

ABSTRACT
Human cytomegalovirus (HCMV) immediate early protein IE1 and the tegument protein pp71 are required for efficient infection. These proteins have some functional similarities with herpes simplex virus 1 (HSV-1) immediate early protein ICP0, which stimulates lytic HSV-1 infection and derepresses quiescent HSV-1 genomes. All three proteins counteract antiviral restriction mediated by one or more components of promyelocytic leukemia (PML) nuclear bodies, and IE1 and pp71, acting together, almost completely complement ICP0 null mutant HSV-1. Here, we investigated whether ICP0 might substitute for IE1 or pp71 during HCMV infection. Using human fibroblasts that express ICP0, IE1, or pp71 in an inducible manner, we found that ICP0 stimulated replication of both wild-type (wt) and pp71 mutant HCMV while IE1 increased wt HCMV plaque formation and completely complemented the IE1 mutant. Although ICP0 stimulated IE2 expression from IE1 mutant HCMV and increased the number of IE2-positive cells, it could not compensate for IE1 in full lytic replication. These results are consistent with previous evidence that both IE1 and IE2 are required for efficient HCMV gene expression, but they also imply that IE2 functionality is influenced specifically by IE1, either directly or indirectly, and that IE1 may include sequences that have HCMV-specific functions. We discovered a mutant form of IE1 (YL2) that fails to stimulate HCMV infection while retaining 30 to 80% of the activity of the wt protein in complementing ICP0 null mutant HSV-1. It is intriguing that the YL2 mutation is situated in the region of IE1 that is shared with IE2 and which is highly conserved among primate cytomegaloviruses.
IMPORTANCE Herpesvirus gene expression can be repressed by cellular restriction factors, one group of which is associated with structures known as ND10 or PML nuclear bodies (PML NBs). Regulatory proteins of several herpesviruses interfere with PML NB-mediated repression, and in some cases their activities are transferrable between different viruses. For example, the requirement for ICP0 during herpes simplex virus 1 (HSV-1) infection can be largely replaced by ICP0-related proteins expressed by other alphaherpesviruses and even by a combination of the unrelated IE1 and pp71 proteins of human cytomegalovirus (HCMV). Here, we report that ICP0 stimulates gene expression and replication of wt HCMV but cannot replace the need for IE1 during infection by IE1-defective HCMV mutants. Therefore, IE1 includes HCMV-specific functions that cannot be replaced by ICP0.
INTRODUCTION
Human cytomegalovirus (HCMV) and herpes simplex virus 1 (HSV-1) are important human pathogens belonging to the beta and alpha subfamilies of the herpesviruses, respectively. As with all herpesviruses, they are prevalent in the population because, after an initial infection, the viruses establish latent infections, in the case of HCMV principally in the myeloid cell compartment and in the case of HSV-1 in sensory neurons. Both viruses retain the ability to reactivate from latency (clinically or subclinically), causing recurrent disease and enabling viral transmission among the population. All herpesviruses share a lytic gene expression strategy, with virion components acting to stimulate immediate early (IE) gene expression and then products of the IE genes acting to stimulate the expression of later classes of viral genes (for general reviews of all the above aspects of HSV-1 and HCMV infection, see references 1 to 3). The control of viral gene expression by viral proteins can operationally be seen as two distinct phenomena. One is conducted by viral proteins (such as VP16 and ICP4 of HSV-1) that engage with the cellular transcriptional apparatus and directly stimulate transcription from the viral genome (4, 5). The other concerns the ability to counteract the effects of cellular inhibitory factors that would otherwise repress transcription from the viral genome. The latter process of antiviral restriction is one arm of a general concept known as intrinsic antiviral resistance or intrinsic immunity (6), and it is conducted by constitutively expressed cellular proteins that (in the case of the initial stages of herpesvirus infections) result in the inhibition of viral gene expression. During a normal wild-type (wt) herpesvirus infection, the effects of intrinsic resistance may be slight because the viruses express proteins that counteract the cellular restriction factors. Infection with mutant viruses that lack such functional viral regulatory molecules reveals severe defects in viral gene expression and productive infection (reviewed in references 7 to 11).
Previous work has established that one aspect of antiviral intrinsic resistance is mediated by components of cellular nuclear substructures known as promyelocytic leukemia nuclear bodies (PML NBs, also known as ND10). These dynamic structures include several proteins that are involved in the regulation of cellular gene expression or chromatin modification and assembly, including PML itself, Sp100, human Daxx (hDaxx), and ATRX (12). Both PML and Sp100 are heavily modified by covalent conjugation to the SUMO family of ubiquitin-like proteins, and both PML and its sumoylation are essential for the assembly of authentic PML NBs (12). Over the years, it has become clear that many viruses, and herpesviruses in particular, have intimate connections with PML NBs (8, 9, 11, 13). These cellular proteins are recruited to the sites of HSV-1 genomes as soon as they enter the nucleus (14), and several regulatory herpesviral tegument or IE proteins engage with one or more PML NB proteins in order to counteract their inhibitory effects (see below). In HSV-1, these functions are achieved by the IE protein ICP0, which is a RING finger E3 ubiquitin ligase that, among other activities (15, 16), targets PML and the sumoylated forms of Sp100 for degradation, causing the disruption of PML NBs. ICP0 also changes the behavior of other PML NB components (including hDaxx and ATRX) so that their recruitment to HSV-1 genomes is inhibited (17–21; reviewed in references 7 and 22). In HCMV, the tegument protein pp71 interacts with hDaxx and disrupts the hDaxx/ATRX complex (23–28) while the IE protein IE1 (also known as IE72) causes desumoylation of PML and Sp100 and the disruption of PML NBs (28–35). The biological significance of these phenomena is supported by several studies in which PML, Sp100, hDaxx, and ATRX have been depleted using RNA interference (RNAi), with consequent improvement in replication efficiency of mutant viruses that do not express the relevant viral protein (21, 24–27, 34, 36–41).
It follows from these studies that if PML NB components have a general inhibitory effect on gene expression of different herpesviruses and if major functions of ICP0, pp71, and IE1 counteract this inhibition, then it is possible that these viral proteins could be functionally interchangeable between different viruses. We along with others have previously found that members of the ICP0 family of proteins expressed by other alphaherpesviruses (which also affect PML NBs in one or more ways) can substitute, at least in part, for ICP0 to stimulate HSV-1 infection (42, 43). We have also reported that both pp71 and IE1 can stimulate ICP0 null mutant HSV-1 infection and that the combination of both together does so almost as well as ICP0 itself (44). These findings are consistent with early work reporting that HCMV infection stimulates ICP0 null mutant HSV-1 plaque formation to wt levels (45). In this study, we have conducted the converse experiment to investigate whether ICP0 can functionally substitute for either pp71 or IE1 during HCMV infection. We found that ICP0 increases both wt and pp71 mutant HCMV plaque formation, but while ICP0 augments IE2 expression during IE1 mutant HCMV infection, it could not stimulate the whole HCMV replicative cycle in the absence of IE1. Because the IE1/pp71 combination is so effective at stimulating ICP0 null mutant HSV-1 infection, these results indicate that IE1 includes a function which is HCMV specific. We discuss the possibility that this reflects a specific and substantial requirement for the presence of IE1 to enable the efficient function of another critical HCMV protein, most likely IE2, in the context of HCMV infection.
MATERIALS AND METHODS
Viruses and cells.
HSV-1 wild-type (wt) strain 17+ was used, from which the ICP0 null mutant dl1403 was derived (46). Virus dl1403/CMV lacZ is a derivative of dl1403 that contains the lacZ gene under the control of the HCMV promoter/enhancer inserted into the tk gene (gift from Chris Preston). These viruses were grown in BHK cells and titrated in U2OS cells. The wt HCMV isolate used (designated TNwt) was derived from a bacmid of strain Towne, and it includes a simian virus 40 (SV40)-driven enhanced green fluorescent protein (EGFP) marker gene inserted into a nonessential region of the short unique (US) segment of the viral genome (47). An IE1 deletion mutant (TNdlIE1) that had been derived from the same bacmid was kindly provided by Michael Nevels (48). Stocks of HCMV wt strain TBE40 (described as a clinical isolate [49]) and its IE1 mutant derivative (TBdlIE1) (49) were also very kindly provided by Michael Nevels. The HCMV pp71 deletion mutant ADsubUL82 (50) was kindly provided by Tom Shenk and was used in conjugation with the AD169 parental wt strain. HFT cells expressing IE1 were for preparation and titration of stocks of TNwt, TNdlIE1, TBwt, and TBdlIE1 HCMV, and HFT cells expressing ICP0 were used for the growth of stocks of ADsubUL82. The cells were treated with doxycycline to induce IE1 or ICP0 expression and then infected with the relevant HCMV virus at a multiplicity of infection (MOI) of 1 PFU per cell the following day in the presence of doxycycline. The medium was replaced with doxycycline-free medium the following day; the medium was harvested after a further 12 days and clarified by low-speed centrifugation to remove cell debris, and then the supernatants were stored as virus stocks.
U2OS and HEK-293T cells and human diploid fibroblasts immortalized by expression of human telomerase from a lentivirus vector (51) (HFT cells; lentivirus and cells kindly provided by David Davido and Chris Boutell, respectively) were grown in Dulbecco's modified Eagles' medium supplemented with 10% fetal calf serum (FCS). BHK cells were grown in Glasgow modified Eagles' medium supplemented with 10% newborn calf serum and 10% tryptose phosphate broth. All cell growth media were supplemented with 100 units/ml penicillin and 0.1 mg/ml streptomycin.
Plasmids and lentiviral vectors.
Lentivirus vector plasmids expressing the tetracycline repressor (TetR) linked to a nuclear localization signal (NLS) and enhanced green fluorescent protein (pLKOneo.EGFPnlsTetR) and ICP0 from a tetracycline-inducible promoter (pLDT.cICP0) have been described previously (52), as have derivatives expressing IE1, pp71, and various mutant forms of IE1 (44). We also constructed vectors that have the same tetracycline-inducible expression cassette for ICP0, IE1, and the IE1 mutants of interest but which also include the tetracycline repressor coding sequence downstream of the human phosphoglycerate kinase promoter, followed by an internal ribosome entry site (IRES) element and the puromycin resistance coding region (Fig. 1) (the empty vector was kindly provided by Chris Boutell). Vectors of this type enable inducible expression of a protein following a single transduction of HFT cells. Cells expressing IE1 or ICP0 were isolated after transduction with these single inducible vectors and also by a two-step approach involving the initial isolation of HFT cells expressing EGFPnlsTetR, followed by a second transduction with lentiviruses derived from the previously described pLDT vectors. Cells expressing pp71 were isolated solely by the double-transduction procedure using a blasticidin-resistant pp71 vector, as described previously (44). HFT cells that inducibly express both pp71 and IE1 were isolated by a sequential triple-transduction method using EGFPnlsTetR (neomycin selection), IE1 (puromycin selection), and pp71 (blasticidin selection) lentiviral vectors (44).
FIG 1.
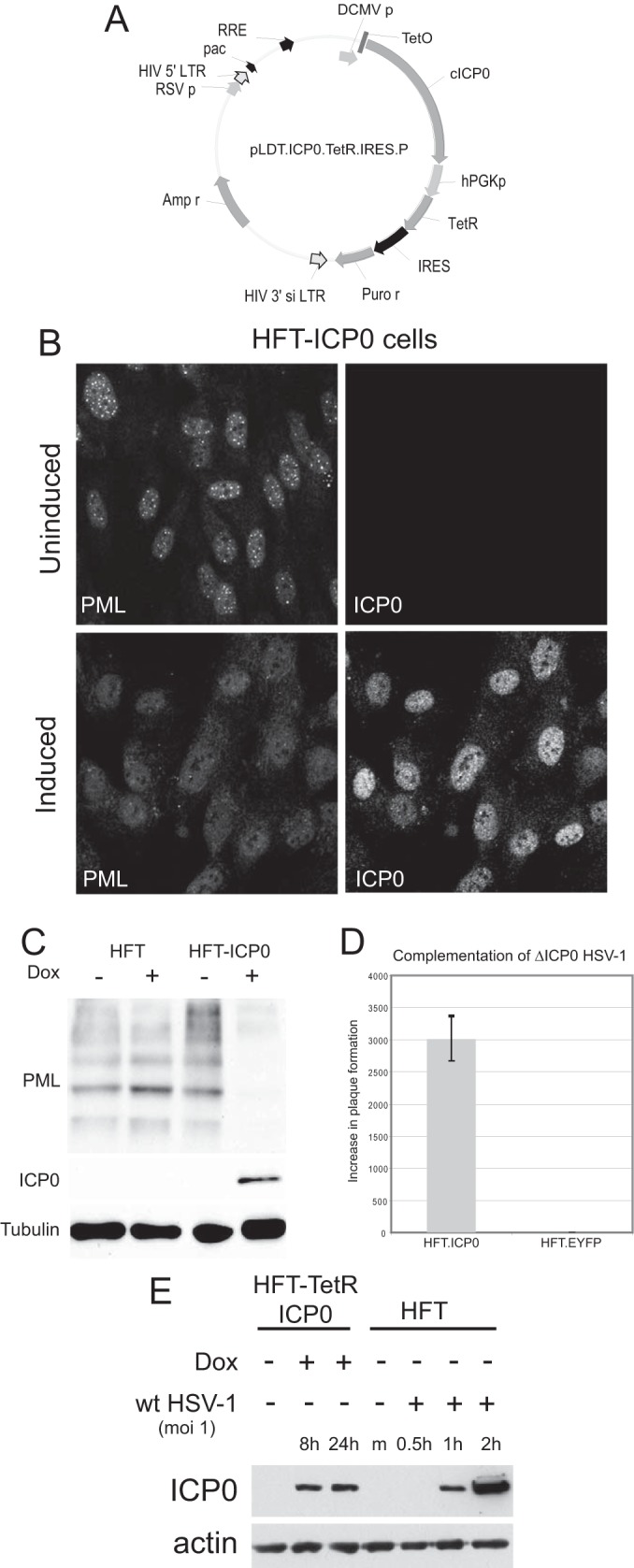
Characterization of a system for inducible expression of ICP0 in human fibroblasts. (A) A map of lentivirus plasmid vector pLDT.ICP0.TetR.IRES.P. The key features of the lentivirus vector are noted: pac, HIV packaging sequence; RSV p, RSV promoter; RRE, Rev response element; hPGK, human phosphoglycerate kinase promoter; IRES, internal ribosome binding site; Puro r, puromycin resistance coding sequence; LTR, long terminal repeat. (B) Immunofluorescence staining of ICP0 and PML in HFT-ICP0 cells before and after induction of ICP0 expression. (C) Western blot analysis of HFT.ICP0 cells, probing for PML, ICP0, and tubulin. (D) Complementation of ICP0 null mutant HSV-1 in plaque formation assays. HFT.ICP0 and HFT.EYFP cells were treated with doxycycline (100 ng/ml) for 24 h before infection with dl1403/CMV lacZ, an ICP0 deletion virus carrying a β-galactosidase marker gene. Plaques were counted 24 h after infection by staining for β-galactosidase. The results are expressed as fold increase in PFU per ml in each cell type compared to HFT cells; error bars are ± standard deviations. EYFP, enhanced yellow fluorescent protein. (E) Comparison of the level of ICP0 expressed after 8 and 24 h of induction of HFT-TetR-ICP0 cells (made by sequential transduction with TetR and ICP0 lentiviral vectors) with that expressed during the early stages of wt HSV-1 infection of HFT cells (MOI of 1; based on the titer of wt HSV-1 on HFs). Dox, doxycycline; m, mock infected.
Lentivirus transductions and induction of protein expression.
Lentivirus transduction, selection of transduced cells, and maintenance of cell lines were as described previously (36). Selection during routine culture used puromycin at 500 ng/ml, G418 at 0.5 mg/ml, and blasticidin (Invitrogen) at 1 μg/ml. The antibiotics were omitted from cells seeded for and during experimentation. For induction of protein expression, cells were treated with medium containing doxycycline (BD Biosciences) at 100 ng/ml for various times, as indicated in the text, and throughout the duration of an experiment.
Virus plaque and reactivation assays.
For determination of relative plaque-forming efficiencies of ICP0 null mutant HSV-1, cells were seeded for plaque assays into 24-well dishes at 1 × 105 cells per well and then infected the following day with appropriate sequential 3-fold dilutions of dl1403/CMV lacZ. After virus adsorption, the cells were overlaid with medium containing 1% human serum; then the cells were stained for β-galactosidase-positive plaques 24 h later (53). For assay of HCMV plaque formation, HFT-based cells were seeded into 24-well dishes, treated or not with doxycycline the following day, and then infected with the relevant HCMV virus at appropriate multiplicities. At 3 h after virus adsorption, the virus inoculum was removed and replaced with fresh medium. Plaques were stained at 10 days after infection by immunological detection of UL44. The cells were fixed with formaldehyde and treated with NP-40 as for immunofluorescence (IF) staining and then washed twice with phosphate-buffered saline (PBS) containing 0.1% Tween 20 (PBST). The cells were then treated with PBST containing 5% dried milk for 30 min and incubated for 2 h at room temperature with anti-UL44 monoclonal antibody (see below). The cells were then washed three times with PBST before being incubated with horseradish peroxidase-conjugated goat anti-mouse secondary antibody for 1 h. The cells were washed with PBST three times again and then incubated with 0.2 ml of True Blue solution (product code 50-7802; Insight Biotechnology) for 10 min.
Infections and Western blot analysis.
Cells were seeded into 24-well dishes at 1 × 105 cells per well. At 3 h after virus adsorption, the virus inoculum was removed and replaced with fresh medium. After the relevant experimental manipulations, the cells were washed twice with PBS before being harvested in SDS-PAGE loading buffer. Proteins were resolved on 7.5% SDS gels and then transferred to nitrocellulose membranes by Western blotting. The following antibodies were used: anti-IE1/2 mouse monoclonal antibody (MAb) E13 (Serotec), anti-IE1 MAb 1B12 (a gift from Tom Shenk), anti-myc tag MAb 9E10 (Santa Cruz), anti-tubulin MAb T4026 (Sigma-Aldrich), anti-PML rabbit polyclonal antibody (rAb) A301-167A (Bethyl Laboratories) or MAb 5E10 (54), anti-UL44 MAb ab6501 (Abcam) or MAb 10D8 (Santa Cruz), anti-pp28 MAb ab6502 (Abcam), anti-pp71 MAb 2H10-9 (a gift from Tom Shenk), and anti-ICP0 MAb 11060 and anti-EGFP rAb ab290 (Abcam).
Immunofluorescence and confocal microscopy.
Cells on 13-mm glass coverslips were fixed and prepared for immunofluorescence as described previously (14). PML was detected using rAb ABD030 (Jena) or MAb 5E10, IE1 was detected with MAb E13 or IB12, and IE2 was detected with MAb 8140 (Merck-Millipore). The secondary antibodies used were Alexa Fluor 555-conjugated goat anti-mouse IgG and Alexa Fluor 633-conjugated goat anti-rabbit IgG (Invitrogen). The samples were examined using a Zeiss LSM 710 confocal microscope, with 488-nm, 561-nm, and 633-nm laser lines, scanning each channel separately under image capture conditions that eliminated channel overlap. The images were exported as TIFF files, minimally adjusted using Photoshop, and then assembled into figures using Illustrator.
RESULTS
Development of inducible expression systems for ICP0 and IE1 in human HFs.
In order to determine whether ICP0 of HSV-1 could complement IE1 mutant HCMV, it was necessary to develop an inducible expression cell line system in cells permissive for HCMV infection. We modified the system used previously based on human hepatocyte HepaRG cells (44, 52) by using human fibroblasts (HFs) that had been immortalized by transduction with a telomerase-expressing lentivirus (HFT cells) and then transducing these cells with a second lentivirus based on our previous inducible ICP0 expression vector but which integrates simultaneous expression of the tetracycline repressor (TetR) and IRES-enabled puromycin resistance within the TetR transcript (Fig. 1A). After treatment with doxycycline, these HFT-ICP0 cells expressed ICP0 in amounts that were sufficient to disrupt PML NBs (Fig. 1B), degrade PML (Fig. 1C), and complement plaque formation by ICP0 null mutant HSV-1 by over 3 orders of magnitude (similar to that expected from the plaque-forming defect of the mutant in HFs) (Fig. 1D). In addition, we constructed ICP0-expressing cells by sequential transduction with EGFPnlsTetR- and ICP0-expressing lentiviruses as described previously (44). These cells, named HFT-TetR-ICP0, expressed ICP0 in amounts equivalent to those at the early stages of wt HSV-1 infection after induction (Fig. 1E). We also constructed a single inducible lentivirus vector expressing HCMV IE1. Cells transduced with this vector produced uniformly large amounts of IE1 after induction, which dispersed PML NBs (Fig. 2A) and caused a reduction in the sumoylated species of PML with a concomitant increase in the major unsumoylated band (Fig. 2B). Furthermore, comparison of HFT-IE1 cells after induction with HFT cells infected with wt HCMV strain Towne (TNwt; MOI of 1) at 24 h and 48 h after infection indicated that the levels of IE1 expression were comparable (Fig. 2C). These results were as expected on the basis of previous observations (48), and they illustrate that the inducible expression system enables ectopic expression of ICP0 and IE1 at physiologically relevant levels in a very high proportion of the HF cell populations. Prior to induction, IE1 expression was detectable in a higher proportion of HFT cells (Fig. 2A) than was observed previously in the HepaRG cell system (44). Although easily detectable by fluorescence in aggregated foci that colocalized with PML (Fig. 2A), this amount of IE1 was not detectable by Western blotting and had no noticeable effect on PML sumoylation in the bulk of the cell culture (Fig. 2B). The HFT-ICP0 and HFT-IE1 cells formed the basis for the following parts of the study.
FIG 2.

Analysis of HFT-IE1 cells before and after induction of IE1 expression. (A) Cells were stained for IE1 and PML before and after induction of IE1 expression. (B) HFT and HFT-IE1 cells were treated with doxycycline for 24 h or left untreated; then samples were analyzed by Western blotting for PML, IE1, and tubulin as indicated. (C) Comparison of IE1 expression in induced HFT-IE1 cells with that in wt HCMV-infected HFT cells at 24 and 48 h, as indicated. m, mock infected.
ICP0 and IE1 enhance gene expression of wt HCMV.
We investigated the effect of expression of ICP0 and IE1 on the replication of wt HCMV in induced and uninduced cultures of the various cell lines. To allow for the possibility that there might be HCMV strain-dependent differences in the results, we compared three wt strains of virus, namely, HCMV TNwt (an isolate derived from a strain Towne bacmid that includes an EGFP reporter gene driven from the SV40 promoter/enhancer) (47), AD169, and the more clinically related isolate TBE40 (49). Compared to uninduced and HFT control cells, we found that the plaque formation efficiency of all three wt HCMV strains was increased by about 10- to 30-fold by induced expression of IE1 and by 10- to 25-fold by ICP0 (Fig. 3A to C) although the plaques were smaller in the ICP0-expressing cells. The TBE40 strain plaques were generally much smaller than those of the Towne-based viruses (data not shown). The effect of expression of IE1 on the plaque-forming efficiency of HCMV TNwt in a similar system has been noted before (48), and it appears that ICP0 expression causes a similar effect. The increase in plaque formation of TBE40 wt in ICP0- and IE1-expressing cells was mirrored by an increase in IE2- and UL44-positive cells at 2 days after infection (Fig. 3D). Western blot analysis of the expression of IE1 and representative early (UL44) and late (pp28) proteins during HCMV TNwt infection of induced and uninduced HFT-ICP0 cells also demonstrated the stimulatory effect of ICP0. These results indicated that EGFP was being expressed from the SV40 promoter with the kinetics of a late, rather than IE or early, gene (Fig. 3E). That the EGFP marker acts as a surrogate for late gene expression is relevant to some subsequent parts of the paper. Western blot analysis of HCMV TNwt infection (MOI of 1) of induced and uninduced HFT-IE1 cells gave similar levels of UL44 expression, but that of EGFP was enhanced in the IE1 cells (data not shown).
FIG 3.

ICP0 and IE1 augment infection of wt HCMV. (A to C). Stocks of wt HCMV strains TNwt (A), AD169 (B), and TBE40 wt (C) were titrated on HFT, HFT-ICP0, and HFT-IE1 cells. In panel A the results are presented as the mean of the average of three independent experiments, with the error bars indicating the range of values obtained, plotted on a log10 scale. In panels B and C, the results are presented as fold increases above the titers in HFT cells (mean ± standard deviation). (D) Comparison of the proportion of IE2- and UL44-positive cells in HFT, HFT-IE1, and HFT-ICP0 cells with TBE40 as indicated. p.i., postinfection. (E) Western blot analysis of HCMV TNwt infection on HFT.ICP0 cells with or without induction of ICP0 expression 24 h before infection. Samples were collected at 24, 48, 72, and 96 h after infection then probed with anti-IE1 MAb E13, anti-pp28 MAb, anti-UL44 MAb, anti-EGFP rAb, anti-ICP0 MAb 11060, and anti-tubulin MAb, as indicated. hpi, hours postinfection.
Because this study involves the use of HCMV IE1 mutants, which of necessity were grown and titrated in IE1-expressing cells, subsequent experiments were based on titers for both wt and mutant viruses that were determined in induced HFT-IE1 cells to ensure equivalent inputs. As a consequence and following from the results shown in Fig. 3A to C, when control HFT cells are infected with wt HCMV at a given MOI, the degree of infection is considerably reduced compared to what would be expected on the more conventional basis of titers determined on HFs (i.e., a stated MOI of 1 for wt HCMV strains equates to approximately an MOI of 0.1 or less if inputs were based on plaque-forming titers on HFs).
Immunofluorescence analysis of IE2 and EGFP expression after infection of the various cells at varied MOIs with HCMV TNwt also gave results that were consistent with the plaque formation efficiency data (Fig. 4). At a high MOI, a high proportion of cells of all cell lines expressed both IE2 and EGFP, but as the MOI was reduced, the relative proportion of positive HFT cells (Fig. 4A) was reduced compared to the ICP0 and IE1 cells (Fig. 4B and C).
FIG 4.
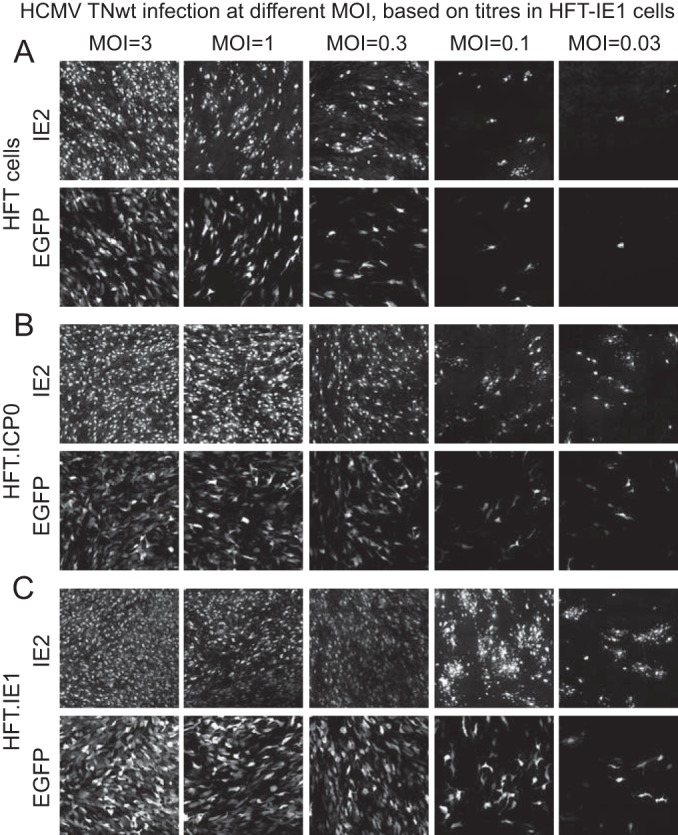
Analysis of HCMV TNwt infection in HFT, HFT-ICP0, and HFT-IE1 cells by immunofluorescence. Cells were infected at decreasing multiplicities, as indicated, and then examined for IE2 and EGFP expression at 5 days after infection.
IE1 but not ICP0 complements IE1 mutant HCMV.
Having established that both IE1 and ICP0 enhance the efficiencies of plaque formation and gene expression by wt HCMV, the next obvious question was whether ICP0 could complement IE1-defective HCMV. We analyzed two IE1 mutants: HCMV TNdlIE1, which has the same background as the HCMV TNwt strain but includes a deletion that inactivates IE1 but not IE2 (48), and TBdlIE1, which is derived from strain TBE40 (described as a clinical isolate [49]). Stocks of HCMV TNdlIE1 were generated on HFT-IE1 cells (changing the medium the day after infection so that the harvested virus did not contain doxycycline) and then titrated on HFT-IE1, HFT-ICP0, and HFT cells in parallel. The results indicated that the defect in plaque formation of HCMV TNdlIE1 in human fibroblasts was at least 3 orders of magnitude. A similar very low number of plaques were observed in HFT-ICP0 cells, demonstrating that ICP0 does not compensate for the lack of IE1 in terms of the ability to stimulate plaque formation (raw data not shown). Immunofluorescence analysis during TNdlIE1 infection of the three cell lines showed that IE2 expression was detectable in a proportion of control HFT cells (consistent with previous analyses of the HCMV IE1 mutant CR208) (55, 56) and that this proportion was increased by expression of ICP0 and even more so by IE1 (Fig. 5A). However, HFT- or HFT-ICP0-infected cells that express EGFP from the inserted marker gene were extremely rare, whereas progression to EGFP expression was efficient in HFT-IE1 cells (Fig. 5A). Western blot analysis confirmed that early and late TNdlIE1 gene expression was readily detectable in induced but not uninduced HFT-IE1 cells and that EGFP was detectable by Western blotting only at later times (Fig. 5B). A direct comparison of TNwt and TNdlIE1 in HFT cells demonstrated expression of UL44, pp28, and EGFP in the wt infection while none of these were detectable in the equivalent TNdlIE1 infection (Fig. 5C). As might be expected from the immunofluorescence results (Fig. 5A) and plaque assay data, UL44, pp28, and EGFP were undetectable in TNdlIE1-infected induced HFT-ICP0 cells, while IE2 was expressed in slightly larger amounts and over a more sustained time course in induced HFT-ICP0 cells than in the uninduced cells (Fig. 5D). These findings were not restricted to the high-passage-number laboratory strain Towne-derived virus as the TBE40 IE1 mutant also exhibited highly defective UL44 expression in HFT-ICP0 compared to HFT-IE1 cells (Fig. 5E). Indeed, we found that compared to that in HFT-IE1 cells, plaque formation efficiency of TBdlIE1 in both HFT and HFT-ICP0 cells was reduced by at least 5 orders of magnitude. A similar lack of complementation of TNdlIE1 and TBdlIE1 was observed in the independently derived HFT-TetR-ICP0 cells (raw data not shown). We conclude that ICP0 stimulates HCMV IE2 gene expression in the absence of IE1 and increases the proportion of IE2-positive cells but that IE1 is required for the infection to progress to the full viral gene expression program, even in the presence of IE2. Given the ability of ICP0 to stimulate expression of all viral genes during HSV-1 infection (providing that other HSV-1 IE proteins, especially ICP4, are also expressed), this is perhaps a surprising result. Although it was originally suggested that a major role of IE1 during HCMV infection is the stimulation of IE2 expression (57), our results are consistent with and strengthen the later studies that concluded that IE1 and IE2 cooperate to stimulate the expression of later classes of HCMV genes (55, 56).
FIG 5.

IE1 but not ICP0 complements IE1 mutant HCMV infection. (A) Analysis of IE2 and EGFP expression in HFT, HFT-ICP0, and HFT-IE1 cells infected with HCMV TNdlIE1. p.i., postinfection. (B) Western blot analysis of pp28, UL44, and EGFP expression at the indicted time points after HCMV TNdlIE1 infection of uninduced and induced HFT.IE1 cells. Expression of these proteins from the viral genome was not detectable in induced HFT-ICP0 cells (data not shown). Tubulin provides the loading control. (C) Comparison of viral gene expression by TNdlIE1 and TNwt after infection of HFT cells. (D) Analysis of viral gene expression after infection of uninduced and induced HFT-ICP0 cells with TNdlIE1. (E) Comparison of UL44 expression by TBdlIE1, the IE1 mutant derived from HCMV strain TBE40, in induced HFT, HFT-IE1, and HFT-ICP0 cells as detected by immunohistochemistry staining of infected cell cultures.
ICP0 augments replication of pp71 mutant HCMV.
Given that ICP0 stimulates infection by wt HCMV strains but not that by the IE1 mutants TNdlIE1 and TBdlIE1, it was of interest to determine whether ICP0 could stimulate pp71 mutant HCMV. This point arises because pp71 functions at least in part by targeting hDaxx and disrupting the hDaxx/ATRX complex (23–27). Although ICP0 does not disrupt this complex, it eliminates the recruitment of these proteins to HSV-1 genome sites after their entry into the nucleus (21). Furthermore, depletion of either hDaxx or ATRX improves the plaque formation efficiency of ICP0 null mutant HSV-1 (21). Therefore, it seemed feasible that ICP0 might substitute for the effects of pp71 on hDaxx/ATRX. We constructed HFT cells that express myc-tagged pp71 (HFT-mycpp71) and found that expression of pp71 after induction was at physiologically relevant levels, similar to that found between 3 and 4 days after wt HCMV infection (Fig. 6A). Titration of HCMV pp71 deletion mutant ADsubUL82 (Δpp71 HCMV) on cells expressing myc-tagged pp71 or ICP0 showed that ICP0 stimulated Δpp71 mutant HCMV infection by around 4,000-fold, which is about five times more effective than pp71 itself (Fig. 6B). The finding that ICP0 is more active in this assay than pp71 is probably explained by the stimulation of plaque formation of wt HCMV by ICP0 (see above). Thus, ICP0 overcomes the inhibitory effects of hDaxx/ATRX on Δpp71 HCMV infection and then provides a further stimulus, as observed on wt HCMV infection. HFT-ICP0 cells therefore provide an effective method of making stocks of Δpp71 HCMV without the complicating feature of packaging of pp71 into the virus particle during growth in pp71-complementing cell lines.
FIG 6.

Complementation of pp71 HCMV mutant ADsubUL82 by ICP0. (A) Analysis of expression of myc-tagged pp71 before and after induction of HFT-mycpp71 cells with doxycycline for 24 h compared to control HFT cells and wt HCMV TNwt-infected HFT cells, as indicated. The difference in mobility of pp71 in the induced and infected cells is due to the presence of the myc tag in the former. m, mock infected. (B) Comparison of the relative plaque-forming efficiencies of HCMV ADsubUL82 in induced HFT, HFT-mycpp71, and HFT-ICP0 cells, expressed as fold increase over that in control HFT cells. (C). Comparison of IE1, IE2, pp71, UL44, and pp28 expression during wt HCMV strain AD169 and ADsubUL82 (Δpp71) HCMV infection of induced HFT-ICP0 cells, as indicated, confirming the absence of pp71 expression by ADsubUL82 and illustrating similar levels of expression of UL44. (D) Western blot analysis of IE1, pp71, and UL44 expression after infection by HCMV ADsubUL82 in uninduced and induced HFT-ICP0 cells.
Interestingly, in a separate experiment, plaque formation by ADsubUL82 was stimulated 4.7-fold ± 1.8-fold in HFT-TetR-ICP0 cells over that in HFT-TetR-pp71 cells (consistent with the results shown in Fig. 6B) but was at least 250-fold lower in HFT-TetR-IE1 cells than in HFT-TetR-ICP0 cells (raw data not shown). This implies that IE1 is much less able to stimulate major immediate early promoter activity from parental viral genomes than both pp71 and ICP0. This is probably explained because IE1 would not be expected to overcome hDaxx-ATRX-mediated repression. The effectiveness of ICP0-mediated complementation was further illustrated by the comparable levels of expression of IE1, IE2, pp71, UL44, and pp28 by the parental wt AD169 strain and the Δpp71 mutant derivative virus in induced HFT-ICP0 cells (Fig. 6C). In contrast, expression of IE1 and UL44 by the Δpp71 mutant virus was highly defective in uninduced compared to induced HFT-ICP0 cells (Fig. 6D).
A potential virus-specific function of IE1.
It is possible that IE1 but not ICP0 complements IE1 mutant HCMV strains because IE1 has an important function that requires the presence of another HCMV protein (or vice versa) to stimulate progression from the IE stage to later stages of HCMV infection. This would be consistent with the observation that ICP0 stimulates IE2 expression by the HCMV IE1 mutants but not efficient progress to early or late gene expression (Fig. 5). If so, it is possible that mutation of IE1 might reveal a portion of the protein that is required for stimulation of HCMV infection but not that of HSV-1. Therefore, we analyzed a series of mutations in IE1 that were previously characterized in the HepaRG cell system (44) (Fig. 7A). These mutations were designed on the basis of being in motifs that are locally highly conserved between different primate cytomegaloviruses; Fig. 7B illustrates an example of this for mutant YL2. Following transduction of HFT cells with IE1 lentiviral vectors, we found that IE1 mutant proteins YL1 to YL4 and K450R were all expressed, albeit to different levels (Fig. 7C, upper panels). Expression of the YL7 mutant was not detectable by the antibody used here (MAb 1B12) although it was detectable at wt levels using a different antibody (MAb E13) that later became unavailable (Fig. 7C, bottom panel). These cells were infected with HCMV TNdlIE1 (MOI of 1), and then samples were harvested for analysis of IE1, UL44, and pp28 expression at 5 days after infection. UL44 and pp28 were expressed with similar efficiencies in cells expressing wt, YL1, K450R, and YL7 IE1 proteins, whereas YL2, YL3, and YL4 failed to stimulate HCMV early and late gene expression at this time point and multiplicity (Fig. 7C). These results were reflected in plaque assays, with plaque formation by HCMV TNdlIE1 being of similar efficiency in cells expressing wt, YL1, K450R, and YL7 proteins and undetectable in the YL2, YL3, and YL4 cell lines (Fig. 7D). The complementation of IE1 mutant HCMV to wt levels by the sumoylation-deficient IE1 mutant K450R is consistent with previous studies illustrating that this mutation did not compromise IE1 activity in a variety of functional assays (32, 58). This is in apparent contrast to reduced efficiency of infection when the mutation is present in the viral genome (59). Efficient complementation by YL7, which removes the chromatin tethering domain, is also consistent with the findings of two recent studies (60, 61).
FIG 7.

Effects of mutations in IE1 on its ability to complement HCMV TNdlIE1. (A) A map of IE1, showing the region in common with IE2 (exons 2 and 3), the IE1-specific exon 4, the acidic region, the C-terminal domain, and the positions of the mutations analyzed in this study (modified from reference 44, in which details of all the mutations are described). The amino acid residues mutated are as follows: YL1, residues 14 and 16; YL2, residues 53, 55, and 56; YL3, residues 130, 132, and 133; YL4, residues 316 to 318; YL5, residues 372 and 374; YL6, residues 417 to 419; YL7, deletion of residues 480 to 491. (B) Conservation of the region mutated in the mutant YL2 in human, chimpanzee, green monkey, and rhesus CMVs (modified from reference 44). (C) Western blot analysis of the expression of IE1, UL44, and pp28 during HCMV TNdlIE1 infection of cells expressing wt IE1 and mutants YL1, YL2, YL3, YL4, K450R, and YL7 (MOI of 1 at 10 days postinfection). The IE1 monoclonal antibody used to detect IE1 in the experiment shown in the main panel is unable to detect YL7 because the epitope is in the deleted region. The lowermost panel shows IE1 in the same samples detected by monoclonal E13, which recognizes a different epitope. (D) Relative plaque-forming efficiencies of HCMV TNdlIE1 on HFT cells expressing wt IE1 and mutants YL1, YL2, YL3 YL4, K450R, and YL7, expressed as a percentage of the titer in HFT-IE1 cells.
In a previous study mutants YL3 and YL4 had been found unable to stimulate ICP0 null mutant HSV-1 infection and were also defective for sumoylation, PML NB localization and disruption, and the ability to stimulate desumoylation of PML (44). Given that the YL3 and YL4 proteins are expressed at reduced levels in this system, these mutations might induce major structural changes that limit further analysis. Indeed, on the basis of the recently published structural analysis of IE1 (62), the YL3 mutations lie within a long helix which is part of an elongated helical bundle that forms the core of the IE1 structure, while the YL4 mutations lie near the tip of another helix that makes up the bundle. On the other hand, in our previous study YL2 was sumoylated; it disrupted PML NBs, induced desumoylation of PML, and retained about 20% of the complementing activity of wt IE1 on plaque formation by ICP0 null mutant HSV-1 (44). These data were therefore consistent with the hypothesis that YL2 might define a region of IE1 that is specifically or preferentially required for stimulating HCMV infection although the relatively weak expression of YL2 in these particular HF-based cells weakens this evidence. The YL2 mutations are less likely to disrupt the overall structure of IE1 because they lie in a less structured loop region that extends from the core helical bundle (62).
We analyzed the phenotype of mutant YL2 in more detail by constructing another set of cells, this time using sequential transduction of HFT cells with EGFPnlsTetR and IE1-expressing lentiviruses. These cells showed higher relative levels of YL2 expression (albeit still slightly reduced compared to the wt protein) and both sumoylation of YL2 and desumoylation of PML (albeit again slightly less efficiently that the wt protein) (Fig. 8A). The slight difference in gel mobilities of the YL2 and wt proteins must be caused by the YL2 point mutations themselves as sequencing the whole IE1 region in the expression vector revealed no additional changes (data not shown). YL2 also localized to PML NBs when expressed in small amounts and disrupted these structures after high-level expression was induced (again, with a slight defect compared to the wt protein [data not shown]). Plaque formation efficiency of ICP0 mutant HSV-1 was stimulated about 14-fold following induction of wt IE1 expression in these cells, with YL2 being about 30% as effective (Fig. 8B). Because these stimulation levels were less than those observed for wt IE1 in our previous HepaRG cell system (44), we transduced the IE1 wt and YL2 cells a subsequent time with a vector expressing pp71 (Fig. 8C) as the combination of IE1 and pp71 stimulates ICP0 null mutant plaque formation almost as well as ICP0 itself in HepaRG cells (44). In these HFT-derived cells, pp71 by itself and in combination with wt IE1 and YL2 stimulated ICP0 null mutant plaque formation by about 9-, 710-, and 600-fold, respectively (Fig. 8D). These data clearly show that YL2 retains substantial biological activity in a wide range of assays, yet again in these independently derived cells, it failed to stimulate UL44 and pp28 gene expression by TNdlIE1 whether assayed by Western blotting (Fig. 8E) or IF (Fig. 8F). These data exclude the possibility that the lack of complementation of IE1 mutant HCMV by YL2 is simply due to the protein being grossly misfolded and nonfunctional, and the data greatly strengthen the hypothesis that the region of IE1 affected by the YL2 point mutations is required specifically or preferentially for stimulating HCMV infection. It is interesting that the motif mutated in YL2 is in a region that is in common with IE2 and one of the most highly conserved among the IE1 proteins of the primate cytomegaloviruses (Fig. 7B) (44).
FIG 8.
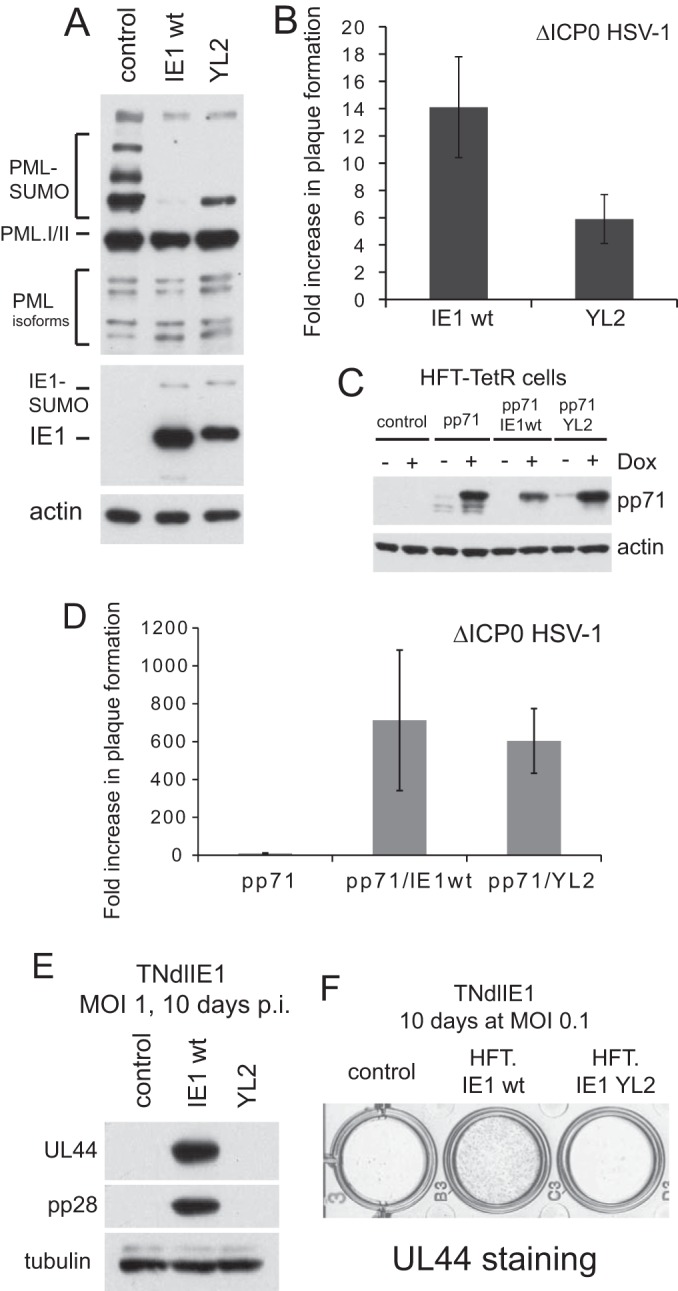
Confirmation of the phenotype and further analysis of mutant YL2. (A) Western blot analysis of PML, IE1, and actin expression in induced HFT-TetR-IE1 wt and HFT-TetR-YL2 cells compared with HFT-TetR control cells. (B) Fold increases in plaque formation of ICP0 null mutant HSV-1 in HFT-TetR-IE1 wt and HFT-TetR-YL2 cells compared to control HFT-TetR cells. The mean of two independent experiments is presented, with the error bars showing the range of values obtained in the two experiments. (C) Expression of pp71 in HFT-TetR, HFT-TetR-IE1 wt, and HFT-TetR-YL2 cells after additional transduction of these cells with a lentiviral vector expressing myc pp71. (D) Fold increases in plaque formation of ICP0 null mutant HSV-1 in HFT-TetR-pp71/IE1 wt and HFT-TetR-pp71/YL2 cells compared to control HFT-TetR-pp71 cells. The mean of two independent experiments is presented, with the error bars showing the range of values obtained in the two experiments. The increase in plaque formation of the mutant virus in HFT-TetR-pp71 cells over control HFT-TetR cells was 9.5-fold ± 3.1-fold. (E) Expression of UL44 and pp28 after infection of induced control (HFT-TetR), HFT-TetR-IE1 wt, and HFT-TetR-YL2 cells with TNdlIE1. (F) Immunohistochemical staining of UL44 expression in cultures of induced control (HFT-TetR), HFT-TetR-IE1 wt, and HFT-TetR-YL2 cells with TNdlIE1. p.i., postinfection.
IE1-mediated derepression of HCMV gene expression.
Infection of HFT cells with HCMV TNdlIE1 results in a population of cells that express IE2, of which only a very small number are able to express EGFP (which acts as a marker for later classes of viral proteins) (Fig. 5). Therefore, in the absence of IE1, HCMV gene expression is restricted, with a minority of cells able to express IE2 while the majority fails to express viral proteins at detectable levels. This situation has some analogies with the models of quiescent infection using HSV-1 mutants that are defective for IE gene functions, in which gene expression from the quiescent viral genomes can be reactivated (or derepressed) by provision of ICP0 (63, 64). Therefore, it was of interest to determine whether IE1 could have a similar effect at some point after an initial infection with the IE1 mutant virus. HFT-IE1 cells were infected with HCMV TNdlIE1 (MOI of 1), and then 4 days later IE1 expression was induced with doxycycline. The samples were examined for expression of IE2 and EGFP at various time points thereafter. As before, we observed that a proportion of the TNdlIE1 mutant-infected cells were expressing IE2 even without induction of IE1 expression (Fig. 9A, 2 days after induction). We were unable to test whether this is related to the leakiness of IE1 expression prior to induction (Fig. 2), but in any event the amounts of IE1 and IE2 expressed in such cells are insufficient to allow the infection to progress to the stage of EGFP expression. Induction of IE1 expression induced a clear increase in the proportion of IE2-positive cells (Fig. 9A), with the infection later progressing to EGFP marker gene expression at 4 days after induction (Fig. 9B). At even later times after induction, all the cells expressed abundant amounts of IE2 and EGFP (which may have been partly due to production of progeny virus and its spread through the cell monolayer), whereas the uninduced cells remained in a state similar to that at the initial time points (data not shown).
FIG 9.

Stimulation of HCMV gene expression after delayed expression of IE1 during HCMV TNdlIE1 infection of HFT-IE1 cells. Cells were infected with HCMV TNdlIE1 (MOI of 1, based on the titer in HFT-IE1 cells) prior to induction and then incubated for a further 4 days. At that point, doxycycline was added to 100 ng/ml to induce expression of IE1; then samples were analyzed for expression of IE2 and EGFP at various times thereafter. The results shown in panels A and B illustrate the basal level of IE2 expression in the absence of IE1, the increase in the number of cells expressing IE2 after induction, and the progression of these cells to EGFP expression in the presence of induced IE1 (B). Panels C and D show images from an analogous experiment using HFT-ICP0 cells. The images are composites of 9 tiled images captured with a 40× lens at a zoom factor of 1.
Figure 9A illustrates that IE1 has the capability of converting IE2-negative HCMV TNdlIE1-infected cells into IE2-positive cells that later progress to active infection. These IE2-negative cells can be considered to be those in which HCMV gene expression has been repressed but which retain the potential to engage in viral gene expression after subsequent expression of IE1. This infection model has some parallels with quiescent infections of cultured cells by HSV-1 mutants, in which expression from the repressed viral genomes may be reactivated by expression of ICP0 (63, 64). While there are complicating features of this HCMV system, such as the proportion of cells expressing IE2 and the background IE1 expression in some cells prior to induction, at the least these results indicate that potentially active HCMV genomes can be retained within human diploid fibroblasts for a number of days in a form that is available for lytic gene expression once IE1 has been provided.
In an analogous series of experiments, we found that induction of ICP0 expression at 4 days after an initial HCMV TNdlIE1 infection also caused an increase in the proportion of IE2-positive cells (Fig. 9C), but in this case these cells were unable to progress to EGFP expression in subsequent days (Fig. 9D). These results are as expected from the data shown in Fig. 5, but they underline that IE2 is insufficient to drive HCMV infection into the full lytic cycle in the absence of IE1, even in the presence of the very strong activating function of ICP0. Induction of ICP0 expression at 4 days postinfection was also unable to stimulate UL44 expression in TBdlIE1-infected HFT-TetR-ICP0 cells although in this case the increase in proportion of IE2-positive cells was only about 2-fold (compared to at least 4-fold in the corresponding TNdlIE1 infection) (data not shown).
DISCUSSION
We report here that preexpression of both ICP0 of HSV-1 and the HCMV protein IE1 enhances wt HCMV replication in cultured human fibroblasts. In contrast, although ICP0 augments IE2 expression from IE1 mutant HCMV, it cannot substitute for IE1 for full productive replication. Thus, while both IE1 and pp71 can stimulate ICP0 null mutant HSV-1 infection and while the two HCMV proteins acting together can almost completely replace the requirement for ICP0 (44), some aspect of IE1 is specifically required during the HCMV lytic cycle. These results raise several issues concerning the replication of HCMV in cultured HFs.
The advantages and limitations of using an inducible cell system.
There are some limitations to the use of an inducible cell system rather than engineered viruses for this type of study, including limited control over the level and time course of protein expression. Clearly, these factors will be different in inducible cells from the situation expected if reconstructed viruses were used. However, we have shown in this particular system that the expression of ICP0, IE1, and pp71 in the various cell lines is at physiologically relevant levels and in a very high proportion of the cells and that the complementation of mutant virus plaque formation by exogenous expression is obviously as efficient as one could expect by, for example, restoring IE1 sequences into the IE1 mutant backbones. The advantages of the use of inducible cells include (i) that it is the only way one could realistically study the effect of ICP0 on HCMV infection in a well-defined system, (ii) that it is a powerful method of studying the effect of delaying the time of expression of IE1 (for example, in the experiments shown in Fig. 9), and (iii) that it is also a very powerful approach to study the effects of mutations such as YL2 as introduction of mutations into this region of the viral genome would also affect IE2. Thus, the inducible cell system adds capabilities to the study that extend beyond what may be possible using viral mutants and certainly far beyond what is possible using transient expression methods. Inducible cell lines have also been used very effectively to study IE2 mutants (65).
The plaque-forming efficiency of wt HCMV.
It may be initially surprising that the plaque formation efficiency of even wt HCMV can be enhanced by preexpression of either ICP0 or IE1. In the case of IE1, our data are consistent with a previous study (48). It is clear that stocks of wt HCMV contain many more potentially infectious particles than is apparent from plaque assays on normal HFs. A simple way of interpreting this is to posit that a PFU titer reflects not only the number of infectious virus particles but also the probability that a given cell will proceed to lytic infection after being infected with a viable virus particle. In this view, a PFU titer reflects a probability that can be altered depending on the status of the cell or cell type, and it may reflect the outcome of the competing effects of viral stimulatory and cellular inhibitory factors. This hypothesis is consistent with previous work on the phenotype of ICP0 null mutant HSV-1 (66), on the increase in the number of IE protein-positive cells after wt HCMV infection of cells depleted of PML NB components (40, 41), and on the effects of pp71 on hDaxx during infection at multiplicities that are substantially below 1 PFU per cell (26). It follows that many cultured cells infected with wt HCMV at a low MOI do not enter active, lytic infection, and at least a proportion of the input viral genomes must be repressed by cellular factors. That prior expression of IE1 or ICP0 enhances wt HCMV infection implies that these viral proteins compromise the ability of the cell to restrict the virus. Since both proteins affect the integrity of PML NBs and since depletion of PML NB components also increases the probability that cells infected with wt HCMV infection will proceed to lytic infection (39–41), these results are consistent with the hypothesis that repression by PML NB components influences the efficiency of herpesvirus infection. Although it is formally possible that preexpression of IE1 or ICP0 increases nuclear uptake of viral DNA, given the lack of previous evidence or a mechanism to suggest that this might be the case, we consider this to be an unlikely explanation of the observed increase in wt HCMV plaque formation.
Cell type differentials in IE1 function.
Taken with our previous work (44), this study enabled the comparison of the magnitude of the stimulatory effect of IE1 on ICP0 null mutant HSV-1 in two different cell types. Although IE1, by itself, augmented plaque formation efficiency of ICP0 null mutant HSV-1 by around 2 orders of magnitude in HepaRG-based cells (44), this effect was only about 14-fold in HF-derived cells (Fig. 8B). Nonetheless, in both the HepaRG and HF systems, the addition of pp71 increased complementation of the HSV-1 mutant to within a few fold of that achieved by ICP0 itself. An overall current paradigm is that the defect in ICP0 null mutant plaque formation is caused by inhibitory cellular factors. The existing data indicate that PML NBs contribute at least a part of that inhibition, which is comprised of the additive effects of several inhibitory components (38). Since IE1 affects PML and Sp100 while pp71 disrupts hDaxx/ATRX function, it appears that targeting of PML and Sp100 has a less pronounced effect in HFs than in HepaRG cells, indicating a more major role in HFs for hDaxx/ATRX (or some other inhibitory factor not studied here). Whatever the explanation, these data reveal the potential for differential effects of cellular intrinsic factors that influence herpesvirus infections depending on cell type and the importance of targeting combinations of such factors for fully efficient viral gene expression.
A potential specific requirement for IE1 for HCMV lytic replication.
Given the very powerful effects of ICP0 in stimulating HSV-1 infection and the parallels between the effects of IE1 and ICP0 on PML NBs, we were surprised to find that ICP0 could not substitute for IE1 during HCMV infection. This is particularly the case because ICP0 stimulates the HCMV major immediate early promoter (MIEP)-driven expression in the context of both quiescent HSV-1 infection (52, 63) and HCMV infection (Fig. 5 and 9). Therefore, some specific aspect of IE1 that is not provided by ICP0 is required for the efficient progression to full HCMV gene expression. Because IE2 is expressed in a number of IE1 mutant-infected cells (55, 56) (Fig. 5 and 9), a model according to which IE2 functionality has a specific requirement for IE1 is attractive. In other words, it is not just that IE1 and IE2 can cooperate to stimulate abundant transcription from other HCMV gene promoters, it appears that IE2 must cooperate specifically with IE1 for this to happen efficiently. In this regard, it is interesting that the original analyses of IE1 activity in plasmid transfection transcription reporter assays found that IE1 and IE2 exhibited a strong synergistic ability to activate certain HCMV early gene promoters (67–69).
A region of IE1 required preferentially for function in the context of HCMV infection.
Although ICP0 stimulates the efficiency of all classes of HSV-1 gene expression during a normal wt virus infection, it cannot activate early gene expression efficiently in the absence of ICP4 (64). Conversely, in the absence of ICP0, many infected cells express detectable (albeit low) levels of ICP4, which by itself drives infection into the lytic cycle inefficiently (66, 70). Thus, both ICP0 and ICP4 are required for efficient HSV-1 infection, but because the combination of IE1 and pp71 can substitute for ICP0, it follows that ICP4 has no requirement for ICP0 specifically; ICP4 can work independently as long as ICP0's function is provided by other means. The situation in HCMV is clearly distinct. Both IE1 and IE2 (often viewed as the functional analogues of ICP0 and ICP4, respectively) are required for efficient HCMV infection (47, 55, 56), yet in this case the function of IE1 cannot be replaced by ICP0. Thus, despite commonalities in many aspects of function, ICP0 lacks sequences that would enable it to substitute for IE1. It follows that IE1 might include sequences that are required for stimulating HCMV infection that may be dispensable for stimulating HSV-1 infection. Analysis of a number of available IE1 mutants identified mutations that had no effect on stimulating either HSV-1 or HCMV (e.g., YL7 and K450R) and some that eliminated stimulation of both viruses (YL3 and YL4). The YL2 mutant, however, failed completely to stimulate HCMV infection but retained at least 30% of the activity of the wt protein on ICP0 null mutant HSV-1 (Fig. 8).
It is intriguing that the YL2 mutation lies in the region of IE1 (within exon 3) that is in common with IE2 and which is completely conserved among the primate cytomegaloviruses (Fig. 7B). It may be relevant that exon 3 is required for function by both IE1 and IE2 (67, 71), and it may be that this region in some manner (not necessarily a direct interaction) enables the two proteins to work in concert. A previous study characterized an HCMV mutant virus with a deletion of residues 30 to 77 in both IE1 and IE2 (72). This virus was highly defective in plaque assays, with up to 1,000-fold fewer plaques than expected on the basis of cells positive for IE proteins detected by immunofluorescence. It also exhibited delayed kinetics of viral yields in growth curves and an approximate 500-fold decreased yield at 9 days after infection at a low MOI (72). Since the defect in viral yield could be corrected by growth in IE1-expressing cells, it was concluded that exon 3 of IE1 is important for IE1 function in the context of HCMV infection. However, the deletion of residues 33 to 77 caused the IE1 protein to be very unstable and unable to disperse PML NBs (72). The defined point mutations of YL2 (which are within the region of residues 33 to 77) allow the protein to accumulate to almost wt levels (Fig. 8) and do not inactivate the PML NB disruption activity of IE1 (data not shown). Therefore, the YL2 mutation not only defines a small conserved motif of IE1 in exon 3 that is highly important for stimulating HCMV infection but also overcomes interpretational limitations arising from the use of more drastic deletion mutations.
Potential biological implications.
The work presented here and previously illustrates the theoretical potential of HCMV to influence HSV-1 infection in a clinical setting and vice versa. Perhaps of greatest interest is the possibility that expression of ICP0 might simulate the activity of the major immediate early promoter in a cell latently infected with HCMV and thus enable reactivation to lytic infection (as in this case both IE1 and IE2 would be expressed). The converse is also conceivable, i.e., that pp71 and IE1 expressed from superinfecting HCMV might reactivate HSV-1 from a quiescent or latent infection, an outcome which has precedent in cultured cells (44, 45). Although it remains to be seen if such clinical scenarios actually occur in practice, the ability of the viral IE proteins to cross-complement illustrates that there is no mechanistic barrier to these possibilities.
ACKNOWLEDGMENTS
The work in R.D.E.'s laboratory is supported by the Medical Research Council; Y.L. is supported by the China Scholarship Council and the University of Glasgow.
We thank Anne Orr for technical assistance, Michael Nevels, Tom Shenk, Chris Boutell, Steven McFarlane, David Davido, and Roel van Driel for generous gifts of essential materials, and members of the laboratories of R.D.E. and Chris Boutell for helpful discussions.
REFERENCES
- 1.Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Strauss SE (ed). 2007. Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Reddehase MJ. 2013. Cytomegaloviruses: from molecular pathogenesis to intervention. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 3.Weller SK. 2011. Alphaherpesviruses: molecular virology. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 4.DeLuca NA. 2011. Functions and mechanism of action of the herpes simplex virus regulatory protein, ICP4, p 17–38 InWeller SK. (ed), Alphaherpesviruses: molecular virology. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 5.Wysocka J, Herr W. 2003. The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem Sci 28:294–304. doi: 10.1016/S0968-0004(03)00088-4. [DOI] [PubMed] [Google Scholar]
- 6.Yan N, Chen ZJ. 2012. Intrinsic antiviral immunity. Nat Immunol 13:214–222. doi: 10.1038/ni.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boutell C, Everett RD. 2013. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol 94:465–481. doi: 10.1099/vir.0.048900-0. [DOI] [PubMed] [Google Scholar]
- 8.Everett RD, Boutell C, Hale BG. 2013. Interplay between viruses and host sumoylation pathways. Nat Rev Microbiol 11:400–411. doi: 10.1038/nrmicro3015. [DOI] [PubMed] [Google Scholar]
- 9.Everett RD, Chelbi-Alix MK. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819–830. doi: 10.1016/j.biochi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Schreiner S, Wodrich H. 2013. Virion factors that target Daxx to overcome intrinsic immunity. J Virol 87:10412–10422. doi: 10.1128/JVI.00425-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tavalai N, Stamminger T. 2008. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim Biophys Acta 1783:2207–2221. doi: 10.1016/j.bbamcr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Bernardi R, Pandolfi PP. 2007. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8:1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 13.Geoffroy MC, Chelbi-Alix MK. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res 31:145–158. doi: 10.1089/jir.2010.0111. [DOI] [PubMed] [Google Scholar]
- 14.Everett RD, Murray J. 2005. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J Virol 79:5078–5089. doi: 10.1128/JVI.79.8.5078-5089.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hagglund R, Roizman B. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J Virol 78:2169–2178. doi: 10.1128/JVI.78.5.2169-2178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith MC, Boutell C, Davido DJ. 2011. HSV-1 ICP0: paving the way for viral replication. Future Virol 6:421–429. doi: 10.2217/fvl.11.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boutell C, Cuchet-Lourenco D, Vanni E, Orr A, Glass M, McFarlane S, Everett RD. 2011. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog 7:e1002245. doi: 10.1371/journal.ppat.1002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuchet-Lourenco D, Boutell C, Lukashchuk V, Grant K, Sykes A, Murray J, Orr A, Everett RD. 2011. SUMO pathway dependent recruitment of cellular repressors to herpes simplex virus type 1 genomes. PLoS Pathog 7:e1002123. doi: 10.1371/journal.ppat.1002123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cuchet-Lourenco D, Vanni E, Glass M, Orr A, Everett RD. 2012. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J Virol 86:11209–11222. doi: 10.1128/JVI.01145-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J Virol 72:6581–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukashchuk V, Orr A, Everett RD. 2010. Regulation of ICP0 null mutant HSV-1 infection by ND10 components ATRX and hDaxx. J Virol 84:4026–4040. doi: 10.1128/JVI.02597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Everett RD. 2011. The role of ICP0 in counteracting intrinsic cellular resistance to virus infection, p 39–50 InWeller SK. (ed), Alphaherpesviruses: molecular virology. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 23.Cantrell SR, Bresnahan WA. 2006. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J Virol 80:6188–6191. doi: 10.1128/JVI.02676-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lukashchuk V, McFarlane S, Everett RD, Preston CM. 2008. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J Virol 82:12543–12554. doi: 10.1128/JVI.01215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Preston CM, Nicholl MJ. 2006. Role of the cellular protein hDaxx in human cytomegalovirus immediate-early gene expression. J Gen Virol 87:1113–1121. doi: 10.1099/vir.0.81566-0. [DOI] [PubMed] [Google Scholar]
- 26.Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woodhall DL, Groves IJ, Reeves MB, Wilkinson G, Sinclair JH. 2006. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J Biol Chem 281:37652–37660. doi: 10.1074/jbc.M604273200. [DOI] [PubMed] [Google Scholar]
- 28.Hofmann H, Sindre H, Stamminger T. 2002. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J Virol 76:5769–5783. doi: 10.1128/JVI.76.11.5769-5783.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korioth F, Maul GG, Plachter B, Stamminger T, Frey J. 1996. The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp Cell Res 229:155–158. doi: 10.1006/excr.1996.0353. [DOI] [PubMed] [Google Scholar]
- 30.Muller S, Dejean A. 1999. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J Virol 73:5137–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkinson GW, Kelly C, Sinclair JH, Rickards C. 1998. Disruption of PML-associated nuclear bodies mediated by the human cytomegalovirus major immediate early gene product. J Gen Virol 79:1233–1245. [DOI] [PubMed] [Google Scholar]
- 32.Xu Y, Ahn JH, Cheng M, apRhys CM, Chiou CJ, Zong J, Matunis MJ, Hayward GS. 2001. Proteasome-independent disruption of PML oncogenic domains (PODs), but not covalent modification by SUMO-1, is required for human cytomegalovirus immediate-early protein IE1 to inhibit PML-mediated transcriptional repression. J Virol 75:10683–10695. doi: 10.1128/JVI.75.22.10683-10695.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang H, Kim ET, Lee HR, Park JJ, Go YY, Choi CY, Ahn JH. 2006. Inhibition of SUMO-independent PML oligomerization by the human cytomegalovirus IE1 protein. J Gen Virol 87:2181–2190. doi: 10.1099/vir.0.81787-0. [DOI] [PubMed] [Google Scholar]
- 34.Kim YE, Lee JH, Kim ET, Shin HJ, Gu SY, Seol HS, Ling P, Lee CH, Ahn JH. 2011. Human cytomegalovirus infection causes degradation of Sp100 proteins that suppress viral gene expression. J Virol 85:11928–11937. doi: 10.1128/JVI.00758-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HR, Huh YH, Kim YE, Lee K, Kim S, Ahn JH. 2007. N-terminal determinants of human cytomegalovirus IE1 protein in nuclear targeting and disrupting PML-associated subnuclear structures. Biochem Biophys Res Commun 356:499–504. doi: 10.1016/j.bbrc.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 36.Everett RD, Parada C, Gripon P, Sirma H, Orr A. 2008. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J Virol 82:2661–2672. doi: 10.1128/JVI.02308-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol 80:7995–8005. doi: 10.1128/JVI.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glass M, Everett RD. 2013. Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection. J Virol 87:2174–2185. doi: 10.1128/JVI.02950-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tavalai N, Adler M, Scherer M, Riedl Y, Stamminger T. 2011. Evidence for a dual antiviral role of the major nuclear domain 10 component Sp100 during the immediate-early and late phases of the human cytomegalovirus replication cycle. J Virol 85:9447–9458. doi: 10.1128/JVI.00870-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tavalai N, Papior P, Rechter S, Leis M, Stamminger T. 2006. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J Virol 80:8006–8018. doi: 10.1128/JVI.00743-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tavalai N, Papior P, Rechter S, Stamminger T. 2008. Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J Virol 82:126–137. doi: 10.1128/JVI.01685-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Everett RD, Boutell C, McNair C, Grant L, Orr A. 2010. Comparison of the biological and biochemical activities of several members of the alphaherpesvirus ICP0 family of proteins. J Virol 84:3476–3487. doi: 10.1128/JVI.02544-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kyratsous CA, Silverstein SJ. 2009. Components of nuclear domain 10 bodies regulate varicella-zoster virus replication. J Virol 83:4262–4274. doi: 10.1128/JVI.00021-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Everett RD, Bell AJ, Lu Y, Orr A. 2013. The replication defect of ICP0-null mutant herpes simplex virus 1 can be largely complemented by the combined activities of human cytomegalovirus proteins IE1 and pp71. J Virol 87:978–990. doi: 10.1128/JVI.01103-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stow EC, Stow ND. 1989. Complementation of a herpes simplex virus type 1 Vmw110 deletion mutant by human cytomegalovirus. J Gen Virol 70:695–704. doi: 10.1099/0022-1317-70-3-695. [DOI] [PubMed] [Google Scholar]
- 46.Stow ND, Stow EC. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J Gen Virol 67:2571–2585. doi: 10.1099/0022-1317-67-12-2571. [DOI] [PubMed] [Google Scholar]
- 47.Marchini A, Liu H, Zhu H. 2001. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J Virol 75:1870–1878. doi: 10.1128/JVI.75.4.1870-1878.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knoblach T, Grandel B, Seiler J, Nevels M, Paulus C. 2011. Human cytomegalovirus IE1 protein elicits a type II interferon-like host cell response that depends on activated STAT1 but not interferon-gamma. PLoS Pathog 7:e1002016. doi: 10.1371/journal.ppat.1002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zalckvar E, Paulus C, Tillo D, Asbach-Nitzsche A, Lubling Y, Winterling C, Strieder N, Mucke K, Goodrum F, Segal E, Nevels M. 2013. Nucleosome maps of the human cytomegalovirus genome reveal a temporal switch in chromatin organization linked to a major IE protein. Proc Natl Acad Sci U S A 110:13126–13131. doi: 10.1073/pnas.1305548110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bresnahan WA, Hultman GE, Shenk T. 2000. Replication of wild-type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J Virol 74:10816–10818. doi: 10.1128/JVI.74.22.10816-10818.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith MC, Goddard ET, Perusina Lanfranca M, Davido DJ. 2013. hTERT extends the life of human fibroblasts without compromising type I interferon signaling. PLoS One 8:e58233. doi: 10.1371/journal.pone.0058233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Everett RD, Parsy ML, Orr A. 2009. Analysis of the functions of herpes simplex virus type 1 regulatory protein ICP0 that are critical for lytic infection and derepression of quiescent viral genomes. J Virol 83:4963–4977. doi: 10.1128/JVI.02593-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jamieson DRS, Robinson LH, Daksis JI, Nicholl MJ, Preston CM. 1995. Quiescent viral genomes in human fibroblasts after infection with herpes simplex virus Vmw65 mutants. J Gen Virol 76:1417–1431. doi: 10.1099/0022-1317-76-6-1417. [DOI] [PubMed] [Google Scholar]
- 54.Stuurman N, de Graaf A, Floore A, Josso A, Humbel B, de Jong L, van Driel R. 1992. A monoclonal antibody recognizing nuclear matrix-associated nuclear bodies. J Cell Sci 101:773–784. [DOI] [PubMed] [Google Scholar]
- 55.Gawn JM, Greaves RF. 2002. Absence of IE1 p72 protein function during low-multiplicity infection by human cytomegalovirus results in a broad block to viral delayed-early gene expression. J Virol 76:4441–4455. doi: 10.1128/JVI.76.9.4441-4455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greaves RF, Mocarski ES. 1998. Defective growth correlates with reduced accumulation of a viral DNA replication protein after low-multiplicity infection by a human cytomegalovirus IE1 mutant. J Virol 72:366–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mocarski ES, Kemble GW, Lyle JM, Greaves RF. 1996. A deletion mutant in the human cytomegalovirus gene encoding IE1491aa is replication defective due to a failure in autoregulation. Proc Natl Acad Sci U S A 93:11321–11326. doi: 10.1073/pnas.93.21.11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spengler ML, Kurapatwinski K, Black AR, Azizkhan-Clifford J. 2002. SUMO-1 modification of human cytomegalovirus IE1/IE72. J Virol 76:2990–2996. doi: 10.1128/JVI.76.6.2990-2996.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nevels M, Brune W, Shenk T. 2004. SUMOylation of the human cytomegalovirus 72-kilodalton IE1 protein facilitates expression of the 86-kilodalton IE2 protein and promotes viral replication. J Virol 78:7803–7812. doi: 10.1128/JVI.78.14.7803-7812.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mucke K, Paulus C, Bernhardt K, Gerrer K, Schon K, Fink A, Sauer EM, Asbach-Nitzsche A, Harwardt T, Kieninger B, Kremer W, Kalbitzer HR, Nevels M. 2014. Human cytomegalovirus major immediate early 1 protein targets host chromosomes by docking to the acidic pocket on the nucleosome surface. J Virol 88:1228–1248. doi: 10.1128/JVI.02606-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shin HJ, Kim YE, Kim ET, Ahn JH. 2012. The chromatin-tethering domain of human cytomegalovirus immediate-early (IE) 1 mediates associations of IE1, PML and STAT2 with mitotic chromosomes, but is not essential for viral replication. J Gen Virol 93:716–721. doi: 10.1099/vir.0.037986-0. [DOI] [PubMed] [Google Scholar]
- 62.Scherer M, Klingl S, Sevvana M, Otto V, Schilling EM, Stump JD, Muller R, Reuter N, Sticht H, Muller YA, Stamminger T. 2014. Crystal structure of cytomegalovirus IE1 protein reveals targeting of TRIM family member PML via coiled-coil interactions. PLoS Pathog 10:e1004512. doi: 10.1371/journal.ppat.1004512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Preston CM, Nicholl MJ. 1997. Repression of gene expression upon infection of cells with herpes simplex virus type 1 mutants impaired for immediate-early protein synthesis. J Virol 71:7807–7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samaniego LA, Neiderhiser L, DeLuca NA. 1998. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J Virol 72:3307–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Burgdorf SW, Clark CL, Burgdorf JR, Spector DH. 2011. Mutation of glutamine to arginine at position 548 of IE2 86 in human cytomegalovirus leads to decreased expression of IE2 40, IE2 60, UL83, and UL84 and increased transcription of US8-9 and US29-32. J Virol 85:11098–11110. doi: 10.1128/JVI.05315-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Everett RD, Boutell C, Orr A. 2004. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J Virol 78:1763–1774. doi: 10.1128/JVI.78.4.1763-1774.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Malone CL, Vesole DH, Stinski MF. 1990. Transactivation of a human cytomegalovirus early promoter by gene products from the immediate-early gene IE2 and augmentation by IE1: mutational analysis of the viral proteins. J Virol 64:1498–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stenberg RM, Fortney J, Barlow SW, Magrane BP, Nelson JA, Ghazal P. 1990. Promoter-specific trans activation and repression by human cytomegalovirus immediate-early proteins involves common and unique protein domains. J Virol 64:1556–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Depto AS, Stenberg RM. 1989. Regulated expression of the human cytomegalovirus pp65 gene: octamer sequence in the promoter is required for activation by viral gene products. J Virol 63:1232–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yao F, Schaffer PA. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J Virol 69:6249–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sommer MH, Scully AL, Spector DH. 1994. Transactivation by the human cytomegalovirus IE2 86-kilodalton protein requires a domain that binds to both the TATA box-binding protein and the retinoblastoma protein. J Virol 68:6223–6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.White EA, Spector DH. 2005. Exon 3 of the human cytomegalovirus major immediate-early region is required for efficient viral gene expression and for cellular cyclin modulation. J Virol 79:7438–7452. doi: 10.1128/JVI.79.12.7438-7452.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]