

Abstract
In the present study, in continuation of our previous experiment in order to investigate the mode of action (MOA) of ethyl tertiary-butyl ether (ETBE) hepatotumorigenicity in rats, we aimed to examine alterations in cell proliferation, that are induced by short-term administration of ETBE. F344 rats were administered ETBE at doses of 0, and 1,000 mg/kg body weight twice a day by gavage for 3, 10, 17 and 28 days. It was found that the previously observed significant increase of P450 total content and hydroxyl radical levels after 7 days of ETBE administration, and 8-OHdG formation at day 14, accompanied by accumulation of CYP2B1/2B2, CYP3A1/3A2, CYP2C6, CYP2E1 and CYP1A1 and downregulation of DNA oxoguanine glycosylase 1, was preceded by induction of cell proliferation at day 3. Furthermore, we observed an increase in regenerative cell proliferation as a result of ETBE treatment at day 28, followed by induction of cell cycle arrest and apoptosis by day 14. These results indicated that short-term administration of ETBE led to a significant early increase in cell proliferation activity associated with induction of oxidative stress, and to a regenerative cell proliferation as an adaptive response, which could contribute to the hepatotumorigenicity of ETBE in rats.
Keywords: ethyl tertiary-butyl ether, cell proliferation, oxidative stress, mode of action
Introduction
Ethyl tertiary-butyl ether (ETBE, CAS RN 637-92-3) is a well-known chemical and gasoline oxygenate synthesized from bioethanol and isobutene and used as a gasoline blending component to decrease exhaust emissions such as carbon monoxide, unburned hydrocarbons, polycyclic aromatics, oxides of nitrogen and particulate carbon1. It is accepted that, nevertheless, humans are at risk of exposure to oxygenates not only by inhalation while fueling automobiles but also orally when drinking contaminated water1,2,3.
ETBE has been shown to be not genotoxic1, 2; however, recently it has been reported that ETBE administered to male F344 rats by inhalation at a dose of 5,000 ppm for 2 years induced the development of liver preneoplastic lesions (eosinophilic and basophilic foci) and hepatocellular adenomas4, 5. In other studies, the promoting effect of ETBE on rat hepatocarcinogenesis, when administered by gavage at a dose of 1,000 mg/kg body weight/day, was detected in a multiorgan carcinogenesis bioassay and in an initiation/promotion carcinogenicity assay using N-ethyl-N-(2-hydroxyethyl)nitrosamine (EHEN) as an initiator6, 7.
In our previous study, the possible mode of action (MOA) for ETBE hepatotumorigenicity in rats was investigated after 7 and 14 days of ETBE application by gavage at a dose of 1,000 mg/kg body weight twice a day to F344 rats8. The results indicated that the ETBE MOA in rats could be related to induction of CYP2B1/2B2, CYP3A1/3A2, CYP2C6, CYP2E1 and CYP1A1 isoenzymes of cytochrome P450 and as a result, an increase in 8-OHdG formation, subsequent cell cycle arrest, apoptosis, predominantly via activation of CAR and PXR nuclear receptors by a mechanism similar to that of non-genotoxic carcinogen phenobarbital (PB), and differentially by activation of peroxisome proliferating receptors (PPARs)8. However, the distinct alterations in cell proliferation, that occur during short-term ETBE administration are still unclear, but they are, however, very important for analysis of the ETBE hepatotumorigenicity MOA.
To further elucidate the mechanisms of ETBE hepatotumorigenicity in rats, in the present study we aimed to focus on alterations in liver cell proliferation during short-term time-dependent application of high-dose ETBE to F344 rats.
Materials and Methods
Chemicals
All reagents were from Wako Pure Chemicals Industries (Osaka, Japan) or Sigma (St. Louis, MO, USA). ETBE was manufactured by Nippon Refine Co., Ltd. (Ogaki, Gifu, Japan), and had the following properties: lot no., L-506251; appearance, colorless transparent liquid; vapor pressure, 17 kPa (25°C); boiling point, 70°C; solubility, slightly soluble in water (1.2 g/100 g, 20°C); purity, >99% (measured by Toray Research Center Inc., Tokyo, Japan). The chemical structure of ETBE is shown in Fig. 1. The stability was determined by gas chromatography (Agilent HP-5890A Agilent Technologies, Santa Clara, CA, USA) before the beginning and at the end of the treatment period. A single batch of ETBE was used in this study. There were no differences between the results obtained at these two time points, indicating that the test substance was stable throughout the examination.
Fig. 1.
ETBE chemical structure.
Animals and treatment
All experimental procedures were conducted under the guidelines set by the National Institutes of Health, Public Health Service Policy on the Humane Use and Care of Laboratory Animals. Five-week-old male Fisher F344/DuCrlCrlj rats (Charles River Laboratories Japan Inc., Yokohama, Japan) were quarantined for 1 week before the start of the experiment. The rats were housed in an animal facility maintained on a 12 h (8:00–20:00) light/dark cycle, at a constant temperature of 22 ± 3°C and a relative humidity of 55 ± 15% and were given free access to tap water and food (Oriental MF powder diet, Oriental Yeast Co., Ltd., Tokyo, Japan). Rat body weights were measured on days 2, 7, 9, 14, 16, 21 and 27, and on days 3, 10, 17 and 28 after the fasting overnight. Water and food intakes were recorded once weekly.
Experimental design
Before the start of the experiment, 36 male 6-week-old rats were randomized into 2 groups (18 rats each). ETBE was administered by intragastric gavage (i.g.) at concentrations of 0 (olive oil control), and 1,000 mg/kg body weight in olive oil twice a day (1,000 mg/kg b.w. ×2/day) with a 6-hour interval for 3, 10, 17 and 28 days. The dose level and route of ETBE exposure were the same as those applied in our previous short-term MOA study8, which demonstrated significant oxidative stress induction and related molecular changes in the rat liver. Furthermore, in a preliminary study, F344 rats well tolerated 1,000 mg/kg b.w. ETBE was administered by gavage twice a day with a 6-hour interval, and no abnormalities were found in their general conditions.
One hour prior to euthanasia, 5-bromo-2′-deoxyuridine (BrdU, Lot No.: SLBF0632V; Sigma-Aldrich Chemical Co., St.Louis, MO, USA) solution was i.p. injected at a dose of 100 mg/kg body weight. Two hours after BrdU injection, rats were euthanized under isoflurane, and a systemic macroscopic pathological examination was performed. Liver, kidneys, thyroids, urinary bladders and small intestines were excised, and the organ weights of the liver and kidneys were measured.
Histopathology
Separate portions of the liver (left lateral lobe, median lobe, and right lateral lobe, caudal part) and small intestine were fixed in 10% buffered formalin and routinely embedded in paraffin, sectioned, stained with hematoxylin and eosin solution, and examined histopathologically under a light microscope.
Immunohistochemistry
Livers from all rats were examined by BrdU immunohistochemistry. Serial sections (3 μm) were cut from paraffin-embedded liver tissues and mounted on poly-l-lysine-coated slides. Established procedures for immunohistochemical staining with the avidin-biotin-peroxidase complex (ABC) method were used. Paraffin sections were deparaffinized and rehydrated through graded alcohols. Endogenous peroxidase activity was blocked with 0.3% H2O2 in distilled water for 5 min, and then antigen retrieval was performed by microwaving at 98°C for 20 min in 0.01 M citrate buffer (pH 6.0). After blocking nonspecific binding with normal horse serum at 37°C for 30 min, sections were incubated with mouse monoclonal anti-BrdU antibody (Dako Japan, Kyoto, Japan) at 1:500 dilution overnight at 4°C. Immunoreactivity was detected using a Vectastain Elite ABC Kit (PK-6102; Vector Laboratories, Burlingame, CA, USA) and 3,3′-diaminobenzidine hydrochloride (Sigma Chemical Co., St Louis, MO, USA) followed by counterstaining with Mayer’s hematoxylin. For BrdU immunohistochemistry, small intestine formalin-fixed paraffin-embedded sections were used as positive controls. A negative control was also included with each staining procedure by omitting the primary antibody.
At least 3,000 hepatocyte nuclei were counted in each liver; labeling indices were calculated as the percentage of cells positive for BrdU.
Statistical analysis
The significance of differences for each parameter (excluding general conditions) was analyzed and evaluated at P<0.05. Statistical comparisons between control and ETBE groups for numerical data were assessed using the F test. If homogeneous, the data were analyzed with the Student’s t-test (two-sided), and if not, they were analyzed with Welch’s test. The significance of intergroup differences in incidences of findings from gross pathology was analyzed using the Fisher’s exact probability test (two-sided) or the Wilcoxon test.
Results
Survival and general observations
No animals died during the experiment, and no abnormalities of general condition were found in any animal of the control or ETBE group, which received a dose two times higher than that resulting in a promoting effect on rat liver carcinogenesis7. The mean final body weights in the ETBE group were comparable to those of the control at every time point (Table 1). No significant differences in food and water intakes were observed between ETBE-treated and control rats during the treatment period (data not shown).
Table 1. Relative Liver and Kidney Weights and Immunohistochemical BrdU Labeling Indices in Hepatocytes.
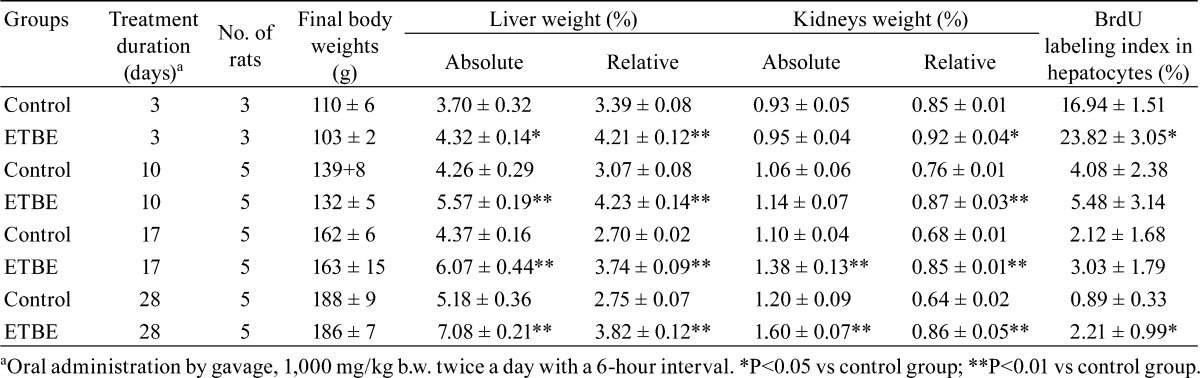
The mean absolute and relative liver weights in rats given ETBE were significantly higher as compared with those of controls at all examined time points (Table 1). The observed increase in liver weights was similar to that of previously reported studies when ETBE was administered by inhalation or by gavage1, 5,6,7, 9. In addition, significant increases in the mean relative kidney weights at all experimental time points and absolute kidney weights at days 17 and 28 in rats given ETBE as compared with control rats were found (Table 1). Kidneys were not examined microscopically as this was not in the purpose of this study. However, in our previous 13-week oral treatment study, deposition of eosinophilic bodies, indicating the presence of α 2u-globulin, was observed and was considered severe (unpublished data).
Macroscopic and histopathological changes of the liver
No treatment-related macroscopic changes were found in the ETBE and control rats at all time points except in the liver. The livers of the rats in the ETBE group were swollen at all experimental time points. Similar to our previous findings8, at days 3 (1 of 3 rats (33%)), 10 (3 of 5 rats (60%)), 17 (5 of 5 rats (100%)) and 28 (5 of 5 rats (100%)) slight and/or moderate centrilobular hypertrophy was detected in the liver in ETBE treated rats (Fig. 2A-D). The shift from slight to moderate liver hypertrophy was observed with the increase in time of ETBE treatment from day 3 to day 28. An increase in the number of apoptotic cells was found by histological examination in the livers of rats administered ETBE at day 17 (data not shown). Furthermore, higher numbers of mitotic figures in the liver of ETBE-treated rats were detected as compared with the control animals at days 3 and 28 (data not shown).
Fig. 2.

Histopathological examination (A–D) and immunohistochemical analysis of BrdU (E–H) in the livers of rats treated with ETBE for 3 (control (A, E) and ETBE-treated rats (B, F)) and 28 (control (C, G) and ETBE-treated rats (D, H)) days. Note the centrilobular hypertrophy of hepatocytes and increases of BrdU-labeled hepatocyte nuclei in ETBE-treated rat livers at days 3 and 28.
Alterations of cell proliferation (BrdU) in the rat liver
Significant increases in the BrdU-positive cell ratios for hepatocytes were observed in ETBE-treated rats, first at day 3 (1.5-folds increase) and then, at day 28 (2.5-fold increase) as compared with the control rats (Table 1 and Fig. 2E–H). The values at days 10 and 17 were slightly higher than those of the control group. The control levels of the rat liver BrdU indices detected in the present study at each time point fell into the range of historical background values for data obtained in our laboratory10,11,12. At the start of the experiment on day 3, the BrdU index levels in young rats of both the control and ETBE-treated groups were much higher than those observed in rats at days 10, 17 and 28 (Table 1).
Discussion
The present study demonstrated a range of changes in rat liver cell proliferation dependent on the period of ETBE exposure. Significant enhancements of the BrdU labeling index in rat hepatocytes at 3 and 28 days after starting ETBE application were evident.
Previously, we observed a trend towards an increase in the PCNA-positive cell ratio for hepatocytes in rats treated with 1,000 mg/kg b.w. ×2 /day ETBE at day 7 (1.3-fold) followed by significant inhibition (2.0-fold) at day 14 after starting the treatment8. The changes in the PCNA and BrdU labeling indices of the liver at day 3 (1.5-fold) in our previous8 and present studies, respectively, appeared to be comparable. If we combine the data of both MOA studies, cell proliferation in the ETBE treatment group changed according to the following scheme: a significant increase at day 3 was followed by decline at days 7 and 10 (values were slightly higher than in the control) and significant inhibition (cell cycle arrest) at day 14. Thereafter, we observed its recovery at day 17 (values were slightly higher than in the control) and a significant increase at day 28 (regenerative proliferation).
The short-term increase in cell proliferation observed here in ETBE-treated rat livers indicates that the peak of 8-OHdG generation, followed by cell cycle arrest and induction of apoptosis, is likely to occur at day 14, as we reported previously5, 8. Early elevation of cell proliferation as an acute response to treatment with a non-genotoxic chemical was previously demonstrated with PB10, 13, 14. Thus, PB administered to F344 rats at a dose of 500 ppm was shown to induce time-dependent elevation of cell proliferation starting at day 1 or 2 and reaching its peak at day 6, followed by a decrease at day 8, which was associated with induction of 8-OHdG formation and apoptosis10. According to the results of the present investigation and our previous ETBE tumorigenicity and MOA studies5, 8, the MOA of PB appeared to be similar to that of ETBE except for the activation of PPARs. The increase of cell proliferation by PB was shown to be due to induction of hydroxyl radicals and, as a result, 8-OHdG generation mostly by cytochrome P450 isoenzymes CYP2B1/2 and CYP3A2, which are downstream of CAR and PXR nuclear receptors. Our previous results indicated that ETBE induced not only CYP2B1/2 and CYP3A1/2, but also CYP2E1 (an isoenzyme induced by ethanol), CYP1A1 and CYP4A2, which is dependent on the activation of PPARs8. Furthermore, we observed induction of xenobiotic metabolism enzymes and enzymes involved in fatty acid metabolism in mitochondria including glutathione S-transferase (GST) alpha 1 (GSTA1), alpha 3 (GSTA3) and alpha 5 (GSTA5); GST mu 5 (GSTM5); glutathione peroxidase 1 (GPX1); epoxide hydrolase 1, microsomal (EPHX1); carboxylesterase 3 (CES3); cytochrome P450 oxidoreductase (POR); UGT2B1, UGT2B5 and UDP-glucose 6-dehydrogenase (UGDH); acyl-CoA oxidase 1, palmitoyl (ACOX1); enoyl CoA hydratase 1, peroxisomal (ECH1); solute carrier family 27, member 2 (SLC27A2); solute carrier family 27, member 5 (SLC27A5); PPARα; and PPARγ. Moreover, ETBE was found to inhibit Ugt1a1, Sult1d1 and 8-OHdG repair enzyme Ogg1 expression.
The significant increase of 8-OHdG formation in the DNA of hepatocytes, cell cycle arrest and significant increase in the number of apoptotic cells localized in the centrilobular region previously detected8 at day 14 were found to be followed by increase of cell proliferation at day 28 in the present study. Regenerative cell proliferation after day 14, which is known to occur as an adaptive response after induction of apoptosis, is strongly suggested by the present results.
In a 2-year carcinogenicity test in F344 rats, ETBE inhalation at dose of 5,000 ppm (equal to 4,222 mg/kg/day) resulted in induction of preneoplastic lesions (eosinophilic and basophilic foci) and liver tumors (hepatocellular adenomas; 9 of 50 rats (18%))5. Furthermore, PB administered to male B6C3F1 mice and male F344 rats at doses of 100 and 500 mg/kg diet has been also suggested to promote focal hepatic lesion growth both by increasing DNA synthesis and cell proliferation and by decreasing the rate of apoptosis15. Thus, the mechanism of ETBE tumorigenicity in the rat liver could be bound to its ability to induce cell proliferation.
Other than ETBE, non-genotoxic chemicals inducing oxidative stress, such as peroxisome proliferators, were also found to be able cause early induction of cell proliferation in the rat liver after short-term administration and in primary rat hepatocytes, and this was linked to bioactivation of TNF-alpha, which requires p38 MAP kinase activity and other processes, i.e., interference of Gap junctions coupled with a proliferative stimulus16,17,18. Furthermore, in support of the present results, recent studies examining the effects of a compound closely related to ETBE, methyl tert-butyl ether (MTBE), administered by inhalation in toxicity and carcinogenicity tests found that cell proliferation activity of hepatocytes was significantly increased at days 3–5, but returned to normal levels at days 21–28, which was suggested to be the adaptive response of the liver10, 19, 20. Time-dependent treatment with ETBE was continued for 4 weeks, and combination of the present results with those of our previous experiment on the MOA of ETBE, gave us an opportunity to reveal the correlative changes in cell proliferation, oxidative stress, apoptosis, DNA damage and DNA repair, resulting in formation of preneoplastic lesions and hepatocellular adenomas, which occur in the rat liver during continuous ETBE exposure (Fig. 3).
Fig. 3.

Graphically illustrated changes induced by short-term ETBE treatment in the rat liver.
In conclusion, in the present study, administration of ETBE to rats was found to result in early induction of cell proliferation activity at day 3 and regenerative cell proliferation at day 28. This study proved that the MOA of ETBE hepatotumorigenicity basically is likely to be the same as the MOA of PB, in terms of short-term induction of cell proliferation, which strongly contributes to the hepatotumorigenicity of ETBE in the rat liver, and the mechanisms of induction of cell proliferation are likely to be due to induction of oxidative stress and DNA modifications, which are dependent on activation of CAR, PXR and PPARs.
Declaration of Conflicting Interests: We have no conflicts of interest to be declared.
Acknowledgments
We thank Keiko Sakata and Yuko Hisabayashi for their technical assistance. This research was supported in part by Grants-in-Aid for Chemical Risk Research from the Ministry of Health, Labour and Welfare (MHLW) of Japan and by Petroleum Industry Technology and Research Institute, Inc., Japan.
References
- 1.McGregor D. Ethyl tertiary-butyl ether: a toxicological review. Crit Rev Toxicol. 37: 287–312 2007. [DOI] [PubMed] [Google Scholar]
- 2.ACGIH Ethyl tert-butyl ether. American Conference of Governmental Industrial Hygienists. 2012.
- 3.Ahmed FE. Toxicology and human health effects following exposure to oxygenated or reformulated gasoline. Toxicol Lett. 123: 89–113 2001. [DOI] [PubMed] [Google Scholar]
- 4.Nagano K, Nishizawa T, Yamazaki K, Noguchi T, Hagiwara A, Nishimaki F, Iida S, Komiya K, and Fukushima S. Hepatotumorigenicity of ethyl tertiary-butyl ether (ETBE) by inhalation exposure but not oral administration in F344 rats. The Toxicologist. Washington DC. 2011. [Google Scholar]
- 5.Saito A, Sasaki T, Kasai T, Katagiri T, Nishizawa T, Noguchi T, Aiso S, Nagano K, and Fukushima S. Hepatotumorigenicity of ethyl tertiary-butyl ether with 2-year inhalation exposure in F344 rats. Arch Toxicol. 87: 905–914 2013. [DOI] [PubMed] [Google Scholar]
- 6.Hagiwara A. Threshold level for tumor promotion by ethyl tertiary-butyl ether (ETBE) of hepatic carcinogenesis in rats. Society of Toxicologic Pathology. Mechanisms of Toxicity. Annual Symposium. 2012.
- 7.Hagiwara A, Doi Y, Imai N, Nakashima H, Ono T, Kawabe M, Furukawa F, Tamano S, Nagano K, and Fukushima S. Medium-term multi-organ carcinogenesis bioassay of ethyl tertiary-butyl ether in rats. Toxicology. 289: 160–166 2011. [DOI] [PubMed] [Google Scholar]
- 8.Kakehashi A, Hagiwara A, Imai N, Nagano K, Nishimaki F, Banton M, Wei M, Fukushima S, and Wanibuchi H. Mode of action of ethyl tertiary-butyl ether hepatotumorigenicity in the rat: evidence for a role of oxidative stress via activation of CAR, PXR and PPAR signaling pathways. Toxicol Appl Pharmacol. 273: 390–400 2013. [DOI] [PubMed] [Google Scholar]
- 9.Medinsky MA, Wolf DC, Cattley RC, Wong B, Janszen DB, Farris GM, Wright GA, and Bond JA. Effects of a thirteen-week inhalation exposure to ethyl tertiary butyl ether on fischer-344 rats and CD-1 mice. Toxicol Sci. 51: 108–118 1999. [DOI] [PubMed] [Google Scholar]
- 10.Kinoshita A, Wanibuchi H, Imaoka S, Ogawa M, Masuda C, Morimura K, Funae Y, and Fukushima S. Formation of 8-hydroxydeoxyguanosine and cell-cycle arrest in the rat liver via generation of oxidative stress by phenobarbital: association with expression profiles of p21(WAF1/Cip1), cyclin D1 and Ogg1. Carcinogenesis. 23: 341–349 2002. [DOI] [PubMed] [Google Scholar]
- 11.Kinoshita A, Wanibuchi H, Wei M, Yunoki T, and Fukushima S. Elevation of 8-hydroxydeoxyguanosine and cell proliferation via generation of oxidative stress by organic arsenicals contributes to their carcinogenicity in the rat liver and bladder. Toxicol Appl Pharmacol. 221: 295–305 2007. [DOI] [PubMed] [Google Scholar]
- 12.Yano Y, Yano T, Kinoshita A, Matoba A, Hasuma T, Wanibuchi H, Morimura K, Otani S, and Fukushima S. Sensitive quantitative assay for point mutations in the rat H-ras gene based on single nucleotide primer extension. Exp Ther Med. 1: 657–661 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones HB, and Clarke NA. Assessment of the influence of subacute phenobarbitone administration on multi-tissue cell proliferation in the rat using bromodeoxyuridine immunocytochemistry. Arch Toxicol. 67: 622–628 1993. [DOI] [PubMed] [Google Scholar]
- 14.Murkofsky RL, Glover SE, Miller RT, Popp JA, and Cattley RC. Effect of regeneration and hyperplasia on levels of DNA base oxidation in rat liver. Cancer Lett. 70: 51–56 1993. [DOI] [PubMed] [Google Scholar]
- 15.Kolaja KL, Stevenson DE, Walborg EF, Jr , and Klaunig JE. Dose dependence of phenobarbital promotion of preneoplastic hepatic lesions in F344 rats and B6C3F1 mice: effects on DNA synthesis and apoptosis. Carcinogenesis. 17: 947–954 1996. [DOI] [PubMed] [Google Scholar]
- 16.Cosulich S, James N, and Roberts R. Role of MAP kinase signalling pathways in the mode of action of peroxisome proliferators. Carcinogenesis. 21: 579–584 2000. [DOI] [PubMed] [Google Scholar]
- 17.Eldridge SR, Tilbury LF, Goldsworthy TL, and Butterworth BE. Measurement of chemically induced cell proliferation in rodent liver and kidney: a comparison of 5-bromo-2′-deoxyuridine and [3H]thymidine administered by injection or osmotic pump. Carcinogenesis. 11: 2245–2251 1990. [DOI] [PubMed] [Google Scholar]
- 18.Mally A, and Chipman JK. Non-genotoxic carcinogens: early effects on gap junctions, cell proliferation and apoptosis in the rat. Toxicology. 180: 233–248 2002. [DOI] [PubMed] [Google Scholar]
- 19.Bird MG, Burleigh-Flayer HD, Chun JS, Douglas JF, Kneiss JJ, and Andrews LS. Oncogenicity studies of inhaled methyl tertiary-butyl ether (MTBE) in CD-1 mice and F-344 rats. J Appl Toxicol. 17(Suppl 1): S45–S55 1997. [DOI] [PubMed] [Google Scholar]
- 20.Ryoo HD, and Bergmann A. The role of apoptosis-induced proliferation for regeneration and cancer. Cold Spring Harb Perspect Biol. 4: a008797 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]