PubMed] [Google Scholar]
- 31.Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest. 1999;104:73–81. doi: 10.1172/JCI5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Novak J, Tomana M, Matousovic K, Brown R, Hall S, Novak L, Julian BA, Wyatt RJ, Mestecky J. IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int. 2005;67:504–13. doi: 10.1111/j.1523-1755.2005.67107.x. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki H, Fan R, Zhang Z, Brown R, Hall S, Julian BA, Chatham WW, Suzuki Y, Wyatt RJ, Moldoveanu Z, Lee JY, Robinson J, Tomana M, Tomino Y, Mestecky J, Novak J. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest. 2009;119:1668–77. doi: 10.1172/JCI38468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hashimoto A, Suzuki Y, Suzuki H, Ohsawa I, Brown R, Hall S, Tanaka Y, Novak J, Ohi H, Tomino Y. Determination of severity of murine IgA nephropathy by glomerular complement activation by aberrantly glycosylated IgA and immune complexes. Am J Pathol. 2012;181:1338–47. doi: 10.1016/j.ajpath.2012.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moldoveanu Z, Wyatt RJ, Lee JY, Tomana M, Julian BA, Mestecky J, Huang WQ, Anreddy SR, Hall S, Hastings MC, Lau KK, Cook WJ, Novak J. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. 2007;71:1148–54. doi: 10.1038/sj.ki.5002185. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki H, Moldoveanu Z, Hall S, Brown R, Vu HL, Novak L, Julian BA, Tomana M, Wyatt RJ, Edberg JC, Alarcón GS, Kimberly RP, Tomino Y, Mestecky J, Novak J. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J Clin Invest. 2008;118:629–3. doi: 10.1172/JCI33189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishie T, Miyaishi O, Azuma H, Kameyama A, Naruse C, Hashimoto N, Yokoyama H, Narimatsu H, Wada T, Asano M. Development of immunoglobulin A nephropathy-like disease in β-1,4-galactosyltransferase-I-deficient mice. Am J Pathol. 2007;170:447–456. doi: 10.2353/ajpath.2007.060559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otani M, Nakata J, Kihara M, Leroy V, Moll S, Wada Y, Izui S. O-glycosylated IgA rheumatoid factor induces IgA deposits and glomerulonephritis. J Am Soc Nephrol. 2012;23:438–446. doi: 10.1681/ASN.2011070701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kihara M, Ito K, Nakata J, Otani M, Tran NL, Morito N, Takahashi S, Wada Y, Izui S. O-linked glycosylation determines the nephritogenic potential of IgA rheumatoid factor. J Am Soc Nephrol. 2014;25:1282–1290. doi: 10.1681/ASN.2013070771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moldoveanu Z, Suzuki H, Satake K, Suzuki Y, Novak L, Huang ZQ, Winstead CJ, O'Quinn DB, Julian BA, Weaver CT, Mestecky J, Tomino Y, Novak J. IgA Nephropathy: A murine model that displays typical IgAN pathology after passive administration of immune complexes. J Am Soc Nephrol. 2012;23:519A. [Google Scholar]
- 41.Smith AC, Molyneux K, Feehally J, Barratt J. O-glycosylation of serum IgA1 antibodies against mucosal and systemic antigens in IgA nephropathy. J Am Soc Nephrol. 2006;17:3520–8. doi: 10.1681/ASN.2006060658. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki H, Raska M, Yamada K, Moldoveanu Z, Julian BA, Wyatt RJ, Tomino Y, Gharavi AG, Novak J. Cytokines alter IgA1 O-glycosylation by dysregulating C1GalT1 and ST6GalNAc-II enzymes. J Biol Chem. 2014;289:5330–9. doi: 10.1074/jbc.M113.512277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki H, Suzuki Y, Narita I, Aizawa M, Kihara M, Yamanaka T, Kanou T, Tsukaguchi H, Novak J, Horikoshi S, Tomino Y. Toll-like receptor 9 affects severity of IgA nephropathy. J Am Soc Nephrol. 2008;19:2384–2395. doi: 10.1681/ASN.2007121311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sato D, Suzuki Y, Kano T, Suzuki H, Matsuoka J, Yokoi H, Horikoshi S, Ikeda K, Tomino Y. Tonsillar TLR9 expression and efficacy of tonsillectomy with steroid pulse therapy in IgA nephropathy patients. Nephrol Dial Transplant. 2012;27:1090–1097. doi: 10.1093/ndt/gfr403. [DOI] [PubMed] [Google Scholar]