

Abstract
To solve the problem of synthesized magnetic nanoparticles in cancer therapy, a new drug delivery system synthesized from bacteria was used to load cytosine arabinoside (Ara-C). Genipin (GP) and poly-l-glutamic acid (PLGA) were selected as dual cross-linkers. The preparation and characterization of Ara-C-loaded GP-PLGA-modified bacterial magnetosomes (BMs) (ABMs-P), as well as their in vitro antitumor effects, were all investigated. Transmission electron micrographs (TEM) and Fourier transform infrared (FTIR) spectroscopy suggested that Ara-C could be bound to the membrane of BMs modified by GP-PLGA. The diameters of the BMs and ABMs-P were 42.0±8.6 nm and 74.9±8.2 nm, respectively. The zeta potential revealed that the nanoparticles were stable. Moreover, this system exhibited optimal drug-loading properties and long-term release behavior. The optimal encapsulation efficiency and drug-loading were 64.1%±6.6% and 38.9%±2.4%, respectively, and ABMs-P could effectively release 90% Ara-C within 40 days, without the release of an initial burst. In addition, in vitro antitumor experiments elucidated that ABMs-P is cytotoxic to HL-60 cell lines, with an inhibition rate of 95%. The method of coupling drugs on BMs using dual cross-linkers is effective, and our results reveal that this new system has potential applications for drug delivery in the future.
Keywords: magnetosomes, dual cross-linkers, nanoparticle, drug delivery
Introduction
Cytosine arabinoside (Ara-C) is a pyrimidine antimetabolic chemotherapeutic agent used to treat acute myelogenous leukemia.1 However, high doses of Ara-C are cytotoxic and cause serious side effects, which especially include hepatotoxicity and neurotoxicity,2–4 thus greatly limiting its application. To solve these problems, drug delivery systems have been used. In recent years, magnetic drug targeting has been one of the most active fields in cancer therapy. Magnetic targeted drug delivery systems and magnetic nanoparticles are widely studied,5–10 but most research has addressed the synthesis of magnetic nanoparticles by pure chemical or physical methods. The application of these magnetic nanoparticles has been hampered by various problems, such as dispersion difficulties, poor control of shape or size, and low drug-loading capability. Recently, new natural magnetic nanoparticle carriers called magnetosomes, derived from bacteria, have aroused interest.
Bacterial magnetosomes (BMs) are membrane-bound nanocrystals with a magnetic iron oxide or iron sulfide core, synthesized by magnetotactic bacteria.11,12 The particle size is generally distributed between 25 and 55 nm. As the nanocarrier of a targeting drug, BMs have many advantages in medical applications, including narrow size distribution, superparamagnetism, and excellent biocompatibility, compared with artificial magnetic nanoparticles.13,14 Because BMs possess a natural membrane consisting of lipids and abundant amino groups, a previous study has found that BMs can be used as drug carriers after introducing coupling reagents, such as glutaraldehyde.15 Moreover, in order to improve the drug-loading ratio, a novel cross-linking method has also been investigated, through the surface modification of BMs by poly-l-glutamic acid (PLGA).16
Because of the potential toxicity of glutaraldehyde, we have successfully prepared a BM drug delivery system that relies on genipin (GP), instead of glutaraldehyde, for cross-linking.17,18 GP is isolated from the fruit of the gardenia plant and has conspicuous therapeutic effects that are antitumor, antithrombus, anti-inflammation, and antidiabetes. In considering the effect of PLGA in this study, we tried to combine GP and PLGA as dual cross-linkers to increase the binding sites of Ara-C. The preparation and characterization as well as in vitro antitumor effects of Ara-C-loaded GP-PLGA-modified BMs (ABMs-P) were all investigated in this study.
Materials and methods
Materials
Magnetospirillum magneticum strain AMB-1 was purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA); Ara-C was purchased from Sunray Pharmaceutical Co., Ltd (Suzhou, People’s Republic of China); GP was obtained from Zhixin Biotechnology Company (Fuzhou, People’s Republic of China); and PLGA (3,000–15,000 Da) was provided by Sigma-Aldrich Corp (St Louis, MO, USA). All other chemicals were purchased from Sinopharm Chemical Reagent Co., Ltd. (Beijing, People’s Republic of China), unless otherwise specified. The human acute promyelocytic leukemia cell line, HL-60, was from the China Academy Typical Culture Preservation Committee Cell Library (Shanghai, People’s Republic of China). The cell culture medium was composed of Iscove’s Modified Dulbecco’s Medium (IMDM) (Gibco; Life Technologies Corp, Carlsbad, CA, USA) supplemented with 20% fetal bovine serum (Gibco; Life Technologies Corp). All cells were incubated at 37°C in humidified air with 5% CO2.
Preparation of the ABMs-P
Figure 1 shows the procedure for preparing ABMs-P. Firstly, AMB-1 cells were collected by centrifugation (10,000 rpm, 4°C, 10 minutes), then were suspended in phosphate-buffered saline (PBS) (pH 7.4) and disrupted by an ultrasonic cell crusher (300 W; on for 3 seconds with an interval of 5 seconds, repeated 100 times). The BMs were collected by a magnet bound to the bottom of the tubes, and cell debris was removed. The BM sediments were collected after being resuspended in PBS (pH 7.4) and treated with ultrasonic cleaning (40 W, on for 3 seconds with an interval of 5 seconds, repeated 50 times); this process was repeated five to ten times. The purification level could be evaluated using the following formula:
| (1) |
where A is the amount of protein (g/L), OD280 is, and OD260C is. The value was measured by ultraviolet-visible (UV-vis) spectrophotometer.
Figure 1.

Synthesis of ABMs-P.
Abbreviations: ABMs-P, cytosine arabinoside-loaded genipin-poly-l-glutamic acid-modified bacterial magnetosomes; Ara-C, cytosine arabinoside; BMs, bacterial magnetosomes; BMs-P, genipin-poly-L-glutamic acid-modified bacterial magnetosomes; GP, genipin; PLGA, poly-l-glutamic acid.
Secondly, 0.1 mg purified BMs were suspended in PBS (pH 7.4). The same amount of PLGA solution (1 mg/mL) was added to the BM suspension and then was treated with ultrasonic bathing (50 W, 5 minutes). Following the addition of GP solution (the concentration of GP in the mixture was 0.05%, 0.1%, 0.5%, and 0.8% respectively), the mixture was distributed by ultrasonic bathing (50 W, on for 1 minute with an interval of 5 minutes, repeated ten times). Then the mixture was placed in an incubator shaker at 60 rpm at 37°C for 12, 24, 48, and 72 hours to react completely.
Finally, the PLGA-modified BMs were then resus-pended in PBS (pH 7.4), with the same addition of Ara-C solution (1 mg/mL) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) solution (0.2 mg/mL), through sonication (50 W, on for 1 minute with an interval of 5 minutes, repeated ten times). The mixture was then incubated in a shaker at 60 rpm at 37°C for 12, 24, 48, and 72 hours. The resultant ABMs-P were collected and sterilized before use.
Morphology of the ABMs-P
The ABMs-P were washed by PBS and distilled water until no colors were observed, and then they were suspended in distilled water, using an ultrasonic bath. The BMs and the previous ABMs-P supernatant (15–20 μL) were dropped on cropper grids and examined by TEM. The zeta potential was measured by a Zetasizer (ZEN3600; Malvern Instruments Ltd, Malvern, UK). All assays were performed in triplicate. DTS version 5.00 for Windows (Malvern Instruments Ltd) was used for statistical analysis.
Examination of the ABMs-P by Fourier transform infrared (FTIR) spectroscopy
FTIR spectra of the Ara-C, ABMs-P, and BM samples were pressed using KBr, and the pellets were recorded with a FTIR spectrometer, in the wavenumber range of 4,000–400 cm−1 (FTIR 8400S; Shimadzu Corp, Kyoto, Japan).
The encapsulation efficiency and drug-loading of ABMs-P
To eliminate superfluous Ara-C, GP, and PLGA, the ABMs-P were washed with PBS, in conjunction with magnetic absorption, until the supernatant solution revealed no color. The ABMs-P were evenly distributed in 5 mL PBS by ultrasonic bathing (50 W, 5 minutes). Then, 200 μL ABMs-P suspensions were removed and dissolved in 4.8 mL membrane-breaking liquid composed of 1.2 mL 36%–38% hydrochloric acid and 3.6 mL 70% ethanol solution, for 1 hour. The absorbance was calculated at the maximum absorption wave (283.5 nm), with the blank group treated with membrane-breaking liquid. The amount of Ara-C coupled to the BMs was represented, with the standard curve in 1:3 hydrochloric acid and ethanol solution, through the equation:
| (2) |
where the value y is the concentration of Ara-C (μg/mL) and the value x is the absorbance of Ara-C in membrane-breaking liquid at 283.5 nm.
In accordance with China Pharmacopoeia (2010 version),19 drug-loading and encapsulation efficiency were calculated by the following equation:
| (3) |
| (4) |
where W1 is the amount of Ara-C loaded on ABMs-P, W2 is the amount of ABMs-P, and W3 is the whole amount of Ara-C in the solution.
In vitro Ara-C-release studies
The Ara-C-release studies were tested in tubes placed in an incubator shaker at 60 rpm and 37°C. The Ara-C concentration in the supernatant of each tube was evaluated by UV-vis spectrometer after incubation for 0.5, 1, 12, 24, 36, or 48 hours. And then the same amount of PBS (2 mL) was added into the tubes. The concentration of Ara-C in the PBS was calculated through the following standard curve:
| (5) |
where the value y is the amount of Ara-C (μg/mL) and value x is the absorption of Ara-C in PBS at 271.4 nm. Thus, in vitro release studies were conducted. All assays were performed in triplicate.
Cell-Counting Kit-8 (CCK-8) assays
CCK-8 assays were used to determine the cytotoxicity response to different drug concentrations and incubation times. The BMs and purified ABMs-P were harvested, as described above. The samples of Ara-C, BMs, and ABMs-P were sterilized by Co60 irradiation (7.5 kGy) before use. The Ara-C content with adequate IMDM medium for control was 0.2, 2, 10, and 50 μg/mL, respectively. BMs and ABMs-P were performed CCK-8 assay. The effects of drug dose and incubation time were investigated, respectively, as follows.
Dose–response study
First, 100 μL of the different Ara-C solutions was added sequentially to each well containing 100 μL cell suspension (the concentration of Ara-C was 0.2, 2, 10, and 50 μg/mL, respectively, so the final concentration in each well was 0.1, 1, 5, and 25 μg/mL). Then, the IMDM medium (the negative control group) and ABMs-P and BMs solutions were added in the same way, then incubated at 37°C for 72 hours in a humidified environment containing 5% CO2.
Incubation-time study
First, 100 μL of Ara-C solution (the concentration of Ara-C was 10 μg/mL, so the final concentration of Ara-C was 5 μg/mL) was placed in each well, and the same volume was added of the IMDM medium (the negative control group) and the ABMs-P and BMs solutions. Then, they were incubated at 37°C for 24, 48, 72, 120, or 168 hours in a humidified environment containing 5% CO2. Six duplicates were performed of all assays. The absorption of each well was measured at 450 nm. The cell inhibition rate was calculated through the following formula:
| (6) |
where ODex is the absorbance of experimental wells, ODsb is the absorbance of sample blank wells, ODctrl is the absorbance of control wells, and ODb is the absorbance of blank wells.
Acridine orange/ethidium bromide (AO/EB) assays
First, 1 mL of the HL-60 cell suspensions (2×105 cells) was added to a 24-well plate and incubated for 4–6 hours at 37°C in a humidified environment containing 5% CO2. Then, 1 mL Ara-C solution (10 μg/mL) was placed in each well and incubated for 24 and 48 hours. IMDM medium, ABMs-P, and BMs solutions were treated in the same manner. The resultant cells were washed and resuspended with 100 μL PBS at 24 and 48 hours. The cell suspensions were mixed with 2 μL mixed dyes reagent (v1/v2 =1), followed by staining for 2–3 minutes in the dark, and then they were recorded by the laser scanning confocal microscope (LSM710; Carl Zeiss Meditec, Jena, Germany).
Results and discussion
The characterization of ABMs-P
BMs that were isolated from the AMB-1 cells possessed a uniform size distribution (Figure 2A), and a clearly identified lipid membrane could be found around the surface of the BMs (Figure 2B). As Figure 3A and B show, ABMs-P was surrounded by thick nebulous material and appeared to have aggregated, which was consistent with the diameter of ABMs-P (74.9±8.2 nm) being larger than that of BMs (42.0±8.6 nm). Compared with previous studies,16–18 these findings suggested that Ara-C might attach to the PLGA on the BM lipid membrane. The zeta potentials of the BMs and ABMs-P were −28.8±7.6 mV and −30.5±6.6 mV, respectively, which indicated that both the BMs and ABMs-P were stable and could distribute well in the next drug release experiment.
Figure 2.

TEM images of BMs with different magnifications.
Abbreviations: BMs, bacterial magnetosomes; TEM, transmission electron micrograph.
Figure 3.

TEM images of ABMs-P with different magnifications.
Abbreviations: ABMs-P, cytosine arabinoside-loaded genipin-poly-l-glutamic acid-modified bacterial magnetosomes; TEM, transmission electron micrograph.
FTIR spectra
The FTIR spectra are presented in Figure 4. The main peaks for ABMs-P were similar to those of the BMs, with the typical Fe-O stretching vibration appearing to be at around 582 cm−1. It is noteworthy that a weaker absorbance at 3,439, 1,541, and 1,522 cm−1 was found in ABMs-P compared with the BMs. The cause of this is likely to be the reaction of amino groups on the ABMs-P. Furthermore, a stronger absorbance peak of ABMs-P at 1,734 cm−1 may have been due to the carbonyl on the structures of PLGA and Ara-C. Compared with the main peaks of Ara-C, absorption peaks at 1,273 and 1,281 cm−1 were assigned to ABMs-P and Ara-C respectively, while the BMs showed no absorbance peak. These data imply that Ara-C was successfully grafted onto the surface of ABMs-P.
Figure 4.
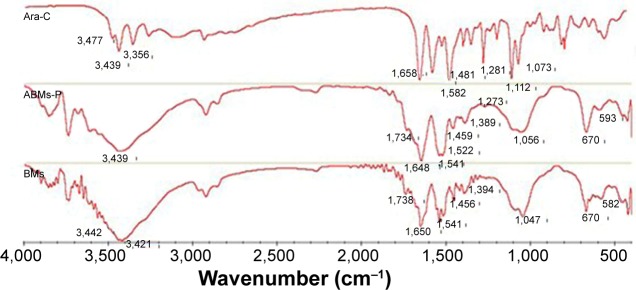
The FTIR spectrum of Ara-C, ABMs-P, and BMs.
Abbreviations: ABMs-P, cytosine arabinoside-loaded genipin-poly-l-glutamic acid-modified bacterial magnetosomes; Ara-C, cytosine arabinoside; BMs, bacterial magnetosomes; FTIR, Fourier transform infrared.
Drug-loading and encapsulation efficiency of ABMs-P
It is well known that increasing the drug-loading can facilitate the therapy. Figure 5 shows that a different cross-linking reaction time greatly influenced the drug-loading and encapsulation efficiency of ABMs-P. Figure 5A shows that both the drug-loading and encapsulation efficiency attained an optimal value, of 38.9%±2.4% and 64.1%±6.6%, respectively, when the first-step cross-linking reaction lasted for 72 hours and the second for 24 hours. A tendency for lower drug-loading and encapsulation efficiency was obtained when the second-step cross-linking reaction time was longer, which might be because the Ara-C was coupled to BMs through the chemical cross-linking reaction and physical absorption, and further small amounts of Ara-C were released because of surface absorption.20 As Figure 5B shows, as in the case of the second-step cross-linking reaction time of 24 hours, both the drug-loading and encapsulation efficiency reached a maximum value when the first-step cross-linking reaction time was 72 hours. However, when the second-step cross-linking reaction lasted for 48 hours, both the drug-loading and encapsulation efficiency had a decreasing trend. This is because PLGA forms a gel on the surface of BMs through spontaneous coupling, and this further covers the function positions used to load the drugs originally.16
Figure 5.
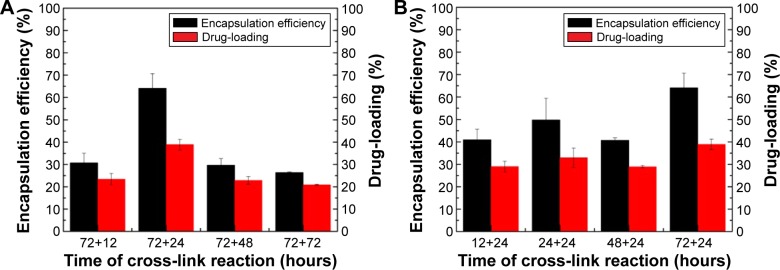
Drug-loading and encapsulation efficiency of ABMs-P prepared with different cross-linking reaction time: (A) the second-step cross-linking reaction and (B) the first-step cross-linking reaction.
Abbreviation: ABMs-P, cytosine arabinoside-loaded genipin-poly-l-glutamic acid-modified bacterial magnetosomes.
In Figure 6, an enhancement of the drug-loading and encapsulation efficiency can be seen with the increasing GP concentration, especially when the GP concentration increased from 0.05% to 0.1%. When the concentration reached 0.5%, it seems that the drug-loading process reached a balance. In a previous report, we found that better drug-loading, of about 50%, was obtained by introducing LGA with the chemical coupling reagent glutaraldehyde.16 In comparison with that work, the preparation parameters we used were lower than those that had been reported. So we are considering increasing the drug-loading by further adjusting the concentration and ratio of GP and PLGA.
Figure 6.
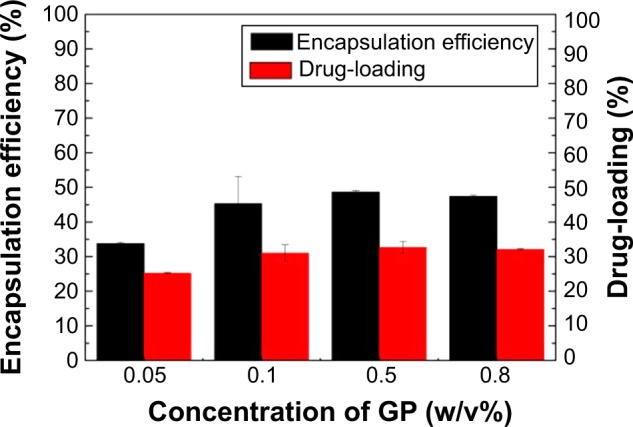
Drug-loading and encapsulation efficiency of ABMs-P preparation with different GP concentrations.
Abbreviations: ABMs-P, cytosine arabinoside-loaded genipin-poly-l-glutamic acid-modified bacterial magnetosomes; GP, genipin.
In vitro release of Ara-C from ABMs-P
As Figure 7 shows, the free Ara-C was released almost completely within 1 hour at 37°C, whereas ABMs-P compounds were significantly stable in PBS (pH 7.4) prepared at different cross-linking times. Figure 8A shows that the effect of the duration time on the release of Ara-C from ABMs-P in the second-step reaction was more obvious. When the first-step cross-linking reaction lasted for 72 hours, a slower release behavior could be observed, with the second-step cross-linking reaction lasting longer. The reason for this is that the longer the cross-linking reaction time is, the more thorough the chemical cross-linking reaction will be; therefore, Ara-C can link to BMs more closely, which extends the release period accordingly.
Figure 7.
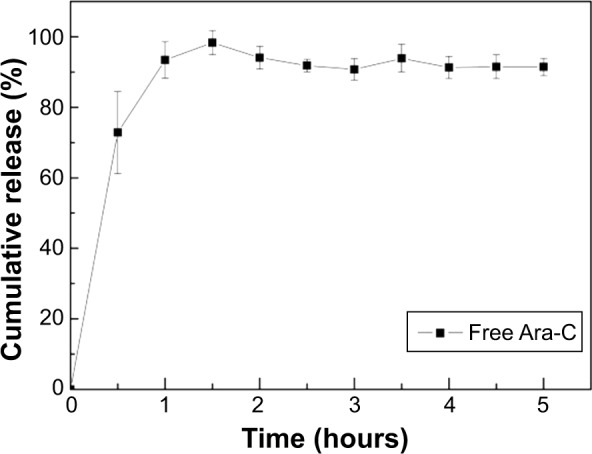
Cumulative release of Ara-C in vitro.
Abbreviation: Ara-C, cytosine arabinoside.
Figure 8.
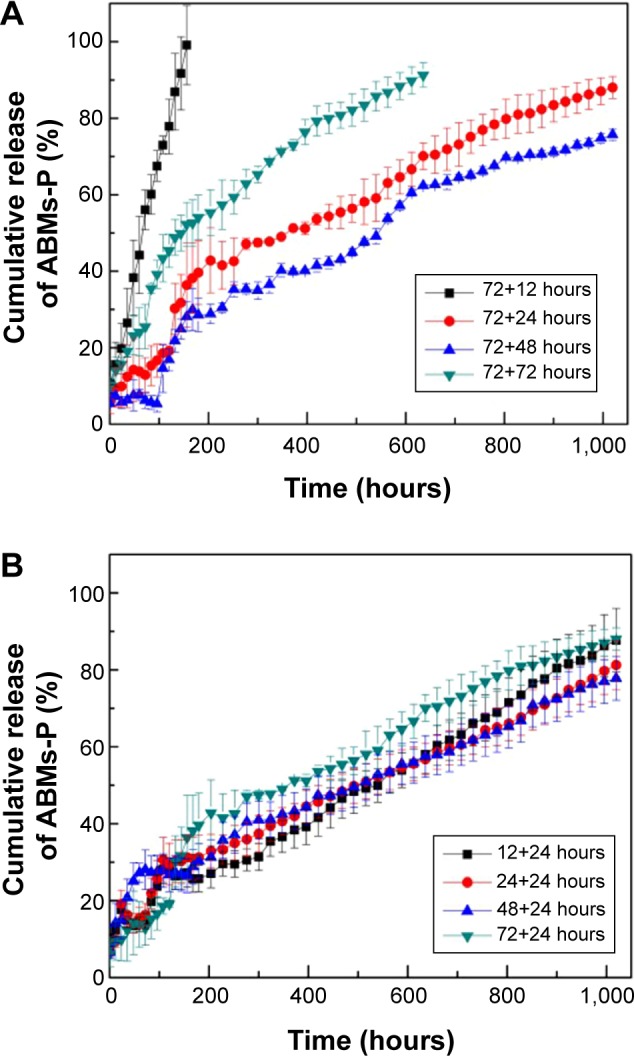
Cumulative release of ABMs-P preparation with different cross-linking reaction times. (A): second-step reaction time; (B): first-step reaction time.
Abbreviation: ABMs-P, cytosine arabinoside-loaded genipin-poly-l-glutamic acid-modified bacterial magnetosomes.
Figure 8B demonstrates that the first-step reaction time had no obvious influence on the release property, as more than 85% of the Ara-C was released within 40 days. Different GP concentrations also enhanced the release property (Figure 9). Furthermore, drug burst release behavior was not found in the initial 0.5 hours. When the concentration of GP was 0.5% and 0.8%, ABMs-P had similar release properties, with 90% of Ara-C released from ABMs-P within 40 days. The reason for this is presumed to be the same as that for the second-step cross-linking time. Little investigation of the long-term release properties of BMs coupled with drugs was found in an earlier relevant research report, which mainly focused on short-term release.16 In this report, we successfully constructed a magnetic targeting antitumor drug delivery system with long-term release behavior and no burst release effect.
Figure 9.
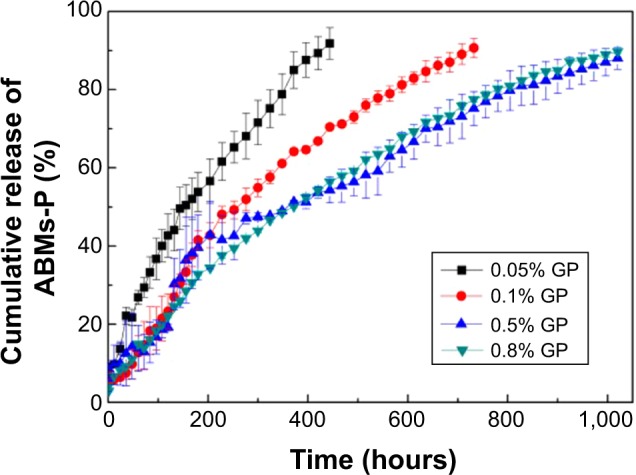
Cumulative release of ABMs-P preparation with different GP concentrations.
Abbreviations: ABMs-P, cytosine arabinoside-loaded genipin-poly-l-glutamic acid- modified bacterial magnetosomes; GP, genipin.
Influence of ABMs-P on HL-60 cells in response to drug concentration
Structural modification may change drug efficacy significantly. In this report, we obtained the ABMs-P by cross-linking PLGA and GP. This is the key to evaluating the medical effects of the drug when coupled with BMs in the study, for example, to evaluating the inhibition of tumor cells. Figure 10 shows the influence of ABMs-P on HL-60 cells in response to drug concentration. In the 0.1–25 μg/mL concentration range, the inhibition rate of Ara-C, ABMs-P, and BMs showed a rising trend with an increase of drug concentration. In the case of low concentration (≤5 μg/mL), the inhibition rate of ABMs-P was lower than that of Ara-C, except at 0.1 μg/mL. This is because at the same concentration, the drug-loading of the ABMs-P complex was less than Ara-C, and the release time was limited; therefore, the release of the drug was insufficient to inhibit cell proliferation significantly. When the drug concentration reached 25 μg/mL, the inhibition rates of Ara-C and ABMs-P were similar; the effect of ABMs-P on the cell growth inhibition rate increased obviously to 95%, which was higher than that of the contrast group (BMs). Here, the inhibition rate of BMs was 48%, which is close to the findings of a previous study.15
Figure 10.
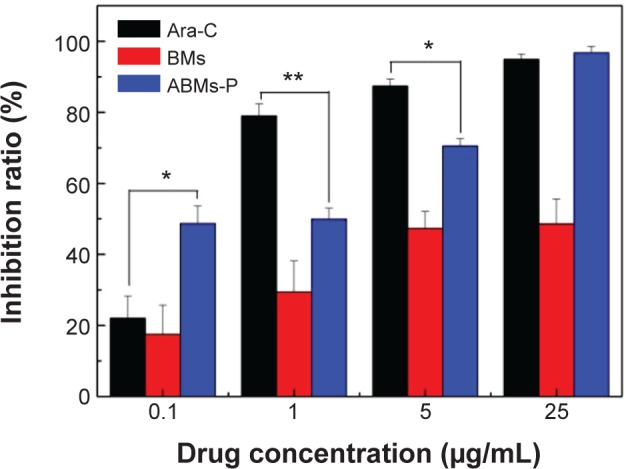
The effect of concentrations of Ara-C, ABMs-P, and BMs on the growth of HL-60 cells.
Notes: *P<0.05, **P<0.01.
Abbreviations: ABMs-P, cytosine arabinoside-loaded genipin-poly-l-glutamic acid-modified bacterial magnetosomes; Ara-C, cytosine arabinoside; BMs, bacterial magnetosomes.
As Figure 11A shows, the control group, when incubated for 48 hours, showed a rapid proliferation, with characteristic round, translucent particles, moderate density, and good dispersion. Ara-C and ABMs-P both suppressed cell growth (Figure 11B–D). The HL-60 cells cocultured with Ara-C and ABMs-P exhibited a propensity for aggregating, with a mass of debris, while BMs and the control group showed no significant change, which indicated that 5 μg/mL ABMs-P had a certain cytotoxicity to HL-60 cells.
Figure 11.

Optical micrograph of HL-60 cells cultured for 72 hours (×100) with (A) fresh medium (control); (B) 5 μg/mL Ara-C; (C) 5 μg/mL ABMs-P; and (D) 5 μg/mL BMs.
Abbreviations: ABMs-P, cytosine arabinoside-loaded genipin-poly-l-glutamic acid-modified bacterial magnetosomes; Ara-C, cytosine arabinoside; BMs, bacterial magnetosomes.
Influence of ABMs-P on HL-60 cells in response to incubation time
The BMs group maintained a high level of cell activity throughout 7 days (Figure 12). Both the Ara-C and ABMs-P group showed obvious cytotoxicity. The inhibition rate of ABMs-P was strengthened with the increase of incubation time. The activity of cells decreased to 32% on day 7 and was greater than that of the raw Ara-C drug. The reason for this is that the drug content released from ABMs-P was less than that from free Ara-C within 7 days and failed to meet the requirements of entire cell inhibition. While ABMs-P had a long-term release property, it can be predicted that the ABMs-P would continue to suppress the cell proliferation with the extension of time. The viability of the HL-60 cells treated by Ara-C increased after 5 days. The reason for this may be that the initial high concentration of Ara-C inhibited cell proliferation significantly, but failed to kill the cells completely. After 5 days, the concentration of the residual Ara-C was insufficient to exert an inhibitory effect on the surviving cells, which resulted in a new round of proliferation of the cancer cells.
Figure 12.
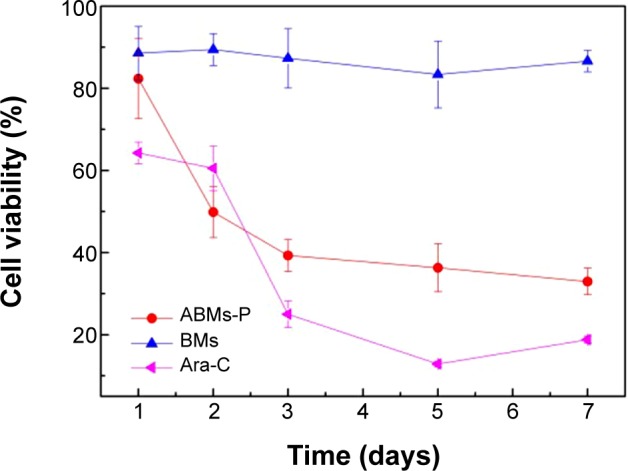
The effect of treatment times of Ara-C, ABMs-P, and BMs on the growth of HL-60 cells.
Abbreviations: ABMs-P, cytosine arabinoside-loaded genipin-poly-l-glutamic acid- modified bacterial magnetosomes; Ara-C, cytosine arabinoside; BMs, bacterial magne-tosomes.
Influence of ABMs-P on cell apoptosis
Apoptosis is physiological cell death controlled by a gene. At present, it is regarded as one of the most important indexes for evaluating the drug efficacy in inhibition of tumors. In this experiment, cell apoptosis of the tested samples cocultured with HL-60 cells for 24 hours (A, C, E, G) and 48 hours (B, D, F, H), respectively, was observed by double staining. As Figure 13 shows, rapid cell proliferation was present in the control group, and almost no apoptotic cells could be found at 24 hours and 48 hours, and only a few appeared to be in early apoptosis. In the Ara-C group, most cells showed apoptosis by 24 hours, and we observed cell proliferation with the phenomenon of serious necrosis and apoptosis. Some cells also showed obvious lyses and had broken after 48 hours. In the ABMs-P group, parts of the cells were tending toward apoptosis at 24 hours, and when the coculture time was prolonged to 48 hours, cell proliferation increased, and the number of cells at the stage of late apoptosis and necrosis increased significantly. However, in the HL-60 cells treated with BMs, only a small number of cells showed late apoptosis, with a normal cell proliferation. These results are consistent with previous results. Due to the sustained-release property, the necrosis and apoptosis phenomena of the HL-60 cells cocultured with ABMs-P were no more obvious than those cocultured with the Ara-C.
Figure 13.

AO-EB dual staining of HL-60 cells after culturing with (A and B) fresh medium (control), (C and D) 5 μg/mL Ara-C, (E and F) 5 μg/mL ABMs-P, and (G and H) 5 μg/mL BMs, for 24 and 48 hours.
Abbreviations: ABMs-P, cytosine arabinoside-loaded genipin-poly-l-glutamic acid-modified bacterial magnetosomes; AO-EB, acridine orange/ethidium bromide; Ara-C, cytosine arabinoside; BMs, bacterial magnetosomes.
In conclusion, because ABMs-P had long-term stability and sustained release behavior, it can reduce the frequency of drug administrations to a certain extent compared with Ara-C. In addition, 25 μg/mL ABMs-P achieved similar a inhibition rate with to the equivalent Ara-C, which suggests that less drug would be necessary to treat the disease. Therefore, the magnetic drug targeting system constructed in this study can have the desired drug effect and is expected to reduce adverse side effects as well as achieve sustained release in treatment.
Conclusion
This investigation demonstrated that the modification of BMs, using the natural cross-linking reagent GP to introduce PLGA, was successful. This modification provides more coupled sites to facilitate the construction of a magnetic targeting antitumor drug delivery system. The drug-loading and encapsulation efficiency of ABMs-P were 38.9%±2.4% and 64.1%±6.6%, respectively. Furthermore, ABMs-P exhibited a long-term release property: 90% of the loaded Ara-C released effectively from ABMs-P within 40 days, without an initial burst release. Moreover, the ABMs-P prepared in this study showed strong cytotoxicity to HL-60 cells, similar to Ara-C, which could make this an appropriate controlled long-term drug release system to ease the severe side effects of the drug. The introduction of dual cross-linkers would be a new approach for conjugating other drugs. However, it is necessary to conduct further investigations for a comprehensive understanding of this system. It is essential to evaluate the biocompatibility of BMs, and the pharmacokinetics and administration routes of ABMs-P. As well, some problems still need to be solved, for example, ABMs-P has a propensity to aggregate, which might increase the difficulty of drug absorption. This new system shows potential for promising applications in future drug delivery systems, especially in magnetic targeting treatment.
Acknowledgments
Financial support from the following is gratefully acknowledged: Natural Science Foundation of China (grant numbers 31000441 and 31170939); China Postdoctoral Science Foundation (grant number 2014M551833); the Natural Science Foundation of Fujian Province (grant number 2013J01189); Program for Prominent Young Talent in Fujian Province University (grant number JA12004); Science and Technology Project of Fujian Province, China (grant number 2013Y2002); the Scientific Research Foundation for Returned Overseas Chinese Scholars (State Education Ministry); and the Promotion Programme for Young and Middle-Aged Teachers in Science and Technology Research of Huaqiao University (grant number ZQN-PY108).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood. 2005;106(4):1154–1163. doi: 10.1182/blood-2005-01-0178. [DOI] [PubMed] [Google Scholar]
- 2.Elli M, Aydin O, Bilge S, et al. Protective effect of vitamin A on ARA-C induced intestinal damage in mice. Tumori. 2009;95(1):87–90. doi: 10.1177/030089160909500114. [DOI] [PubMed] [Google Scholar]
- 3.Keime-Guibert F, Napolitano M, Delattre JY. Neurological complications of radiotherapy and chemotherapy. J Neurol. 1998;245(11):695–708. doi: 10.1007/s004150050271. [DOI] [PubMed] [Google Scholar]
- 4.Kwong YL, Yeung DY, Chan JC. Intrathecal chemotherapy for hematologic malignancies: drugs and toxicities. Ann Hematol. 2009;88(3):193–201. doi: 10.1007/s00277-008-0645-y. [DOI] [PubMed] [Google Scholar]
- 5.Yamashita F, Hashida M. Pharmacokinetic considerations for targeted drug delivery. Adv Drug Deliv Rev. 2013;65(1):139–147. doi: 10.1016/j.addr.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Pilapong C, Keereeta Y, Munkhetkorn S, Thongtem S, Thongtem T. Enhanced doxorubicin delivery and cytotoxicity in multidrug resistant cancer cells using multifunctional magnetic nanoparticles. Colloids Surf B Biointerfaces. 2014;113:249–253. doi: 10.1016/j.colsurfb.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Mu Q, Yang L, Davis JC, et al. Biocompatibility of polymer grafted core/shell iron/carbon nanoparticles. Biomaterials. 2010;31(19):5083–5090. doi: 10.1016/j.biomaterials.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guardia P, Di Corato R, Lartigue L, et al. Water-soluble iron oxide nanocubes with high values of specific absorption rate for cancer cell hyperthermia treatment. ACS Nano. 2012;6(4):3080–3091. doi: 10.1021/nn2048137. [DOI] [PubMed] [Google Scholar]
- 9.Santhosh PB, Ulrih NP. Multifunctional superparamagnetic iron oxide nanoparticles: promising tools in cancer theranostics. Cancer Lett. 2013;336(1):8–17. doi: 10.1016/j.canlet.2013.04.032. [DOI] [PubMed] [Google Scholar]
- 10.Balasubramanian S, Girija AR, Nagaoka Y, et al. Curcumin and 5-fluorouracil-loaded, folate- and transferrin-decorated polymeric magnetic nanoformulation: a synergistic cancer therapeutic approach, accelerated by magnetic hyperthermia. Int J Nanomedicine. 2014;9:437–459. doi: 10.2147/IJN.S49882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siponen MI, Legrand P, Widdrat M, et al. Structural insight into magnetochrome-mediated magnetite biomineralization. Nature. 2013;502(7473):681–684. doi: 10.1038/nature12573. [DOI] [PubMed] [Google Scholar]
- 12.Lefèvre CT, Menguy N, Abreu F, et al. A cultured greigite-producing magnetotactic bacterium in a novel group of sulfate-reducing bacteria. Science. 2011;334(6063):1720–1723. doi: 10.1126/science.1212596. [DOI] [PubMed] [Google Scholar]
- 13.Stanley S. Biological nanoparticles and their influence on organisms. Curr Opin Biotechnol. 2014;28:69–74. doi: 10.1016/j.copbio.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Tang YS, Wang D, Zhou C, et al. Bacterial magnetic particles as a novel and efficient gene vaccine delivery system. Gene Ther. 2012;19(12):1187–1195. doi: 10.1038/gt.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun JB, Duan JH, Dai SL, et al. Preparation and anti-tumor efficiency evaluation of doxorubicin-loaded bacterial magnetosomes: magnetic nanoparticles as drug carriers isolated from Magnetospirillum gryphiswaldense. Biotechnol Bioeng. 2008;101(6):1313–1320. doi: 10.1002/bit.22011. [DOI] [PubMed] [Google Scholar]
- 16.Guo L, Huang J, Zheng LM. Control generating of bacterial magnetic nanoparticle-doxorubicin conjugates by poly-L-glutamic acid surface modification. Nanotechnology. 2011;22(17):175102. doi: 10.1088/0957-4484/22/17/175102. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Xie M, Wang S, et al. Facile fabrication of high performances MTX nanocomposites with natural biomembrane bacterial nanoparticles using GP. Materials Letters. 2013;100:248–251. [Google Scholar]
- 18.Deng Q, Liu Y, Wang S, et al. Construction of a novel magnetic targeting anti-tumor drug delivery system: Cytosine arabinoside-loaded bacterial magnetosome. Materials. 2013;6(9):3755–3763. doi: 10.3390/ma6093755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chinese Pharmacopoeia Commission . Pharmacopoeia of the People’s Republic of China. Beijing: Ministry of Health, People’s Republic of China; 2010. [Google Scholar]
- 20.Calabresi P, Chabner BA, Goodman LS. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. New York: MacMillan; 1991. [Google Scholar]