

ABSTRACT
Mumps virus (MuV) infection induces formation of cytoplasmic inclusion bodies (IBs). Growing evidence indicates that IBs are the sites where RNA viruses synthesize their viral RNA. However, in the case of MuV infection, little is known about the viral and cellular compositions and biological functions of the IBs. In this study, pulldown purification and N-terminal amino acid sequencing revealed that stress-inducible heat shock protein 70 (Hsp72) was a binding partner of MuV phosphoprotein (P protein), which was an essential component of the IB formation. Immunofluorescence and immunoblotting analyses revealed that Hsp72 was colocalized with the P protein in the IBs, and its expression was increased during MuV infection. Knockdown of Hsp72 using small interfering RNAs (siRNAs) had little, if any, effect on viral propagation in cultured cells. Knockdown of Hsp72 caused accumulation of ubiquitinated P protein and delayed P protein degradation. These results show that Hsp72 is recruited to IBs and regulates the degradation of MuV P protein through the ubiquitin-proteasome pathway.
IMPORTANCE Formation of cytoplasmic inclusion bodies (IBs) is a common characteristic feature in mononegavirus infections. IBs are considered to be the sites of viral RNA replication and transcription. However, there have been few studies focused on host factors recruited to the IBs and their biological functions. Here, we identified stress-inducible heat shock protein 70 (Hsp72) as the first cellular partner of mumps virus (MuV) phosphoprotein (P protein), which is an essential component of the IBs and is involved in viral RNA replication/transcription. We found that the Hsp72 mobilized to the IBs promoted degradation of the MuV P protein through the ubiquitin-proteasome pathway. Our data provide new insight into the role played by IBs in mononegavirus infection.
INTRODUCTION
One of the characteristic features of mononegavirus infection is formation of cytoplasmic inclusion bodies (IBs), which can be observed by light microscopy (1), fluorescence microscopy (2–6), and electron microscopy (7–9). It is well known that IBs contain nucleocapsid-like structures, but the detailed compositions and biological functions of IBs remain to be elucidated. In the case of Ebola virus (family Filoviridae, order Mononegavirales), the IBs have been reported to be the sites of viral RNA replication (6). Similar findings were also reported for other RNA viruses, including rabies virus (RV) (4) and vesicular stomatitis virus (VSV) (5) (both in the family Rhabdoviridae, order Mononegavirales). In regard to the significance of IB formation, it is currently considered that the IBs concentrate the machinery for viral RNA synthesis. In the present study, we studied mumps virus (MuV), which is also known to form IBs.
MuV is the causative agent of mumps, a common childhood illness characterized by fever and swelling of the salivary glands (10). It often causes neurological complications, including aseptic meningitis, encephalitis, and deafness. MuV belongs to the genus Rubulavirus within the family Paramyxoviridae (order Mononegavirales) (11). The viral nonsegmented negative-strand RNA genome encodes eight viral proteins: the nucleocapsid (N), V, phospho- (P), matrix (M), hemagglutinin-neuraminidase (HN), fusion (F), large (L), and small hydrophobic (SH) proteins. The genome is encapsidated by the N protein and forms an active template for RNA replication and transcription, a viral ribonucleoprotein (vRNP), with viral polymerases composed of the P and L proteins (12). The F and HN proteins are envelope glycoproteins, and the M protein is an intravirion protein that associates with the cytoplasmic tails of the envelope glycoproteins and vRNP. The SH protein is also a structural integral membrane protein with unknown function. The V protein is a nonstructural protein that counteracts the host antiviral responses. The V and P proteins are encoded by the same gene using overlapping reading frames. The V protein is translated from the V mRNA, a faithful transcript of the V/P gene, whereas the P protein is translated from the P mRNA possessing two additional nontemplated guanine residues inserted by an RNA-editing mechanism. Therefore, the resulting P and V proteins have identical N-terminal regions and unique C-terminal regions.
Heat shock protein 70 (Hsp70) family proteins are molecular chaperones that comprise a set of abundant cellular machines (13). Under normal, unstressed conditions, Hsp70 proteins play central roles in protein homeostasis, such as assisting in the folding or assembly of newly translated proteins, guiding the intracellular trafficking of client proteins, disassembling oligomeric protein structures, and facilitating the proteolytic degradation of unstable proteins. Under conditions of stress, they prevent abnormal protein aggregation and assist in the renaturation or degradation of misfolded proteins. Human Hsp70 proteins are comprised of at least eight gene products with different amino acid sequences, expression levels, and subcellular localizations (14). Among the major Hsp70 proteins expressed at high levels in a wide range of tissues, stress-inducible heat shock protein 70 (Hsp72) and constitutively expressed heat shock cognate protein 70 (Hsc70) are present in the cytoplasm and nucleus, while glucose-regulated protein 78 (GRP78) and GRP75 are localized in the lumen of the endoplasmic reticulum (ER) and the mitochondrial matrix, respectively. During virus infections, Hsp70 family proteins are frequently mobilized to the viral replication sites and play roles in all steps of the life cycles of many DNA and RNA viruses (4, 15–17). In the case of negative-strand RNA viruses, Hsp70 has been found to have both positive and negative regulatory effects on viral propagation. For example, Hsp72 interacts with the N protein of measles virus (MV), which is another member of the paramyxovirus family, and enhances viral RNA replication (18). Hsp70 is also recruited to the IBs of RV and positively regulates RV infection (19). On the other hand, Hsp70 interferes with the polymerase activity of influenza virus and negatively regulates viral RNA replication (20, 21), highlighting the complexity of the virus-chaperone interaction.
Our data revealed that MuV-infected cells also recruited Hsp72 to the IBs. In the present study, we analyzed the molecular basis and significance of this event in MuV-infected cells.
MATERIALS AND METHODS
Cells and virus.
Vero (African green monkey kidney), 293T (human kidney), and Huh7 (human hepatocellular carcinoma) cells were maintained in Dulbecco's modified Eagle's minimal essential medium (DMEM) (Nacalai Tesque, Kyoto, Japan) supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin, and 10% fetal bovine serum (FBS).
The highly neuropathogenic MuV strain Odate was isolated from a patient who developed aseptic meningitis (22) and used in this study.
Plasmids.
The cDNA of the P protein was amplified from 293T cells infected with MuV Odate by reverse transcription (RT)-PCR and cloned into pCAGGS, pCAGPM-N-HA, and pCAG-MCS2-FOS for expression in mammalian cells as nontagged, hemagglutinin (HA)-tagged, and FLAG-One-STrEP (FOS)-tagged proteins, respectively (23). The resulting plasmids were designated pCAGGS-P, pCAGPM-HA-P, and pCAG-P-FOS, respectively. The cDNAs of the N, L, and V proteins were also amplified and cloned into pCAGGS and/or pCAG-MCS2-FOS, resulting in pCAGGS-N, -L, and -V and pCAG-N-FOS, respectively. The cDNAs of human Hsp72, Hsc70, GRP78, and ubiquitin (Ub) were amplified from 293T cells by RT-PCR and cloned into pcDNA3.1-FLAG or pCAGPM-N-HA for expression in mammalian cells as a FLAG- or HA-tagged protein. The resulting plasmids were designated pcDNA-FLAG-Hsp72, pcDNA-FLAG-Hsc70, pcDNA-FLAG-GRP78, and pCAGPM-HA-Ub, respectively. A series of deletion mutants of the P protein and Hsp72 were generated by PCR-based mutagenesis. All plasmids were confirmed by sequencing with an ABI Prism 3130xl genetic analyzer (Life Technologies Inc., Rockville, MD).
Reagents and antibodies.
MG-132 and cycloheximide (CHX) were purchased from Cell Signaling Technology (Danvers, MA) and Sigma (St. Louis, MO), respectively. Lactacystin and epoxomycin were purchased from Peptide Institute Inc. (Osaka, Japan). Anti-N (23D), -P (57A), -M (79D), -F (170C), and -HN (78) mouse monoclonal antibodies (MAbs) and anti-MuV V (T60), -V/P (T61), and -L (L17) rabbit polyclonal antibodies (PAbs) were prepared as described previously (24–26). Anti-MuV N rabbit PAb was generated with a synthetic peptide derived from the MuV N protein at Sigma. Anti-FLAG (M2) and anti-α-tubulin mouse MAbs were purchased from Sigma. Anti-Hsp70 (C92F3A-5) and anti-Hsc70 (1F2-H5) mouse MAbs were purchased from StressMarq Bioscience Inc. (Victoria, Canada). Anti-HA mouse MAb (HA11), anti-GRP78 rabbit PAb (ab21685), and anti-ubiquitin rabbit PAb (number 3933) were purchased from Covance (Richmond, CA), Abcam (Cambridge, United Kingdom), and Cell Signaling Technology, respectively.
Virus titration.
Virus titers were determined by plaque assay in triplicate using Vero cells in 12-well plates. After 1 to 2 h of virus adsorption, the cells were cultured in DMEM with 5% FBS and 1% agarose. At 6 days postinoculation, the cells were stained with Neutral Red solution (Sigma), and the plaque counts were determined.
Cell extracts, immunoblotting, and immunoprecipitation.
For the preparation of cell extracts, cells were washed twice with cold phosphate-buffered saline (PBS) and then lysed in cell lysis buffer (20 mM Tris-HCl, pH 7.5, 135 mM NaCl, 1% Triton X-100, and protease inhibitor cocktail [Complete Mini; Roche, Mannheim, Germany]). For immunoblotting, the cell lysate was boiled in sodium dodecyl sulfate (SDS) sample buffer and subjected to SDS-polyacrylamide gel electrophoresis (PAGE). The proteins were transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA) and incubated with the appropriate antibodies. Each protein was visualized with SuperSignal West Femto Maximum Sensitivity Substrate (Life Technologies Inc.) and detected by use of an LAS-3000 image analyzer system (Fuji Film, Tokyo, Japan). For immunoprecipitation, the cell lysate was precleaned with protein G-Sepharose (GE Healthcare, Buckinghamshire, United Kingdom). Antibody-protein complexes were purified with protein G beads and washed with cell lysis buffer three times. After boiling in SDS sample buffer, the proteins were separated by SDS-PAGE and processed for immunoblotting.
Immunofluorescence microscopy.
Vero cells were fixed in 4% paraformaldehyde in PBS for 15 min at room temperature. Then, the cells were permeabilized with 0.2% Triton X-100 in PBS for 10 min, blocked with PBS containing 2% bovine serum albumin (BSA) for 30 min at room temperature, and incubated with the appropriate antibodies. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). The samples were examined under an FV1000D confocal laser scanning microscope (Olympus, Tokyo, Japan).
Fluorescence in situ hybridization.
MuV genomic RNA was detected using the QuantiGene ViewRNA ISH cell assay kit (Affymetrix, Santa Clara, CA) and mumps virus probe set (Affymetrix), which can hybridize to nucleotides 13501 to 14518 of the MuV genome. Vero cells infected with MuV were fixed in 4% paraformaldehyde in PBS for 30 min at room temperature. Then, the cells were permeabilized and hybridized according to the manufacturer's protocol. The samples were examined under an FV1000D confocal laser scanning microscope.
FOS-tagged purification and N-terminal amino acid sequencing.
pCAG-N-FOS, pCAG-P-FOS, or empty vector was transfected into 293T cells by use of TransIT LT1 (Mirus, Madison, WI), harvested at 24 h posttransfection, washed twice with ice-cold PBS, suspended in cell lysis buffer, and centrifuged at 14,000 × g for 20 min at 4°C. The supernatant was pulled down using 50 μl of STrEP-Tactin Sepharose (IBA, Gottingen, Germany) equilibrated with cell lysis buffer for 2 h at 4°C. The affinity beads were washed three times with cell lysis buffer and suspended in 2× SDS-PAGE sample buffer. The proteins were subjected to SDS-PAGE and transferred to membranes, followed by Coomassie brilliant blue (CBB) staining using CBB Stain One (Nakalai Tesque). Each band was spliced out and subjected to N-terminal amino acid sequencing (Procise 491cLC; Applied Biosystems).
Gene silencing.
A commercially available small interfering RNA (siRNA) pool targeting Hsp72 (siGenome Smartpool, human Hsp72) and control nontargeting siRNA were purchased from Dharmacon (Buckinghamshire, United Kingdom) and transfected using Lipofectamine RNAiMax (Life Technologies Inc.) according to the manufacturer's protocol.
qRT-PCR.
Total RNA was prepared by use of an RNeasy minikit (Qiagen), and first-strand cDNA was synthesized using PrimeScript II RTase and an oligo(dT) primer (TaKaRa Bio, Shiga, Japan). The amount of each cDNA was measured using the Universal ProbeLibrary and the LightCycler 480 system (Roche) according to the manufacturer's instructions. Primers for quantitative RT-PCR (qRT-PCR) were designed by using the Probe Finder software (Roche). The value of each RNA was normalized to that of hypoxanthine phosphoribosyltransferase 1 (HPRT1) mRNA.
TUNEL staining.
Vero cells were fixed in 4% paraformaldehyde in PBS for 15 min at room temperature. Then, the cells were permeabilized with 0.2% Triton X-100 in PBS for 10 min at room temperature and incubated with terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) reaction mixture (In situ Apoptosis Detection Kit; TaKaRa Bio) for 90 min at 37°C. Nuclei were stained with DAPI. The samples were examined under a BZ-8000 fluorescence microscope (Keyence Co., Osaka, Japan).
Caspase activity and cell viability assays.
Caspase 3/7 activity in Vero cells in 96-well plates was measured by using a Caspase-Glo 3/7 Assay Kit (Promega, Madison, WI) according to the manufacturer's protocol. Cell viability was measured by using a CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega) and used for normalization.
RESULTS
Coexpression of the MuV N and P proteins induces formation of an IB-like structure, where these proteins were concentrated.
A previously reported analysis using electron microscopy suggested that the IBs observed in MuV-infected cells are aggregates of nucleocapsids (7). In order to identify viral components of the MuV-induced IBs, the intracellular localizations of MuV proteins and genomic RNA were analyzed by immunofluorescence microscopy and fluorescence in situ hybridization (Fig. 1A and B). The N, V, P, and L proteins were localized mainly to the IBs, as well as viral genomic RNA, while the M protein was detected not only in the IBs, but also in the nucleoli and the pericellular region. The F and HN proteins were localized mainly in the pericellular region. To further clarify the intracellular localizations of the vRNP components, the N, P, and L proteins were expressed in cells alone or in combination using expression plasmids. Each protein showed a diffuse distribution pattern throughout the cytoplasm when expressed alone (Fig. 1C). Coexpression of the N and P proteins, but not other combinations, led to the formation of IB-like structures, where these proteins were concentrated (Fig. 1D).
FIG 1.

Intracellular localizations of MuV proteins and genomic RNA. (A) Vero cells infected with MuV were immunostained at 24 h postinfection (p.i.) with mouse anti-N (23D), anti-P (57A), anti-M (79D), anti-F (170C), or anti-HN (78) MAb and rabbit anti-N, anti-V/P (T61), anti-V (T60), or anti-L (L17) PAb, followed by AF488-conjugated anti-mouse IgG and AF594-conjugated anti-rabbit IgG, respectively. The cell nuclei were stained with DAPI (blue). (B) Vero cells infected with MuV were immunostained at 24 h p.i. with rabbit anti-V/P PAb and AF594-conjugated anti-rabbit IgG. Then, the cells were subjected to a fluorescence in situ hybridization assay to detect viral genomic RNA. The cell nuclei were stained with DAPI (blue). (C and D) Vero cells transfected with pCAGGS-N, -P, and/or -L alone or in combination were immunostained at 24 h posttransfection with anti-N or anti-P MAb and anti-V/P or anti-L PAb, followed by AF488-conjugated anti-mouse IgG and AF594-conjugated anti-rabbit IgG, respectively. Cell nuclei were stained with DAPI (blue).
MuV P protein associates with Hsp70 family proteins.
Data from immunofluorescence assays showed that the N, P, V, M, and L proteins were concentrated in the IBs and that coexpression of the N and P proteins induced the formation of IB-like structures. As an initial step in the search for host factors involved in the formation of IBs, host proteins associated with the N and P proteins were analyzed by an FOS affinity tag purification method (Fig. 2A). Coexpression of N- and P-FOS proteins led to the formation of IB-like structures similar to the untagged N and P proteins, indicating that placement of the FOS tag at the C termini of the N and P proteins had minimal effects on the function of IB-like structure formation (Fig. 2B). The N- and P-FOS proteins were expressed in 293T cells and purified together with associated proteins. Several polypeptides, including three polypeptides with molecular masses of approximately 72, 73, and 78 kDa, were copurified with the P protein, whereas no N protein-associated host proteins were detected (Fig. 2C). Analysis by immunoblotting confirmed that the purified P-FOS protein was observed at a mass of ∼45 kDa and suggested that several other bands were different forms of the P protein, seemingly corresponding to phosphorylated and cleaved P-FOS products (Fig. 2D). In order to identify the three polypeptides with masses of approximately 72, 73, and 78 kDa, individual bands were isolated and analyzed by N-terminal amino acid sequencing. The 78-kDa polypeptide was identified as GRP78, with a sequence of EEEDKKEDVG (residues 19 to 28), whereas the amino acid sequences of the other two polypeptides of 72 and 73 kDa were not identified by the assay. It was postulated that they could be Hsp72 and Hsc70, because Hsp70 family proteins have conserved domains and the masses of Hsp72 and Hsc70 are 72 kDa and 73 kDa, respectively. We investigated this possibility by immunoblotting using specific antibodies against Hsp72, Hsc70, and GRP78. As shown in Fig. 2E, Hsp72 and Hsc70, as well as GRP78, were clearly detected. These data thus showed that all three Hsp70 family proteins were associated with the P protein.
FIG 2.

Identification of cellular proteins associated with the MuV P protein by FOS-tagged purification. (A) Overview of the FOS-tagged purification of cellular proteins associated with the MuV N and P proteins. (B) Vero cells transfected with pCAG-N-FOS and/or pCAG-P-FOS were immunostained at 24 h posttransfection with mouse anti-N MAb and rabbit anti-V/P PAb, followed by AF488-conjugated anti-mouse IgG and AF594-conjugated anti-rabbit IgG. The cell nuclei were stained with DAPI (blue). (C) Purified products were subjected to SDS-PAGE, followed by CBB staining. The empty vector (EV) was used as a negative control. Lane M is a protein molecular mass marker. (D) Purification of the P-FOS protein was confirmed by immunoblotting using mouse anti-FLAG MAb (M2) and rabbit anti-V/P PAb. (E) Detection of Hsp70 family proteins in the purified P-FOS complexes was performed by immunoblotting using the appropriate antibodies.
Hsp72 is upregulated and recruited to IBs during MuV infection.
The interaction between the P protein and the three Hsp70 family proteins (Fig. 3A) was analyzed by coimmunoprecipitation assay. 293T cells expressing the HA-tagged P protein (HA-P) and FLAG-tagged Hsp70 family proteins were used. As shown in Fig. 3B, a massive amount of FLAG-Hsp72 was coimmunoprecipitated with HA-P, whereas only a small amount of FLAG-GRP78 was coprecipitated. Although the binding affinities between the P protein and Hsp70 family proteins might be different, all three Hsp70 family proteins were indeed capable of associating with the P protein in cell lysates. However, it was still unclear whether all three Hsp70 family proteins interact with the P protein in living cells. To clarify this point, we investigated the intracellular localizations of Hsp70 family proteins in MuV-infected cells by immunofluorescence microscopy. The results showed that Hsp72 expression was upregulated and the protein was redistributed to the IBs in the MuV-infected cells (Fig. 3C). On the other hand, the expression levels and localization of Hsc70 and GRP78 were unchanged (Fig. 3C). These results suggested that Hsp72, but not Hsc70 or GRP78, interacts with the P protein in MuV-infected cells. Data from immunoblotting assays also demonstrated that the expression of Hsp72 was increased by MuV infection (Fig. 3D). To better understand the requirements for the upregulation and localization change of Hsp72, the P protein was expressed in cells alone or in combination with the N protein. While ectopic expression of the P protein alone did not induce the upregulation of Hsp72 and was not colocalized with Hsp72 (Fig. 3E and F), the formation of IB-like structures caused by the coexpression of the N and P proteins dramatically induced the expression of Hsp72 and recruited Hsp72 to the IB-like structures (Fig. 3G). Taken together, the upregulation and recruitment to IBs of Hsp72 occurred with IB formation during MuV infection.
FIG 3.

Hsp72 was upregulated and recruited to the IBs during MuV infection. (A) Summary of Hsp70 family protein candidates for binding partners of the P protein. (B) FLAG-Hsp70 proteins were coexpressed with HA-P in 293T cells, immunoprecipitated (IP) with anti-HA antibody, and immunoblotted (IB) with anti-HA and anti-FLAG antibodies. (C) Vero cells infected with MuV were immunostained at 24 h p.i. with mouse anti-Hsp72 or anti-Hsc70 MAb and rabbit anti-MuV V/P PAb or mouse anti-P MAb and rabbit anti-GRP78 PAb, followed by the appropriate Alexa-conjugated secondary antibodies. The cell nuclei were stained with DAPI (blue). (D) Vero cells were infected with MuV at a multiplicity of infection (MOI) of 1.0. The cell lysates were collected at the indicated times and subjected to immunoblotting with the appropriate antibodies. (E) 293T cells were transfected with pCAGGS-P (P) or empty vector (EV). At 24 h posttransfection, the cell lysates were collected and subjected to immunoblotting with the appropriate antibodies. (F and G) Vero cells transfected with pCAGGS-P alone (F) or pCAGGS-N and -P (G) were immunostained at 24 h posttransfection with mouse anti-Hsp72 MAb and rabbit anti-V/P PAb, followed by AF488-conjugated anti-mouse IgG and AF594-conjugated anti-rabbit IgG. The cell nuclei were stained with DAPI (blue).
The N-terminal region of the P protein and the C-terminal region of Hsp72 are responsible for their interaction.
To determine the interacting regions of Hsp72 and the P protein, N-terminally FLAG-tagged Hsp72 (FLAG-Hsp72F), HA-tagged P protein (HA-P-Full), and their truncation polypeptides were expressed in cells, and their interaction was analyzed by coimmunoprecipitation assays. FLAG-Hsp72N was comprised of the N-terminal ATPase domain, while FLAG-Hsp72C was comprised of the C-terminal peptide binding domain and variable region (Fig. 4A). HA-P-Full was coprecipitated with FLAG-Hsp72F and FLAG-Hsp72C (Fig. 4A), indicating that the C-terminal region of Hsp72 interacted with the P protein. HA-PΔN was comprised of the oligomerization domain and C-terminal region, while HA-PΔC was comprised of the N-terminal region and the oligomerization domain (Fig. 4B). The coimmunoprecipitation assay showed that FLAG-Hsp72F was coprecipitated with HA-P-Full and HA-PΔC (Fig. 4B). The data, taken together, indicated that the N-terminal region of the P protein and the C-terminal region of Hsp72 were responsible for their interaction. A coimmunoprecipitation assay was also performed for the V protein, as the P and V proteins possess a common N-terminal region. The data showed that Hsp72 was also associated with the V protein (Fig. 4C).
FIG 4.
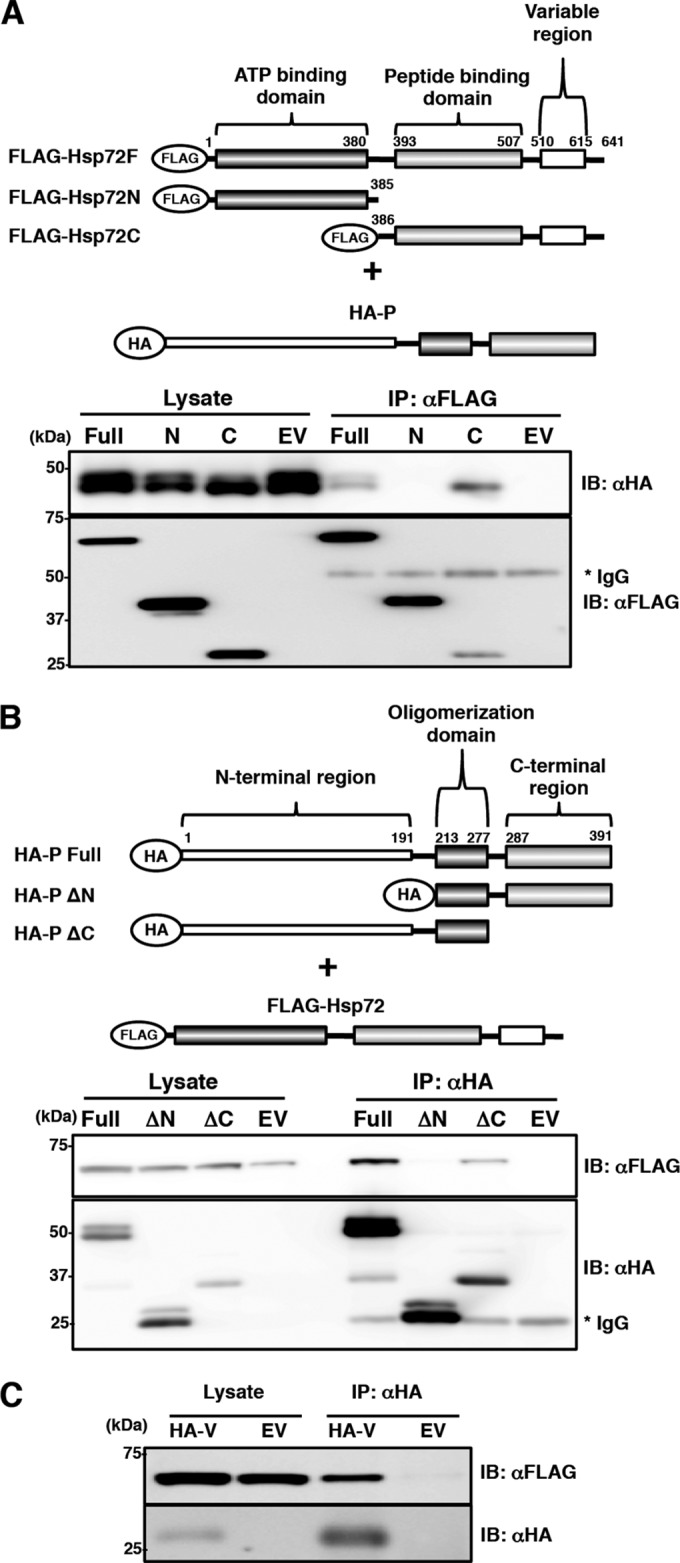
Determination of the regions responsible for the interaction between the P protein and Hsp72. (A) FLAG-Hsp72 and its deletion mutants were coexpressed with HA-P in 293T cells, immunoprecipitated with anti-FLAG antibody, and immunoblotted with anti-FLAG and anti-HA antibodies. (B) HA-P and its deletion mutants were coexpressed with FLAG-Hsp72 in 293T cells, immunoprecipitated with anti-HA antibody, and immunoblotted with anti-HA and anti-FLAG antibodies. (C) FLAG-Hsp72 was coexpressed with HA-V in 293T cells, immunoprecipitated with anti-HA antibody, and immunoblotted with anti-HA and anti-FLAG antibodies.
Hsp72 is nonessential for MuV replication but suppresses apoptotic cell death of MuV-infected cells.
To determine the roles of Hsp72 in MuV infection, the expression of Hsp72 was suppressed by Hsp72-specific siRNAs (siHsp72). Transfection of siHsp72 efficiently knocked down Hsp72 expression (Fig. 5A). However, the levels of viral RNAs, virus production, and IB formation were not affected (Fig. 5B to D), demonstrating that Hsp72 was nonessential for MuV replication. Alternatively, it was noted that ∼10% of Hsp72 knockdown cells infected with MuV were positive for the TUNEL stain, indicating apoptosis induction, while only ∼2.5% of control cells were TUNEL positive (Fig. 6A and B). The induction of apoptosis in Hsp72 knockdown cells was confirmed by the elevated caspase 3/7 activity (Fig. 6C). These data indicated that Hsp72 was needed to suppress apoptotic cell death of MuV-infected cells.
FIG 5.

Effects of Hsp72 knockdown on MuV propagation. (A) At 48 h posttransfection with either siHsp72 or siNC, Vero cells were inoculated with MuV at an MOI of 1.0. The cell lysates were collected at 24 h p.i. and subjected to immunoblotting with the indicated antibodies. (B) At 48 h posttransfection with either siHsp72 or siNC, Vero cells were inoculated with MuV at an MOI of 5.0. Total cellular RNA was extracted at 24 h p.i. and subjected to RT using an oligo(dT) primer. The levels of N mRNA were determined by real-time PCR and calculated as percentages of the control HPRT1 mRNA level. The data are representative of three independent experiments. The error bars indicate the standard deviations of the means. (C) At 48 h posttransfection with either siHsp72 or siNC, Vero or Huh7 cells were infected with MuV at an MOI of 0.01. The supernatants were collected at 24, 48, 72, and 96 h p.i., and the infectious titers were determined by plaque assay in Vero cells. The results shown are from three independent experiments, with the error bars representing the standard deviations. (D) At 48 h posttransfection with either siHsp72 or siNC, Vero cells were inoculated with MuV at an MOI of 1.0. At 24 h p.i., the cells were fixed, permeabilized, and immunostained with mouse anti-Hsp72 MAb and rabbit anti-V/P PAb, followed by AF488-conjugated anti-mouse IgG and AF594-conjugated anti-rabbit IgG. The cell nuclei were stained with DAPI (blue).
FIG 6.
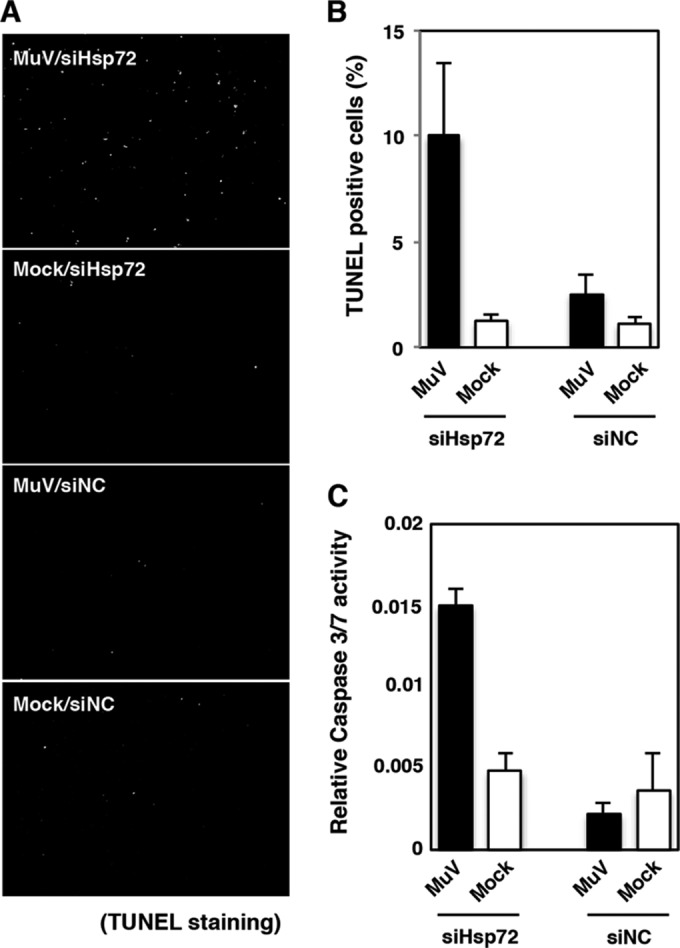
Increased apoptosis in MuV-infected Hsp72 knockdown cells. (A) At 48 h posttransfection with either siHsp72 or siNC, Vero cells were infected with MuV at an MOI of 5.0. At 24 h p.i., the cells were fixed, permeabilized, and stained with TUNEL stain. (B) TUNEL-positive cells were counted and expressed as percentages of the total number of cells (n > 1,000). The total number of cells was calculated by DAPI staining. (C) At 48 h posttransfection with either siHsp72 or siNC, Vero cells were inoculated with MuV at an MOI of 5.0. At 24 h p.i., caspase 3/7 activity was measured, and the relative caspase 3/7 activity was calculated by normalization with cell viability. The results are from three independent assays, with the error bars representing standard deviations.
The P and V proteins were ubiquitinated in MuV-infected cells.
Since IBs are comprised of abundant viral proteins that could lead to deleterious consequences for the cells, Hsp72 may play a role in ubiquitin-mediated degradation of the accumulated viral proteins. To examine this possibility, HA-Ub-expressing 293T cells were infected with MuV and subjected to immunoprecipitation assays. Polypeptides were immunoprecipitated with the anti-MuV V/P antibody (T61) and detected by immunoblotting using an anti-HA antibody. They showed broad size distributions (Fig. 7A). The signals of ubiquitinated proteins were increased in the presence of MG132, a proteasomal inhibitor. Since the T61 antibody detects both the P and V proteins, these data suggested that the V protein, the P protein, or both were ubiquitinated in MuV-infected cells. Furthermore, endogenous ubiquitinated P and V proteins were detected in Vero cells infected with MuV (Fig. 7B). Taken together, these results demonstrated that both the P and V proteins were ubiquitinated in MuV-infected cells.
FIG 7.

Ubiquitination of MuV P and V proteins. (A) 293T cells were transfected with a plasmid encoding HA-Ub and infected with MuV. At 24 h p.i., the cells were treated with 10 μM MG-132 for 5 h, and then the cell lysates were subjected to an immunoprecipitation assay with anti-V/P antibody, followed by immunoblotting with the appropriate antibodies. (B) At 24 h p.i., Vero cells infected with MuV were treated with 10 μM MG-132 for 5 h, and then the cell lysates were subjected to an immunoprecipitation assay with anti-V/P antibody, followed by immunoblotting with the appropriate antibodies.
Hsp72 targets the P protein for degradation through the ubiquitin-proteasome pathway.
To investigate the roles of Hsp72 in ubiquitin-mediated degradation of the P and V proteins, the effects of Hsp72 knockdown were analyzed. 293T cells expressing HA-Ub were transfected with either siHsp72 or control siNC. The cells were then infected with MuV and subjected to immunoprecipitation assays, in which the polypeptides were immunoprecipitated with the T61 antibody and detected by immunoblotting using an anti-HA antibody. The signal levels were clearly higher in the Hsp72 knockdown cells than in control cells (Fig. 8A). In fact, the signal levels were as high as those in MG-132-treated cells (Fig. 8A). These data suggested that the ubiquitinated V protein, P protein, or both were accumulated in Hsp72 knockdown cells. Similar experiments were performed using cells expressing the V or P protein individually. Signals for the ubiquitinated P protein were low in control (siNC-transfected) cells but increased in Hsp72 knockdown (siHsp72-transfected) cells (Fig. 8B). On the other hand, signals for the ubiquitinated V protein were similar between Hsp72 knockdown and control cells (Fig. 8B). These data suggested that Hsp72 was involved in P protein degradation, but not in V protein degradation. Next, the kinetics of P and V protein degradation was analyzed. Hsp72 knockdown and control Vero cells were infected with MuV and cultured for 24 h. Then, these cells were incubated for 0 to 12 h in the presence of CHX. Degradation of the P protein was suppressed in Hsp72 knockdown cells (Fig. 8C, dimethyl sulfoxide [DMSO]-treated lanes). On the other hand, degradation of the V protein was minimally affected. To further confirm the proteasomal degradation of P protein, we treated the MuV-infected cells with specific proteasome inhibitors, lactacystin and epoxomycin, which have higher degrees of specificity than MG-132 (27). Degradation of the P and V proteins was blocked by treatment with the proteasome inhibitors (Fig. 8C). Taken together, these data show that Hsp72 binds to both the V and P proteins but specifically promotes proteasomal degradation of the P protein.
FIG 8.

Hsp72 regulates P protein degradation through the ubiquitin-proteasome system. (A) At 48 h posttransfection with either siHsp72 or siNC, 293T cells were transfected with a plasmid encoding HA-Ub and infected with MuV. At 24 h p.i., the cells were treated with 10 μM MG-132 for 5 h, and then the cell lysates were subjected to an immunoprecipitation assay with anti-V/P antibody, followed by immunoblotting with anti-HA and anti-V/P antibodies. (B) At 48 h posttransfection with either siHsp72 or siNC, 293T cells were transfected with plasmids pCAGPM-HA-Ub and pCAGGS-P or -V, respectively. After 24 h, the cell lysates were subjected to an immunoprecipitation assay with anti-V/P antibody, followed by immunoblotting with anti-HA and anti-V/P antibodies. (C) At 48 h posttransfection with either siHsp72 or siNC, Vero cells were infected with MuV. At 24 h p.i., the cells were treated with MG-132 (10 μM), lactacystin (5 μM), epoxomycin (10 μM), or DMSO for 2 h. Then, 50 μg/ml of CHX was added, and the cell lysates were collected at the indicated times. The expression levels of the P and V proteins were detected by immunoblotting with anti-V/P antibody.
DISCUSSION
Many RNA viruses form IBs. However, the precise functions and complete compositions of IBs remain to be elucidated. Studies of negative-strand RNA viruses have reported that the viral genomic RNA and mRNA were present, along with the machineries for viral RNA synthesis in IBs (3–6). Therefore, IBs are likely the sites of viral RNA replication and transcription (3–6). Also, in the case of MuV infection, the vRNP components were concentrated in the IBs (7). This compartmentalization may facilitate virus replication. In addition to viral proteins, host factors involved in innate immune responses are localized to the IBs. This is thought to be a virus strategy to sequester cellular detectors of viral infections (3, 28).
Our data demonstrated that Hsp72 was recruited to the IBs and interacted with the P protein during MuV infection. Many viruses use cellular chaperones for their genome replication, protein synthesis, and virion assembly (29). In the paramyxovirus family, Hsp72 has been shown to enhance MV RNA replication and transcription through interaction with the C-terminal region of the N protein (18). Hsp72 also associates with polymerase complexes of respiratory syncytial virus (RSV) to positively affect viral RNA synthesis (30). Therefore, it was possible that MuV actively used Hsp72 for its replication. However, we considered this unlikely, since knockdown of Hsp72 showed little, if any, effect on MuV propagation in cultured cells. Recent studies revealed that the MuV P protein forms a unique tetramer structure different from those of other paramyxoviruses (31, 32). Thus, MuV might differ from MV and RSV in the requirements for viral RNA replication and propagation. Further investigation will be required to define the roles of Hsp72 in MuV infection and to explain these differences.
Abnormally accumulated proteins disrupt cellular functions and are implicated in various cellular disorders. Thus, these abnormal aggregates are required for rapid degradation (33, 34). Recruitment of chaperones is an adaptive cellular response to assist in ubiquitin-dependent degradation. Hsp72 is usually expressed at low basal levels and increases in response to cellular stressors; therefore, the upregulation and recruitment to IBs of Hsp72 indicates that the formation of abnormal structures consisting of massive viral proteins is deleterious to cell survival. Indeed, our data revealed that Hsp72 bound to the P protein through its C-terminal peptide binding domain and promoted the ubiquitin-proteasomal degradation of the P protein and suppressed apoptotic cell death of MuV-infected cells. Because the accumulation of the ubiquitinated P protein had no effect on viral propagation, Hsp72 might contribute to the degradation of excess amounts of the P protein. It is unclear why the P protein, but not the N protein, is targeted by Hsp72-mediated degradation. However, these data may suggest a specific role for P protein degradation in cellular stress responses. One possibility is speculated to be as follows: the specific degradation of the MuV P protein may elicit a high-avidity cytotoxic-T-lymphocyte (CTL) response, since the degraded paramyxoviral P protein, but not other viral proteins, has been reported to contribute to effective CTL elicitation via major histocompatibility complex (MHC) class I presentation (35, 36). Although we did not examine the possibility in this study, Hsp72 might play a crucial role in the elicitation of high-avidity CTLs by regulating the ubiquitin-proteasomal degradation of the MuV P protein.
Hsp70 family chaperone proteins play major roles in protein quality control (PQC) and maintenance of protein homeostasis. They not only support correct folding and refolding of client proteins, but also guide the substrates to protein degradation systems when a native folding state cannot be reached (37, 38). Hsp70 is involved in the ubiquitin-proteasome system, which is the main cellular protein degradation system. Recruitment of the carboxyl terminus of Hsp70-interacting protein (CHIP), which serves as an E3 ubiquitin ligase, is a well-known function of Hsp70 in the ubiquitin-proteasome pathway (39). Hsp70 is also involved in multistep processes, such as substrate unfolding, insertion into the proteasomal core, and proteasome biogenesis (40, 41). Accumulation of the ubiquitinated P protein in Hsp72-knocked-down cells suggests that Hsp72 plays a crucial role in degradation of the P protein after the step of ubiquitination.
The V protein was also ubiquitinated and associated with Hsp72. However, Hsp72 was not essential for V protein degradation. The MuV P protein contains a multimerization domain at the unique C-terminal region and forms a homotetramer (31), while the V protein lacks the multimerization domain and functions as a monomer (42, 43). The rate-limiting step of proteasomal degradation is the unfolding of substrates (44). Therefore, degradation of the P protein may require the support of Hsp72 due to the tetrameric structure.
In summary, the formation of IBs is a common strategy of many negative-strand RNA viruses. The present study demonstrated for the first time the functional interaction between viral and host proteins in the IBs of MuV-infected cells. The host chaperone protein Hsp72 interacted with the MuV P protein in IBs and targeted the MuV P protein for ubiquitin-mediated degradation. These data should provide insights into the roles of chaperone proteins in virus-infected cells.
ACKNOWLEDGMENTS
We thank Nozomi Takeda of the Equipment Management Center (Creative Research Institution, Hokkaido University) for technical support. We also thank all the members of the Department of Virology III, NIID, for their technical advice and critical input.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology (Grant-in-Aid for Research Activity Start-up 25893295) and the Ministry of Health, Labor and Welfare of Japan.
We declare no conflict of interest.
REFERENCES
- 1.Baskerville A, Fisher-Hoch SP, Neild GH, Dowsett AB. 1985. Ultrastructural pathology of experimental Ebola haemorrhagic fever virus infection. J Pathol 147:199–209. doi: 10.1002/path.1711470308. [DOI] [PubMed] [Google Scholar]
- 2.Carlos TS, Fearns R, Randall RE. 2005. Interferon-induced alterations in the pattern of parainfluenza virus 5 transcription and protein synthesis and the induction of virus inclusion bodies. J Virol 79:14112–14121. doi: 10.1128/JVI.79.22.14112-14121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lifland AW, Jung J, Alonas E, Zurla C, Crowe JE Jr, Santangelo PJ. 2012. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J Virol 86:8245–8258. doi: 10.1128/JVI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lahaye X, Vidy A, Pomier C, Obiang L, Harper F, Gaudin Y, Blondel D. 2009. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: evidence that NBs are sites of viral transcription and replication. J Virol 83:7948–7958. doi: 10.1128/JVI.00554-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heinrich BS, Cureton DK, Rahmeh AA, Whelan SP. 2010. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog 6:e1000958. doi: 10.1371/journal.ppat.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoenen T, Shabman RS, Groseth A, Herwig A, Weber M, Schudt G, Dolnik O, Basler CF, Becker S, Feldmann H. 2012. Inclusion bodies are a site of ebolavirus replication. J Virol 86:11779–11788. doi: 10.1128/JVI.01525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duc-Nguyen H, Rosenblum EN. 1967. Immuno-electron microscopy of the morphogenesis of mumps virus. J Virol 1:415–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norrby E, Marusyk H, Orvell C. 1970. Morphogenesis of respiratory syncytial virus in a green monkey kidney cell line (Vero). J Virol 6:237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geisbert TW, Jahrling PB. 1995. Differentiation of filoviruses by electron microscopy. Virus Res 39:129–150. doi: 10.1016/0168-1702(95)00080-1. [DOI] [PubMed] [Google Scholar]
- 10.Hviid A, Rubin S, Muhlemann K. 2008. Mumps. Lancet 371:932–944. doi: 10.1016/S0140-6736(08)60419-5. [DOI] [PubMed] [Google Scholar]
- 11.Lamb RA, Parks GD. 2006. Paramyxoviridae: the viruses and their replication, p 1449–1496 InKnipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 12.Whelan SP, Barr JN, Wertz GW. 2004. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr Top Microbiol Immunol 283:61–119. doi: 10.1007/978-3-662-06099-5_3. [DOI] [PubMed] [Google Scholar]
- 13.Bukau B, Horwich AL. 1998. The Hsp70 and Hsp60 chaperone machines. Cell 92:351–366. doi: 10.1016/S0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 14.Daugaard M, Rohde M, Jaattela M. 2007. The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett 581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 15.Burch AD, Weller SK. 2004. Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J Virol 78:7175–7185. doi: 10.1128/JVI.78.13.7175-7185.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown G, Rixon HW, Steel J, McDonald TP, Pitt AR, Graham S, Sugrue RJ. 2005. Evidence for an association between heat shock protein 70 and the respiratory syncytial virus polymerase complex within lipid-raft membranes during virus infection. Virology 338:69–80. doi: 10.1016/j.virol.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Kaufer S, Coffey CM, Parker JS. 2012. The cellular chaperone hsc70 is specifically recruited to reovirus viral factories independently of its chaperone function. J Virol 86:1079–1089. doi: 10.1128/JVI.02662-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carsillo T, Zhang X, Vasconcelos D, Niewiesk S, Oglesbee M. 2006. A single codon in the nucleocapsid protein C terminus contributes to in vitro and in vivo fitness of Edmonston measles virus. J Virol 80:2904–2912. doi: 10.1128/JVI.80.6.2904-2912.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lahaye X, Vidy A, Fouquet B, Blondel D. 2012. Hsp70 protein positively regulates rabies virus infection. J Virol 86:4743–4751. doi: 10.1128/JVI.06501-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirayama E, Atagi H, Hiraki A, Kim J. 2004. Heat shock protein 70 is related to thermal inhibition of nuclear export of the influenza virus ribonucleoprotein complex. J Virol 78:1263–1270. doi: 10.1128/JVI.78.3.1263-1270.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li G, Zhang J, Tong X, Liu W, Ye X. 2011. Heat shock protein 70 inhibits the activity of influenza A virus ribonucleoprotein and blocks the replication of virus in vitro and in vivo. PLoS One 6:e16546. doi: 10.1371/journal.pone.0016546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saito H, Takahashi Y, Harata S, Tanaka K, Sano T, Suto T, Yamada A, Yamazaki S, Morita M. 1996. Isolation and characterization of mumps virus strains in a mumps outbreak with a high incidence of aseptic meningitis. Microbiol Immunol 40:271–275. doi: 10.1111/j.1348-0421.1996.tb03346.x. [DOI] [PubMed] [Google Scholar]
- 23.Katoh H, Okamoto T, Fukuhara T, Kambara H, Morita E, Mori Y, Kamitani W, Matsuura Y. 2013. Japanese encephalitis virus core protein inhibits stress granule formation through an interaction with Caprin-1 and facilitates viral propagation. J Virol 87:489–502. doi: 10.1128/JVI.02186-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takeuchi K, Tanabayashi K, Hishiyama M, Yamada YK, Yamada A, Sugiura A. 1990. Detection and characterization of mumps virus V protein. Virology 178:247–253. doi: 10.1016/0042-6822(90)90400-L. [DOI] [PubMed] [Google Scholar]
- 25.Tanabayashi K, Takeuchi K, Hishiyama M, Yamada A, Tsurudome M, Ito Y, Sugiura A. 1990. Nucleotide sequence of the leader and nucleocapsid protein gene of mumps virus and epitope mapping with the in vitro expressed nucleocapsid protein. Virology 177:124–130. doi: 10.1016/0042-6822(90)90466-5. [DOI] [PubMed] [Google Scholar]
- 26.Tsurudome M, Yamada A, Hishiyama M, Ito Y. 1986. Monoclonal antibodies against the glycoproteins of mumps virus: fusion inhibition by anti-HN monoclonal antibody. J Gen Virol 67:2259–2265. doi: 10.1099/0022-1317-67-10-2259. [DOI] [PubMed] [Google Scholar]
- 27.Meng L, Mohan R, Kwok BH, Elofsson M, Sin N, Crews CM. 1999. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl Acad Sci U S A 96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menager P, Roux P, Megret F, Bourgeois JP, Le Sourd AM, Danckaert A, Lafage M, Prehaud C, Lafon M. 2009. Toll-like receptor 3 (TLR3) plays a major role in the formation of rabies virus Negri bodies. PLoS Pathog 5:e1000315. doi: 10.1371/journal.ppat.1000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayer MP. 2005. Recruitment of Hsp70 chaperones: a crucial part of viral survival strategies. Rev Physiol Biochem Pharmacol 153:1–46. doi: 10.1007/s10254-004-0025-5. [DOI] [PubMed] [Google Scholar]
- 30.Munday DC, Wu W, Smith N, Fix J, Noton SL, Galloux M, Touzelet O, Armstrong SD, Dawson JM, Aljabr W, Easton AJ, Rameix-Welti MA, de Oliveira AP, Simabuco F, Ventura AM, Hughes DJ, Barr JN, Fearns R, Digard P, Eleouet JF, Hiscox JA. 2015. Interactome analysis of the human respiratory syncytial virus RNA polymerase complex identifies protein chaperones as important cofactors that promote L-protein stability and RNA synthesis. J Virol 89:917–930. doi: 10.1128/JVI.01783-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cox R, Green TJ, Purushotham S, Deivanayagam C, Bedwell GJ, Prevelige PE, Luo M. 2013. Structural and functional characterization of the mumps virus phosphoprotein. J Virol 87:7558–7568. doi: 10.1128/JVI.00653-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cox R, Pickar A, Qiu S, Tsao J, Rodenburg C, Dokland T, Elson A, He B, Luo M. 2014. Structural studies on the authentic mumps virus nucleocapsid showing uncoiling by the phosphoprotein. Proc Natl Acad Sci U S A 111:15208–15213. doi: 10.1073/pnas.1413268111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bence NF, Sampat RM, Kopito RR. 2001. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 34.Li X, Li H, Li XJ. 2008. Intracellular degradation of misfolded proteins in polyglutamine neurodegenerative diseases. Brain Res Rev 59:245–252. doi: 10.1016/j.brainresrev.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gray PM, Parks GD, Alexander-Miller MA. 2001. A novel CD8-independent high-avidity cytotoxic T-lymphocyte response directed against an epitope in the phosphoprotein of the paramyxovirus simian virus 5. J Virol 75:10065–10072. doi: 10.1128/JVI.75.21.10065-10072.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gray PM, Parks GD, Alexander-Miller MA. 2004. Modulation of CD8+ T cell avidity by increasing the turnover of viral antigen during infection. Cell Immunol 231:14–19. doi: 10.1016/j.cellimm.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Hartl FU, Hayer-Hartl M. 2009. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol 16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 38.Kettern N, Dreiseidler M, Tawo R, Hohfeld J. 2010. Chaperone-assisted degradation: multiple paths to destruction. Biol Chem 391:481–489. doi: 10.1515/BC.2010.058. [DOI] [PubMed] [Google Scholar]
- 39.Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C. 1999. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol 19:4535–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esser C, Alberti S, Hohfeld J. 2004. Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim Biophys Acta 1695:171–188. doi: 10.1016/j.bbamcr.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 41.Rosenzweig R, Glickman MH. 2008. Chaperone-driven proteasome assembly. Biochem Soc Trans 36:807–812. doi: 10.1042/BST0360807. [DOI] [PubMed] [Google Scholar]
- 42.Motz C, Schuhmann KM, Kirchhofer A, Moldt M, Witte G, Conzelmann KK, Hopfner KP. 2013. Paramyxovirus V proteins disrupt the fold of the RNA sensor MDA5 to inhibit antiviral signaling. Science 339:690–693. doi: 10.1126/science.1230949. [DOI] [PubMed] [Google Scholar]
- 43.Li T, Chen X, Garbutt KC, Zhou P, Zheng N. 2006. Structure of DDB1 in complex with a paramyxovirus V protein: viral hijack of a propeller cluster in ubiquitin ligase. Cell 124:105–117. doi: 10.1016/j.cell.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 44.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. 2000. Recognition of the polyubiquitin proteolytic signal. EMBO J 19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]