

ABSTRACT
Hepatitis C virus (HCV) is a serious global health problem and establishes chronic infection in a significant number of infected humans worldwide. Interferon (IFN) and IFN-stimulated genes (ISGs) are amplified during HCV infection but fail to eliminate virus from the liver in a large number of infected patients, and the mechanism is not fully understood. MicroRNAs (miRNAs) have been implicated in the control of many biological processes, including IFN signaling. To gain more insights into the role of cellular miRNAs in possible countermeasures of HCV for suppression of the host antiviral response, a miRNA array was performed by using primary human hepatocytes infected with in vitro cell culture-grown HCV. A group of miRNAs were modulated in HCV-infected primary human hepatocytes. We focused on miR-373, as this miRNA was significantly upregulated in HCV-infected primary human hepatocytes. Here, we analyzed the function of miR-373 in the context of HCV infection. HCV infection upregulates miR-373 expression in hepatocytes and HCV-infected liver biopsy specimens. Furthermore, we discovered that miR-373 directly targets Janus kinase 1 (JAK1) and IFN-regulating factor 9 (IRF9), important factors in the IFN signaling pathway. The upregulation of miR-373 by HCV also inhibited STAT1 phosphorylation, which is involved in ISG factor 3 (ISGF3) complex formation and ISG expression. The knockdown of miR-373 in hepatocytes enhanced JAK1 and IRF9 expression and reduced HCV RNA replication. Taken together, our results demonstrated that miR-373 is upregulated during HCV infection and negatively regulated the type I IFN signaling pathway by suppressing JAK1 and IRF9. Our results offer a potential therapeutic approach for antiviral intervention.
IMPORTANCE Chronic HCV infection is one of the major causes of end-stage liver disease worldwide. Although the recent introduction of direct-acting antiviral (DAA) therapy is extremely encouraging, some infected individuals do not respond to this therapy. Furthermore, these drugs target HCV nonstructural proteins, and with selective pressure, the virus may develop a resistant strain. Therefore, understanding the impairment of IFN signals will help in designing additional therapeutic modalities. In this study, we provide evidence of HCV-mediated upregulation of miR-373 and show that miR-373 impairs IFN signaling by targeting JAK1/IRF9 molecules. The knockdown of miR-373 inhibited HCV replication by upregulating interferon-stimulating gene expression. Together, these results provided new mechanistic insights into the role of miR-373 in HCV infection and suggest a new potential target against HCV infection.
INTRODUCTION
Hepatitis C virus is a member of the Hepacivirus genus of the Flaviviridae family and is represented by seven major genotypes. The virion contains a 9.6-kb single-stranded RNA genome of positive polarity, with highly invariant 5′ and 3′ untranslated regions (UTRs) flanking a long open reading frame (ORF) that is translated via an internal ribosome entry site (IRES) (1). The resulting polyprotein is processed by viral and cellular proteases to yield structural (core, E1, and E2/p7) and nonstructural (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins.
HCV infection is one of the main causes of chronic liver disease. It is estimated that ∼200 million people are chronically infected with HCV worldwide (2, 3). Chronic HCV infection is linked to a deregulation of innate and adaptive immune signaling mechanisms (4). Despite progress in understanding immunity against HCV infection and its pathogenesis along with the development of highly effective direct-acting antivirals (DAAs), a prophylactic anti-HCV vaccine is still lacking, and at least 2 million new HCV infections occur each year (5).
MicroRNAs (miRNAs) are a class of ∼18- to 22-nucleotide-long noncoding RNA molecules that function through posttranscriptional regulation of gene expression. The production of miRNAs requires several processing steps. First, primary miRNAs are processed by the enzymes Drosha and DGCR8 into hairpin-loop-containing pre-miRNAs, which are then subject to nuclear export via exportin 5. Enzymatic processing of the pre-miRNAs by Dicer leads to a mature miRNA duplex (6, 7). Once synthesized, mature miRNA associates with the RNA-induced silencing complex (RISC) together with Argonaute/EIF2C. On the basis of the degree of homology, target mRNA recognition is mediated through interactions between the 5′ end (“seed” region) of miRNA and sites within the coding and 3′ UTRs of mRNAs, leading to mRNA destabilization (8). Deregulation of miRNA occurs frequently in a variety of diseases, including liver disease (9).
Innate and adaptive antiviral immune responses are essential for host survival during viral infection. Upon the recognition of viral components, host cells are activated to produce type I interferon (IFN) and proinflammatory cytokines, thereby upregulating a family of IFN-stimulated genes (ISGs) that exert inhibitory effects on viral replication (10). These pathways are tightly regulated by the host to prevent an inappropriate cellular response. Viruses have also developed strategies to evade the host immune response and resist the antiviral actions of IFN. HCV infection is known to alter the miRNA expression profile of the host cell in facilitating replication or escape from the immune system (11, 12). We have shown previously that miR-130a expression is upregulated in HCV-infected cells, which facilitates HCV replication by inhibiting the interferon-induced transmembrane protein IFITM1 (13). Other miRNAs, such as miR-21, miR-122, miR-146a, and miR-155, also participate in innate and adaptive immune responses to HCV infection (14–17).
In the present study, we demonstrated that miR-373 is upregulated upon HCV infection in human hepatocytes. The upregulation of miR-373 suppresses Janus kinase 1 (JAK1) and IFN-regulating factor 9 (IRF9) expression in hepatocytes, which subsequently represses the type I IFN-mediated antiviral response. In addition, we observed that the knockdown of miR-373 restricts HCV replication by upregulating interferon-stimulating gene expression. To the best of our knowledge, this report is the first to show that miR-373 is upregulated upon RNA virus infection and acts as a negative regulator of type I IFN signaling by targeting JAK1 and IRF9.
MATERIALS AND METHODS
Cells and viruses.
Primary human hepatocytes were procured from a commercially available source (Lonza, MD) and maintained in SABM medium (Lonza, MD) supplemented with 5% fetal bovine serum (FBS). The generation of immortalized human hepatocytes (IHH) was described previously (18). IHH were maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma, MO) containing 10% FBS, 100 U of penicillin G/ml, and 100 μg of streptomycin/ml. Huh7.5 cells harboring an HCV genotype 2a genome-length replicon with a Renilla luciferase reporter gene (Rep2a-Rluc; kindly provided by Hengli Tang) were cultured in DMEM containing 10% FBS with 1% antibiotics. All cells were maintained at 37°C in a humidified 5% CO2 atmosphere.
HCV genotype 1a (clone H77) was grown in IHH, or HCV genotype 2a (clone JFH1) was grown in IHH or Huh7.5 cells, as previously described (19). The virus released into the cell culture supernatant was filtered through a 0.45-μm-pore-size cellulose acetate membrane. For infection, IHH were incubated with HCV genotype 1a/2a (multiplicity of infection of 0.5) for 72 to 96 h.
Patient samples.
Liver biopsy specimens from 6 patients with unrelated liver diseases (other than HCV or hepatitis B virus [HBV]) and 8 adult patients with chronic HCV infection were used for our study. Sample collection and use were approved by the Saint Louis University Institutional Review Board, and written informed consent was obtained from all subjects.
RNA quantitation and reverse transcription-quantitative PCR (qRT-PCR).
Total RNA was isolated by using TRIzol reagent (Invitrogen, CA). cDNA was synthesized by using miR-373- or U6-specific primers with a TaqMan microRNA reverse transcription kit and a random hexamer with Superscript III reverse transcriptase for gene expression studies. Real-time PCR was performed for quantitation by using TaqMan universal PCR master mix and 6-carboxyfluorescein (FAM)-MGB probes for miR-373 (assay identification number 000561), ISG56 (assay identification number Hs00356631), and HCV (assay identification number AI6Q1GI). U6 (assay identification number 001973) or 18S rRNA (assay identification number Hs03928985_g1) was used as an endogenous control for microRNA or gene expression, respectively. The relative expression levels were normalized to the endogenous control by using the 2−ΔΔCT formula (ΔΔCT = ΔCT of the sample − ΔCT of the untreated control).
Luciferase reporter assays.
IHH were transfected with plasmid DNAs (200 ng) harboring the firefly luciferase gene under the control of the interferon-stimulated response element (ISRE) promoter (ISRE-luc) or the ISG56 promoter (ISG56-luc) along with control miRNA (control-miR), miR-373, and miR-10b (as an additional control miRNA) by using Lipofectamine 2000 (Invitrogen, CA). Cells were stimulated by using poly(I-C) at 1 μg/ml by transfection. Cell extracts were prepared after 48 h of transfection, and relative luciferase activity was determined as previously described (20).
The 3′-UTR luciferase reporter constructs of JAK1 and IRF9 were generated by cloning the PCR-amplified human JAK1 and IRF9 mRNA 3′ UTRs (miR-373 target sites) into the MluI/HindIII site of the pMIR-Report miRNA expression luciferase reporter plasmid (Ambion). Mutant 3′ UTRs of JAK1 or IRF9 were used as a control in parallel. IHH were cotransfected with the luciferase reporter plasmid and control-miR or different doses of mimic miR-373, and luciferase activity was determined. Primers used for the JAK1 3′ UTR and the IRF9 3′ UTR are listed in Table 1.
TABLE 1.
Primer sequences used in this study

Rep2a-Rluc cells were transfected with either anti-miR-373 or anti-control-miR, and HCV replication were determined by qRT-PCR or relative Renilla luciferase activity assays (Promega) after 48 h of transfection.
Western blot analysis.
Cell lysates were subjected to polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. The membranes were blocked with 5% nonfat dried milk and incubated with specific antibodies. The membrane was probed with antibodies to JAK1, IRF9 (Santa Cruz), phosphorylated STAT1 (pSTAT1), STAT1 (Cell Signaling), ISG56, or HCV NS3 (Thermo Scientific). Proteins were detected by using enhanced chemiluminescence (ECL). The membrane was reprobed with actin as an internal control.
Ago2 RNA coimmunoprecipitation.
The mRNA target-associated RNA-induced silencing complex (RISC) and miR-373 were precipitated as described previously (21). Briefly, control-miR mimic- or miR-373 mimic-transfected IHH were lysed with lysis buffer (150 mM KCl, 25 mM Tris-HCl [pH 7.4], 5 mM EDTA, 1% Triton X-100, 5 mM dithiothreitol [DTT], protease inhibitor mixture, and 100 U/ml RNaseOUT [Invitrogen]). Cell lysates were clarified, incubated with an anti-Ago2 monoclonal antibody (MAb) (11A9; Sigma) or isotype control IgG2a at 4°C overnight, and mixed with protein G-Sepharose beads (GE Healthcare) for 2 h. The beads were washed four times, and RNA was isolated by using the RNeasy minikit (Qiagen). JAK1, IRF9, and miR-373 expression levels were quantitated by qRT-PCR.
Statistical analysis.
The results are presented as means ± standard deviations. Data were analyzed by Student's t test. A P value of <0.05 was considered statistically significant.
RESULTS
HCV infection enhances miR-373 expression.
The major targets and functions of a specific miRNA vary under different physiological or pathological conditions and in different cell types. Human hepatocytes are the natural host of HCV, and a miRNA array was performed by using RNA from mock-infected or HCV (genotype 1a)-infected primary human hepatocytes from two different donors (21). The differential expression of miRNAs following HCV infection is shown in Table 2. A significant upregulation of miR-373 (∼2-fold) was observed in the miRNA array. We next validated the expression of miR-373 in primary human hepatocytes infected with HCV genotype 1a (Fig. 1A). Similar results were obtained for the expression of miR-373 in IHH infected with both HCV genotypes 1a and 2a (Fig. 1B and C). We did not observe an upregulation of miR-373 in IHH exposed to UV-inactivated HCV. These results indicated that both HCV genotypes enhance miR-373 expression in hepatocytes and that HCV replication is needed for the upregulation of miR-373. RNA from HCV genotype 1a-infected liver biopsy specimens also displayed higher miR-373 expression levels than did control liver biopsy specimens (not infected with HCV or HBV) (Fig. 1D and E). Both groups had similar liver pathologies and aspartate transaminase (AST)/alanine aminotransferase (ALT) levels, and specimens were gender matched. The expression of miR-373 did not correlate with HCV load in sera of infected individuals (Fig. 1E). Together, these results suggested that HCV infection upregulates miR-373 expression.
TABLE 2.
Differential expression of miRNAs following HCV infection in primary human hepatocytesa
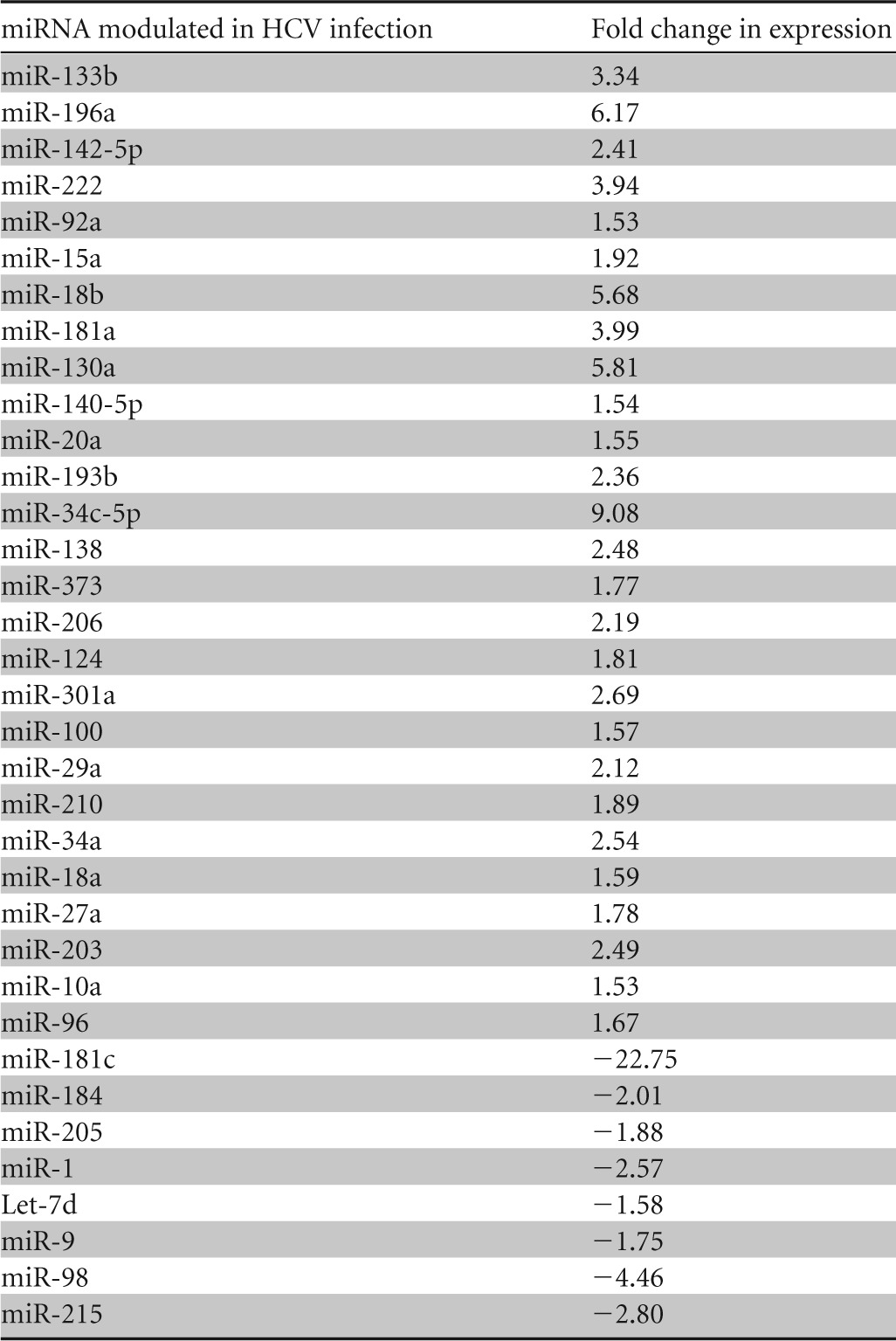
A miRNA array was performed by using RNA from mock-infected or HCV (genotype 1a)-infected primary human hepatocytes from two different donors, and means of array data are presented. Positive values indicate fold upregulation, and negative values indicate fold downregulation.
FIG 1.

HCV infection upregulates miR-373 expression. (A) Primary human hepatocytes (PHH) were mock treated or HCV infected for 96 h. Total RNA from mock- or virus-infected cells was extracted. The expression of miR-373 was analyzed by qRT-PCR and normalized to the expression of U6. (B and C) IHH were mock treated or infected with HCV genotype 1a (clone H77) or 2a (clone JFH1) (multiplicity of infection of 0.5) for 72 h. IHH were also incubated with UV-inactivated HCV genotype 2a. Total RNA from mock- or virus-infected cells was extracted. The expression of miR-373 was analyzed and normalized to the expression of U6. Data are presented as the means and standard deviations from three independent experiments. (D) Total RNA was extracted from non-HCV-infected (infected with HBV or other liver diseases) and HCV-infected liver biopsy specimens. miR-373 expression was analyzed by real-time PCR, using U6 RNA as an internal control. A comparison of the miR-373 levels in non-HCV-infected (n = 6) and HCV-infected (n = 8) liver tissues is shown. (E) Results from individual liver biopsy specimens described above for panel D. HCV titers are shown at the top. *, P < 0.05; ***, P < 0.001.
HCV-induced miR-373 suppresses JAK1 and IRF9 and disrupts the type I IFN signaling pathway.
We next analyzed the consequences of HCV-mediated induction of miR-373. In silico analysis using Targetscan and microRNA.org algorithms suggested that JAK1 and IRF9 of the IFN signaling pathway are potential targets of miR-373. JAK1 is a human tyrosine kinase protein essential for transducing type I and type II interferon signaling and activating certain interferon-stimulating genes (22). JAK1 phosphorylates STAT1 in tyrosine residues, which then dimerizes with STAT2 for the activation of IFN signaling (23). We observed a significant downregulation of JAK1 expression in IHH transfected with a miR-373 mimic (Fig. 2A). To examine the role of JAK1 in the phosphorylation of STAT1, we introduced poly(I-C), which mimics intracellular double-stranded RNA (dsRNA) during viral replication, into miR-373 mimic-transfected IHH. A significant inhibition of the poly(I-C)-induced phosphorylation of STAT1 was observed with miR-373 (Fig. 2A and B). To further verify the specificity of miR-373, we used an unrelated miRNA, miR-10b, as a negative control and did not observe any significant alteration of STAT1 phosphorylation. The expression of total STAT1 was not altered. To rescue the expression levels of JAK1 and pSTAT1, IHH infected with HCV genotype 2a were transfected with anti-miR-373. The expression level of JAK1 and the phosphorylation status of STAT1 were significantly downregulated in HCV-infected cells compared to mock-treated controls, which were rescued by a knockdown of miR-373 (Fig. 2C). Relative expression levels of JAK1, pSTAT1, and STAT1 were determined by densitometry scanning (Fig. 2D). An in silico analysis suggested putative binding sites of miR-373 in the 3′ UTR of JAK1 with a high mirSVR score (Fig. 2E). To verify binding, the 3′ UTRs of JAK1 with a potential binding site for miR-373 and the mutant fragments without the binding sites were cloned into the pMIR-Report luciferase vector. Cells were cotransfected with the pMIR-Report luciferase vector containing the 3′ UTR of JAK1 and the miR-373 mimic, and luciferase activity was measured. The results demonstrated a dose-dependent inhibition of wild-type JAK1 3′-UTR expression by the miR-373 mimic (Fig. 2F). The mutant 3′ UTR did not exhibit inhibition in the presence of the miR-373 mimic (Fig. 2G).
FIG 2.

miR-373 inhibits JAK1 expression and STAT1 phosphorylation status. (A) Hepatocytes were transfected with the control-miR mimic, the miR-373 mimic, or the miR-10b mimic (unrelated miRNA as a negative control) for 24 h, followed by poly(I-C) transfection. Cell extracts were prepared after 48 h posttransfection and analyzed for JAK1, pSTAT1, and total STAT1 expression by Western blotting using specific antibodies. The blot was reprobed with an antibody to actin for normalization. (B) Fold changes in the expression levels of all the proteins after normalization to actin or normalization to total STAT1 to pSTAT1 expression from three independent experiments, determined by densitometry scanning. (C) Hepatocytes were mock treated or infected with HCV genotype 2a for 24 h, followed by transfection with anti-control-miR or anti-miR-373. Cell lysates were prepared after 96 h postinfection, and JAK1, pSTAT1, and total STAT1 expression levels were analyzed by Western blotting using specific antibodies. The blot was reprobed with an antibody to actin for normalization. (D) Fold changes in expression levels of all the proteins after normalization to actin or normalization to total STAT1 to pSTAT1 expression from three independent experiments, determined by densitometry scanning. (E) In silico analysis of the JAK1 3′ UTR reveals a single putative miR-373 binding site. The miR-373 target region of JAK1 (GenBank accession number NM_002227) is indicated. (F) Hepatocytes were cotransfected with the 3′ UTR of JAK1 cloned into the pMIR-Report luciferase vector and different doses of the miR-373 mimic (10, 50, or 100 nM). Relative luciferase activity was measured after 48 h of transfection. (G) Luciferase activity of the mutant 3′ UTR of JAK1 and the miR-373 mimic (50 nM) was not altered compared to the wild-type 3′ UTR of JAK1. The results are shown as means and standard deviations from three independent experiments. **, P < 0.01; ***, P < 0.001.
IRF9 is another major component of the IFN signaling pathway. It forms a multisubunit complex with pSTAT1 and pSTAT2, called interferon-stimulated gene factor 3 (ISGF3), and translocates from the cytoplasm to the nucleus. ISGF3 interacts with the IFN-stimulated response element present in the promoters of ISGs (24). The overexpression of the miR-373 mimic in IHH suppressed IRF9 and its downstream signaling molecule ISG56 (Fig. 3A). Relative expression levels of IRF9 and ISG56 were determined by densitometry scanning (Fig. 3B). We also examined whether miR-373 knockdown enhances IRF9 and/or ISG56 expression in hepatocytes. Transfection of anti-miR-373 in IHH significantly upregulated the expression of IRF9 and ISG56 compared to control anti-miR (Fig. 3C and D). To further examine the effect of anti-miR-373 on HCV-infected hepatocytes, IHHs were infected with HCV genotype 2a, followed by the introduction of the control-anti-miRNA or anti-miR-373, and the expression levels of IRF9 and ISG56 were analyzed. The expression of IRF9 and ISG56 was significantly downregulated in HCV-infected cells compared to mock-treated controls, which was rescued by a knockdown of miR-373 (Fig. 3E). Relative expression levels of IRF9 and ISG56 were determined by densitometry scanning (Fig. 3F). Our in silico analysis also suggested a putative binding site of miR-373 in the 3′ UTR of IRF9 (Fig. 3G). The 3′ UTR of IRF9 with a potential binding site for miR-373 and the mutant fragments without the binding sites were cloned into the pMIR-Report luciferase vector. The reporter assay demonstrated a dose-dependent inhibition of wild-type IRF9 3′-UTR expression by the miR-373 mimic (Fig. 3H). The mutant 3′ UTR did not exhibit an effect in the presence of the miR-373 mimic (Fig. 3I). Together, these results suggested that miR-373 downregulates JAK1 and IRF9 and suppresses the type I IFN signaling pathway in HCV-infected hepatocytes.
FIG 3.

Enhanced expression of miR-373 inhibits IRF9 and ISG56 expression. (A) Introduction of the miR-373 mimic in IHH reduces IRF9 and ISG56 expression. The blot was reprobed with an antibody to actin for comparison of protein loads. (B) Fold changes of IRF9 and ISG56 expression levels after normalization to actin levels from three independent experiments, determined by densitometry scanning. (C) Hepatocytes were transfected with anti-control-miR or anti-miR-373. Cell extracts were prepared after 48 h posttransfection, and IRF9 and ISG56 expression levels were analyzed by Western blotting using specific antibodies. The blot was reprobed with an antibody to actin for normalization. (D) Fold changes of IRF9 and ISG56 expression levels after normalization to actin from three independent experiments, determined by densitometry scanning. (E) Hepatocytes were mock treated or infected with HCV genotype 2a, followed by transfection with anti-control-miR or anti-miR-373 after 24 h postinfection. Cell lysates were analyzed for IRF9 and ISG56 expression by Western blotting using specific antibodies. The blot was reprobed with an antibody to actin for normalization. (F) Fold changes in expression levels of all the proteins after normalization to actin from three independent experiments, determined by densitometry scanning. (G) In silico analysis of the IRF9 3′ UTR reveals a single putative miR-373 binding site. The miR-373 target region of IRF9 (GenBank accession number NM_006084) is indicated. (H) Hepatocytes were cotransfected with the 3′ UTR of IRF9 cloned into the pMIR-Report luciferase vector and different doses of the miR-373 mimic (10 or 50 nM). Relative luciferase activity was measured after 48 h of transfection. (I) Luciferase activity decreased with the 3′ UTR of the IRF9 reporter after cotransfection with the miR-373 mimic (50 nM) but not with the mutant 3′ UTR of IRF9. The results are shown as means and standard deviations from three independent experiments. *, P < 0.05; ***, P < 0.001.
We further examined poly(I-C)-induced interferon-stimulated response element (ISRE) and ISG56 promoter activities by overexpressing miR-373 in IHH using in vitro reporter assays. The poly(I-C)-induced enhancement of ISRE and ISG56 promoter activities was inhibited following the overexpression of the miR-373 mimic (Fig. 4A and B). To verify this effect, we silenced miR-373 using an miR-373 inhibitor (anti-miR-373) in IHH, and ISG56 transcripts were examined by qRT-PCR. As anticipated, the knockdown of miR-373 significantly upregulated the expression of ISG56 mRNA (Fig. 4C) in hepatocytes. Together, these results further suggested that miR-373 acts as a negative regulator of the type I IFN signaling pathway.
FIG 4.
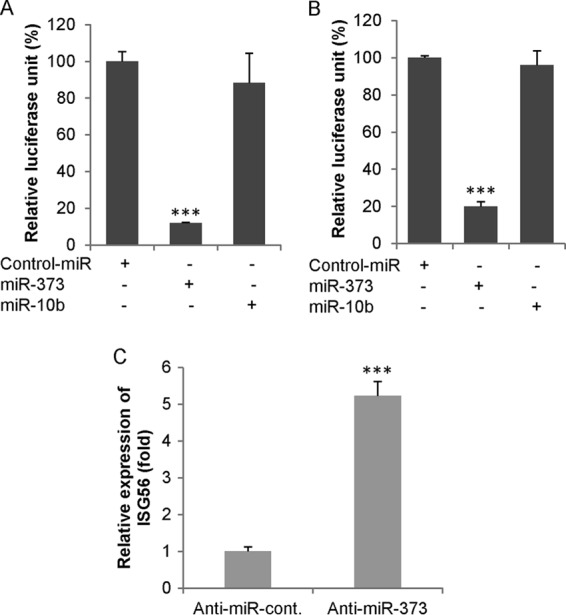
miR-373 inhibits ISRE and ISG56 promoter activities. (A and B) Hepatocytes were cotransfected with either ISRE-luc (A) or ISG56-luc (B) reporter plasmid DNA along with the control-miR mimic, miR-373, and miR-10b (unrelated miRNA as a negative control). After 24 h, cells were transfected with poly(I-C). Cell extracts were prepared after 48 h, and relative luciferase activity was measured. (C) Hepatocytes were transfected with either anti-control-miR or anti-miR-373. After 48 h, total RNA was extracted, and the transcriptional expression of ISG56 was measured by qRT-PCR using cellular 18S RNA as an internal control for normalization. Data are presented as the means and standard deviations from three independent experiments. ***, P < 0.001.
miR-373 binds to JAK1 and IRF9 in vivo.
We further examined the in vivo association of miR-373 with JAK1 and IRF9. For this, RISC RNA immunoprecipitation (RISC-RIP) with Ago2 antibody or control IgG was performed to isolate JAK1 or IRF9 mRNAs associated with the RISC of miR-373. miR-373-transfected cell lysates were immunoprecipitated with an Ago2-specific monoclonal antibody or the isotype control. The miR-373-bound mRNA expression of JAK1 or IRF9 was significantly upregulated in Ago2 immunoprecipitates compared to the control (Fig. 5A). To study specificity, we examined the expression of IFIH1 (a nontargeted mRNA) from the immunoprecipitates and did not observe a significant change in the Ago2 immunoprecipitates compared to the isotype control (Fig. 5A). We further transfected IHH with control-miR or a miR-373 mimic and immunoprecipitated the RISC with Ago2 antibody. We observed a greater association of JAK1/IRF9 in miR-373-transfected cells than in control-miR-transfected cells (Fig. 5B). A higher expression level of miR-373 in the immunoprecipitate was also observed (Fig. 5C). Together, these in vitro reporter assays and in vivo Ago2 IP assays suggested that miR-373 directly binds to the 3′ UTRs of JAK1 and IRF9.
FIG 5.

miR-373 binds directly to JAK1 and IRF9. (A) IHH lysates were immunoprecipitated with an Ago2-specific monoclonal antibody or an unrelated IgG2a isotype control. RNA was isolated from immunoprecipitates by using an RNeasy kit, and JAK1, IRF9, and IFIH1 (as a nontargeted mRNA) expression levels were analyzed by qRT-PCR. (B) IHH transfected with control-miR or miR-373 were immunoprecipitated with an Ago2-specific monoclonal antibody. RNA was isolated from immunoprecipitates, and JAK1 and IRF9 expression levels were analyzed by qRT-PCR. (C) Relative expression levels of miR-373 from Ago2 immunoprecipitates, analyzed by qRT-PCR. The results are shown as means and standard deviations from three independent experiments. *, P < 0.05; **, P < 0.01.
Knockdown of miR-373 suppresses HCV replication.
IHH were infected with HCV genotype 2a and transfected with anti-control-miR or anti-miR-373 to determine whether anti-miR-373 treatment blocks HCV replication and expression. Intracellular HCV RNA levels were determined by qRT-PCR, and HCV NS3 protein expression levels were determined by Western blotting. The results showed that the knockdown of miR-373 significantly suppressed HCV replication (Fig. 6A) and HCV NS3 protein expression (Fig. 6B) in virus-infected hepatocytes. We next examined whether the overexpression of the miR-373 mimic in anti-miR-373-treated HCV-infected cells can alter HCV RNA replication. Our results suggested that the miR-373 mimic could inhibit anti-miR-373-induced HCV RNA replication (Fig. 6A). Rep2a-Rluc cells were also transfected with anti-control-miR or anti-miR-373, and HCV replication was measured by qRT-PCR or a luciferase reporter assay after 48 h of transfection. A significant inhibition of HCV RNA expression was observed for miR-373 knockdown Rep2a-Rluc cells compared to the control cells (Fig. 6C and D). Together, these results indicated the antiviral role of anti-miR-373 in HCV replication. To rule out additional miR-373-dependent effects on the regulation of HCV replication, we knocked down STAT1 expression by using small interfering RNA (siRNA) and then infected cells with HCV for 72 h. Viral RNA and miR-373 expression levels were higher in STAT1 knockdown cells than in control-miR-treated virus-infected cells (data not shown). Since Huh7.5 cells are RIG-I defective, these cells were treated with IFN-α for 2 h, and pSTAT1 expression was examined. As expected, pSTAT1 expression was enhanced after treatment with IFN-α, suggesting that the JAK/STAT pathway is active in this cell line (Fig. 6E). We also observed the upregulation of ISG56 in miR-373 knockdown Rep2a-Rluc cells compared to cells treated with anti-control-miR (Fig. 6F), further suggesting that the JAK/STAT pathway is active in Huh7.5 cells harboring the HCV genome-length replicon.
FIG 6.

Knockdown of miR-373 suppresses HCV replication. (A) IHHs were infected with HCV genotype 2a, followed by transfection with anti-control-miR or anti-miR-373. RNA was isolated, and HCV RNA replication was measured by qRT-PCR after 48 h of transfection. For the reciprocal experiment, anti-miR-373-treated HCV-infected IHH were transfected with miR-373, and expression of HCV RNA was measured by qRT-PCR. (B) Hepatocytes were mock treated or infected with HCV genotype 2a, followed by transfection with anti-control-miR or anti-miR-373 after 24 h of infection. Cell lysates were prepared after 96 h postinfection, and HCV NS3 expression levels were analyzed by Western blotting using specific antibodies. The blot was reprobed with an antibody to actin for normalization. (C) Rep2a-Rluc cells were transfected with anti-control-miR or anti-miR-373. HCV genomic RNA replication was analyzed by qRT-PCR after 48 h of transfection. Cellular 18S RNA was used as an internal control for normalization. (D) Rep2a-Rluc cells were transfected with anti-control-miR or anti-miR-373, and relative luciferase activity was measured by a Renilla luciferase assay after 48 h of transfection. Results are presented as the means and standard deviations from three independent experiments. ***, P < 0.001. (E) Huh7.5 cells were treated with IFN-α for 2 h, and cell lysates were analyzed for pSTAT1 and STAT1 expression using specific antibodies. The blot was reprobed with actin for comparison of protein loads. (F) Rep2a-Rluc cells were transfected with anti-control-miR or anti-miR-373 for 48 h, and RNA was extracted for determination of the expression of ISG56 by qRT-PCR. 18S RNA was used for normalization. Results are presented as the means and standard deviations from three independent experiments. ***, P < 0.001.
DISCUSSION
In this study, we have demonstrated a novel regulation of JAK1-STAT1 signaling by miR-373 in HCV infection. First, we found that HCV infection upregulated miR-373 expression in hepatocytes. Second, we demonstrated that miR-373 suppresses the transcription of ISGs by blocking ISRE and ISG56 promoter activities, thus demonstrating a novel mechanism for viral immune evasion. Third, we validated that both JAK1 and IRF9 are targets of miR-373. Fourth, we demonstrated that the inhibition of miR-373 upregulates IFN response gene expression. Finally, we showed that the suppression of miR-373 blocks HCV replication and may serve as a potential therapeutic target for antiviral intervention. In summary, we have identified a host-virus interaction pathway where HCV infection results in the stimulation of a particular cellular miRNA, which in turn disrupts the type I IFN signaling pathway (Fig. 7). To our knowledge, this is the first report showing the HCV-mediated upregulation of miR-373 interfering with the IFN signaling pathway in hepatocytes.
FIG 7.
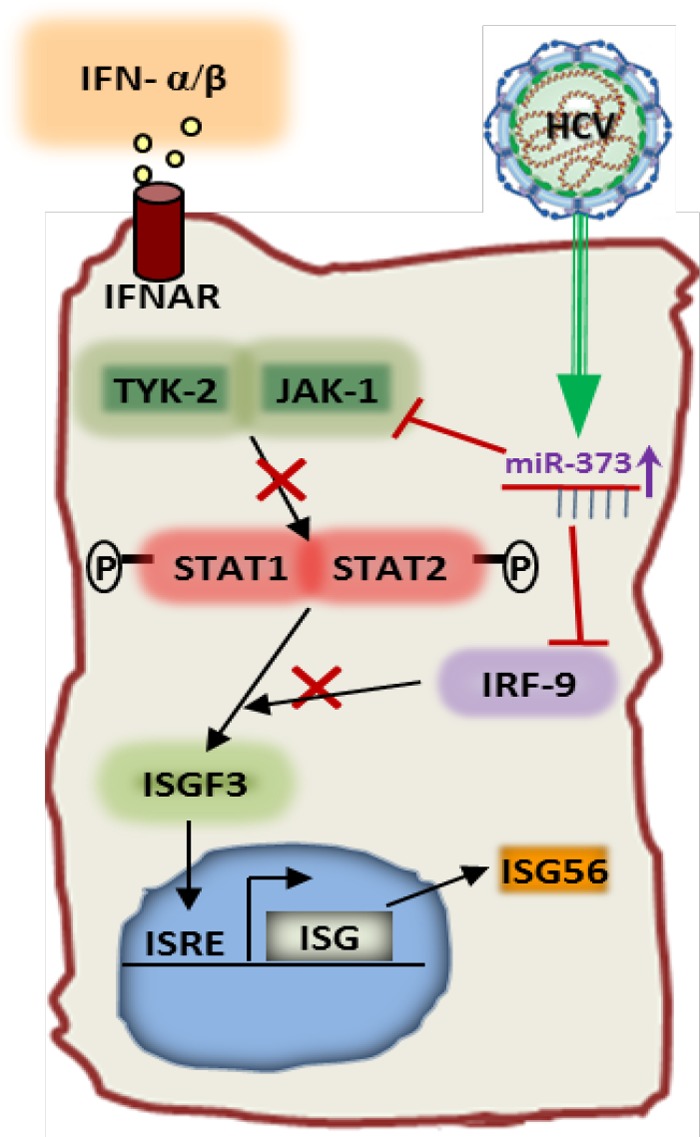
Schematic diagram showing the cross talk between HCV and miR-373 inhibiting IFN signaling. HCV infection upregulates miR-373 in hepatocytes, which in turn inhibits JAK1 and IRF9 expression. Inhibition of JAK1 and IRF9 restricts STAT1 phosphorylation and blocks ISGF3 complex formation, respectively. This results in the inhibition of ISRE promoter activity. Arrows denote upregulation, and blunt arrows denote inhibition.
We have demonstrated that the overexpression of miR-373 blocks ISRE and ISG56 promoter activities, whereas the knockdown of miR-373 upregulates ISG56 expression, which limits HCV replication. This observation corroborates our previous observations that the overexpression of ISG56 displays anti-HCV activity (25). The activation of IFN-stimulated genes occurs primarily through the JAK/STAT pathway. Here, we evaluated the expression status of IRF9 and JAK1 and the phosphorylation status of STAT1 and found that miR-373 overexpression reduced the protein expression levels of all these molecules but had little effect on the total STAT1 protein level. Our results furthermore confirmed that JAK1 and IRF9 are the direct targets of miR-373. Several RNA viruses, such as measles virus or human metapneumovirus, have been reported to counteract JAK/STAT and their downstream signaling events (26, 27). HCV infection evades the host response through multifaceted processes, including signaling interference, effector modulation, and continuous genetic variation, in establishing acute to chronic infection (28). The JAK/STAT signaling pathway plays a critical role in interferon-stimulating gene transcription and the initiation of the antiviral response. HCV infection modulated the JAK/STAT signaling pathway (29–31); however, the precise mechanism was not fully understood. Therefore, identification of the role of miR-373 by which HCV regulates the JAK/STAT signaling pathway provides a new mechanistic insight into dampening host innate immune responses to virus infection.
We observed a significant upregulation of miR-373 in HCV-infected liver biopsy specimens. Interestingly, miR-373 upregulation does not correlate with HCV load in patient sera. miR-373 was upregulated in HBV-infected liver tissues, and miR-373 expression promotes HBV expression by involving the NFIB transcription factor (32), although modulation of the JAK/STAT signaling pathway was not studied. Since miR-373 expression is upregulated in chronic HCV infection and modulates the innate immune system, it will be important to investigate the status of miR-373 in other chronic infections.
In conclusion, we have demonstrated that HCV infection upregulates miR-373 expression, which in turn suppresses the expression of JAK1, IRF9, and their downstream molecules involved in the interferon signaling pathway. The knockdown of miR-373 exerts anti-HCV activity by inhibiting viral replication and the upregulation of IFN-stimulating gene expression. Thus, the induction of miR-373 can interrupt the JAK/STAT pathway in HCV infection.
ACKNOWLEDGMENTS
We thank Hengli Tang for providing the Rep2a-Rluc cell line. We also thank Joydip Bhanja Chowdhury, Shubham Shrivastava, and Robert Steele for their contributions to this work.
This work was supported by research grant DK081817 (R.B.R.) from the National Institutes of Health and by the Doisy Research Fund of Saint Louis University.
REFERENCES
- 1.Choo QL, Richman KH, Han JH, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby R, Barr PJ. 1991. Genetic organization and diversity of the hepatitis C virus. Proc Natl Acad Sci U S A 88:2451–2455. doi: 10.1073/pnas.88.6.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gravitz L. 2011. Introduction: a smouldering public-health crisis. Nature 474:S2–S4. doi: 10.1038/474S2a. [DOI] [PubMed] [Google Scholar]
- 3.Thomas DL. 2013. Global control of hepatitis C: where challenge meets opportunity. Nat Med 19:850–858. doi: 10.1038/nm.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saha B, Szabo G. 2014. Innate immune cell networking in hepatitis C virus infection. J Leukoc Biol 96:757–766. doi: 10.1189/jlb.4MR0314-141R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. 2014. Hepatitis C. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs164/en/. [Google Scholar]
- 6.Meister G, Tuschl T. 2004. Mechanisms of gene silencing by double-stranded RNA. Nature 431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 7.Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. 2009. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 9.Roberts AP, Lewis AP, Jopling CL. 2011. The role of microRNAs in viral infection. Prog Mol Biol Transl Sci 102:101–139. doi: 10.1016/B978-0-12-415795-8.00002-7. [DOI] [PubMed] [Google Scholar]
- 10.Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 11.Shrivastava S, Mukherjee A, Ray RB. 2013. Hepatitis C virus infection, microRNA and liver disease progression. World J Hepatol 5:479–486. doi: 10.4254/wjh.v5.i9.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cullen BR. 2013. MicroRNAs as mediators of viral evasion of the immune system. Nat Immunol 14:205–210. doi: 10.1038/ni.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhanja Chowdhury J, Shrivastava S, Steele R, Di Bisceglie AM, Ray R, Ray RB. 2012. Hepatitis C virus infection modulates expression of interferon stimulatory gene IFITM1 by upregulating miR-130A. J Virol 86:10221–10225. doi: 10.1128/JVI.00882-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Chen J, Wang H, Shi J, Wu K, Liu S, Liu Y, Wu J. 2013. HCV-induced miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1. PLoS Pathog 9:e1003248. doi: 10.1371/journal.ppat.1003248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li A, Song W, Qian J, Li Y, He J, Zhang Q, Li W, Zhai A, Kao W, Hu Y, Li H, Wu J, Ling H, Zhong Z, Zhang F. 2013. miR-122 modulates type I interferon expression through blocking suppressor of cytokine signaling 1. Int J Biochem Cell Biol 45:858–865. doi: 10.1016/j.biocel.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 16.Yoshikawa T, Takata A, Otsuka M, Kishikawa T, Kojima K, Yoshida H, Koike K. 2012. Silencing of microRNA-122 enhances interferon-α signaling in the liver through regulating SOCS3 promoter methylation. Sci Rep 2:637. doi: 10.1038/srep00637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Ekiaby N, Hamdi N, Negm M, Ahmed R, Zekri AR, Esmat G, Abdelaziz AI. 2012. Repressed induction of interferon-related microRNAs miR-146a and miR-155 in peripheral blood mononuclear cells infected with HCV genotype 4. FEBS Open Bio 2:179–186. doi: 10.1016/j.fob.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ray RB, Meyer K, Ray R. 2000. Hepatitis C virus core protein promotes immortalization of primary human hepatocytes. Virology 271:197–204. doi: 10.1006/viro.2000.0295. [DOI] [PubMed] [Google Scholar]
- 19.Kanda T, Basu A, Steele R, Wakita T, Ryerse JS, Ray R, Ray RB. 2006. Generation of infectious hepatitis C virus in immortalized human hepatocytes. J Virol 80:4633–4639. doi: 10.1128/JVI.80.9.4633-4639.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shrivastava S, Bhanja Chowdhury J, Steele R, Ray R, Ray RB. 2012. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J Virol 86:8705–8712. doi: 10.1128/JVI.00616-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mukherjee A, Shrivastava S, Bhanja Chowdhury J, Ray R, Ray RB. 2014. Transcriptional suppression of miR-181c by hepatitis C virus enhances homeobox A1 expression. J Virol 88:7929–7940. doi: 10.1128/JVI.00787-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aaronson DS, Horvath CM. 2002. A road map for those who don't know JAK-STAT. Science 296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 23.Mowen K, David M. 2000. Regulation of STAT1 nuclear export by Jak1. Mol Cell Biol 20:7273–7281. doi: 10.1128/MCB.20.19.7273-7281.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rani MRS, Ransohoff MR. 2005. Alternative and accessory pathways in the regulation of IFN-β-mediated gene expression. J Interferon Cytokine Res 25:788–798. doi: 10.1089/jir.2005.25.788. [DOI] [PubMed] [Google Scholar]
- 25.Raychoudhuri A, Shrivastava S, Steele R, Kim H, Ray R, Ray RB. 2011. ISG56 and IFITM1 proteins inhibit hepatitis C virus replication. J Virol 85:12881–12889. doi: 10.1128/JVI.05633-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Devaux P, Priniski L, Cattaneo R. 2013. The measles virus phosphoprotein interacts with the linker domain of STAT1. Virology 444:250–256. doi: 10.1016/j.virol.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ren J, Kolli D, Liu T, Xu R, Garofalo RP, Casola A, Bao X. 2011. Human metapneumovirus inhibits IFN-β signaling by downregulating Jak1 and Tyk2 cellular levels. PLoS One 6:e24496. doi: 10.1371/journal.pone.0024496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horner SM, Gale M Jr. 2013. Regulation of hepatic innate immunity by hepatitis C virus. Nat Med 19:879–888. doi: 10.1038/nm.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shanker V, Trincucci G, Heim HM, Duong HT. 2013. Protein phosphatase 2A impairs IFNα-induced antiviral activity against the hepatitis C virus through the inhibition of STAT1 tyrosine phosphorylation. J Viral Hepat 20:612–621. doi: 10.1111/jvh.12083. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L, Jilg N, Shao RX, Lin W, Fusco DN, Zhao H, Goto K, Peng LF, Chen WC, Chung RT. 2011. IL28B inhibits hepatitis C virus replication through the JAK-STAT pathway. J Hepatol 55:289–298. doi: 10.1016/j.jhep.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raychoudhuri A, Shrivastava S, Steele R, Dash S, Kanda T, Ray R, Ray RB. 2010. Hepatitis C virus infection impairs IRF-7 translocation and alpha interferon synthesis in immortalized human hepatocytes. J Virol 84:10991–10998. doi: 10.1128/JVI.00900-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo H, Liu H, Mitchelson K, Rao H, Luo M, Xie L, Sun Y, Zhang L, Lu Y, Liu R, Ren A, Liu S, Zhou S, Zhu J, Zhou Y, Huang A, Wei L, Guo Y, Cheng J. 2011. MicroRNAs-372/373 promote the expression of hepatitis B virus through the targeting of nuclear factor I/B. Hepatology 54:808–819. doi: 10.1002/hep.24441. [DOI] [PubMed] [Google Scholar]