

ABSTRACT
Protection from lethality by postchallenge administration of brincidofovir (BCV, CMX001) was studied in normal and immune-deficient (nude, nu/nu) BALB/c mice infected with vaccinia virus (VACV). Whole-body bioluminescence imaging was used to record total fluxes in the nasal cavity, lungs, spleen, and liver and to enumerate pox lesions on tails of mice infected via the intranasal route with 105 PFU of recombinant IHD-J-Luc VACV expressing luciferase. Areas under the flux curve (AUCs) were calculated for individual mice to assess viral loads. A three-dose regimen of 20 mg/kg BCV administered every 48 h starting either on day 1 or day 2 postchallenge protected 100% of mice. Initiating BCV treatment earlier was more efficient in reducing viral loads and in providing protection from pox lesion development. All BCV-treated mice that survived challenge were also protected from rechallenge with IHD-J-Luc or WRvFire VACV without additional treatment. In immune-deficient mice, BCV protected animals from lethality and reduced viral loads while animals were on the drug. Viral recrudescence occurred within 4 to 9 days, and mice succumbed ∼10 to 20 days after treatment termination. Nude mice reconstituted with 105 T cells prior to challenge with 104 PFU of IHD-J-Luc and treated with BCV postchallenge survived the infection, cleared the virus from all organs, and survived rechallenge with 105 PFU of IHD-J-Luc VACV without additional BCV treatment. Together, these data suggest that BCV protects immunocompetent and partially T cell-reconstituted immune-deficient mice from lethality, reduces viral dissemination in organs, prevents pox lesion development, and permits generation of VACV-specific memory.
IMPORTANCE Mass vaccination is the primary element of the public health response to a smallpox outbreak. In addition to vaccination, however, antiviral drugs are required for individuals with uncertain exposure status to smallpox or for whom vaccination is contraindicated. Whole-body bioluminescence imaging was used to study the effect of brincidofovir (BCV) in normal and immune-deficient (nu/nu) mice infected with vaccinia virus, a model of smallpox. Postchallenge administration of 20 mg/kg BCV rescued normal and immune-deficient mice partially reconstituted with T cells from lethality and significantly reduced viral loads in organs. All BCV-treated mice that survived infection were protected from rechallenge without additional treatment. In immune-deficient mice, BCV extended survival. The data show that BCV controls viral replication at the site of challenge and reduces viral dissemination to internal organs, thus providing a shield for the developing adaptive immunity that clears the host of virus and builds virus-specific immunological memory.
INTRODUCTION
Smallpox was eradicated following a global immunization program using live vaccinia virus (VACV) vaccine implemented by the World Health Organization (WHO). Routine smallpox vaccination was subsequently discontinued due to a low but significant risk of severe adverse reactions. As a result, the current population has low or nonexistent immunity to smallpox, creating a serious public health concern should variola virus, the virus that causes smallpox, be used as a biological weapon (1). Monkeypox virus (MPXV) is related to variola virus and can be transmitted to humans. MPXV induces a disease in humans similar to smallpox but with lower mortality (2). MPXV remains endemic in parts of Africa and was accidentally imported to the United States, where it caused a limited outbreak in 2003 (3, 4).
Protection from infection caused by variola virus or MPXV can be achieved by immunization with smallpox vaccine, historically, Dryvax in the United States. Dryvax, however, has a risk of causing serious side effects in some vaccine recipients (e.g., eczema vaccinatum, myocarditis, and progressive vaccinia) (reviewed in references 5 and 6). More recently, a nonreplicating vaccine prepared from a highly attenuated modified Ankara vaccinia virus (MVA), Imvamune, which has a lower risk of producing adverse reactions, was acquired for the Strategic National Stockpile. Although mass vaccination continues to be a key part of the public health response to a smallpox outbreak, the need remains for smallpox therapeutics that can be used in patients with uncertain exposure status or for whom vaccination is contraindicated (7).
Currently, only intravenous vaccinia immunoglobulin (VIGIV; Cangene Corporation), obtained from the plasma of healthy donors previously vaccinated with Dryvax, is licensed for treatment of complications following smallpox vaccination, and it has been suggested that VIGIV might also be effective in unvaccinated persons exposed to variola virus (8). However, no therapeutic is currently licensed by the Food and Drug Administration (FDA) for the treatment of smallpox. A case report described an army recruit diagnosed with acute mylogenous leukemia M0 (AML M0) following vaccination with the ACAM2000 smallpox vaccine that resulted in life-threatening progressive vaccinia (9). The case management demonstrated that even high doses of VIGIV (6 to 24,000 U/kg) together with the investigational antiviral drug tecovirimat (ST-246) failed to ameliorate disease in an immune-deficient patient. As a result, a second investigational antiviral drug, brincidofovir (previously known as CMX001; see below), was added to the regimen. The patient subsequently cleared the virus and was discharged from the hospital. This case highlights the need for safe, orally bioavailable antiviral drugs for postexposure prophylaxis or treatment of poxvirus infections in addition to vaccines.
The acyclic cytidine analogue cidofovir (CDV), licensed under the name Vistide for the treatment of cytomegalovirus (CMV) retinitis in AIDS patients, is protective in murine models of VACV, cowpox virus, herpes simplex virus, and CMV (10–14). It was also shown to protect monkeys from lethal infection with MPXV (15). Inside cells, CDV is phosphorylated to cidofovir diphosphate (CDV-PP), the active antiviral (16). CDV-PP has a long intracellular half-life, but CDV has poor oral absorption that necessitates administration by the intravenous route and is associated with a high risk of severe nephrotoxicity (16, 17).
Alkoxyalkyl ester prodrugs of CDV have demonstrated greatly increased potency against a wide range of double-stranded DNA (dsDNA) viruses, including poxviruses, relative to CDV (18, 19). Brincidofovir (BCV) is a lipid conjugate of CDV with improved in vitro potency and oral bioavailability as well as decreased risk of nephrotoxicity (20). Inside cells, brincidofovir is metabolized to CDV and subsequently phosphorylated to CDV-PP. Brincidofovir has potent broad-spectrum antiviral activity, including against poxviruses in vitro, and was shown to provide protection from lethality in several animal models, including mouse intranasal infections with ectromelia virus, cowpox virus, and VACV and a rabbit/rabbitpox virus intradermal challenge model (21–23). Because BCV's mechanism of action involves inhibition of viral replication, its potential effect on the elicitation of protective immunity by live smallpox vaccines is of interest. Coadministration of BCV with smallpox vaccine (Dryvax or ACAM2000) reduced vaccine-associated lesion development but did not interfere with vaccine-induced protection of mice from lethal infection with ectromelia virus (24). However, whether BCV can protect immune-deficient mice from VACV-induced lethality and whether BCV has an effect on replication of the VACV at the site of infection have not been investigated.
Previous publications including data from our laboratory have shown that whole-body bioluminescence imaging (BLI) provides an advantage over more traditional methods for assessing the effectiveness of vaccines and immunoglobulin-based treatments in mouse models of respiratory challenge with VACV (25–29). In the current study, we explored the effects of brincidofovir on protection of normal and immune-deficient mice from lethality following intranasal challenge with IHD-J-Luc VACV. We assessed viral loads at the site of challenge and internal organs by calculating the area under the flux curve (AUC). Using this approach, we evaluated survival and virus dissemination in mice treated with BCV beginning 24 or 48 h postchallenge. In normal BALB/c mice and in nude (nu/nu) mice reconstituted with limited numbers of T cells, three doses of BCV beginning 24 h postchallenge conferred 100% protection from lethality, significantly reduced viral loads in the upper respiratory tract and internal organs, prevented pox lesion development, and protected immunocompetent animals from rechallenge with VACV with no further treatment.
MATERIALS AND METHODS
Viruses.
The thymidine kinase-positive (TK+) recombinant International Health Department J (IHD-J) VACV expressing the luciferase gene (IHD-J-Luc) inserted at a truncated host range gene locus equivalent to the cowpox gene, CPX077 (under the control of the synthetic E/L promoter), was kindly provided by Clement Meseda (30). The TK+ recombinant Western Reserve (WR) VACV expressing the luciferase reporter gene under the control of a synthetic E/L promoter (WRvFire) was kindly provided by Bernard Moss (NIAID, NIH) (31). WRvFire was propagated in HeLa cells, and titers were determined in BSC-1 cells. Single stocks of WRvFire and of IHD-J-Luc vaccinia viruses containing 1.0 × 108 and 3.9 × 109 PFU/ml, respectively, were used in the study. The numbers of particles in sucrose gradient-purified viral stocks were determined by transmission electron microscopy (TEM) and were 2.4 × 109 and 3.3 × 1010 particles/ml for WRvFire and IHD-J-Luc, respectively; thus both stocks contained similar ratios of PFU/particles (20:1 and 10:1, respectively).
Mice and protocols for in vivo treatments.
Five-week-old female BALB/c mice or 7- to 8-week-old female BALB/c nu/nu mice (National Cancer Institute, Frederick, MD) were used in all experiments. Immediately prior to challenge, mice were anesthetized using 1.5%, vol/vol, Avertin solution (2, 2, 2-tribromoethanol dissolved in tertiary amyl alcohol and diluted in sterile phosphate-buffered saline [PBS] according to the manufacturer's instructions) at 20 μl per gram of body weight by intraperitoneal (i.p.) injection. Normal BALB/c mice were challenged with 105 PFU (two times the 50% lethal dose [LD50]) while nude mice were challenged with 104 PFU (2 LD50s) of IHD-J-Luc VACV via the intranasal route (i.n.) in a 10-μl volume delivered in one nostril.
For postchallenge treatments, BCV at 20 mg/kg (or as specified in the study) or vehicle was administered in 100-μl volumes via oral gavage using 38.1-mm animal feeding needles (Cadence Science, Inc., Cranston, RI) starting 24 h postchallenge or as specified in the study. Selection of the 20 mg/kg BCV dose for evaluation was based on the results of previous studies conducted in the lab of Mark Buller at St. Louis University (21, 32; also unpublished data) as well as unpublished studies conducted by Chimerix, Inc. The regimen of three doses every 48 h (q48h) was based on the duration of disease in the model and the anticipated half-life of the active antiviral metabolite, CDV-PP, in mice and in humans as well as on the plasma pharmacokinetics of BCV in both mice and humans. Due to the favorable metabolic and pharmacokinetic profile of BCV in humans, systemic exposure (maximum concentration of drug in serum [Cmax] and AUC) in mice given a dose of 20 mg/kg BCV is lower than that achieved in humans at the doses currently proposed for treatment of smallpox and under evaluation for treatment of other diseases caused by dsDNA viruses.
For adoptive transfer experiments, T cells were obtained from the spleens of normal female BALB/c mice at 6 to 8 weeks of age using a mouse Pan-T Cell Isolation Kit II (Milenyi Biotec, Inc., Auburn, CA) according to the manufacturer's instructions. The T cells were 99% pure as verified by flow cytometry. Purified T cells in 400-μl volumes in PBS were injected into the tail vein of nude mice 24 h before challenge using a 1-ml insulin syringe with a 28-gauge, 0.5-inch needle (Becton Dickinson). For the sham adoptive transfer, mice were injected with 400 μl of PBS. Treatment of mice and experimental procedures were approved by the Center for Biologics Evaluation and Research (CBER) animal study review committee. Group sample sizes for each experiment are provided in figure legends.
In vivo measurements of luciferase activity.
The details of whole-body imaging using an IVIS (in vivo imaging system) 50 instrument (PerkinElmer, Waltham, MA) were previously described (33). In brief, 10 to 15 min prior to imaging, mice received a single i.p. injection of 150 μg/g of body weight of d-luciferin (PerkinElmer). Mice were anesthetized in an oxygen-rich induction chamber with 2% isoflurane and imaged daily on days 1 to 10 postchallenge or as specified in the study. Images were analyzed with Living Image, version 3.02, software (PerkinElmer) according to the manufacturer's instructions. A single region of interest (ROI) was established for each organ and used throughout analysis to quantify the amount of light emission in specific organs. The amount of light within designated ROIs was quantified as photon fluxes with normalization for the imaging area and exposure time (photons/s/cm2 or photons/s), as recommended by the manufacturer. The background bioluminescence was determined using images of d-luciferin-injected animals acquired 1 day prior to infection. Pox lesions were counted on mouse tails using dorsal images, as previously described (34).
Statistical analysis.
Kaplan-Meier survival curves of time to death following infection were plotted using GraphPad Prism, version 5, software and compared using a log rank (Mantel-Cox) test. In all tests, P values of ≤0.05 were considered statistically significant. The area under the flux curve (AUC; log10 photons/s × day) was calculated for each mouse for the number of days specified in the study; mean AUCs of various groups were compared using two-sample t tests. For each set of comparisons, the flux curves were truncated at the last time point at which all mice had complete data to ensure fair comparisons between groups. We also used two-sample t tests to compare the number of pox lesions between groups. All results were considered statistically significant at a P value of ≤0.05 (two-tailed).
RESULTS
Effect of BCV dose and treatment start day postchallenge on protection of BALB/c mice from lethal challenge with IHD-J-Luc VACV.
To determine the minimal dose of BCV that confers protection of immunocompetent mice from lethal challenge with VACV, BALB/c mice were infected with 105 PFU of IHD-J-Luc VACV i.n. and were treated with either vehicle or BCV at doses of 2.5, 5, or 20 mg/kg on days 1, 3, and 5 postchallenge (Fig. 1A). All mice that received vehicle succumbed by day 8 postchallenge (Fig. 1A). Administration of BCV at doses of 5 and 20 mg/kg protected 100% of mice, while a dose of 2.5 mg/kg BCV provided minimal protection (16% survival) (Fig. 1A). To determine the effect of delayed treatment initiation, mice received vehicle or 5 or 20 mg/kg BCV on days 2, 4, and 6 postchallenge (Fig. 1B). All vehicle-treated mice succumbed by day 8, and all mice that received 20 mg/kg BCV on days 2, 4, and 6 survived (Fig. 1B). BCV at 5 mg/kg on days 2, 4, and 6 provided partial protection (33% survival) (Fig. 1B). Further delay of 20 mg/kg BCV treatment initiation to day 3 resulted in loss of protection (data not shown). No significant differences in the weights of mice that received BCV at 2.5, 5, or 20 mg/kg and that survived infection were observed (data not shown).
FIG 1.

The effect of BCV initiated 24 or 48 h postchallenge with IHD-J-Luc VACV on mortality and viral load in various tissues in BALB/c mice. BALB/c mice were infected with IHD-J-Luc VACV at 105 PFU i.n. Mice received vehicle or 2.5 mg/kg, 5 mg/kg, or 20 mg/kg BCV on days 1, 3, and 5 (A and C to F) or 5 mg/kg or 20 mg/kg BCV on days 2, 4, and 6 postinfection (B and G to J). Mice were observed for survival (A and B) and were subjected to daily whole-body bioimaging (C to F and G to J). Total fluxes in the nasal cavity, lungs, spleen, and liver, as indicated, in individual mice were determined and used to calculate mean total flux ± SD using a t test and data from three to four independent experiments. Mean background levels of fluxes (photons/s) ± SD were recorded in BALB/c mice prior to infection: nasal cavity, 23.0 × 103 ± 4.2 × 103; lungs, 40.3 × 103 ± 9.3 × 103; spleen, 24.0 × 103 ± 2.1 × 103; liver, 37.2 × 103 ± 1.9 × 103. Values are represented as dotted horizontal lines (means only) in panels C to J. Data were combined from four and three experiments for treatment on days 1, 3, and 5 and for treatment on days 2, 4, and 6, respectively. Treatments on days 1, 3, and 5 were as follows: vehicle, 23 animals per group; BCV 20 mg/kg, 21 animals per group; BCV 2.5 and 5 mg/kg, 6 animals/group. Treatments on day 2, 4, 6 were as follows: vehicle, 17 animals per group; BCV 20 mg/kg, 12 animals per group; 5 mg/kg of BCV, 6 animals per group. All BCV treatment groups demonstrated a significant increase in median survival time compared to that of vehicle-treated mice (P ≤ 0.0001).
Images of infected mice were acquired using an IVIS 50 instrument, and means of total photon fluxes emitted by infected organs in individual mice were calculated using Living Image software as previously described (27). Mean total fluxes increased in all groups of BCV-treated mice and reached a plateau on days 4 to 6 postchallenge, after which they gradually declined (Fig. 1C to J). Bioluminescence returned to control levels by days 14 and 21 (nasal cavity) or by days 8 and 21 (all other organs) in mice that received 20 mg/kg BCV on days 1, 3, and 5 or days 2, 4, and 6, respectively. Viral clearance was slower in surviving mice that received 2.5 or 5 mg/kg BCV than in mice given 20 mg/kg in both experiments.
To gain further insight into the effect of BCV on viral loads in surviving mice, we subjected acquired photon fluxes to biostatistical analysis; a direct correlation between the acquired photon fluxes per area per second and viral loads (PFU per gram of tissue) was previously described (27, 29). Fluxes acquired between days 1 and 21 were used to calculate AUCs for the individual mice in the groups where BCV conferred 100% protection from lethal challenge (Fig. 2). In mice that received BCV on days 1, 3, and 5, the mean AUCs in all organs were significantly lower for the group that received 20 mg/kg BCV than for mice given 5 mg/kg BCV (Fig. 2A). Similarly, mean AUCs were significantly lower in all organs in mice that received 20 mg/kg BCV on days 1, 3, and 5 than in those that received 20 mg/kg BCV on days 2, 4, and 6 (Fig. 2B).
FIG 2.
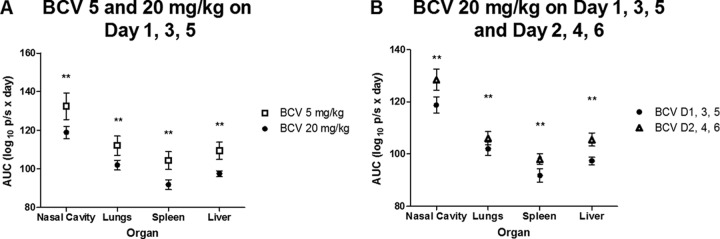
Statistical analyses of the areas under the flux curve (AUC) generated based on bioimaging of IHD-J-Luc VACV-infected mice that survived infection following treatment with BCV (Fig. 1). AUCs were calculated for fluxes acquired from individual mice between days 1 and 21 postchallenge for mice that were treated with BCV at doses of 5 mg/kg or 20 mg/kg on days 1, 3, 5 (A) or with 20 mg/kg of BCVs on day 2, 4, and 6 (B). Mean AUCs ± SD were compared between mice that received BCV at 5 versus 20 mg/kg on days 1, 3, and 5 (A) and between mice that received 20 mg/kg BCV on days 1, 3, and 5 versus days 2, 4, and 6 (B) using a t test (**, P ≤ 0.001). D, day.
Dorsal images of the same mice were used to monitor pox lesion development (Table 1). No pox lesions were detected during the first 2 days postchallenge (data not shown). In vehicle-treated mice, the maximal numbers of pox lesions were observed on days 5 and 6 postchallenge. BCV treatment initiated on day 1 either prevented (20 mg/kg) or significantly reduced (5 mg/kg) pox lesion development on days 5 and 6 compared with results in vehicle-treated mice (Table 1). In mice that received 2.5 mg/kg BCV from day 1, pox lesions were significantly reduced on day 5 only and were present at low numbers in surviving animals until day 21, the scheduled termination of the study. BCV treatments started on day 2 either significantly reduced (20 mg/kg) or did not affect (5 mg/kg) pox lesion development (Table 1). These data demonstrate that early initiation of treatment (24 h postchallenge) with BCV using an optimal dose of 20 mg/kg provided the most effective protection from lethality, reduced virus replication in multiple organs, including the site of infection, and prevented pox lesion formation. Lower doses and/or delayed initiation of treatment (48 h postchallenge) was less effective in controlling virus replication/dissemination and pox lesion formation, including in animals that survived the challenge.
TABLE 1.
Pox lesion counts in mice that were infected with IHD-J-Luc VACV and treated with BCV
| Treatment regimen | Survival (%) | Pox lesion count by day postinfection (mean ± SD)a |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 14 | 21 | ||
| Days 1, 3, and 5 | |||||||||||
| Vehicle | 0 | 1.4 ± 1.6 | 4.0 ± 2.8 | 6.2 ± 2.6 | 6.7 ± 2.2 | NA | NA | NA | NA | NA | NA |
| BCV | |||||||||||
| 2.5 mg/kg | 17 | 0.2 ± 0.4 | 1.7 ± 1.2 | 2.7 ± 1.4** | 5.2 ± 2.5 | 4.2 ± 2.1 | 5.3 ± 2.3 | 2.8 ± 2.7 | 1.6 ± 1.3 | 1.0 | 0.0 |
| 5 mg/kg | 100 | 0.7 ± 0.8 | 2.2 ± 2.4 | 3.7 ± 2.3* | 4.2 ± 2.3* | 5.0 ± 4.2 | 3.3 ± 2.6 | 3.0 ± 2.6 | 2.5 ± 2.4 | 0.5 ± 1.2 | 0 ± 0 |
| 20 mg/kg | 100 | 0 ± 0 | 0 ± 0* | 0 ± 0** | 0 ± 0** | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| Days 2, 4, and 6 | |||||||||||
| Vehicle | 0 | 1.2 ± 0.7 | 5.5 ± 2.2 | 7.5 ± 2.5 | 7.5 ± 1.1 | NA | NA | NA | NA | NA | NA |
| BCV | |||||||||||
| 5 mg/kg | 33 | 3.7 ± 2.0 | 6.8 ± 2.5 | 8.7 ± 1.9 | 9.2 ± 0.9 | 8.8 ± 3.7 | 6.0 ± 3.2 | 5.3 ± 4.6 | 2.5 ± 1.8 | 0 ± 0 | 0 ± 0 |
| 20 mg/kg | 100 | 2.3 ± 0.9 | 2.7 ± 1.2* | 4.8 ± 0.7* | 4.8 ± 1.3** | 2.2 ± 1.1 | 1.5 ± 0.5 | 1.7 ± 1.4 | 0.2 ± 0.4 | 0.2 ± 0.4 | 0 ± 0 |
Mice were infected at day 0. Pox lesions on mouse tails were counted using dorsal images of individual mice and were used to calculate average pox numbers ± SD per surviving animal on each day shown. Asterisks denote significant differences between vehicle- and BCV-treated groups of mice in mean daily pox counts. *, P ≤ 0.05; **, P ≤ 0.001. NA, not applicable.
BCV-treated mice that survived challenge with IHD-J-Luc VACV also survived rechallenge with IHD-J-Luc or with WRvFire VACV.
To determine whether the VACV-specific immune response developed during the primary infection under the protection of BCV treatment was sufficient to protect mice from rechallenge with VACV, new groups of BALB/c mice were infected with IHD-J-Luc VACV and treated with vehicle or 20 mg/kg BCV on days 1, 3, and 5. An additional group of animals was left uninfected and was treated with 20 mg/kg BCV on days 1, 3, and 5. All mice were subjected to bioimaging for 40 days. All infected and vehicle-treated mice [IHDJ(V)] succumbed by day 7, while all BCV-treated uninfected mice [BCV/IHDJ and BCV/WRvFire] and BCV-treated infected mice [IHDJ(BCV)/IHDJ and IHDJ(BCV)/WRvFire] survived (Fig. 3A). After the challenge, total fluxes in BCV-treated infected animals [IHDJ(BCV)/IHDJ and IHDJ(BCV)/WRvFire] were lower than those in the vehicle-treated animals and were further decreased to baseline by day 21 in the nasal cavity and by day 8 in the lungs, spleen, and liver, after which no increase in bioluminescence was detected in any organs of BCV-treated infected mice through day 40 (Fig. 3B, C, D, and E). In uninfected BCV-treated mice, no bioluminescence was detected above background between days 1 and 40 (data not shown).
FIG 3.

IHD-J VACV-infected BALB/c mice treated with BCV were protected from rechallenge with IHD-J and WRvFire VACV. BALB/c mice were infected with IHD-J-Luc VACV as described in the legend of Fig. 1 and treated with vehicle (filled circles) or 20 mg/kg BCV (filled triangles and squares) on days 1, 3, and 5. Control mice were left uninfected and were treated with BCV at 20 mg/kg on days 1, 3, and 5 (open triangles and squares). Mice were observed for mortality (A) and imaged daily for 10 days and on days 14, 21, and 40 postchallenge (B to E). On day 41, surviving mice were infected with 105 PFU of IHD-J-Luc (triangles) or with 105 PFU of WRvFire (squares) and were imaged on days 42 to 48. All mice were observed for mortality until day 62 (A), at which time the experiment was terminated. Total fluxes in the nasal cavity, lungs, spleen, and liver were determined and used to calculate mean total fluxes ± SD. Arrows in panels indicate day 41 when mice were rechallenged. Dotted horizontal lines in panels B to E depict mean background levels of fluxes (photons/s) ± SD that were recorded in mice prior to infection. Five mice per group were used in the experiment. BCV treatment of IHD-J-Luc-infected mice significantly increased the median survival time compared with infected vehicle-treated mice or with BCV-treated not infected mice (P = 0.0027 and P = 0.0035, respectively). The experiment was performed twice with similar results.
After bioluminescence was reduced to background levels in all organs of BCV-treated infected mice, all mice (including the group of BCV-treated uninfected mice) were rechallenged with 105 PFU of IHD-J-Luc or WRvFire VACV on day 41. As expected, after infection with either IHD-J-Luc or WRvFire VACV on day 41, all control mice that were initially treated with BCV in the absence of the primary infection succumbed on day 49 (Fig. 3A). In contrast, all mice that were infected with IHD-J-Luc in the primary challenge and treated with BCV on days 1, 3, and 5 survived rechallenge with either IHD-J-Luc or WRvFire VACV on day 41 without further BCV treatment (Fig. 3A). The BCV-treated uninfected control mice demonstrated rapid increases in fluxes in all organs after the day 41 challenge (Fig. 3B to E, BCV/IHDJ and BCV/WRvFire). In contrast, no bioluminescence signals were recorded after IHD-J-Luc or WRvFire VACV rechallenge in mice that were initially infected with IHD-J-Luc VACV and treated with BCV. Similar results were observed in BALB/c mice rechallenged with WRvFire or with IHD-J-Luc VACV following initial infection with WRvFire VACV and treatment with three doses of 20 mg/kg BCV (data not shown).
Vaccinia virus-induced pox lesions were scored on tails of the mice used in this experiment (Table 2). The uninfected animals that were treated with 20 mg/kg BCV on days 1, 3, and 5 developed pox lesions following challenge with IHD-J-Luc or WRvFire VACV on day 41, as expected. In contrast, all mice that had primary infection on day 0 and were treated with BCV on days 1, 3, and 5 did not exhibit any pox lesions after rechallenge with IHD-J-Luc or with WRvFire VACV on day 41. Similar results were observed in mice that were infected with WRvFire on day 0 and treated with BCV and rechallenged with WRvFire or with IHD-J-Luc VACV on day 41 (data not shown).
TABLE 2.
Pox lesion counts in mice that were infected with IHD-J-Luc VACV, treated with BCV, and rechallenged with IHD-J-Luc or with WRvFire on day 41
| Treatmenta | Survival (%) | Pox lesion count by day postinfection (mean ± SD)b |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 5 | 6 | 7 | 42 | 44 | 45 | 46 | 49 | 51 | ||
| IHDJ(V) | 0 | 2 ± 1.9 | 3.8 ± 1.6 | 4.2 ± 1.9 | 5.2 ± 1.9 | NA | NA | NA | NA | NA | NA | NA |
| IHDJ(BCV)/IHDJ | 100 | 0 ± 0* | 0 ± 0** | 0 ± 0* | 0 ± 0** | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0† | 0 ± 0† | 0 ± 0 |
| IHDJ(BCV)/WRvFire | 100 | 0 ± 0 | 0 ± 0* | 0 ± 0* | 0 ± 0* | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0† | 0 ± 0† | 0 ± 0 |
| BCV/IHDJ | 0 | NA | NA | NA | NA | NA | 0 ± 0 | 0 ± 0 | 1.2 ± 1.3 | 3.4 ± 2.9 | 4.4 ± 2.7 | NA |
| BCV/WRvFire | 0 | NA | NA | NA | NA | NA | 0 ± 0 | 0 ± 0 | 1.6 ± 2.1 | 2.2 ± 1.8 | 1.4 ± 1.1 | NA |
IHDJ(V), IHDJ-infected and vehicle-treated group; IHDJ(BCV)/IHDJ, IHDJ-infected and BCV-treated group rechallenged with IHDJ; IHDJ(BCV)/WRvFire, IHDJ-infected and BCV-treated group rechallenged with WRvFire; BCV/IHDJ, BCV-treated and IHDJ-infected group; BCV/WRvFire, BCV-treated and WRvFire-infected group.
Pox lesions on mouse tails were counted using dorsal images of individual mice and were used to calculate average pox numbers ± SD per surviving animal on each day shown. Significant differences in mean daily pox counts between groups of mice that received BCV or vehicle on days 1, 3, and 5 following primary infection on day 0 for IHDJ(BCV)/IHDJ and IHDJ(BCV)/WRvFire versus IHDJ(V) are indicated as follows: *, P ≤ 0.05; **, P ≤ 0.001. Significant differences between groups of mice that were infected with IHD-J-Luc and treated with BCV on days 1, 3, and 5 and rechallenged with IHD-J-Luc or WRvFire on day 42 versus mice that were treated with BCV on days 1, 3, and 5 and challenged with IHD-J-Luc or WRvFire on day 42, respectively, for IHDJ(BCV)/IHDJ versus BCV/IHDJ and IHDJ(BCV)/WRvFire versus BCV/WRvFire are also indicated (†, P ≤ 0.05). NA, not applicable.
These data showed that in normal BALB/c mice, primary infection with VACV followed by three doses of orally administered BCV protected animals from lethality, cleared the virus from internal organs, and allowed the generation of an immune response that provided complete protection against subsequent rechallenge with two different VACV strains.
BCV treatment extended survival in immune-deficient BALB/c nu/nu mice following lethal challenge with IHD-J-Luc VACV.
Individuals with severe immune deficiency are highly sensitive to smallpox infection and to complications following smallpox vaccination. Therefore, we evaluated the activity of BCV in athymic nude mice that lack all T cells as a model of poxvirus infection in patients with severe immune deficiency. BALB/c nu/nu mice were infected with 104 PFU of IHD-J-Luc VACV on day 0 and treated with vehicle or 20 mg/kg BCV on days 1, 3, and 5 or on days 1, 3, 5, 7, 10, 14, 17, 21, and 24 postchallenge (Fig. 4A to F). All vehicle-treated nude mice lost weight between days 3 and 8 and succumbed to infection by day 8 (Fig. 4A and B). Mice treated with BCV on days 1, 3, and 5 maintained their weights initially but started to lose weight beginning on day 9 and succumbed between days 24 and 29 postchallenge, indicating the durability of response to treatment with BCV (Fig. 4A and B). Animals that received extended BCV treatment (last treatment on day 24) started to lose weight after day 30, and all succumbed by day 48 (Fig. 4A and B). Importantly, both short and extended treatment with BCV significantly increased median survival time compared with results in vehicle-treated mice (P = 0.0025 and P = 0.0091, respectively) (Fig. 4A). Bioluminescence signals in the organs of mice that received vehicle on days 1, 3, and 5 increased 3 to 4 logs in the nasal cavity, lungs, spleen, and liver between day 1 and days 4 to 6 postchallenge (Fig. 4C to F). In contrast, nude mice that were treated with 20 mg/kg BCV on days 1, 3, and 5 exhibited dramatic reductions in bioluminescence in the lungs, spleen, and liver and a modest reduction in the nasal cavity compared with vehicle-treated mice. BCV continued to control viral loads up to day 10 postinfection in these organs after treatment was stopped. After day 14, viral loads increased and reached levels comparable to those of the vehicle-treated mice by day 21 (Fig. 4C to F). In the extended treatment group, bioluminescence signals remained low in all organs for the duration of treatment and started to go up after day 28 postinfection with the same kinetics seen in the animals that received a short course of treatment (Fig. 4C to F).
FIG 4.

Treatment with BCV extended survival of nude mice following lethal challenge with IHD-J-Luc VACV. BALB/c nu/nu mice were infected with 104 PFU of IHD-J-Luc VACV and treated with vehicle or 20 mg/kg BCV on days 1, 3, and 5 or on days 1, 3, 5, 7, 10, 14, 17, 21, and 24, as indicated. Mice were observed for mortality (A) and weight loss (B) and were imaged daily for the first 10 days and then twice weekly for 45 days to calculate mean fluxes ± SD in the nasal cavity, lungs, spleen, and liver. Mean background levels of fluxes (photons/s) ± SD were recorded in nude mice prior to infection: nasal cavity, 23.3 × 103 ± 4.5 × 103; lungs, 36.3 × 103 ± 11.6 × 103; spleen, 10.9 × 103 ± 2.5 × 103; liver, 30.2 × 103 ± 2.1 × 103. Values are represented as dotted horizontal lines (means only) in panels C to F. Four mice per group were used in the experiment. Short and extended treatment with BCV significantly increased median survival time compared with vehicle-treated mice (P = 0.0025 and P = 0.0091, respectively). The experiment was performed twice with similar results.
To determine whether the BCV short course or extended treatment significantly reduced viral loads in nude mice, AUCs were calculated for individual mice that received vehicle or three doses or nine doses of BCV (Table 3). Mean AUCs were significantly lower in mice that received BCV on days 1, 3, and 5 than in vehicle-treated mice for lungs, liver, and spleens but not the nasal cavity between days 1 and 6, while all mice were alive in both groups (Table 3). We also compared the AUCs of mice that received the extended treatment course of BCV with those that received the three-dose treatment. Mean AUCs were significantly lower in mice that received extended BCV treatment in all organs than in mice that received the three-dose treatment course of BCV between days 1 and 21 while all mice were alive in both groups.
TABLE 3.
AUCs for nude mice infected with IHD-J-Luc VACV and treated with a short or extended course of 20 mg/kg BCV
| Organ | Mean AUC by day and regimen (log10 photons/s × day)a |
|||
|---|---|---|---|---|
| Day 6 |
Day 21 |
|||
| Vehicle | BCV short courseb | BCV short course | BCV extended coursec | |
| Nasal cavity | 42.3 ± 1.4 | 41.4 ± 1.1 | 174.3 ± 1.6 | 162.1 ± 3.5* |
| Lungs | 32.2 ± 1 | 27.3 ± 1.5† | 120.0 ± 3.4 | 104.2 ± 1.8** |
| Liver | 36.4 ± 2.3 | 27.8 ± 2.5† | 115.2 ± 3.0 | 99.8 ± 1.8** |
| Spleen | 35.2 ± 2.7 | 26.2 ± 3.1† | 109.8 ± 2.0 | 92.0 ± 4.3* |
Mean AUCs (log10 photons/s × day) ± SD were calculated for fluxes in mice for days 1 to 6 for the vehicle-treated group and the BCV short course group and for days 1 to 21 for the BCV short course and extended course. Significant differences in mean AUCs between the groups receiving vehicle and the short course of BCV treatment are indicated as follows: †, P ≤ 0.001. Significant differences in mean AUCs between the groups receiving the short course and extended course of BCV treatment are indicated as follows *, P ≤ 0.05; **, P ≤ 0.001. Significance was determined with a two-sample t test.
Treatment on days 1, 3, and 5.
Treatment on days 1, 3, 5, 7, 10, 14, 17, 21, and 24.
These data demonstrated that a short or extended course of BCV treatment did not rescue nude mice from lethality but prolonged survival of the animals for approximately 10 to 20 days after treatment termination for both short (three doses) and extended (nine doses) regimens. While on treatment, mice showed significant reduction in viral loads (AUCs) in internal organs compared with vehicle-treated mice. Viral loads in the lungs, liver, and spleen remained at low levels for an additional 4 to 9 days after treatment termination. Thus, efficient control of virus dissemination to internal organs was afforded by BCV, even in the absence of adaptive immunity, and provided a survival advantage in the immune-deficient animal model.
Nude mice reconstituted with low numbers of T cells and treated with BCV generate VACV-specific immunological memory.
Since the majority of immunocompromised individuals retain some functional T cells and since in some individuals immune deficiency status may be a result of temporary treatment with immune-modulating therapies, we modeled less severe immunocompromised conditions by partially reconstituting nu/nu mice with T cells from normal mice as previously described (29). BALB/c nu/nu mice received an adoptive transfer with 104, 105, or 106 T cells (104 Tc, 105 Tc, or 106 Tc, respectively) isolated from the spleens of normal BALB/c mice, or mice were sham reconstituted with PBS (Fig. 5A). At 1 day postreconstitution, all mice were infected with 104 PFU of IHD-J-Luc VACV and received either no treatment or treatment with vehicle or BCV (20 mg/kg) on days 1, 3, and 5 postchallenge. Mice were observed for mortality and subjected to BLI daily for the first 10 days and then twice weekly or as specified on Fig. 5 for the duration of the experiment. All mice that were sham reconstituted and treated with vehicle or were reconstituted with 106 T cells without any treatment (106 T/NC) succumbed by day 8 (Fig. 5A). All sham-reconstituted mice that received BCV on days 1, 3, and 5 succumbed between days 25 and 30, consistent with the previous study. In contrast, an adoptive transfer of T cells followed by challenge and BCV treatment conferred 100% survival in mice that received 105 T cells and 57% survival in nude mice that received 104 T cells (Fig. 5A). The differences in the median survival times were not statistically significant between mice that received an adoptive transfer with 104 or 105 T cells (P = 0.1514) (Fig. 5).
FIG 5.

Transfer of T cells from normal BALB/c mice to nude animals infected with IHD-J-Luc VACV and treated with BCV resulted in virus clearance and protection from a second high-dose challenge. Nude mice received an adoptive transfer with 104 or 105 T cells or were sham transferred with PBS. All mice were infected with 104 PFU of IHD-J-Luc VACV on day 0 and were treated with vehicle (PBS/vehicle) or 20 mg/kg BCV on days 1, 3, and 5 (purple circles, blue squares, and green triangles); control mice received an adoptive transfer with 106 T cells, were infected with 104 PFU of IHD-J-Luc VACV, and were left untreated (106 Tc/NT) (A and C to F). Mice were observed for mortality (A) and imaged as shown (C to F). On day 55, all mice that survived the initial infection (mice that received an adoptive transfer of 104 or 105 T cells followed by treatment with 20 mg/kg BCV on days 1, 3, and 5) and a control group of naive nude mice (Rechallenge control) were infected with 105 PFU of IHD-J-Luc VACV (B and C to F). Total fluxes in the nasal cavity, lungs, liver, and spleen were determined and used to calculate mean total fluxes ± SD. Mortality curves in panels A and B show survival of mice between day 0 and day 55 and between days 55 and 81, respectively. Fluxes in C, D, E, and F show bioluminescence recorded in all mice used in the experiment between days 0 and 81. Dotted vertical and horizontal lines in panels C to F show day of rechallenge (D55) and mean background levels of fluxes (photons/s) as described in the legend to Fig. 4, respectively. Groups were as follows: PBS/Vehicle, 5 mice; PBS/BCV, 3 mice per group; 104 Tc/BCV, 7 mice per group; 105 Tc/BCV, 4 mice per group; 106 Tc, 2 mice per group; rechallenge control mice (mice that were not infected during primary infection), 2 mice per group. The PBS/BCV treatment group demonstrated a significant increase in median survival time compared to that of PBS/vehicle group (P = 0.0082). Transfer of 104 or of 105 T cells prior to challenge significantly increased median survival time in BCV-treated mice compared with the PBS/BCV treatment group (P = 0.0008 and P = 0.01, respectively); there were no significant increases in median survival times between mice that received 104 or 105 T cells prior to challenge (P = 0.1514). Following rechallenge, median survival time was significantly higher in T cell-reconstituted mice treated with BCV that survived primary infection than in rechallenge control mice (P = 0.0177). The experiment was performed twice with similar results.
There were no differences in bioluminescence signals in all four organs between sham-reconstituted/vehicle-treated mice and mice that were reconstituted with 106 T cells without any treatment, and the kinetics of mortality in these groups were similar. In nonreconstituted mice that were treated with 20 mg/kg BCV, viral replication was controlled in the lungs, liver, and spleen until day 11 and increased thereafter (Fig. 5C to F). In contrast, animals that were reconstituted with 105 T cells and treated with 20 mg/kg BCV after challenge started to clear the virus between days 8 and 11 in the lungs, spleen, liver, and nasal cavity (Fig. 5C to F). In surviving animals that received 104 T cells prior to challenge and treatment with 20 mg/kg BCV, the kinetics of viral clearance was slower than that in animals that received 105 T cells. However, even in this group bioluminescence signals were reduced to background levels by day 55 in surviving animals (Fig. 5C to F).
We next evaluated whether partially reconstituted nude mice that survived the IHD-J-Luc VACV challenge after a short course of BCV treatment (days 1, 3, and 5) also developed vaccinia-specific protective immunity, as was observed in normal BALB/c mice (Fig. 3). To that end, all T cell-reconstituted mice that cleared the first infection (all 4 mice that received an adoptive transfer with 105 T cells and 4/7 mice that received an adoptive transfer with 104 T cells prior to challenge) were rechallenged on day 55 with a higher dose (105 PFU) of IHD-J-Luc VACV, which is lethal in immunocompetent mice. In addition, a separate control group of age-matched nude mice (n = 2) that was not infected during the primary infection was infected with 105 PFU of IHD-J-Luc VACV (rechallenge control) (Fig. 5B). As expected, the rechallenge control nude mice succumbed 10 days after challenge (Fig. 5B). In contrast, all mice that received an adoptive T cell transfer and survived the primary infection also survived the rechallenge with 105 PFU of IHD-J-Luc VACV on day 55 without any weight loss (Fig. 5B and data not shown). A moderate increase in bioluminescence signals was observed in the nasal cavity only on days 2 to 3 postrechallenge, which subsequently decreased to background levels on day 65 in mice that received either 104 or 105 T cells (Fig. 5C). Importantly, in the lungs, spleen, and liver, no major increase in bioluminescence after the day 55 rechallenge was observed (Fig. 5D to F).
For statistical analyses, the AUCs for fluxes in individual mice were calculated for groups of mice when all mice in these groups were alive, as follows: days 1 to 6 for all groups, days 1 to 22 for BCV-treated mice only, days 1 to 29 for T cell-reconstituted and BCV-treated mice only, days 56 to 60 for T cell-reconstituted mice treated with BCV and rechallenge control, and days 56 to 81 for T cell-reconstituted rechallenged mice. Values were then used to calculate mean AUCs ± standard deviations (SD) (Table 4). A transfer of 106 T cells alone did not significantly reduce the mean AUC compared with that of vehicle-treated mice. In contrast, BCV treatment with or without 104 or 105 T cells significantly reduced mean AUCs compared with those of vehicle-treated mice on day 6. At this time point, there were no significant differences between mice that received BCV with or without T cells (data not shown), suggesting that early after infection BCV played the major role in reducing viral replication. By day 22 a significant reduction in mean AUCs in the nasal cavity was observed in T cell-reconstituted compared with nonreconstituted BCV-treated mice and also in the spleens and livers of BCV-treated nude mice reconstituted with 105 but not 104 T cells. In the lungs, mean AUCs between BCV-treated T cell-reconstituted and BCV-treated nonreconstituted mice were not significantly different due to large variability in bioluminescence signals between animals in this organ. By day 29, all mice that were reconstituted with 104 or with 105 T cells and treated with BCV were alive, and at this time point, mean AUCs in all organs except for lungs were significantly lower in mice that were reconstituted with 105 T cells than in those who received 104 T cells. After rechallenge with 105 PFU of IHD-J-Luc VACV, mean AUCs were significantly lower in all four organs of T cell-reconstituted and BCV-treated mice than in control infected and untreated mice between days 56 and 60. In addition, there were no differences in mean AUCs in all four organs between mice reconstituted with 105 or 104 T cells after rechallenge between days 56 and 81 (end of observation period).
TABLE 4.
AUCs for nude mice reconstituted with T cells, infected with IHD-J-Luc VACV, treated with BCV, and rechallenged with IHD-J-Luc VACV on day 55
| Tissue and treatmenta | Mean AUC (log10 photons/s × day) for:b |
||||
|---|---|---|---|---|---|
| Days 1–6 | Days 1–22 | Days 1–29 | Days 56–60 | Days 56–81 | |
| Nasal cavity | |||||
| PBS/vehicle | 41.3 ± 1.5 | ||||
| 106 Tc/NT | 41.4 ± 0.2 | ||||
| PBS/BCV | 38.2 ± 1.1* | 176.9 ± 2.7 | |||
| 104 Tc/BCV | 39.5 ± 1.1* | 167.0 ± 6.9* | 211.9 ± 12.8 | 22.7 ± 5.0* | 46.6 ± 7.5 |
| 105 Tc/BCV | 40.6 ± 0.9* | 141.8 ± 2.8** | 172.4 ± 2.6** | 18.6 ± 1.5** | 42.2 ± 1.9 |
| Rechallenge control | 34.2 ± 0.7 | ||||
| Lungs | |||||
| PBS/vehicle | 34.7 ± 1.5 | ||||
| 106 Tc/NT | 33.5 ± 0.5 | ||||
| PBS/BCV | 29.7 ± 3.9* | 131.7 ± 16.7 | |||
| 104 Tc/BCV | 30.7 ± 3.2* | 126.5 ± 11.6 | 163.7 ± 14.7 | 20.0 ± 1.0* | 43.6 ± 2.3 |
| 105 Tc/BCV | 32.7 ± 0.5* | 119.1 ± 4.4 | 151.5 ± 4.6 | 19.3 ± 0.7* | 43.8 ± 1.3 |
| Rechallenge control | 26.2 ± 2.3 | ||||
| Liver | |||||
| PBS/vehicle | 35.6 ± 0.7 | ||||
| 106 Tc/NT | 34.5 ± 0.4 | ||||
| PBS/BCV | 27.1 ± 2.7** | 132.0 ± 19.2 | |||
| 104 Tc/BCV | 28.4 ± 1.5** | 123.7 ± 11.2 | 162.0 ± 16.0 | 18.8 ± 0.6** | 42.3 ± 2.2 |
| 105 Tc/BCV | 29.7 ± 0.8** | 110.7 ± 2.1* | 143.2 ± 2.2* | 18.6 ± 0.2** | 43.3 ± 1.6 |
| Rechallenge control | 27.4 ± 0.7 | ||||
| Spleen | |||||
| PBS/vehicle | 34.7 ± 0.9 | ||||
| 106 Tc/NT | 33.3 ± 0.1 | ||||
| PBS/BCV | 25.0 ± 2.6** | 128.3 ± 18.8 | |||
| 104 Tc/BCV | 26.4 ± 1.4** | 119.8 ± 11.5 | 156.1 ± 14.7 | 17.6 ± 0.6** | 39.9 ± 2.5 |
| 105 Tc/BCV | 27.5 ± 0.5 ** | 102.0 ± 2.8* | 132.4 ± 3.1* | 17.2 ± 0.2** | 40.2 ± 1.6 |
| Rechallenge control | 26.3 ± 0.5 | ||||
PBS/vehicle, PBS-reconstituted mice treated with vehicle postchallenge; PBS/BCV, PBS-reconstituted mice treated with BCV postchallenge; 104 Tc/BCV, nude mice reconstituted with 104 T cells and treated with BCV postchallenge; 105 Tc/BCV, nude mice reconstituted with 105 T cells and treated with BCV postchallenge; 106 Tc, nude mice reconstituted with 106T cells without BCV treatment; rechallenge control, naive nude mice; NT, not treated.
Mean AUCs ± SD were calculated for fluxes in mice for the groups of mice where all mice survived during the time frame. Significant differences were determined for the following groups: for days 1 to 6, between mean AUCs in PBS/vehicle versus 106 Tc/NT, PBS/BCV, 104 Tc/BCV, and 105 Tc/BCV groups; for days 1 to 22, between PBS/BCV and 104 Tc/BCV and 105 Tc/BCV groups; for days 1 to 29, between 104 Tc/BCV and 105 Tc/BCV groups; for days 56 to 60, between the rechallenge control and 104 Tc/BCV and 105 Tc/BCV groups. *, P ≤ 0.05; **, P ≤ 0.001.
The comparison of mean AUCs between groups of reconstituted mice suggested that reconstitution with 105 T cells was more efficient than 104 T cells in curtailing viral replication in BCV-treated mice after primary infection. However, memory response in mice that survived the primary infection was similarly effective in controlling viral dissemination following rechallenge in mice that initially were reconstituted with either 105 or with 104 T cells.
Dorsal images of the same mice were used to monitor pox lesion development. No poxes were observed in mice on days 1 to 3 postchallenge (data not shown). As expected, T cells alone did not significantly reduce pox numbers compared with results in sham-reconstituted mice (Table 5). In contrast, BCV treatment, regardless of whether mice were sham reconstituted or reconstituted with T cells, significantly reduced pox lesions on days 4 and 6 postchallenge compared with vehicle. In all BCV-treated groups, irrespective of T cell reconstitution, the numbers of poxes did not differ significantly between days 4 and 11 (data not shown). Starting from day 19, no pox lesions were scored in mice that were reconstituted with 105 T cells prior to the first challenge/BCV treatment. Importantly, the same mice did not develop any pox lesions after rechallenge on day 55, and minimal numbers of lesions were observed in mice that were reconstituted with 104 T cells before the first challenge. At the end of the observation period on day 81, all surviving mice were pox lesion free.
TABLE 5.
Pox lesion counts for nude mice reconstituted with T cells, infected with IHD-J-Luc VACV, treated with BCV postchallenge, and then rechallenged with IHD-J-Luc VACV on day 55a
| Treatment group | Survival (%) | Pox lesion count by day postinfection (mean ± SD)b |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 6 | 7 | 11 | 19 | 26 | 33 | 46 | 55 | 60 | 65 | 81 | ||
| PBS/vehicle | 0 | 1.0 ± 0.7 | 4.8 ± 3.6 | 12.0 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 106 Tc/NT | 0 | 1.0 ± 1.4 | 6.0 ± 1.4 | 8.5 ± 0.7 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| PBS/BCV | 0 | 0* | 0* | 1.3 ± 1.2 | 2.3 ± 2.5 | 1.3 ± 2.3 | 1.0 ± 1.0 | NA | NA | NA | NA | NA | NA |
| 104 Tc/BCV | 43 | 0* | 0.3 ± 0.5* | 0.4 ± 0.5 | 0.9 ± 1.9 | 0.6 ± 0.5 | 1.3 ± 1.5 | 0.8 ± 1.0 | 0.5 ± 1.0 | 0.3 ± 0.6 | 0.3 ± 0.6 | 0 | 0 |
| 105 Tc/BCV | 100 | 0.2 ± 0.4* | 1.2 ± 0.5* | 0.8 ± 0.8 | 0.2 ± 0.4 | 0 | 0† | 0 | 0 | 0 | 0‡ | 0 | 0 |
| Rechallenge Control | 0 | NA | NA | NA | NA | NA | NA | NA | NA | 0 | 1.5 ± 0.7 | 2 | NA |
Mice were treated with 20 mg/kg BCV on days 1, 3, and 5 postchallenge.
Pox lesions on mouse tails were counted using dorsal images of individual mice and were used to calculate average pox numbers ± SD per surviving animal for each day. *, P ≤ 0.05 for differences between PBS/vehicle versus 106 Tc/NT, PBS/BCV, 104 Tc/BCV, and 105 Tc/BCV groups of mice in mean daily pox counts; †, P ≤ 0.05 for differences between PBS/BCV versus 104 Tc/BCV and 105 Tc/BCV groups of mice; ‡, P ≤ 0.05 for differences between rechallenge control mice versus 104 Tc/BCV and 105 Tc/BCV groups of mice. NA, not applicable.
Together, the data demonstrate that partial reconstitution of nude mice 1 day prior to lethal challenge with VACV, in combination with BCV treatment postchallenge, protected animals from lethality and significantly reduced viral loads, as measured by bioimaging in multiple organs. In addition, T cell-reconstituted mice developed strong adaptive immune responses that protected them from subsequent high-dose rechallenge with no requirement for additional antiviral treatment.
DISCUSSION
BCV (brincidofovir) is an orally bioavailable lipid conjugate of cidofovir with in vitro broad-spectrum activity and an improved safety profile, most notably a reduced risk of nephrotoxicity. In this study, we used normal BALB/c mice, athymic nude BALB/c mice, and nude mice partially reconstituted with T cells from normal BALB/c mice to assess the effects of postchallenge BCV administration on protection of mice from lethal infection with IHD-J-Luc VACV. Infected mice were subjected to bioluminescence imaging (BLI), and the efficacy of BCV was determined by comparing AUCs calculated from daily fluxes acquired from individual mice. The main results of the study were as follows: (i) 3 days of treatment every 48 h with 20 mg/kg BCV starting 1 day postchallenge protected 100% of normal BALB/c mice from lethality, significantly reduced viral loads (mean AUCs) in the spleen, liver, lungs, and nasal cavity, and prevented pox lesion development; (ii) BALB/c mice that were infected and treated with BCV and that survived challenge were also protected from lethality following rechallenge with IHD-J-Luc or with WRvFire VACV on day 55; (iii) following rechallenge, bioluminescence in internal organs was at background levels, suggesting that a strong VACV-specific immune response was developed in mice following primary infection and treatment with BCV; (iv) 20 mg/kg BCV administered either three or nine times postchallenge prolonged survival of nude mice up to 19 days after treatment termination; (v) viral loads were significantly reduced in the lungs, spleens, and livers of BCV-treated nude mice up to 7 days after treatment termination; (vi) partial reconstitution with T cells 1 day before challenge, followed by BCV treatment on days 1, 3, and 5, protected nude mice from lethality, significantly reduced viral loads in internal organs, and prevented the development of pox lesions; and (vii) importantly, T cell-reconstituted and BCV-treated nude mice that survived primary infection also survived a subsequent IHD-J-Luc rechallenge with a 10-times-larger viral inoculum on day 41 without additional BCV treatment.
Several previous studies demonstrated the efficacy of BCV against lethal orthopoxvirus infection in animal models of smallpox. In rabbits, a single dose of 20 mg/kg BCV administered up to 4 days postchallenge rescued animals from lethality after intradermal infection with rabbitpox virus, with additional doses providing added therapeutic benefit (23). In mice, five daily administrations of BCV at 6.7 mg/kg, 8 mg/kg, and 10 mg/kg were sufficient to protect mice from lethality induced by infection with cowpox virus, ectromelia virus, and VACV, respectively (22, 35). In the current study, three doses of 20 mg/kg or 5 mg/kg BCV administered every other day starting 1 day postchallenge protected mice from lethality. In addition, mice treated with 20 mg/kg BCV beginning 2 days postchallenge were also protected from lethality.
The advantage of bioimaging is the ability to monitor viral replication at the site of challenge (nasal cavity) and its subsequent dissemination to the lungs and other internal organs of living mice as the infection progresses. The analysis of the mean AUCs showed that 20 mg/kg BCV starting on day 1 was superior to 5 mg/kg starting on day 1 and to 20 mg/kg starting on day 2 in reducing viral loads in organs in spite of complete protection from lethality in all three regimens. In addition, 20 mg/kg of BCV starting on day 1 completely prevented pox lesion development. Delayed treatment initiation was less efficient, suggesting that there might be a 24-h window when BCV can prevent dissemination of VACV to the skin following intranasal challenge.
Mice that survived lethal infection with IHD-J-Luc VACV also survived rechallenge with IHD-J-Luc or with WRvFire VACV. The WRvFire and IHD-J-Luc VACV that were generated from WR and IHD-J strains of VACV, respectively, are very similar but not identical. They were previously shown to produce similar amounts of mature virion (MV) particles but different quantities of released enveloped virion (EV) particles (36). Our data from rechallenge experiments show that a primary infection with VACV followed by BCV three-dose treatment induced a strong VACV-specific immunity that does not discriminate between strains of VACV with different ratios of MV to EV particles. It is also interesting that in our previous studies, initial replication of WRvFire was noted by BLI in the nasal cavity and lungs of BALB/c mice immunized with Dryvax vaccine via the peritoneal route (27). In the current study, no bioluminescence was detected in any organ above background in mice after rechallenge with WRvFire or with IHD-J-Luc VACV (i.e., sterilizing immunity). We speculate that BCV administered orally in mice infected with VACV via the intranasal route reduced virus replication but allowed for sufficient exposure of the immune system to drive the differentiation of VACV-specific immunity.
In our study we also assessed the effects of BCV on protection of immune-deficient mice from lethality in a VACV challenge model for the first time. The data showed that a three-dose or nine-dose regimen of BCV extended survival of nude mice. These data are in agreement with previous reports on another VACV inhibitor, ST-246, including our own studies, demonstrating that the virostatic drug protected immune-deficient mice from lethality while they were on treatment (29, 37). As expected, nude mice succumbed after BCV was stopped. However, it is important that the survival of these animals was extended for about 10 to 20 days after the last dose of BCV was administered, which may be attributed to the long intracellular half-life of CDV-PP, the active antiviral metabolite of brincidofovir (38). Daily monitoring of viral loads in nude mice showed that BCV significantly reduced VACV replication in the lungs, spleen, and liver but not in the nasal cavity (no significant difference in AUCs between mice treated with vehicle compared to those treated with BCV on days 1, 3, and 5) (Table 3). It is possible that as a result of oral administration, BCV concentrates and efficiently controls viral replication in the liver, spleen, and lungs, while control of viral replication in the nasal cavity might depend more on T cells (see below).
In addition to nude mice, we used nude mice that were reconstituted with T cells purified from naive BALB/c mice in order to better mimic patient groups with various levels of T cell immune deficiencies. In agreement with our previous studies, 106 T cells adoptively transferred into nude mice failed to protect them from infection or to reduce viral loads (29). In contrast, BCV treatment in combination with an adoptive transfer of 105 or 104 T cells conferred survival to 100% or 60% of mice, respectively. This differs from ST-246, where only mice that received 105 T cells survived (29). The lower pool of T cells that was sufficient to induce partial protection in combination with BCV in the current study can possibly be attributed to its longer half-life in cells that allows expansion of low-frequency VACV-specific T cells and their differentiation into effector T cells under the umbrella of drug treatment.
Bioimaging of infected nude mice showed that early after initiation of treatment with BCV (day 6), all BCV-treated mice, with or without adoptive T cell transfer, exhibited significantly lower viral loads in all four organs. Later in the infection (day 22), the contribution of T cells to the protective effect from BCV could be noted since mean AUCs were significantly lower in at least three organs (nasal cavity, spleen, and liver) in BCV-treated mice reconstituted with 105 T cells than in mice treated with BCV alone. As expected, transfer of 104 T cells resulted in a smaller impact on survival and viral loads than 105 T cells, suggesting a threshold effect in this model. Namely, a combination of the three-dose BCV treatment with transfer of 104 T cells protected only ∼60% of nude mice challenged with a lethal dose of VACV. Similarly, on day 29 (when all mice that received adoptive transfer with 104 or 105 T cells were alive) 104 T cells were less efficient than 105 T cells in controlling viral loads in all organs (Table 4, day 29). Importantly, unlike nonreconstituted nude mice, where virus reemerged about 4 to 9 days after the last dose of BCV, in all surviving nude mice that were reconstituted with 104 or 105 T cells and treated with BCV, bioluminescence was reduced to background levels 50 or 10 days after the last dose of BCV, respectively. Interestingly, there were no significant differences in mean AUCs after rechallenge between mice that were reconstituted with 104 or 105 T cells before the primary infection (Table 4, days 56 to 81). These data demonstrate that the quality of generated VACV-specific immune memory as a result of the primary infection in the presence of BCV was similar between mice that were reconstituted with either 104 or 105 T cells and was sufficient to confer protection from a rechallenge with 105 PFU of IHD-J-Luc VACV, which is a lethal dose for normal BALB/c mice.
VACV has a substantial genome (>200 open reading frames) coding for a large number of potential T cell epitopes identified in mice and humans (reviewed in reference 39). Recent studies have shown that some T cell epitopes elicit more protective immunity than others. For example, of the 49 different CD8+ T cell epitopes studied in C57BL/6 mice, 2 epitopes conferred 100% protection from lethal intranasal infection with VACV (40). In another study, five immune-dominant epitopes out of the predicted 258 open reading frames accounted for half of the VACV-specific CD8+ T cells in C57BL/6 mice (41). Furthermore, immune dominance was greatly affected by the route of VACV infection (41). In our experiments all nude mice that received 105 T cells survived lethal challenge, and of the nude mice that received 104 T cells, ∼60% survived. Based on the calculated precursor frequency of VACV-specific CD8+ T cells of 1:1,500 (42), it could be predicted that at least 22 and 2.2 VACV-specific naive CD8+ T cells were present in the pool of 105 and 104 transferred T cells, respectively. Yet these low numbers of VACV-specific T cells combined with BCV were sufficient to protect 100 and 60% of mice from lethal challenge. Though the specificities of VACV-specific precursors in the pools of transferred 105 and 104 T cells were not identified in our study, it is possible to speculate that the intranasal route of infection in the presence of BCV provided an optimal environment for expansion and differentiation of T cell clones that were responsible for clearance of the virus. Our current study emphasized the role of T cells in protection of mice from lethal challenge with VACV, and the role of neutralizing antibodies (Abs) was not directly addressed. However, in our previous study, neutralizing Abs were detected at 1 and 2 months postchallenge in 33 to 50% of nude mice reconstituted with 105 T cells and treated with ST-246 postchallenge with IHD-J VACV (29). Based on these data, it is conceivable that at the time of rechallenge in the current experiments, neutralizing Abs and VACV-specific memory B cells most likely contributed to the complete protection from viral dissemination. Future studies are planned to explore the effects of BCV on the kinetics of generation of neutralizing Abs and also the role of memory B cells in protection from lethal infection with VACV of nude mice partially reconstituted with T cells.
It is unethical to intentionally infect humans with variola virus in order to conduct studies of potential smallpox therapeutics. Therefore, developments of animal models of smallpox that mimic key aspects of the disease are essential in order to evaluate their efficacy. The intranasal vaccinia virus model in normal and immune-deficient nude BALB/c mice is one such model. The ability to model disease and the efficacy of potential therapeutics under various immune-deficient/compromised conditions is highly useful and relevant to the anticipated use of smallpox antivirals in an outbreak. Although vaccination is expected to be the primary response to a smallpox outbreak, vaccination is contraindicated in immunocompromised patients; hence, it is these patients who are most likely to receive smallpox antivirals. Application of bioimaging to a traditional model of lethal challenge with VACV in both normal and immune-deficient mice allowed us to gain insight on the protection from lethality conferred by BCV. The ability of BCV to control viral replication at the site of challenge and to successfully reduce viral dissemination to internal organs provides a critical shield for the development of an adaptive immune response that clears the host of VACV and builds VACV-specific immunological memory.
ACKNOWLEDGMENTS
We are grateful to Clement Meseda and Alonso Garcia for careful reading of the manuscript.
This project has been funded in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under IAA 224-06-1322 and by FDA's Medical Countermeasure Initiative fund.
REFERENCES
- 1.Breman JG, Henderson DA. 1998. Poxvirus dilemmas–monkeypox, smallpox, and biologic terrorism. N Engl J Med 339:556–559. doi: 10.1056/NEJM199808203390811. [DOI] [PubMed] [Google Scholar]
- 2.Zaucha GM, Jahrling PB, Geisbert TW, Swearengen JR, Hensley L. 2001. The pathology of experimental aerosolized monkeypox virus infection in cynomolgus monkeys (Macaca fascicularis). Lab Invest 81:1581–1600. doi: 10.1038/labinvest.3780373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention. 2003. Update: multistate outbreak of monkeypox: Illinois, Indiana, Kansas, Missouri, Ohio, and Wisconsin, 2003. MMWR Morb Mortal Wkly Rep 52:642–646. [PubMed] [Google Scholar]
- 4.Parker S, Nuara A, Buller RM, Schultz DA. 2007. Human monkeypox: an emerging zoonotic disease. Future Microbiol 2:17–34. doi: 10.2217/17460913.2.1.17. [DOI] [PubMed] [Google Scholar]
- 5.Lane JM, Goldstein J. 2003. Adverse events occurring after smallpox vaccination. Semin Pediatr Infect Dis 14:189–195. doi: 10.1016/S1045-1870(03)00032-3. [DOI] [PubMed] [Google Scholar]
- 6.Handley L, Buller RM, Frey SE, Bellone C, Parker S. 2009. The new ACAM2000 vaccine and other therapies to control orthopoxvirus outbreaks and bioterror attacks. Expert Rev Vaccines 8:841–850. doi: 10.1586/erv.09.55. [DOI] [PubMed] [Google Scholar]
- 7.Engler RJ, Kenner J, Leung DY. 2002. Smallpox vaccination: risk considerations for patients with atopic dermatitis. J Allergy Clin Immunol 110:357–365. doi: 10.1067/mai.2002.128052. [DOI] [PubMed] [Google Scholar]
- 8.Wittek R. 2006. Vaccinia immune globulin: current policies, preparedness, and product safety and efficacy. Int J Infect Dis 10:193–201. doi: 10.1016/j.ijid.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Lederman ER, Davidson W, Groff HL, Smith SK, Warkentien T, Li Y, Wilkins KA, Karem KL, Akondy RS, Ahmed R, Frace M, Shieh WJ, Zaki S, Hruby DE, Painter WP, Bergman KL, Cohen JI, Damon IK. 2012. Progressive vaccinia: case description and laboratory-guided therapy with vaccinia immune globulin, ST-246, and CMX001. J Infect Dis 206:1372–1385. doi: 10.1093/infdis/jis510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Clercq E, Holy A. 1991. Efficacy of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine in various models of herpes simplex virus infection in mice. Antimicrob Agents Chemother 35:701–706. doi: 10.1128/AAC.35.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neyts J, Sobis H, Snoeck R, Vandeputte M, De Clercq E. 1993. Efficacy of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)-cytosine and 9-(1,3-dihydroxy-2-propoxymethyl)-guanine in the treatment of intracerebral murine cytomegalovirus infections in immunocompetent and immunodeficient mice. Eur J Clin Microbiol Infect Dis 12:269–279. doi: 10.1007/BF01967257. [DOI] [PubMed] [Google Scholar]
- 12.Neyts J, De Clercq E. 1993. Efficacy of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine for the treatment of lethal vaccinia virus infections in severe combined immune deficiency (SCID) mice. J Med Virol 41:242–246. doi: 10.1002/jmv.1890410312. [DOI] [PubMed] [Google Scholar]
- 13.Bray M, Martinez M, Smee DF, Kefauver D, Thompson E, Huggins JW. 2000. Cidofovir protects mice against lethal aerosol or intranasal cowpox virus challenge. J Infect Dis 181:10–19. doi: 10.1086/315190. [DOI] [PubMed] [Google Scholar]
- 14.Roy CJ, Baker R, Washburn K, Bray M. 2003. Aerosolized cidofovir is retained in the respiratory tract and protects mice against intranasal cowpox virus challenge. Antimicrob Agents Chemother 47:2933–2937. doi: 10.1128/AAC.47.9.2933-2937.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stittelaar KJ, Neyts J, Naesens L, van Amerongen G, van Lavieren RF, Holy A, De Clercq E, Niesters HG, Fries E, Maas C, Mulder PG, van der Zeijst BA, Osterhaus AD. 2006. Antiviral treatment is more effective than smallpox vaccination upon lethal monkeypox virus infection. Nature 439:745–748. doi: 10.1038/nature04295. [DOI] [PubMed] [Google Scholar]
- 16.Cundy KC. 1999. Clinical pharmacokinetics of the antiviral nucleotide analogues cidofovir and adefovir. Clin Pharmacokinet 36:127–143. doi: 10.2165/00003088-199936020-00004. [DOI] [PubMed] [Google Scholar]
- 17.Neyts J, De Clercq E. 2003. Therapy and short-term prophylaxis of poxvirus infections: historical background and perspectives. Antiviral Res 57:25–33. doi: 10.1016/S0166-3542(02)00197-3. [DOI] [PubMed] [Google Scholar]
- 18.Kern ER, Hartline C, Harden E, Keith K, Rodriguez N, Beadle JR, Hostetler KY. 2002. Enhanced inhibition of orthopoxvirus replication in vitro by alkoxyalkyl esters of cidofovir and cyclic cidofovir. Antimicrob Agents Chemother 46:991–995. doi: 10.1128/AAC.46.4.991-995.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beadle JR, Hartline C, Aldern KA, Rodriguez N, Harden E, Kern ER, Hostetler KY. 2002. Alkoxyalkyl esters of cidofovir and cyclic cidofovir exhibit multiple-log enhancement of antiviral activity against cytomegalovirus and herpesvirus replication in vitro. Antimicrob Agents Chemother 46:2381–2386. doi: 10.1128/AAC.46.8.2381-2386.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciesla SL, Trahan J, Wan WB, Beadle JR, Aldern KA, Painter GR, Hostetler KY. 2003. Esterification of cidofovir with alkoxyalkanols increases oral bioavailability and diminishes drug accumulation in kidney. Antiviral Res 59:163–171. doi: 10.1016/S0166-3542(03)00110-4. [DOI] [PubMed] [Google Scholar]
- 21.Parker S, Siddiqui AM, Oberle C, Hembrador E, Lanier R, Painter G, Robertson A, Buller RM. 2009. Mousepox in the C57BL/6 strain provides an improved model for evaluating anti-poxvirus therapies. Virology 385:11–21. doi: 10.1016/j.virol.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quenelle DC, Collins DJ, Wan WB, Beadle JR, Hostetler KY, Kern ER. 2004. Oral treatment of cowpox and vaccinia virus infections in mice with ether lipid esters of cidofovir. Antimicrob Agents Chemother 48:404–412. doi: 10.1128/AAC.48.2.404-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rice AD, Adams MM, Wallace G, Burrage AM, Lindsey SF, Smith AJ, Swetnam D, Manning BR, Gray SA, Lampert B, Foster S, Lanier R, Robertson A, Painter G, Moyer RW. 2011. Efficacy of CMX001 as a post exposure antiviral in New Zealand White rabbits infected with rabbitpox virus, a model for orthopoxvirus infections of humans. Viruses 3:47–62. doi: 10.3390/v3010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parker S, Crump R, Foster S, Hartzler H, Hembrador E, Lanier ER, Painter G, Schriewer J, Trost LC, Buller RM. 2014. Co-administration of the broad-spectrum antiviral, brincidofovir (CMX001), with smallpox vaccine does not compromise vaccine protection in mice challenged with ectromelia virus. Antiviral Res 111:42–52. doi: 10.1016/j.antiviral.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez JF, Rodriguez D, Rodriguez JR, McGowan EB, Esteban M. 1988. Expression of the firefly luciferase gene in vaccinia virus: a highly sensitive gene marker to follow virus dissemination in tissues of infected animals. Proc Natl Acad Sci U S A 85:1667–1671. doi: 10.1073/pnas.85.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luker KE, Hutchens M, Schultz T, Pekosz A, Luker GD. 2005. Bioluminescence imaging of vaccinia virus: effects of interferon on viral replication and spread. Virology 341:284–300. doi: 10.1016/j.virol.2005.06.049. [DOI] [PubMed] [Google Scholar]
- 27.Zaitseva M, Kapnick SM, Scott J, King LR, Manischewitz J, Sirota L, Kodihalli S, Golding H. 2009. Application of bioluminescence imaging to the prediction of lethality in vaccinia virus-infected mice. J Virol 83:10437–10447. doi: 10.1128/JVI.01296-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zaitseva M, Kapnick SM, Meseda CA, Shotwell E, King LR, Manischewitz J, Scott J, Kodihalli S, Merchlinsky M, Nielsen H, Lantto J, Weir JP, Golding H. 2011. Passive immunotherapies protect WRvFire and IHD-J-Luc vaccinia virus-infected mice from lethality by reducing viral loads in the upper respiratory tract and internal organs. J Virol 85:9147–9158. doi: 10.1128/JVI.00121-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zaitseva M, Shotwell E, Scott J, Cruz S, King LR, Manischewitz J, Diaz CG, Jordan RA, Grosenbach DW, Golding H. 2013. Effects of postchallenge administration of ST-246 on dissemination of IHD-J-Luc vaccinia virus in normal mice and in immune-deficient mice reconstituted with T cells. J Virol 87:5564–5576. doi: 10.1128/JVI.03426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meseda CA, Campbell J, Kumar A, Garcia AD, Merchlinsky M, Weir JP. 2013. Effect of the deletion of genes encoding proteins of the extracellular virion form of vaccinia virus on vaccine immunogenicity and protective effectiveness in the mouse model. PLoS One 8:e67984. doi: 10.1371/journal.pone.0067984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Townsley AC, Weisberg AS, Wagenaar TR, Moss B. 2006. Vaccinia virus entry into cells via a low-pH-dependent endosomal pathway. J Virol 80:8899–8908. doi: 10.1128/JVI.01053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parker S, Touchette E, Oberle C, Almond M, Robertson A, Trost LC, Lampert B, Painter G, Buller RM. 2008. Efficacy of therapeutic intervention with an oral ether-lipid analogue of cidofovir (CMX001) in a lethal mousepox model. Antiviral Res 77:39–49. doi: 10.1016/j.antiviral.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaitseva M, Kapnick S, Golding H. 2012. Measurements of vaccinia virus dissemination using whole body imaging: approaches for predicting of lethality in challenge models and testing of vaccines and antiviral treatments. Methods Mol Biol 890:161–176. doi: 10.1007/978-1-61779-876-4_10. [DOI] [PubMed] [Google Scholar]
- 34.Golden JW, Zaitseva M, Kapnick S, Fisher RW, Mikolajczyk MG, Ballantyne J, Golding H, Hooper JW. 2011. Polyclonal antibody cocktails generated using DNA vaccine technology protect in murine models of orthopoxvirus disease. Virol J 8:441. doi: 10.1186/1743-422X-8-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hostetler KY, Beadle JR, Trahan J, Aldern KA, Owens G, Schriewer J, Melman L, Buller RM. 2007. Oral 1-O-octadecyl-2-O-benzyl-sn-glycero-3-cidofovir targets the lung and is effective against a lethal respiratory challenge with ectromelia virus in mice. Antiviral Res 73:212–218. doi: 10.1016/j.antiviral.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blasco R, Moss B. 1992. Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J Virol 66:4170–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grosenbach DW, Berhanu A, King DS, Mosier S, Jones KF, Jordan RA, Bolken TC, Hruby DE. 2010. Efficacy of ST-246 versus lethal poxvirus challenge in immunodeficient mice. Proc Natl Acad Sci U S A 107:838–843. doi: 10.1073/pnas.0912134107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ho HT, Woods KL, Bronson JJ, De Boeck H, Martin JC, Hitchcock MJ. 1992. Intracellular metabolism of the antiherpes agent (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine. Mol Pharmacol 41:197–202. [PubMed] [Google Scholar]
- 39.Moutaftsi M, Tscharke DC, Vaughan K, Koelle DM, Stern L, Calvo-Calle M, Ennis F, Terajima M, Sutter G, Crotty S, Drexler I, Franchini G, Yewdell JW, Head SR, Blum J, Peters B, Sette A. 2010. Uncovering the interplay between CD8, CD4 and antibody responses to complex pathogens. Future Microbiol 5:221–239. doi: 10.2217/fmb.09.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moutaftsi M, Salek-Ardakani S, Croft M, Peters B, Sidney J, Grey H, Sette A. 2009. Correlates of protection efficacy induced by vaccinia virus-specific CD8+ T-cell epitopes in the murine intranasal challenge model. Eur J Immunol 39:717–722. doi: 10.1002/eji.200838815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tscharke DC, Karupiah G, Zhou J, Palmore T, Irvine KR, Haeryfar SM, Williams S, Sidney J, Sette A, Bennink JR, Yewdell JW. 2005. Identification of poxvirus CD8+ T cell determinants to enable rational design and characterization of smallpox vaccines. J Exp Med 201:95–104. doi: 10.1084/jem.20041912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seedhom MO, Jellison ER, Daniels KA, Welsh RM. 2009. High frequencies of virus-specific CD8+ T-cell precursors. J Virol 83:12907–12916. doi: 10.1128/JVI.01722-09. [DOI] [PMC free article] [PubMed] [Google Scholar]