

ABSTRACT
Recently, interferon-induced transmembrane proteins (IFITMs) have been identified to be key effector molecules in the host type I interferon defense system. The invasion of host cells by a large range of RNA viruses is inhibited by IFITMs during the entry step. However, the roles of IFITMs in DNA virus infections have not been studied in detail. In this study, we report that human cytomegalovirus (HCMV), a large human DNA virus, exploits IFITMs to facilitate the formation of the virion assembly compartment (vAC) during infection of human fibroblasts. We found that IFITMs were expressed constitutively in human embryonic lung fibroblasts (MRC5 cells). HCMV infection inhibited IFITM protein accumulation in the later stages of infection. Overexpression of an IFITM protein in MRC5 cells slightly enhanced HCMV production and knockdown of IFITMs by RNA interference reduced the virus titer by about 100-fold on day 8 postinfection, according to the findings of a virus yield assay at a low multiplicity of infection. Virus gene expression and DNA synthesis were not affected, but the typical round structure of the vAC was not formed after the suppression of IFITMs, thereby resulting in defective virion assembly and the production of less infectious virion particles. Interestingly, the replication of herpes simplex virus, a human herpesvirus that is closely related to HCMV, was not affected by the suppression of IFITMs in MRC5 cells. These results indicate that IFITMs are involved in a specific pathway required for HCMV replication.
IMPORTANCE HCMV is known to repurpose the interferon-stimulated genes (ISGs) viperin and tetherin to facilitate its replication. Our results expand the range of ISGs that can be exploited by HCMV for its replication. This is also the first report of a proviral function of IFITMs in DNA virus replication. In addition, whereas previous studies showed that IFITMs modulate virus entry, which is a very early stage in the virus life cycle, we identified a new function of IFITMs during the very late stage of virus replication, i.e., virion assembly. Virus entry and assembly both involve vesicle transport and membrane fusion; thus, a common biochemical activity of IFITMs is likely to be involved. Therefore, our findings may provide a new platform for dissecting the molecular mechanism of action of IFITMs during the blocking or enhancement of virus infection, which are under intense investigation in this field.
INTRODUCTION
Human cytomegalovirus (HCMV), a ubiquitous opportunistic pathogen that belongs to the Betaherpesviridae subfamily, is a major cause of morbidity and mortality in immunocompromised individuals (1). HCMV infection induces the expression of type I interferon and a set of interferon (IFN)-stimulated genes (ISGs) early upon infection (2, 3). However, HCMV encodes multiple proteins, including IE1 (4–6), IE2 (7–9), pp65 (10, 11), and TRS1 and IRS1 (12, 13), to antagonize the innate immune responses. Moreover, several IFN-induced host restriction factors are repurposed by HCMV to facilitate its replication. Viperin is relocated from the endoplasmic reticulum to the mitochondria by vMIA, where it enhances HCMV replication by modulating cellular lipid metabolism (14). The antiviral protein BST2/tetherin has also been shown to promote the entry of HCMV in BST2-overexpressing cells (15). Thus, HCMV has developed mechanisms for combating the host antiviral response but also to exploit antiviral molecules for its own benefit.
Recently, interferon-induced transmembrane proteins (IFITMs) have been identified to be antiviral restriction factors that inhibit the replication of influenza A virus and flaviviruses, including West Nile virus and dengue virus (16). IFITMs belong to a family of small ISGs. They contain a conserved CD225 domain, which is related to their antiviral activity (17, 18). The broad-spectrum antiviral activity of IFITMs has been elucidated, and it has been reported that IFITMs are able to restrict infections by severe acute respiratory syndrome coronavirus, filoviruses, HIV-1, bunyaviruses, reoviruses, vesicular stomatitis virus (VSV), and hepatitis C virus (HCV) (19–24).
The majority of the viruses restricted by IFITMs are enveloped RNA viruses whose entry is mediated through clathrin-mediated endocytosis (18). However, the precise mechanism that allows IFITMs to inhibit virus replication remains elusive. It has been hypothesized that IFITMs may interfere with a membrane fusion step of virus entry either at the cell surface or in the endosome/lysosome compartments. This is supported by the subcellular localization of IFITMs. While IFITM1 is primarily located on the cell surface, IFITM2 and IFITM3 are mainly present in late endosomes and lysosomes (25). In addition, IFITMs have been shown to function in stabilization of v-ATPase complexes in intracellular membranes and thus facilitate the proper subcellular localization of clathrin (26). Recently, IFITM3 has been reported to contain an endocytic signal which is essential for its antiviral activity. IFITM3 is likely to interact with the μ2 subunit of the AP-2 complex through which IFITM3 undergoes endocytosis (27). Finally, posttranslational modifications also play an important role in regulating the function of IFITMs (28–31).
Despite their relatively broad spectrum of antiviral activities, IFITMs do not restrict lymphocytic choriomeningitis virus (LCMV), Lassa virus (LASV), Machupo virus (MACV), or murine leukemia virus (MLV) (20). It has also been reported that cells infected by DNA viruses, such as herpes simplex virus and cytomegalovirus, can induce the expression of ISGs, including IFITMs (32, 33). However, the role of IFITMs in the life cycles of DNA viruses is largely unknown. A recent study reports that IFITM protein overexpression fails to inhibit the entry of three DNA viruses, including HCMV (34), but detailed mechanistic investigations have not been performed. In the present study, we found that IFITMs fail to inhibit HCMV replication in human fibroblasts, and they are actually required for the correct formation of the virion assembly compartment (vAC) and virion particle assembly.
HCMV profoundly reorganizes the cellular secretory apparatus to facilitate virus assembly and egress during the later stage of its life cycle. This event is mediated by dynamic interactions among large numbers of viral and host proteins (35–44). The vAC assembles at a vesicular structure, which is formed around the microtubule organization center in juxtaposition to the concave side of the nucleus, where the vesicles with endosomal markers occupy the central position and the vesicles with the Golgi apparatus marker are wrapped around it to form a circle (42, 45, 46). The HCMV vAC is a unique structure, even among the members of the herpesvirus family (35, 47). The viral nucleocapsids are formed inside the nucleus and then bud through the two layers of the nuclear membrane. During this process, viral nucleocapsids undergo primary envelopment and deenvelopment. Subsequently, the naked nucleocapsids are thought to migrate through the vAC adjacent to the nuclear membrane, where they acquire the tegument proteins and complete their secondary envelopment by budding into the vesicles in the vAC. We found that IFITMs are required for the correct formation of the vAC and virion assembly. In cells where IFITMs were suppressed by RNA interference (RNAi), the Golgi stacks failed to rearrange into a typical circular structure that wrapped around the vAC, the virus structure proteins pp28, pp150, and gB failed to concentrate in the vAC, and the secreted virion particles were less infectious. Our findings expand the range of ISGs that can be exploited by HCMV for its own benefit.
MATERIALS AND METHODS
Plasmids and reagents.
Plasmid pIPFLAG2-IFITM3, containing human IFITM3 cDNA, was constructed as described previously (30). A Flag tag-coding sequence was introduced into the IFITM3 cDNA at the 5′ end by PCR amplification. The DNA fragment was then cloned into pLKO.DCMV.TetO, a pLKO-based lentiviral vector under the control of a tetracycline-inducible CMV-TetO2 promoter (48), to generate pLKO-Flag-IFITM3. To create a short hairpin RNA (shRNA)-resistant IFITM3 expression construct, mismatch mutations in the IFITM-targeting shRNA sh8025 target sequence were introduced into IFITM3 cDNA by overlapping PCR with primer pair 5′-ACGCGTCGACCATGGACTACAAGGACGACGATGACAAGATGAATCACACTGTCCAAACCTTC-3′ and 5′-GACTTCACGGAATAAGCGAAAGCTATAAAGCCCAGGCAGCAGG-3′ and primer pair 5′-CCTGCTGCCTGGGCTTTATAGCTTTCGCTTATTCCGTGAAGTC-3′ and 5′-GGAATTCCTATCCATAGGCCTGGAAGATC-3′. The shRNA-resistant IFITM3 cDNA fragment was then cloned into plasmid pLKO.DCMV.TetO. Two IFITM-targeting shRNA (sh8024 or sh8025)-expressing lentiviral vectors were constructed on the basis of the pLKO.1 vector. The shRNA targeting sequences were as follows: 5′-CAACAAGATGAAGAGCACCAA-3′ (control shRNA [shC]), 5′-CCTCATGACCATTCTGCTCAT-3′ (sh8024), and 5′-GCTTCATAGCATTCGCCTACT-3′ (sh8025).
The primary antibodies used in this study included anti-tubulin (Proteintech), anti-IFITM1 (mouse monoclonal antibody; Proteintech), anti-IFITM2/3 (mouse monoclonal antibody; Proteintech), anti-GM130 (Cell Signaling), anti-Flag (Abmart), anti-immediate early (anti-IE) protein IE1/2 (a generous gift from Jay Nelson, Oregon Health & Science University), and anti-pp28 and anti-UL38 (gifts from Thomas Shenk, Princeton University).
Viruses and cell lines.
Human embryonic lung fibroblasts (MRC5 cells) and HEK-293T cells were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (FBS). To generate cells that stably express Flag-tagged IFITM3, HEK-293T cells were transfected with the recombinant pLKO-Flag-IFITM3 plasmid together with the packaging plasmids expressing 9.2ΔR and VSV glycoprotein G to produce the lentivirus stocks. Then, 80% confluent MRC-5 cells were transduced with lentivirus supplemented with 5 μg/ml Polybrene and selected with 2 μg/ml puromycin (Sigma-Aldrich).
Two bacterial artificial chromosome clones containing HCMV (pAD-GFP and pAD/Cre) were used in the present study to produce wild-type virus. pAD/Cre carries the whole genome of HCMV lab strain AD169. pAD-GFP is derived from pAD/Cre, but the viral US4-US6 region is replaced by the green fluorescent protein (GFP) gene under the control of a simian virus 40 (SV40) early promoter (49, 50). Herpes simplex virus 1 (HSV-1; ATCC VR1493) was a generous gift from Chiyu Zhang at the Pathogen Diagnostic Center, Institut Pasteur of Shanghai.
shRNA knockdown.
To knock down IFITMs in MRC5 cells, subconfluent cells were transduced with lentiviruses encoding the shRNAs indicated above supplemented with 5 μg/ml Polybrene. Cells were then incubated for 5 h at 37°C, washed with phosphate-buffered saline (PBS), and added to fresh medium. At 24 h posttransfection, cells were either mock infected with medium alone or infected with HCMV at the multiplicities of infection (MOIs) indicated below. At 48 h posttransfection, cell lysates were collected for Western blotting to detect the knockdown efficiencies of the shRNAs.
Virus growth analysis.
Cells in which IFITM was overexpressed or knocked down were seeded in 12-well dishes overnight to produce a subconfluent monolayer. The cells were incubated with HCMV for 1 h at an MOI of 0.3 or 3, as indicated below. The inoculum was removed, the monolayer was rinsed with PBS, and fresh medium was replenished. At various times postinfection (p.i.), cell-free media from infected cultures were collected, and the virus titers in the media were determined by a 50% tissue culture infective dose (TCID50) assay in MRC5 cells.
Western blotting.
Proteins were analyzed by Western blotting as described previously (50). Briefly, cells were washed with phosphate-buffered saline, lysed in sodium dodecyl sulfate (SDS) sample buffer supplemented with a protease inhibitor cocktail, and then scraped and collected by centrifugation. Cell lysates containing equal cell numbers were resolved by electrophoresis on sodium dodecyl sulfate-containing polyacrylamide gels, subsequently transferred to polyvinylidene difluoride membranes, hybridized with primary antibodies, and reacted with the horseradish peroxidase-conjugated secondary antibodies. The membranes were then treated with enhanced chemiluminescence (ECL) reagents (Bio-Rad). Finally, the protein bands were imaged by exposure to X-ray films (Kodak).
Immunofluorescence microscopy.
Cells grown on glass coverslips were fixed in 2% paraformaldehyde (in phosphate-buffered saline) for 20 min at room temperature and permeabilized with 0.1% Triton X-100 (in phosphate-buffered saline) for 15 min at room temperature. Subsequently, the cells were blocked with 5% FBS (in phosphate-buffered saline) for 20 min and incubated with the primary antibodies indicated below for 60 min at room temperature. Primary antibodies to the following (and their dilutions) were used: IFITM1 (1:200, mouse), IFITM2/3 (1:200, mouse), GM130 (1:100, rabbit), and pp28 (1:200, mouse). Cells were then washed with phosphate-buffered saline and stained with either Alexa Fluor 488-conjugated secondary antirabbit antibody (1:1,000; Invitrogen-Molecular Probes) or Alexa Fluor 555-conjugated secondary antimouse antibody (1:1,000; Invitrogen-Molecular Probes). To visualize the nuclei, cells were counterstained with DAPI (4′,6-diamidino-2-phenylindole; 1:5,000; Beyotime) for 10 min. Finally, labeled cells were mounted on slides with Prolong Gold antifade reagent (Invitrogen-Molecular Probes) overnight. Images were captured using a Leica TCS SP5 confocal laser scanning microscope.
Analysis of DNA and RNA.
The intracellular DNA levels of infected cells were examined by real-time PCR as previously described (50). MRC5 cells were infected with wild-type virus at a multiplicity of infection of 0.3, collected at the times postinfection indicated below, resuspended in lysis buffer (10 mM Tris-HCl [pH 8.0], 200 mM NaCl, 50 mM EDTA, 50 μg/ml proteinase K, 1% SDS), and then lysed by incubation at 55°C overnight. Total DNAs were then extracted with phenol-chloroform, treated with 100 μg/ml RNase A, extracted once more with phenol-chloroform, precipitated with ethanol, and finally, resuspended in water. For quantifying virion DNA, virus in the cultured medium was partially purified and concentrated by ultracentrifugation on a 20% d-sorbitol cushion at 64,000 × g for 1 h and then resuspended in PBS. Ten microliters of concentrated virus was treated with DNase I (30 U; Promega) at 37°C for 30 min to remove residual DNA that was not protected by the virus particle and then treated at 75°C for 20 min to inactivate the DNase I. The samples were then incubated at 55°C overnight in lysis buffer (400 mM NaCl, 10 mM Tris [pH 8.0], 10 mM EDTA, 0.1 mg of proteinase K/ml, 0.2% SDS) and extracted with phenol-chloroform. The aqueous phase were collected and treated with 20 μg of RNase A/ml for 1 h at 37°C and then extracted again with phenol-chloroform. The virion DNA in the aqueous phase was precipitated with ethanol and resuspended in water. Viral DNAs and cellular DNAs were quantified with SYBR Premix Ex Taq (TaKaRa) and primers specific for the HCMV IE gene and human β-actin gene (Table 1). The accumulation of viral DNAs was normalized by dividing the number of IE gene equivalents by the number of β-actin gene equivalents.
TABLE 1.
Primers for qPCR
| Primer | Orientationa | Sequence (5′–3′) |
|---|---|---|
| IFITM1 | F | TCTTCTTGAACTGGTGCTGTC |
| R | GTCGCGAACCATCTTCCTGT | |
| IFITM2 | F | CCTTGACCTGTATTCCACT |
| R | GCCATTGTAGAAAAGCGT | |
| IFITM3 | F | TCCCACGTACTCCAACTTCCA |
| R | AGCACCAGAAACACGTGCACT | |
| GAPDH | F | GAGTCAACGGATTTGGTCGT |
| R | GACAAGCTTCCCGTTCTCAG | |
| β-Actin | F | CTCCATCCTGGCCTCGCTGT |
| R | GCTGTCACCTTCACCGTTCC | |
| HCMV IE | F | TCTGCCAGGACATCTTTCTCG |
| R | GGAGACCCGCTGTTTCCAG |
F, forward; R, reverse.
Transcript accumulation was quantified by reverse transcription coupled to real-time PCR, as previously described (51). Briefly, total RNAs were extracted with the TRIzol reagent (Invitrogen) and reverse transcribed using a PrimeScript RT reagent kit (TaKaRa). The cDNAs were then quantified with SYBR Premix Ex Taq (TaKaRa) and with primers specific for the cellular genes IFITM1, IFITM2, IFITM3, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Table 1).
Electron microscopy.
shRNA-expressing lentivirus-transduced cells were infected with HCMV at an MOI of 0.5. At 4 days p.i., cells on the coverslips were fixed with 2.5% glutaraldehyde in PBS for 1 h and postfixed with 2% aqueous OsO4 for 1.5 h. Briefly, the cell monolayers were then dehydrated with ethanol and propylene oxide, embedded in epoxy resin, and polymerized at 60°C for 48 h. Embedded cell monolayers were cut into ultrathin sections of 70 nm and then stained with uranyl acetate for 6 min and lead citrate for 4 min at room temperature. Samples were analyzed by use of an FEI Tecnai G2 Spirit transmission electron microscope.
RESULTS
HCMV infection suppresses IFITM gene expression.
In order to study a potential role of IFITMs in HCMV infection, we first examined IFITM expression in human embryonic lung fibroblasts (MRC5 cells) before or after HCMV infection. As shown in Fig. 1A, the IFITM1 and IFITM2/3 proteins were both expressed constitutively in MRC5 cells before HCMV infection. They remained stable up to 24 h postinfection (hpi) but were reduced significantly at 48 hpi and became undetectable at 72 hpi, according to the results of immunoblotting. To determine whether IFITM gene expression was altered at the mRNA level after HCMV infection, we performed quantitative reverse transcription-PCR. As shown in Fig. 1B, the accumulation of the mRNAs of all three IFITM genes was reduced after HCMV infection. The reduction was obvious from 24 hpi. The mRNA levels of IFITM1 and IFITM2 remained relatively constant after 24 hpi, whereas the IFITM3 mRNA level decreased continuously to less than 20% of that of the mock-infected cells at 72 hpi.
FIG 1.

HCMV infection suppresses IFITM gene expression. (A) MRC5 cells were mock infected or infected with HCMV at an MOI of 3, and the cell lysates were collected at the indicated times after infection. The protein levels of IFITM1 and IFITM2/3 were analyzed by immunoblotting. Commercially available antibodies recognize both IFITM2 and IFITM3 due to the high sequence similarities of the two proteins. Viral proteins IE1/2 were used to indicate HCMV infection, and tubulin was used as the loading control. (B) IFITM gene transcription was reduced by HCMV infection. MRC5 cells were mock infected or infected with HCMV at an MOI of 3. Total RNA was collected at the indicated times after infection. The mRNA levels of the IFITM genes were quantified by reverse transcription-qPCR. The relative gene expression level was normalized against that of GAPDH, and the normalized gene expression in mock-infected cells at 8 h was set equal to 1. (C) MRC5 cells were infected with HCMV (virus), UV-inactivated virus (virus + UV), or HCMV with ganciclovir (virus + GCV) at an MOI of 3 or mock infected (mock). Cell lysates were collected at 48 h after infection. (Left) The protein levels of IFITM1 and IFITM2/3 were analyzed by immunoblotting. Viral proteins IE1/2 and pp28 were used to indicate the effect of UV inactivation and GCV treatment on virus gene expression, and tubulin was used as the loading control. (Right) A longer exposure of anti-IFITM1.
We then tested whether viral protein expression is required to suppress IFITM accumulation. To do this, we infected MRC5 cells with HCMV in the presence or absence of ganciclovir (GCV) or with UV-inactivated virus and then measured IFITM protein accumulation at 48 hpi. As shown in Fig. 1C, IFITM proteins were dramatically increased in cells infected with UV-inactivated HCMV. UV inactivation blocks viral protein expression, as indicated by no detectable IE1/2 or pp28. Therefore, the data suggest that HCMV binding and/or entry into MRC5 cells has the ability to induce IFITM expression, but this effect is suppressed by viral events or viral gene expression after entry. Ganciclovir treatment blocks viral DNA synthesis and late gene expression, as indicated by no pp28 protein accumulation. Under such a condition, IFITM protein accumulation was slightly reduced compared to that with mock infection. In contrast, HCMV infection without GCV treatment greatly suppressed IFITM2/3 accumulation at this time point. The data imply that a maximum inhibition of IFITM expression requires a late viral event or late viral protein expression.
To test whether the subcellular localization of IFITMs was changed after HCMV infection, we examined the distribution of IFITM proteins in MRC5 cells by confocal microscopy. As shown in Fig. 2A, IFITM2/3 located mainly as punctate structures in the cytoplasm before infection. HCMV infection did not change the localization of IFITM2/3, but their protein level was gradually reduced, as indicated by the weaker staining of IFITM2/3 in infected cells (IE1/2- and GFP-positive cells) at 48 and 72 hpi. The localization of IFITM1 before or after HCMV infection is shown in Fig. 2B. IFITM1 was expressed at a low level in mock-infected cells. At 24 hpi, the IFITM1 signal became stronger, probably due to the increase in the level of IFITM1 expression with time, as shown in Fig. 1A. IFITM1 was distributed in both the nucleus and the cytoplasm at this time point, which is different from the previously reported early endosome and plasma membrane localization of IFITM1 (25). We had confirmed the specificity of the IFITM1 antibody by staining cells expressing shRNA targeting IFITM1 (data not shown); thus, the immunofluorescence staining reflected the real IFITM1 localization in the cell type used in this study. At 48 hpi and particularly at 72 hpi, the reduction of the IFITM1 signal in HCMV-infected cells became apparent, indicating the suppression of IFITM1 expression by HCMV, which is consistent with the immunoblotting result presented in Fig. 1A. Overall, our data indicate that the IFITM genes were constitutively expressed at the basal level in MRC5 cells and they were suppressed rather than induced after HCMV infection.
FIG 2.

IFITM genes were suppressed by HCMV infection. (A) MRC5 cells were infected with HCMV (AD-GFP) at an MOI of 0.5 or mock infected. Cells were fixed at the indicated times after infection and stained with antibodies against the HCMV IE1/2 protein and the host IFITM2/3 protein. (B) An experiment similar to that described in the legend to panel A was performed, except that the cells were stained with IFITM1 and HCMV IE1/2 antibodies.
IFITM overexpression fails to inhibit HCMV replication in human fibroblasts.
As the antiviral function of IFITMs has been well documented, we reasoned that the significant reduction of IFITMs at later times after HCMV infection might represent a viral mechanism to escape from the inhibitory effect of these antiviral molecules. If this is the case, we would expect the inhibition of HCMV replication with the stable expression of IFITMs in the host cells. To test this hypothesis, we compared virus growth at both a high and a low multiplicity of infection (MOI) in MRC5 cells stably expressing Flag-tagged human IFITM1, IFITM2, or IFITM3 proteins and control MRC5 cells, which were transduced with an empty lentiviral vector (Fig. 3A). Unexpectedly, when we measured virus growth, we did not observe any inhibition of HCMV replication by IFITM overexpression; instead, virus production was enhanced slightly by about 3- to 5-fold in IFITM-expressing cells compared with that in the control cells at later time points (Fig. 3B and C). We also checked viral protein expression. As shown in Fig. 3E, during infection, viral immediate early protein (IE1/2), early protein (UL38), and late protein (pp28) were almost identically expressed in control or IFITM-overexpressing cells. These results indicate that IFITMs are not restriction factors for HCMV infection in human fibroblasts.
FIG 3.

Overexpression of IFITMs in MRC5 cells fails to inhibit HCMV replication. (A) MRC5 cells were transduced with a lentiviral vector that expressed Flag-tagged IFITM1, IFITM2, or IFITM3 or a control (ctrl) vector, and the expression of IFITMs was analyzed at 48 h after transduction by immunoblotting using Flag tag-specific antibody. Tubulin was used as the loading control. (B) Growth analysis of HCMV infection in control or IFITM-expressing MRC5 cells at an MOI of 0.3. The cell-free virus in the supernatant was collected on the indicated days postinfection (dpi), and the titer was determined using a TCID50 assay. (C) Analysis of HCMV growth in control or IFITM-expressing MRC5 cells infected at an MOI of 3. (D) HCMV infection suppressed endogenous and overexpressed Flag-IFITM3. Control or Flag-IFITM3-expressing MRC5 cells were mock infected or infected with HCMV at an MOI of 3. Expression of IFITM3 was analyzed at 72 hpi by immunoblotting using IFITM3- or Flag tag-specific antibodies. Both Flag-tagged IFITM3 (arrowhead) and endogenous IFITM2/3 (arrow) were detected. (E) Overexpression of IFITMs did not affect HCMV protein expression. MRC5 cells overexpressing control or Flag-tagged IFITMs (left, IFITM1; middle, IFITM2; right, IFITM3) were infected with HCMV at an MOI of 3. Cell lysates were collected at the indicated times after infection. The accumulation of viral proteins with immediate early (IE1/2), early (UL38), and late (pp28) kinetics was examined by immunoblotting.
We noticed a reduction of Flag-tagged IFITM protein expression at a late time of infection (Fig. 3E), reminiscent of the suppression of endogenous IFITMs. We compared Flag-tagged and endogenous IFITM3 expression during HCMV infection as an example (Fig. 3D). The Flag-tagged IFITM3 protein was expressed at a much higher level than the endogenous IFITM2/3 before HCMV infection (Fig. 3D). Interestingly, HCMV infection dramatically suppressed the expression of both endogenous and Flag-tagged IFITMs at 72 hpi. The endogenous level became undetectable, and the level of Flag-tagged IFITM3 after infection was approximately equivalent to the endogenous level before infection (Fig. 3D). The transcription of the Flag-tagged IFITMs was driven by an exogenous promoter that differed from the promoter driving endogenous transcription; thus, we consider that it is highly likely that HCMV infection can inhibit IFITM protein expression at both the transcriptional and posttranscriptional levels, which might suppress IFITM mRNA nuclear export, attenuate translation, or induce protein degradation.
Knockdown of IFITMs by RNAi suppresses HCMV replication but not HSV replication.
Although the difference was subtle, we repeatedly observed enhanced HCMV growth after the stable expression of IFITM3 in MRC5 cells compared with the growth detected in control MRC5 cells. These findings prompted us to examine whether the endogenous IFITMs are required for HCMV replication. To test this hypothesis, we used two different shRNA constructs to knock down IFITM protein expression. As shown in Fig. 4A, construct sh8024 knocked down IFITM2/3 expression almost completely in MRC5 cells, but IFITM1 expression remained intact. Construct sh8025 suppressed the expression of all three IFITMs. During measurements of virus growth, we observed reduced virus replication in both sh8024- and sh8025-expressing cells compared with that in the control shRNA-expressing cells (Fig. 4B and C). In a virus yield assay (Fig. 4B), the virus titer was reduced by more than 100-fold in sh8025-expressing cells and by about 10-fold in sh8024-expressing cells at 8 days p.i. A growth curve at an MOI of 0.3 with more frequent sampling is shown in Fig. 4D. Virus growth was continuously suppressed up to 10 days p.i. after IFITM depletion. We noted that the impairment of HCMV growth was much lower in the sh8024-expressing cells than the sh8025-expressing cells. This may have occurred because IFITM1 was not targeted by sh8024 and the functional redundancy among IFITMs is well-known (18, 20, 22). Nevertheless, we used two different IFITM-targeting shRNA sequences to knock down IFITMs to demonstrate their requirement for efficient HCMV replication. As our findings revealed a novel proviral role of IFITMs in HCMV replication, we wanted to determine whether it was also required for replication of a closely related herpesvirus. To do this, we compared the growth of herpes simplex virus (HSV) in control cells and cells in which IFITM was knocked down. As shown in Fig. 4E, HSV grew equivalently in cells expressing control shRNA and shRNA targeting IFITMs, indicating a selective inhibition of HCMV replication by knocking down IFITM proteins in MRC5 cells. Indeed, propidium iodide (PI) staining of the cells at up to 5 days after shRNA knockdown did not detect a significant difference in cell death between the control cells and cells in which IFITM was knocked down (Fig. 4F), thereby indicating that cell viability was not affected by IFITM suppression.
FIG 4.

shRNA knockdown of IFITMs inhibits HCMV replication. (A) Knockdown efficiency of shRNAs targeting IFITM genes. Cells were transduced with lentivirus vectors expressing shC, sh8024, or sh8025. Cell lysates were collected at 48 h postransduction, and the protein levels of IFITM2/3 or IFITM1 were examined by immunoblotting. (B) Growth analysis of HCMV infection in cells expressing shC, sh8024, or sh8025. The cells were infected at an MOI of 0.3, the cell-free virus in the supernatant was collected at the indicated times after infection, and the titer was determined using a TCID50 assay. (C) Growth analysis of HCMV infection in cells expressing shC, sh8024, or sh8025 at an MOI of 3. The experimental procedures were similar to those described in the legend to panel B. (D) An independent experiment similar to that described in the legend to panel B, except the results were analyzed at more time points. (E) Downregulation of IFITM gene expression did not inhibit HSV replication. MRC5 cells were transduced with lentivirus overexpressing shC, sh8024, or sh8025 and then infected with HSV at an MOI of 0.01 or 0.001. The supernatant was collected at the indicated times postinfection, and titers were determined by a TCID50 assay. (F) Downregulation of IFITM gene expression did not induce elevated cell death. MRC5 cells were transduced with shC, sh8024, or sh8025 and then stained with propidium iodide (PI) and Hoechst at 3, 4, or 5 days after transduction. Hoechst stains the nucleus of all cells, and PI stains only dying cells with a permeable membrane. Cells treated with PBS for 5 h were included as a positive control for cell death.
IFITM protein downregulation minimally affects HCMV gene expression or DNA synthesis but reduces virus infectivity.
In order to determine how IFITMs facilitate HCMV replication, we first analyzed virus gene expression at the protein level by immunoblotting. As shown in Fig. 5A, the levels of immediate early (IE1/2), early (UL38), and late (pp28) virus proteins that accumulated were almost identical in the control cells and cells in which IFITM was knocked down, thereby indicating that virus gene expression was not affected. Next, we measured virus DNA replication by quantitative PCR (qPCR) and observed that the kinetics of virus DNA synthesis were similar in both cell types (Fig. 5B). These data suggest that IFITMs are involved in a very late step of the virus life cycle, i.e., after viral DNA synthesis and late protein accumulation, which could involve virus assembly and/or egress.
FIG 5.
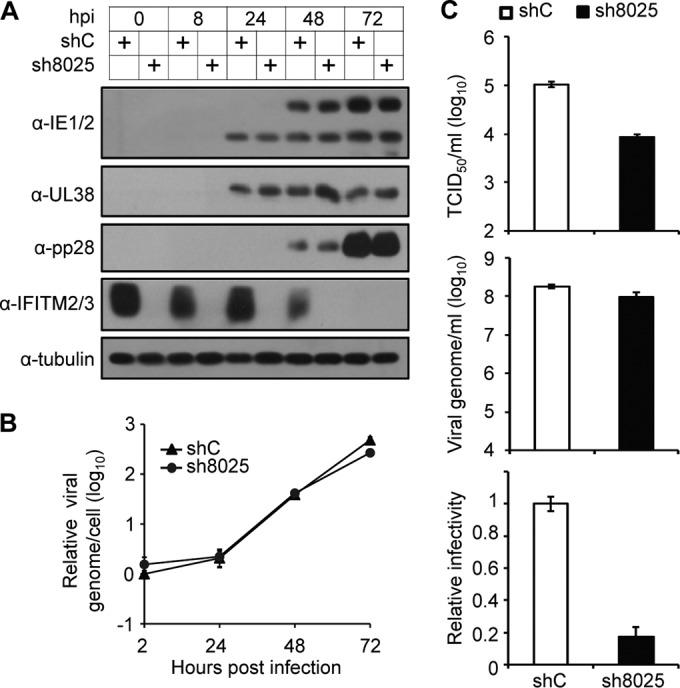
Suppression of IFITM gene expression has a minimal effect on viral gene expression or DNA synthesis but significantly reduces the infectivity of the progeny virus particles. (A) Knockdown of IFITM gene expression by sh8025 had little effect on HCMV protein expression. MRC5 cells transduced with shC or sh8025 were infected with HCMV at an MOI of 3. Cell lysates were collected at the indicated times after infection. The accumulation of viral proteins with immediate early (IE1/2), early (UL38), and late (pp28) kinetics was examined by immunoblotting. (B) Knockdown of IFITM gene expression had a minimal effect on viral DNA synthesis. MRC5 cells transduced with shC or sh8025 were infected with HCMV at an MOI of 0.3. Intracellular DNA was extracted at the indicated times after infection. The accumulation of viral DNA was quantified by qPCR, and the amount was normalized against that of actin. The result for the DNA sample from shC-transduced cells collected at 2 h was set equal to 1. (C) Suppression of IFITM gene expression significantly reduced the infectivity of the progeny virus. shC- or sh8025-expressing cells were infected by HCMV at an MOI of 0.3. The cell-free virus in the culture medium was collected at 4 days p.i., concentrated, and purified by use of a sorbitol cushion. Each virus stock was divided into two to measure the infectious units by a TCID50 assay (top) and the genome copy number by qPCR (middle). (Bottom) The ratio of the TCID50 results relative to the DNA copy numbers was calculated for each sample. The ratio of the TCID50 results relative to the DNA copy number for the progeny virus produced in shC-expressing cells was set equal to 1.
To test if virus assembly or egress was affected by IFITM depletion, we went on to measure the infectivity of progeny virus, represented by the ratio of the infectious unit to the number of genome copies of the secreted virus stock. Since one virus particle contains one viral genome, the number of genome copies equals the number of DNA-containing virus particles. A defect in virus assembly would reduce the infectivity, whereas a defect in egress would reduce the yield of viral particles but not the infectivity. We infected control or sh8025-expressing cells at an MOI of 0.3, and we then concentrated and purified the viruses secreted in the culture supernatant at 4 days p.i. Each virus stock was divided into two to measure the number of infectious units by a TCID50 assay and the genome copy number by qPCR. As shown in Fig. 5C, the number of virus particles, which were represented as genome copy numbers, was reduced slightly in sh8025-expressing cells compared with the number in the control shRNA-expressing cells, but the number of infectious units was reduced much more, by about 10-fold, at 4 days p.i., thereby leading to an approximately 5-fold reduction in the ratio of the number of infectious units relative to the number of genome copies. Thus, the virion particles formed in cells in which IFITM was knocked down were about 5-fold less infectious than those formed in control cells, thereby implying that there was a defect in virus assembly.
IFITMs are required for the proper formation of the vAC.
HCMV virion assembly occurs at a virus-induced perinucleus structure called the vAC, which is formed via the reorganization of the Golgi apparatus and endosomal membrane structures. As virion assembly is likely affected by IFITM depletion, we went on to ask whether the formation of the vAC was defective in the absence of IFITM proteins. As shown in Fig. 6A, the Golgi stacks, which were indicated by the Golgi apparatus marker GM130, formed typical circular structures in cells that expressed the control shRNA. They formed a ring-like structure that wrapped around the vAC, where the virion structure protein pp28 was concentrated. In contrast, the Golgi stacks failed to form a ring-like structure in both the sh8024- and sh8025-expressing cells, and pp28 was diffused throughout the cytoplasm, thereby indicating that the vAC did not form correctly when IFITM protein expression was suppressed. The defect in the proper formation of vAC in the absence of IFITMs was statistically significant (Fig. 6B). To rule out the possibility that the defect was caused by a slower spread of the virus in IFITM-depleted cells after the first round of infection, we quantified vAC maturation at 3 days p.i. (Fig. 6C and D). This allowed us to measure the phenotype within the first round of infection. As shown in Fig. 6D, the percentage of regular vAC was still significantly less in cells in which IFITM was knocked down than in the control cells at 3 days p.i. The localization of two other virus glycoproteins, gB and pp150, was also examined at 3 days p.i. As shown in Fig. 6E, while these two proteins were concentrated in the vAC in control cells, they were relatively diffused throughout the cytoplasm after IFITM suppression. Importantly, expression of an shRNA-resistant IFITM3 almost completely restored the correct formation of the vAC (Fig. 7A and B), further supporting the role of IFITMs in facilitating vAC formation during HCMV infection.
FIG 6.

IFITMs are required for correct vAC formation. (A) MRC5 cells transduced with shC-, sh8024-, or sh8025-expressing lentiviruses were infected with HCMV (AD/Cre) at an MOI of 0.5. Cells were fixed at 96 h postinfection and stained with antibodies against virus structural protein pp28 and the Golgi apparatus marker GM130, both of which are proteins known to be localized to the vAC. DAPI was used to stain the nuclei. (B) Cells from the assay whose results are presented in panel A were counted for the presence of regular vAC and irregular vAC. A typical or regular vAC is characterized by a well-formed circular Golgi apparatus ring which is usually partially surrounded by kidney-like nuclei and a round pp28-positive structure. Irregular vAC displays a malformation of the circular Golgi apparatus ring and a scattered pp28 distribution. Statistical significance was measured with SPSS (version 20) software (SPSS Inc.) using the chi-square test. n, number of infected cells counted; ***, P < 0.002. (C) MRC5 cells transduced with shC- or sh8025-expressing lentiviruses were infected with HCMV (AD/Cre) at an MOI of 0.5. Cells were fixed at 72 h postinfection and stained with antibodies against virus structural protein pp28 and the Golgi apparatus marker GM130. DAPI was used to stain the nuclei. (D) Cells from the assay whose results are presented in panel C were counted for the presence of regular vAC and irregular vAC. n, number of infected cells counted; ***, P < 0.002. (E) MRC5 cells transduced with shC- or sh8025-expressing lentiviruses were infected with HCMV (AD/Cre) at an MOI of 0.5. Cells were fixed at 72 h postinfection and stained with antibodies against virus protein pp150 (left) or gB (right) and the Golgi apparatus marker GM130. DAPI was used to stain the nuclei.
FIG 7.

Overexpression of IFITM3 rescues vAC formation. (A) Control cells or shRNA-resistant IFITM3-expressing cells (sh8025-RE) were transduced with shC- or sh8025-expressing lentiviruses and then infected with HCMV (AD/Cre) at an MOI of 0.5. Cells were fixed at 72 h postinfection and stained with antibodies against virus structural protein pp28 and the Golgi apparatus marker GM130, both of which are proteins known to be localized to the vAC. DAPI was used to stain the nuclei. (B) Cells from the assay whose results are presented in panel A were counted for the presence of regular vAC and irregular vAC. Statistical significance was measured with SPSS (version 20) software (SPSS Inc.) using the chi-square test. n, number of infected cells counted; ns, no significant difference; ***, P < 0.002. (C) Western blotting of sh8025-resistant IFITM3 expression.
Ultrastructural analysis by electron microscopy revealed a defect in virion assembly after IFITM suppression.
We went on to examine the effect of IFITM suppression on HCMV virion assembly by electron microscopy. Capsid formation in the nucleus did not seem to be affected by IFITM knockdown (Fig. 8A). All three forms of nuclear capsids could be detected in the presence or absence of IFITMs, the numbers of A and C capsids were comparable, and the numbers of B capsids were actually increased after IFITM knockdown (Fig. 8B). In contrast, the defect in the formation of vAC and the secondary envelopment of the virion in the cytoplasm were apparent. As shown in Fig. 9, the circular vAC formed clearly at the perinuclear position in the control shRNA-expressing cells (Fig. 9A and C). Different forms of virus particles, including DNA-containing infectious particles, noninfectious enveloped particles (NIEPs) with empty capsids, and dense bodies, were found inside the membranous vesicles (Fig. 9D and E), thereby suggesting successful secondary envelopment. In contrast, the tight circular vAC structure found in the control shRNA-expressing cells was not obvious in IFITM shRNA-expressing cells (Fig. 9B and F), and some Golgi stacks were not reorganized (Fig. 9G, arrow), while the virus particles could be found in a generally diffused area of the cytoplasm (Fig. 9F), but most of the particles were not encapsulated by a membrane structure (Fig. 9G and H). Quantitative analysis of the phenotype confirmed that the defect in virion envelopment was statistically significant (Fig. 9I). These data clearly indicate that there was a defect in the formation of the vAC and a defect during the virus assembly process.
FIG 8.
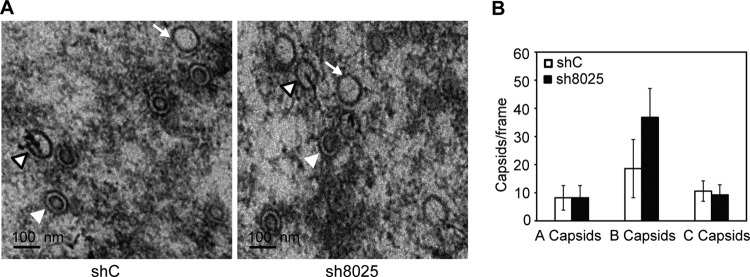
Electron microscopy analysis of the capsid assembly and DNA packaging in the presence or absence of IFITMs. (A) MRC5 cells were transduced with shC- or sh8025-expressing lentiviruses and then infected with HCMV (AD-GFP) at an MOI of 0.5, and cells were fixed at 96 hpi and prepared for electron microscopy. A capsids (white arrow), B capsids (white arrowhead), and C capsids (white arrowhead with a black border) are indicated. A capsids are devoid of viral DNA and a scaffold and presumably result from abortive virus DNA packaging events. B capsids contain a scaffold but lack viral DNA. C capsids contain the viral DNA and are devoid of a scaffold; thus, they likely represent the mature form of nucleocapsids. (B) Quantification of the different capsid types observed in the presence or absence of IFITMs (as average values per frame). The numbers of capsids counted were 187 for shC and 271 for sh8025.
FIG 9.

Electron microscopy analysis of vAC formation and virion assembly reveals a defect in both processes after IFITM downregulation. shC- or sh8025-expressing MRC5 cells were infected with HCMV at an MOI of 0.5, and cells were fixed and stained at 96 hpi. Electron microscopic images of shC-expressing (A) and sh8025-expressing (B) cells infected with HCMV are shown to highlight the morphology of the nucleus (Nuc) and the vAC (circled). The white arrowhead with a black border points to the virion particles in the vAC. (C to E) Images of another infected control shRNA-expressing cell are shown at increasing magnifications to highlight vAC formation and virion assembly. The area in the square box in panel C is shown at a higher magnification in panel D, and the area in the square box in panel D is shown at a further increased magnification in panel E. Different forms of virion particles and dense bodies in panels D and E are indicated with arrows of different styles. (F to H) Images of another infected sh8025-expressing cell are shown at increasing magnifications. The areas in the square boxes labeled G and H in panel F are shown at higher magnifications in panels G and H, respectively. Arrow in panel G, the Golgi stacks; arrow in panel H, incompletely assembled virion particles without envelopment. (I) Quantification of HCMV secondary envelopment in the presence or absence of IFITMs. Enveloped virions are fully formed virions that contain DNA and envelope. Nonenveloped virions include naked particles and budding particles (virion particles attached to membranes). Statistical significance was measured using the chi-square test. n, number of virion particles counted; ***, P < 0.002.
DISCUSSION
IFITMs belong to a family of small ISGs that impede the cytosolic entry of a range of pathogenic viruses. In the present study, however, we were surprised to find a proviral function of IFITMs in HCMV infection. IFITMs were expressed constitutively in human embryonic lung fibroblasts. The suppression of IFITM protein expression by RNAi selectively inhibited HCMV replication but not infection by the closely related human simplex virus. Our detailed characterization of the virus life cycle showed that IFITMs are required for the correct formation of the vAC. Thus, virus secondary envelopment was impaired in the absence of IFITMs, whereas virus gene expression and DNA synthesis were affected only marginally. The known function of IFITMs in virus infection is related to virus entry, which is a very early step of the virus life cycle. IFITMs block the entry of a range of RNA viruses into their host cells (18), but the detailed mechanism has remained elusive, although it probably occurs via inhibition of the membrane fusion step of virus entry. A recent study found that IFITMs promote infection by human coronavirus OC43, where this particular virus utilizes the IFITMs as an entry factor (52). However, our findings are novel because we showed that IFITMs facilitate the replication of a large DNA virus at a very late stage of the viral life cycle.
One counterintuitive result of this study is that HCMV infection drastically suppresses IFITM protein accumulation at the late stage of virus infection (Fig. 1A), regardless of the role of IFITMs in late events of the virus life cycle. How can a host protein promote a late viral event while the protein level is inhibited by virus infection? It is possible that the presence of the IFITMs during the early phase of virus infection primes the cellular environment for a late viral event. This could be tested in the future by depleting IFITMs at late times of the virus life cycle. Alternatively, immunoblotting might not be sufficiently sensitive to detect the trace amounts of IFITMs that remain during the late stages of infection. These trace amounts of IFITMs may contribute to their role in correct vAC formation. The virus-mediated inhibition of IFITMs does not seem to play a role in in vitro infection of HCMV in human fibroblasts, since the stable expression of IFITMs does not suppress HCMV replication (Fig. 3B and C). However, we cannot rule out the possibility that it may play a functional role in certain aspects of HCMV biology or pathology. HCMV has a broad cell tropism in vivo, leading to pathogenesis in multiple organs of immunocompromised patients (53). Other virus infection systems or latency models will be required to fully understand the interplay between IFITMs and HCMV infection.
How can IFITMs promote the formation of the vAC to enhance HCMV replication? First, IFITM3 may promote vAC maturation and HCMV production by increasing intracellular cholesterol accumulation. A recent study has shown that IFITM3 disrupts intracellular cholesterol homeostasis by interfering with the interaction between vesicle-associated membrane protein (VAMP)-associated protein A (VAPA) and oxysterol-binding protein (OSBP). Overexpression of IFITMs induces a marked accumulation of cholesterol in the cells (25). Interestingly, HCMV infection is known to use multiple mechanisms to increase intracellular cholesterol accumulation (54–56), which is required for highly infectious virion particle production (55). Second, the formation of the HCMV vAC involves a drastic reorganization of intracellular organelles, which likely requires highly regulated vesicle budding, trafficking, and tethering events. A recent study reported that a set of HCMV microRNAs attenuates the expression of host secretory pathway genes, including vesicle-associated membrane protein 3 (VAMP3), to facilitate vAC formation (57). VAPA and OSBP are known to mediate lipid transfer and membrane tethering in different intracellular organelles (58, 59). IFITMs could play a role in these processes by interacting with VAPA and therefore promote vAC formation. Finally, IFITMs have been shown to function in stabilization of v-ATPase complexes in intracellular membranes and thus facilitate the proper subcellular localization of clathrin (26), and v-ATPase- and clathrin-mediated vesicle transport are likely involved in vAC maturation (60, 61).
ACKNOWLEDGMENTS
We thank Min-Hua Luo at the Wuhan Institute of Virology, CAS, for critical readings of the manuscript, all the members of the Herpesvirus and Molecular Virology Research Unit for helpful discussions, Tomas Shenk for antibodies, Jay Nelson for the IE1/2 antibody, and Chiyu Zhang at the Institut Pasteur of Shanghai for HSV-1. We thank the National Center for Protein Science Shanghai for the confocal laser scanning microscope, and we are grateful to Yan Wang for her great technical assistance. We thank the National Center for Protein Science Shanghai for performing our electron microscopy, and we are grateful to Mi Cao for his expert technical assistance. We thank Tianqing Zhang and the staff of the Core Facility for Cell Biology at the Institute of Biochemistry and Cell Biology (SIBCB) for performing electron microscopy.
This work was supported by the National Natural Science Foundation of China (grants 81271836 and 81371826 to Z.Q., grant 31300148 to B.X.), the Chinese Academy of Sciences 100 Talents program and the Knowledge Innovation Program of the Chinese Academy of Sciences (grant Y214P1109 to Z.Q.), and the Science and Technology Commission of Shanghai Municipality (grant 13ZR1445500 to B.X.). B.X. was supported by the Youth Innovation Promotion Association, CAS.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Mocarski ES Jr, Shenk T, Griffiths PD, Pass RF. 2013. Cytomegalovirus, p 1960–2014 InKnipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Amsler L, Verweij MC, DeFilippis VR. 2013. The tiers and dimensions of evasion of the type I interferon response by human cytomegalovirus. J Mol Biol 425:4857–4871. doi: 10.1016/j.jmb.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossini G, Cerboni C, Santoni A, Landini MP, Landolfo S, Gatti D, Gribaudo G, Varani S. 2012. Interplay between human cytomegalovirus and intrinsic/innate host responses: a complex bidirectional relationship. Mediators Inflamm 2012:607276. doi: 10.1155/2012/607276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krauss S, Kaps J, Czech N, Paulus C, Nevels M. 2009. Physical requirements and functional consequences of complex formation between the cytomegalovirus IE1 protein and human STAT2. J Virol 83:12854–12870. doi: 10.1128/JVI.01164-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paulus C, Krauss S, Nevels M. 2006. A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc Natl Acad Sci U S A 103:3840–3845. doi: 10.1073/pnas.0600007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reitsma JM, Sato H, Nevels M, Terhune SS, Paulus C. 2013. Human cytomegalovirus IE1 protein disrupts interleukin-6 signaling by sequestering STAT3 in the nucleus. J Virol 87:10763–10776. doi: 10.1128/JVI.01197-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor RT, Bresnahan WA. 2005. Human cytomegalovirus immediate-early 2 gene expression blocks virus-induced beta interferon production. J Virol 79:3873–3877. doi: 10.1128/JVI.79.6.3873-3877.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor RT, Bresnahan WA. 2006. Human cytomegalovirus IE86 attenuates virus- and tumor necrosis factor alpha-induced NFkappaB-dependent gene expression. J Virol 80:10763–10771. doi: 10.1128/JVI.01195-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor RT, Bresnahan WA. 2006. Human cytomegalovirus immediate-early 2 protein IE86 blocks virus-induced chemokine expression. J Virol 80:920–928. doi: 10.1128/JVI.80.2.920-928.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abate DA, Watanabe S, Mocarski ES. 2004. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J Virol 78:10995–11006. doi: 10.1128/JVI.78.20.10995-11006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Browne EP, Shenk T. 2003. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc Natl Acad Sci U S A 100:11439–11444. doi: 10.1073/pnas.1534570100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hakki M, Marshall EE, De Niro KL, Geballe AP. 2006. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J Virol 80:11817–11826. doi: 10.1128/JVI.00957-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marshall EE, Bierle CJ, Brune W, Geballe AP. 2009. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J Virol 83:4112–4120. doi: 10.1128/JVI.02489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seo JY, Yaneva R, Hinson ER, Cresswell P. 2011. Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science 332:1093–1097. doi: 10.1126/science.1202007. [DOI] [PubMed] [Google Scholar]
- 15.Viswanathan K, Smith MS, Malouli D, Mansouri M, Nelson JA, Fruh K. 2011. BST2/tetherin enhances entry of human cytomegalovirus. PLoS Pathog 7:e1002332. doi: 10.1371/journal.ppat.1002332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.John SP, Chin CR, Perreira JM, Feeley EM, Aker AM, Savidis G, Smith SE, Elia AE, Everitt AR, Vora M, Pertel T, Elledge SJ, Kellam P, Brass AL. 2013. The CD225 domain of IFITM3 is required for both IFITM protein association and inhibition of influenza A virus and dengue virus replication. J Virol 87:7837–7852. doi: 10.1128/JVI.00481-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perreira JM, Chin CR, Feeley EM, Brass AL. 2013. IFITMs restrict the replication of multiple pathogenic viruses. J Mol Biol 425:4937–4955. doi: 10.1016/j.jmb.2013.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anafu AA, Bowen CH, Chin CR, Brass AL, Holm GH. 2013. Interferon-inducible transmembrane protein 3 (IFITM3) restricts reovirus cell entry. J Biol Chem 288:17261–17271. doi: 10.1074/jbc.M112.438515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang IC, Bailey CC, Weyer JL, Radoshitzky SR, Becker MM, Chiang JJ, Brass AL, Ahmed AA, Chi X, Dong L, Longobardi LE, Boltz D, Kuhn JH, Elledge SJ, Bavari S, Denison MR, Choe H, Farzan M. 2011. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog 7:e1001258. doi: 10.1371/journal.ppat.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu J, Pan Q, Rong L, He W, Liu SL, Liang C. 2011. The IFITM proteins inhibit HIV-1 infection. J Virol 85:2126–2137. doi: 10.1128/JVI.01531-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mudhasani R, Tran JP, Retterer C, Radoshitzky SR, Kota KP, Altamura LA, Smith JM, Packard BZ, Kuhn JH, Costantino J, Garrison AR, Schmaljohn CS, Huang IC, Farzan M, Bavari S. 2013. IFITM-2 and IFITM-3 but not IFITM-1 restrict Rift Valley fever virus. J Virol 87:8451–8464. doi: 10.1128/JVI.03382-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weidner JM, Jiang D, Pan XB, Chang J, Block TM, Guo JT. 2010. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J Virol 84:12646–12657. doi: 10.1128/JVI.01328-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilkins C, Woodward J, Lau DT, Barnes A, Joyce M, McFarlane N, McKeating JA, Tyrrell DL, Gale M Jr. 2013. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 57:461–469. doi: 10.1002/hep.26066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amini-Bavil-Olyaee S, Choi YJ, Lee JH, Shi M, Huang IC, Farzan M, Jung JU. 2013. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 13:452–464. doi: 10.1016/j.chom.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wee YS, Roundy KM, Weis JJ, Weis JH. 2012. Interferon-inducible transmembrane proteins of the innate immune response act as membrane organizers by influencing clathrin and v-ATPase localization and function. Innate Immun 18:834–845. doi: 10.1177/1753425912443392. [DOI] [PubMed] [Google Scholar]
- 27.Jia R, Xu F, Qian J, Yao Y, Miao C, Zheng YM, Liu SL, Guo F, Geng Y, Qiao W, Liang C. 2014. Identification of an endocytic signal essential for the antiviral action of IFITM3. Cell Microbiol 16:1080–1093. doi: 10.1111/cmi.12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yount JS, Moltedo B, Yang YY, Charron G, Moran TM, Lopez CB, Hang HC. 2010. Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nat Chem Biol 6:610–614. doi: 10.1038/nchembio.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yount JS, Karssemeijer RA, Hang HC. 2012. S-Palmitoylation and ubiquitination differentially regulate interferon-induced transmembrane protein 3 (IFITM3)-mediated resistance to influenza virus. J Biol Chem 287:19631–19641. doi: 10.1074/jbc.M112.362095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shan Z, Han Q, Nie J, Cao X, Chen Z, Yin S, Gao Y, Lin F, Zhou X, Xu K, Fan H, Qian Z, Sun B, Zhong J, Li B, Tsun A. 2013. Negative regulation of interferon-induced transmembrane protein 3 by SET7-mediated lysine monomethylation. J Biol Chem 288:35093–35103. doi: 10.1074/jbc.M113.511949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chesarino NM, McMichael TM, Hach JC, Yount JS. 2014. Phosphorylation of the antiviral protein IFITM3 dually regulates its endocytosis and ubiquitination. J Biol Chem 289:11986–11992. doi: 10.1074/jbc.M114.557694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu H, Cong JP, Shenk T. 1997. Use of differential display analysis to assess the effect of human cytomegalovirus infection on the accumulation of cellular RNAs: induction of interferon-responsive RNAs. Proc Natl Acad Sci U S A 94:13985–13990. doi: 10.1073/pnas.94.25.13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicholl MJ, Robinson LH, Preston CM. 2000. Activation of cellular interferon-responsive genes after infection of human cells with herpes simplex virus type 1. J Gen Virol 81:2215–2218. [DOI] [PubMed] [Google Scholar]
- 34.Warren CJ, Griffin LM, Little AS, Huang IC, Farzan M, Pyeon D. 2014. The antiviral restriction factors IFITM1, 2 and 3 do not inhibit infection of human papillomavirus, cytomegalovirus and adenovirus. PLoS One 9:e96579. doi: 10.1371/journal.pone.0096579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol 20:392–401. doi: 10.1016/j.tim.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buchkovich NJ, Maguire TG, Alwine JC. 2010. Role of the endoplasmic reticulum chaperone BiP, SUN domain proteins, and dynein in altering nuclear morphology during human cytomegalovirus infection. J Virol 84:7005–7017. doi: 10.1128/JVI.00719-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schauflinger M, Fischer D, Schreiber A, Chevillotte M, Walther P, Mertens T, von Einem J. 2011. The tegument protein UL71 of human cytomegalovirus is involved in late envelopment and affects multivesicular bodies. J Virol 85:3821–3832. doi: 10.1128/JVI.01540-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schauflinger M, Landwehr S, Walther P, Mertens T, Einem J. 2008. Insights into the role of pUL71 during HCMV morphogenesis, p 141–142 InEMC 2008 14th European Microscopy Congress. Springer, Berlin, Germany. doi: 10.1007/978-3-540-85228-5_71. [DOI] [Google Scholar]
- 39.Phillips SL, Bresnahan WA. 2012. The human cytomegalovirus (HCMV) tegument protein UL94 is essential for secondary envelopment of HCMV virions. J Virol 86:2523–2532. doi: 10.1128/JVI.06548-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cepeda V, Fraile-Ramos A. 2011. A role for the SNARE protein syntaxin 3 in human cytomegalovirus morphogenesis. Cell Microbiol 13:846–858. doi: 10.1111/j.1462-5822.2011.01583.x. [DOI] [PubMed] [Google Scholar]
- 41.Indran SV, Britt WJ. 2011. A role for the small GTPase Rab6 in assembly of human cytomegalovirus. J Virol 85:5213–5219. doi: 10.1128/JVI.02605-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez V, Greis KD, Sztul E, Britt WJ. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J Virol 74:975–986. doi: 10.1128/JVI.74.2.975-986.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tandon R, Mocarski ES. 2008. Control of cytoplasmic maturation events by cytomegalovirus tegument protein pp150. J Virol 82:9433–9444. doi: 10.1128/JVI.00533-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buchkovich NJ, Maguire TG, Paton AW, Paton JC, Alwine JC. 2009. The endoplasmic reticulum chaperone BiP/GRP78 is important in the structure and function of the human cytomegalovirus assembly compartment. J Virol 83:11421–11428. doi: 10.1128/JVI.00762-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Das S, Pellett PE. 2011. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J Virol 85:5864–5879. doi: 10.1128/JVI.00155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cepeda V, Esteban M, Fraile-Ramos A. 2010. Human cytomegalovirus final envelopment on membranes containing both trans-Golgi network and endosomal markers. Cell Microbiol 12:386–404. doi: 10.1111/j.1462-5822.2009.01405.x. [DOI] [PubMed] [Google Scholar]
- 47.Das S, Vasanji A, Pellett PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol 81:11861–11869. doi: 10.1128/JVI.01077-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Everett RD, Boutell C, McNair C, Grant L, Orr A. 2010. Comparison of the biological and biochemical activities of several members of the alphaherpesvirus ICP0 family of proteins. J Virol 84:3476–3487. doi: 10.1128/JVI.02544-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol 76:2316–2328. doi: 10.1128/jvi.76.5.2316-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Terhune S, Torigoi E, Moorman N, Silva M, Qian Z, Shenk T, Yu D. 2007. Human cytomegalovirus UL38 protein blocks apoptosis. J Virol 81:3109–3123. doi: 10.1128/JVI.02124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xuan B, Qian Z, Torigoi E, Yu D. 2009. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J Virol 83:3463–3474. doi: 10.1128/JVI.02307-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao X, Guo F, Liu F, Cuconati A, Chang J, Block TM, Guo JT. 2014. Interferon induction of IFITM proteins promotes infection by human coronavirus OC43. Proc Natl Acad Sci U S A 111:6756–6761. doi: 10.1073/pnas.1320856111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sinzger C, Jahn G. 1996. Human cytomegalovirus cell tropism and pathogenesis. Intervirology 39:302–319. [DOI] [PubMed] [Google Scholar]
- 54.Seo JY, Cresswell P. 2013. Viperin regulates cellular lipid metabolism during human cytomegalovirus infection. PLoS Pathog 9:e1003497. doi: 10.1371/journal.ppat.1003497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gudleski-O'Regan N, Greco TM, Cristea IM, Shenk T. 2012. Increased expression of LDL receptor-related protein 1 during human cytomegalovirus infection reduces virion cholesterol and infectivity. Cell Host Microbe 12:86–96. doi: 10.1016/j.chom.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu Y, Maguire TG, Alwine JC. 2012. Human cytomegalovirus infection induces adipocyte-like lipogenesis through activation of sterol regulatory element binding protein 1. J Virol 86:2942–2949. doi: 10.1128/JVI.06467-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hook LM, Grey F, Grabski R, Tirabassi R, Doyle T, Hancock M, Landais I, Jeng S, McWeeney S, Britt W, Nelson JA. 2014. Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe 15:363–373. doi: 10.1016/j.chom.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raychaudhuri S, Prinz WA. 2010. The diverse functions of oxysterol-binding proteins. Annu Rev Cell Dev Biol 26:157–177. doi: 10.1146/annurev.cellbio.042308.113334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mesmin B, Bigay J, Moser von Filseck J, Lacas-Gervais S, Drin G, Antonny B. 2013. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell 155:830–843. doi: 10.1016/j.cell.2013.09.056. [DOI] [PubMed] [Google Scholar]
- 60.Pavelin J, Reynolds N, Chiweshe S, Wu G, Tiribassi R, Grey F. 2013. Systematic microRNA analysis identifies ATP6V0C as an essential host factor for human cytomegalovirus replication. PLoS Pathog 9:e1003820. doi: 10.1371/journal.ppat.1003820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moorman NJ, Sharon-Friling R, Shenk T, Cristea IM. 2010. A targeted spatial-temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol Cell Proteomics 9:851–860. doi: 10.1074/mcp.M900485-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]