

ABSTRACT
Kaposi's sarcoma-associated herpesvirus (KSHV) infects many target cells (e.g., endothelial, epithelial, and B cells, keratinocytes, and monocytes) to establish lifelong latent infections. Viral latent-protein expression is critical in inducing and maintaining KSHV latency. Infected cells are programmed to retain the incoming viral genomes during primary infection. Immediately after infection, KSHV transcribes many lytic genes that modulate various cellular pathways to establish successful infection. Analysis of the virion particle showed that the virions contain viral mRNAs, microRNAs, and other noncoding RNAs that are transduced into the target cells during infection, but their biological functions are largely unknown. We performed a comprehensive analysis of the KSHV virion packaged transcripts and the profiles of viral genes transcribed after de novo infections of various cell types (human peripheral blood mononuclear cells [PBMCs], CD14+ monocytes, and telomerase-immortalized vascular endothelial [TIVE] cells), from viral entry until latency establishment. A next-generation sequence analysis of the total transcriptome showed that several viral RNAs (polyadenylated nuclear RNA, open reading frame 58 [ORF58], ORF59, T0.7, and ORF17) were abundantly present in the KSHV virions and effectively transduced into the target cells. Analysis of the transcription profiles of each viral gene showed specific expression patterns in different cell lines, with the majority of the genes, other than latent genes, silencing after 24 h postinfection. We differentiated the actively transcribing genes from the virion-transduced transcripts using a nascent RNA capture approach (Click-iT chemistry), which identified transcription of a number of viral genes during primary infection. Treating the infected cells with phosphonoacetic acid (PAA) to block the activity of viral DNA polymerase confirmed the involvement of lytic DNA replication during primary infection. To further understand the role of DNA replication during primary infection, we performed de novo PBMC infections with a recombinant ORF59-deleted KSHV virus, which showed significantly reduced numbers of viral copies in the latently infected cells. In summary, the transduced KSHV RNAs as well as the actively transcribed genes control critical processes of early infection to establish KSHV latency.
IMPORTANCE Kaposi's sarcoma-associated herpesvirus (KSHV) is the causative agent of multiple human malignancies in immunocompromised individuals. KSHV establishes a lifelong latency in the infected host, during which only a limited number of viral genes are expressed. However, a fraction of latently infected cells undergo spontaneous reactivation to produce virions that infect the surrounding cells. These newly infected cells are primed early to retain the incoming viral genome and induce cell growth. KSHV transcribes a variety of lytic proteins during de novo infections that modulate various cellular pathways to establish the latent infection. Interestingly, a large number of viral proteins and RNA are encapsidated in the infectious virions and transduced into the infected cells during a de novo infection. This study determined the kinetics of the viral gene expression during de novo KSHV infections and the functional role of the incoming viral transcripts in establishing latency.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV), also called human herpesvirus 8 (HHV8), is a double-stranded DNA virus that causes Kaposi's sarcoma, primary effusion lymphomas, and multicentric Castleman's disease (1–3). Like other herpesviruses, KSHV exhibits both latent and lytic modes of infection, persisting predominantly in the latent state in which only a subset of the viral proteins are expressed, including the latency-associated nuclear antigen (LANA) protein (4–8). Although the expression of latent proteins plays a critical role in inducing and maintaining KSHV latency, the infected cells are primed early during the primary infection to retain the viral genomes and induce tumors (9). During the primary infection, KSHV undergoes a short lytic replication cycle that transcribes an array of viral genes, which have been shown to modulate various pathways for establishing the latent infection (9). In addition, a small fraction (1 to 5%) of the infected cells spontaneously undergo lytic reactivation to produce infectious virions, which is likely to be essential for increasing the population of infected cells and inducing viral pathogenesis (10–13).
The infection of target cells with KSHV is a complex multistep process involving a variety of host cell surface receptors and multiple viral glycoproteins. Irrespective of its mechanism of entry, for a successful infection, KSHV must overcome the obstacles it encounters during the transportation of viral capsids from the plasma membrane into the nucleus. The main obstacles include apoptosis triggered by the virus's binding and entry, autophagy, and the induction of various intrinsic, innate, and adaptive immune responses (14, 15). The mechanisms by which KSHV successfully circumvents these obstacles are beginning to be resolved. During de novo infections, KSHV generally establishes latency by 24 h postinoculation (hpi) in cell culture systems (14, 16–20). However, very early, immediately after a de novo infection, KSHV undergoes a limited initial burst of lytic transcript accumulation (14). At this point, the viral gene expression shows a more complex pattern, wherein the latent and lytic genes are expressed concurrently, with an initial moderate proportion of lytic transcripts followed by the onset of accumulation of more latent transcripts (14).
It has been shown that another gamma herpesvirus, Epstein-Barr virus (EBV), exhibits a similar pattern during early infection. The primary infection of B cells by EBV shows an expression of lytic genes (21–23). The expression of the lytic genes before the latent genes during early infection suggests that this initial expression of lytic genes is important for the successful establishment of EBV latency (21, 22). It has also been shown that EBV viral particles contain mRNAs and other nonstructural RNAs that are transduced into the target cells early during infection (22). These mRNAs play crucial roles during infection to establish the EBV latency (22). The detection of lytic transcripts early during de novo KSHV infections, followed by the latent transcripts, suggests that a mechanism similar to that of EBV may be occurring during the primary infection by KSHV. Additionally, the concurrent expression of open reading frame 73 (ORF73) and ORF50 transcripts, detected early during KSHV infection at 2 and 4 hpi (9), respectively, indicates that these transcripts might be transported into the newly infected cell during the virus entry and are involved in altering the cellular environment prior to transcription from the incoming viral genome.
Indeed, several recent studies have shown that herpesviruses, including KSHV particles, contain a variety of viral proteins (24, 25) and diverse RNA species such as viral mRNA, noncoding RNA, viral and cellular microRNAs (miRNAs), and unusual small RNA (usRNA) (26, 27). These RNAs are selectively packaged during virion budding and released into the target cells during de novo infection, and they have been shown to be biologically functional (27). The KSHV ORF59 transcript is also present in the virions and translated very early during primary infection (27). Recent studies have similarly shown that several of the viral and cellular miRNAs are selectively packaged into the virions and released into the target cells during infection (26). However, the exact composition of the virion transcripts in the KSHV particles and their functions remain unclear (27). A recent study reported a higher transient expression of lytic genes at 24 hpi in comparison to latently infected cells. Moreover, the KSHV genome undergoes rapid chromatinization following infection, indicating that the initial burst of lytic gene transcription likely originates from transcriptionally permissive chromatin rather than from incoming naked DNA (28).
This study aimed to analyze the viral genes transcribed early during the primary infection of peripheral blood mononuclear cells (PBMCs), CD14+ and telomerase-immortalized vascular endothelial (TIVE) cells. To differentiate the RNAs present in the virion and transcribed early during primary infection, noninternalized virions were removed from the cell surface by treating the cells with trypsin at 2 hpi, followed by capturing the nascently transcribing RNA after 4 and 24 hpi. Also, the cells were collected at 0, 4, 24, 48, 72, 96, and 120 hpi to extract total RNA for transcriptomic profiling and real-time quantitative PCR (qPCR) analysis. The encapsulated RNA in the virions was determined by RNA-sequencing (RNA-seq) analysis as well as by reverse transcription-quantitative real-time PCR (RT-qPCR) for all of the viral genes. We captured newly synthesized RNA with Click-iT technology during the de novo PBMC infections to identify the actively transcribing genes. The treatment of KSHV-infected human PBMCs with phosphonoacetic acid (PAA) to block the viral DNA polymerase activity determined the involvement of lytic DNA replication during primary infection. To understand the functional significance of the lytic gene transcription during KSHV infection and the establishment of latency, we investigated the role of ORF59, an abundantly packaged mRNA in the virions that is transcribed during primary infection. The infectious recombinant KSHV BAC36WT and its ORF59 deletion mutant (BAC36ΔORF59) were used for de novo PBMC infections. The infected cells showed that virions from the ΔORF59 cells were not able to attain as high a level of latent genomic copies as the wild-type (WT) BAC36 virus, suggesting a defect in the lytic DNA replication. Overall, our data show that a large number of virion transcripts are transduced into the host cells along with the viral genome during infection of the target cells, which helps in amplifying the viral genome copies and latency establishment.
MATERIALS AND METHODS
Cell culture, plasmids, and antibodies.
The KSHV-positive cell line TRExBCBL1-RTA, provided by Jae Jung (University of Southern California), was cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 5 U/ml penicillin, 5 μg/ml streptomycin, and 20 μg/ml hygromycin B. Cells (293L) harboring either BAC36WT or BAC36ΔORF59 were cultured in Dulbecco modified Eagle medium supplemented with 10% FBS, 2 mM l-glutamine, 5 U/ml penicillin, 5 μg/ml streptomycin, and 50 μg/ml hygromycin B. Telomerase-immortalized vascular endothelial (TIVE) cells, kindly provided by Erle S. Robertson, were cultured in endothelial basal medium-2 (Lonza) supplemented with endothelial growth factor supplements (Lonza). Human PBMCs (ReachBio, WA) were cultured in RPMI 1640 medium (HyClone) supplemented with 10% FBS, 2 mM l-glutamine, and penicillin-streptomycin (5 U/ml and 5 μg/ml, respectively). Human CD14+ cells were isolated from cord blood received from the Colorado Cord Blood Bank (University of Colorado). Cells were maintained in Iscove modified Dulbecco medium (HyClone), supplemented with 20% heat-inactivated FBS (HyClone), 50 ng/ml macrophage colony-stimulating factor (M-CSF), 50 ng/ml stem cell factor, 50 ng/ml granulocyte CSF (G-CSF), 50 ng/ml GM-CSF, and 50 ng/ml interleukin-3 (R&D Systems) at a density of 1 × 106 cells/ml on a low-cell-binding plate (Nunc Hydrocell). Protocols to obtain blood-associated cells were approved by the Institutional Review Board and Office of Human Research Protection at the University of Nevada, Reno. The plasmid pA3F-LANA, carrying a Flag-tagged ORF73, has been previously described (29, 30). Commercially available mouse anti-K8.1 and rat anti-LANA antibodies (Advanced Biotechnologies, Inc.), were used in this study.
KSHV virion purification.
KSHV virions were purified as previously described (26, 27). Approximately 100 million TRExBCBL1-RTA cells were induced with doxycycline (1 μg/ml) for 5 days, after which the culture supernatant was collected, centrifuged at 4,000 rpm for 10 min, and filtered through a 0.45-μm filter to remove cellular debris before concentrating the virus by centrifugation at 25,000 rpm for 2 h at 4°C. The concentrated virions were resuspended in 1 ml phosphate-buffered saline (PBS), layered onto a 30 to 50% sucrose gradient, and centrifuged at 70,000 × g for 2 h. The purified virions formed a white opaque ring, which was collected in a syringe by puncturing the tube with the needle at the band site. The collected band was diluted with PBS before centrifuging for 2 h at 25,000 × g to pellet the virions.
Western blotting.
The purified virus particles were resuspended in 100 μl of lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 0.25% sodium deoxycholate, and 1% NP-40) supplemented with protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 10 μg/ml pepstatin, 10 μg/ml leupeptin, and 10 μg/ml aprotinin). The lysates were centrifuged at high speed and mixed with protein sample buffer, and the protein was resolved by 4 to 15% SDS-PAGE and Western blotted using standard protocols (Bio-Rad Laboratories). The KSHV-encoded structural glycoprotein K8.1 was detected using mouse anti-K8.1 antibody followed by incubation with infrared dye-tagged (IR680 and IR800) secondary antibodies and scanning with an Odyssey infrared scanner (Li-Cor Biosciences, Lincoln, NE).
KSHV de novo infection.
Approximately 8 × 107 human PBMCs were infected with KSHV purified from the induced TRExBCBL1-RTA cells. Two hours after infection at 37°C in the presence of 8 μg/ml Polybrene, the cells were washed once with 0.005% trypsin in PBS and three times with PBS to remove the loosely bound virions, after which they were resuspended and maintained in RPMI 1680 medium until harvesting. The de novo KSHV-infected PBMCs (∼5 × 106) were collected at different time points, washed twice with PBS, and processed separately to isolate the DNA and RNA.
Indirect immunofluorescence microscopy.
At 120 hpi, KSHV-infected PBMCs were washed with PBS, spread evenly on coverslips, and air dried. The cells were fixed for 10 min at room temperature with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 in PBS for 10 min, also at room temperature. The cells were blocked by incubating in PBS containing 0.4% fish skin gelatin and 0.05% Triton X-100. The fixed cells were then incubated with primary rat anti-LANA antibody for 1 h at room temperature, washed with PBS, incubated with Alexa Fluor 488 secondary antibody (Molecular Probes) for 45 min at room temperature, and washed with PBS. The nuclear stain TO-PRO-3 (Molecular Probes) was used for counterstaining the nucleus. Images were obtained using a laser scanning confocal microscope (Carl Zeiss, Inc.).
Flow cytometry.
PBMCs de novo infected with KSHV were harvested at 120 hpi and fixed in Streck tissue fixative (STF) (Streck Laboratories) for 30 min. The fixed cells were washed twice in 1× PBS, permeabilized with 0.2% Triton X-100, blocked with 0.4% fish skin gelatin, and incubated with fluorescently tagged mouse anti-CD19 (Alexa Fluor 488), mouse anti-CD3 (Alexa Fluor 488; Rockland Immunochemicals, Inc.), and rat anti-LANA (Advanced Biotechnologies, Inc.), which was detected with a secondary antibody conjugated to Alexa Fluor 555 (Rockland Immunochemicals, Inc.). The data were acquired on a FACSCalibur flow cytometer equipped with CellQuest Pro software and analyzed using FlowJo software.
Viral genome extraction and quantification.
De novo-infected cells were collected by centrifugation (∼2 × 106 cells per sample) and washed twice with PBS before extracting the total DNA using a modified Hirt lysis method (31). The PCR primers used for the KSHV genome quantification were selected from the ORF73 gene as previously described (9, 32). Twofold serial dilutions of the pA3F-LANA plasmid were used as the template in qPCRs to produce a standard curve for the quantifications. The extracted total DNA was resuspended in 50 μl sterile water, and a 5-μl aliquot of the DNA was used for the qPCR amplification of the KSHV-ORF73-specific sequence. The viral DNA copy numbers were calculated with reference to the standard curve.
RNA preparation and sequencing.
The RNA-seq of the KSHV virions and de novo-infected PBMCs was performed on a HiSeq next-generation sequencer (Illumina, Inc.). Total RNA was isolated from the purified KSHV virions after treating them with micrococcal nuclease (NEB) for 30 min at 37°C and terminating the reaction with EGTA. The total RNA was isolated with TRIzol reagent per the manufacturer's instructions (Life Technologies), and viral DNA was eliminated by treating with DNase I (GE Health Care Life Sciences), which was subsequently inactivated. The total RNA from the de novo-infected PBMCs was prepared using an Illustra RNAspin minikit with an in-column DNase treatment per the manufacturer's instructions (GE Health Care Life Sciences). The concentration and purity of the extracted RNA were determined with a NanoDrop 2000c spectrophotometer (NanoDrop Technologies). A TrueSeq RNA sample preparation kit v2 (Illumina, Inc.) was used to prepare cDNA libraries for the RNA-seq according to the manufacturer's instructions. The fragment sizes and purity of the mature libraries were confirmed by analyzing on a Bioanalyzer 2100 (Agilent Technologies). The quantities of the libraries required for RNA-seq were determined by real-time qPCR using a KAPA library quantification kit for the Illumina platform (Kapa Biosystems). The libraries were sequenced using HiSeq (Illumina), and the sequences were mapped to the KSHV reference genome (accession number NC_009333) using the RNA-seq analysis tool in the CLC Genomic Workbench 7 (CLC Bio) software. The relative expression of the viral genes at different time points was determined based on RPKM (reads per kilobase of exon per million mapped reads) values, which were also similarly used to generate heat maps showing the relative expression of those genes at different time points utilizing the hierarchical clustering feature of the CLC Genomic Workbench 7 software.
Nascent RNA capture by Click-iT technology.
To identify the newly synthesized RNA during the de novo infections, a Click-iT RNA capture technology was used as described by the manufacturer (Life technologies, Inc.). Briefly, 8 × 107 human PBMCs were infected with KSHV purified from the induced TRExBCBL1-RTA cells as described above. At 4 hours postinfection, the cells were incubated for 1 h with ethylene uridine (EdU) (33) ribonucleotide homologs containing an alkyne-reactive group. Additionally, a de novo infection of KSHV was performed in the presence of phosphonoacetic acid (PAA), an inhibitor of viral DNA polymerase and lytic DNA replication. The PBMCs were pretreated with 0.5 mM PAA for 1 h prior to the infection, with the treatment continuing until harvesting at 24 hpi, similarly to a previously described method (34, 35). The de novo-infected PBMCs were collected 4 and 24 hpi and washed twice with PBS before isolating total RNA with an Illustra RNAspin minikit per the manufacturer's instructions (GE Health Care). Biotin azide was “clicked on” to the isolated RNA, and the newly synthesized RNA was captured using streptavidin magnetic beads. Dimethyl sulfoxide (DMSO) instead of biotin azide was added as control for the click reaction. The captured RNAs were further utilized for preparing cDNA libraries with a TrueSeq RNA sample preparation kit v2 (Illumina, Inc.) and for the RNA-seq analysis on a HiSeq Illumina sequencer. Relative numbers of copies of the newly synthesized transcripts were determined by the ratios of the biotin-enriched (clicked) versus control, DMSO-enriched (nonclicked) copies of the transcripts identified by RNA sequence analysis.
qPCR.
The real-time quantitative PCRs (qPCRs) were performed in a 96-well plate in a total volume of 20 μl that included 10 μl of SYBR green PCR 2× master mix (Applied Biosystems) and 0.5 μM each KSHV ORF-specific primer. Primers for the human β-actin and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) housekeeping genes were included for normalizing the threshold cycle (CT) values. Purified genomic DNA samples or the virion cDNA samples and KSHV-infected PBMCs were amplified on an ABI StepOnePlus real-time PCR machine (Applied Biosystems). As a standard practice, a no-reverse-transcription (no-RT) control reaction was included for all of the prepared RNA samples to ensure their purity.
Microarray data accession number.
The RNA-seq data were deposited in the NCBI Gene Expression Omnibus (GEO) database, and the accession number for the complete data set is GSE62344.
RESULTS
Purification of the KSHV virions.
We isolated the KSHV particles from doxycycline-induced (1 μg/ml) TRExBCBL1-RTA cells and purified them on a 20 to 50% sucrose gradient. The three white bands observed on the sucrose gradient (Fig. 1A) were consistent with previous reports (26, 36), representing the A-type (empty virus particles), B-type (intermediate virus particles), and C-type (mature virions) KSHV particles. The lower band representing the mature C-type KSHV virion particles was collected with a syringe and diluted before pelleting the virions by ultracentrifugation. These purified virions were tested for the presence of the KSHV envelope glycoprotein K8.1 by immunoblotting with anti-K8.1 antibody (Fig. 1B). The virion DNA was extracted to quantify the number of viral copies in a real-time qPCR assay using ORF73-specific primers (Fig. 1C), which showed approximately 8.0 × 107 virions/ml. This fraction of mature virions, which was confirmed by both analyses, was further used for de novo infections of the indicated target cells.
FIG 1.
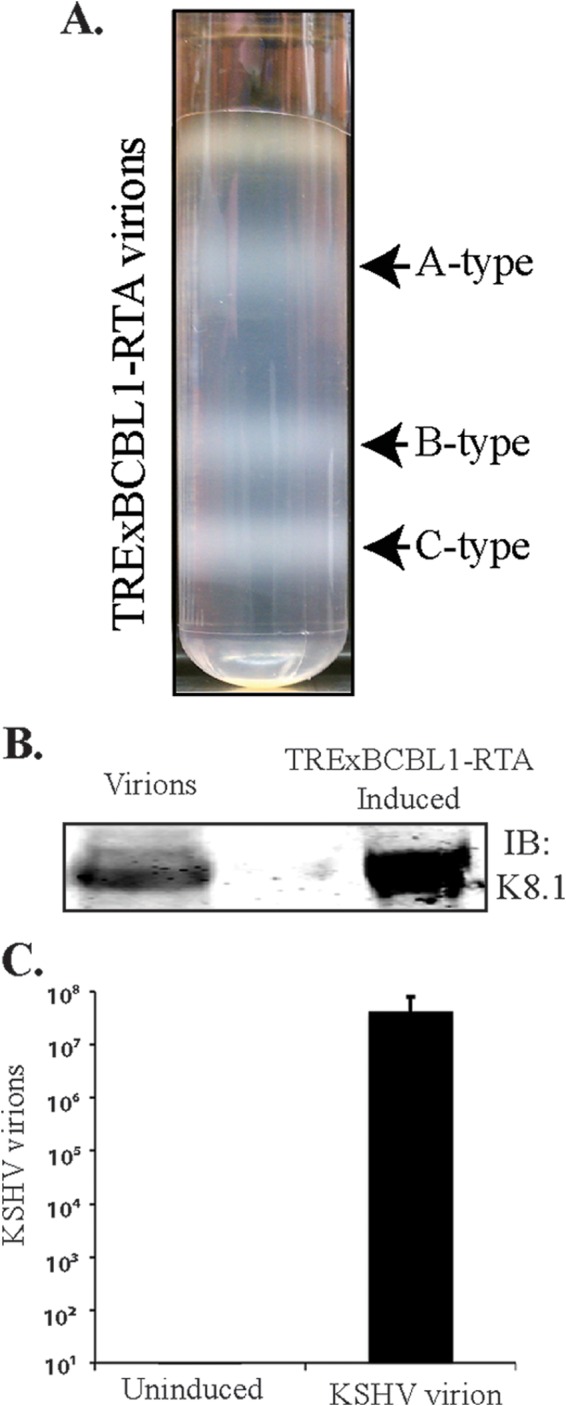
Purification of KSHV virions. (A) Virions were purified from the culture supernatant of approximately 9 × 108 reactivated TRExBCBL1-RTA cells by a20 to 50% sucrose gradient ultracentrifugation. The three white bands as indicated represent A-type (empty), B-type (intermediate), and C-type (mature) KSHV virus particles. (B) Western blotting of purified virions for the presence of KSHV envelope glycoprotein K8.1 by immunoblotting with anti-K8.1 antibody. Total lysate from induced TRExBCBL1-RTA cells was used as a positive control. (C) Supernatants of uninduced or induced TRExBCBL1-RTA cells were used for real-time qPCR quantification of viral genome copy using ORF73-specific primers along with ORF73 plasmid standards. Approximately 8.06 × 107 copies of viral DNA representing C-type virions were detected.
KSHV viral transcripts are abundantly present during the primary infection of human PBMCs, CD14+, and TIVE cells.
To determine the profiles of the viral transcripts expressed during the primary infection of B and endothelial cells (natural target cells of KSHV), purified virions were used for the de novo infection of human PBMCs, CD14+ monocytes, and endothelial (TIVE) cells. An RNA-seq analysis was performed on total RNA extracted from these infected cells at different time points (4, 24, 48, 72, and 120 hpi). Furthermore, 10 pM concentrations of the quantified libraries were sequenced and mapped to the reference KSHV genome (NC_009333). The transcriptome analysis revealed that several of the KSHV lytic and latent transcripts accumulated as early as 4 hpi (Fig. 2A to C and 3). Apart from the ORF50 replication and trans-activator protein (RTA), a number of other lytic transcripts involved in immune modulation, lytic DNA replication, and nucleic acid processing/synthesis were detected in significant amounts early during the infection. These included polyadenylated nuclear (PAN) RNA, ORF58/59, kaposin B, K2, K4, K6, ORF11, ORF17, ORF45, ORF27, ORF37, ORF57, ORF64, ORF65, ORF73, and the recently identified T0.7. Among these, PAN RNA was the most abundant as determined by the relative peak height (Fig. 2A to C) and RPKM values (Fig. 3) in all the three tested cell types. Although the overall patterns of the viral gene expressions were similar, i.e., with a majority of the genes being expressed at 24 hpi in all the three tested cell types, CD14+ monocytes showed complete silencing of viral gene expressions, other than for LANA, after 24 hpi (Fig. 2A to C and 3). However, the human PBMCs and TIVE cells showed detectable levels of PAN RNA and ORF58/59 transcripts along with LANA after 48 hpi (Fig. 2A to C and 3). This suggests cell type-specific viral gene expression patterns during primary de novo infection and latency establishment. The relative abundance of many of the viral transcripts, including ORF1, K2, K4, T1.5, PAN RNA, ORF17.5, ORF26, ORF27, ORF37 ORF38, ORF45, K8, K8.1, ORF52, vIRF-1, ORF58/59, K12/T0.7, and LANA, showed an increase in PBMCs at 4 hpi and 24 hpi compared with their abundance in the virions (Fig. 2A). However, the CD14+ monocyte cells showed increases in the abundance of fewer genes compared to those in the PBMCs (Fig. 2B). Importantly, LANA was the only gene abundantly detected after 72 hpi in these CD14+ cells, representing a true latency model of KSHV infection. The endothelial (TIVE) cells showed detectable levels of viral transcripts, but their relative abundance increased only slightly at 24 hpi (Fig. 2C). The heat maps generated based on the RPKM values showed an increase in their copies from 4 hpi to 24 hpi, suggesting an active transcription, and these included PAN RNA, ORF73, ORF45, ORF58, T0.7, Kaposin B, ORF59, K4, K6, ORF17, ORF65, ORF27, K8.1, ORF11, ORF37, ORF38, ORF57, ORF69, ORF52, ORF54, and T1.5 (Fig. 3). The viral DNA polymerase ORF9, along with other lytic DNA replication proteins, was detected, but their expression levels did not increase (Fig. 3). Also, there were several other detectable transcripts (K1, ORF4, ORF10, K6, ORF43, ORF44, ORF67, ORF68, and K14), but their abundance did not increase, and the RPKM values remained unchanged during the time course tested, suggested that these genes were not transcribed but were present due to their transduction through the virion particles during infection.
FIG 2.



(A) Transcriptome analysis of KSHV during de novo infection of human PBMCs. Total RNAs extracted from de novo-infected PBMCs harvested at 4 h, 24 h, 48 h, 72 h, and 120 h postinfection were subjected to cDNA library preparation using a TrueSeq RNA-seq library kit. The libraries were sequenced using HiSeq, and the sequences were mapped to the reference KSHV genome to determine the relative abundance of viral transcripts. Purified virions treated with micrococcal nuclease to eliminate nucleic acid contamination were subjected to total RNA extraction. RNA treated with DNase was used for sequencing after cDNA library preparation. The number of transcripts mapping to the viral genome is shown in parentheses in each panel. The peak height represents the number of reads for the indicated genes, and the predominant peaks are marked in virion RNA-seq and 4-h-postinfection samples. PAN RNA showed the highest peak in virions as well as KSHV-infected PBMCs. The bottom panel shows the locations of KSHV genes on the coordinates. (B) Transcriptome analysis of de novo-infected CD14+ cells. Total RNAs extracted from de novo-infected CD14+ cells harvested at 4 h, 24 h, 48 h, 72 h, and 96 h postinfection were subjected to cDNA library preparation using a TrueSeq RNA-seq library kit. The libraries were sequenced using HiSeq, and the sequences were mapped to the reference KSHV genome to determine the relative abundance of viral transcripts. The number of transcripts mapping to the viral genome is shown in parentheses in each panel. The peak heights representing the number of reads for the indicated genes are marked in 4-h-postinfection and 24-h-postinfection samples. PAN RNA showed the highest peak until 48 h postinfection, followed by a decline to very low levels. LANA expression progressively increased, showing establishment of latency. The bottom panel shows the locations of KSHV genes on the coordinates. (C) Transcriptome analysis of de novo-infected TIVE cells. Total RNAs extracted from de novo-infected TIVE cells harvested at 4 h, 24 h, 48 h, 72 h, and 120 h postinfection were subjected to cDNA library preparation using a TrueSeq RNA-seq library kit. The libraries were sequenced using HiSeq, and the sequences were mapped to the reference KSHV genome to determine the relative abundance of viral mRNA. The numbers of transcripts mapping to the viral genome are shown in parenthesis on each panel. The peak heights representing the number of reads for the indicated genes are marked in 4-h-postinfection and 24-h-postinfection samples. The sample from 72 h postinfection showed lower peaks because of the lower number of total reads, which were normalized by calculating the RPKM values in the heat maps. The bottom panel shows the locations of KSHV genes on the coordinates.
FIG 3.
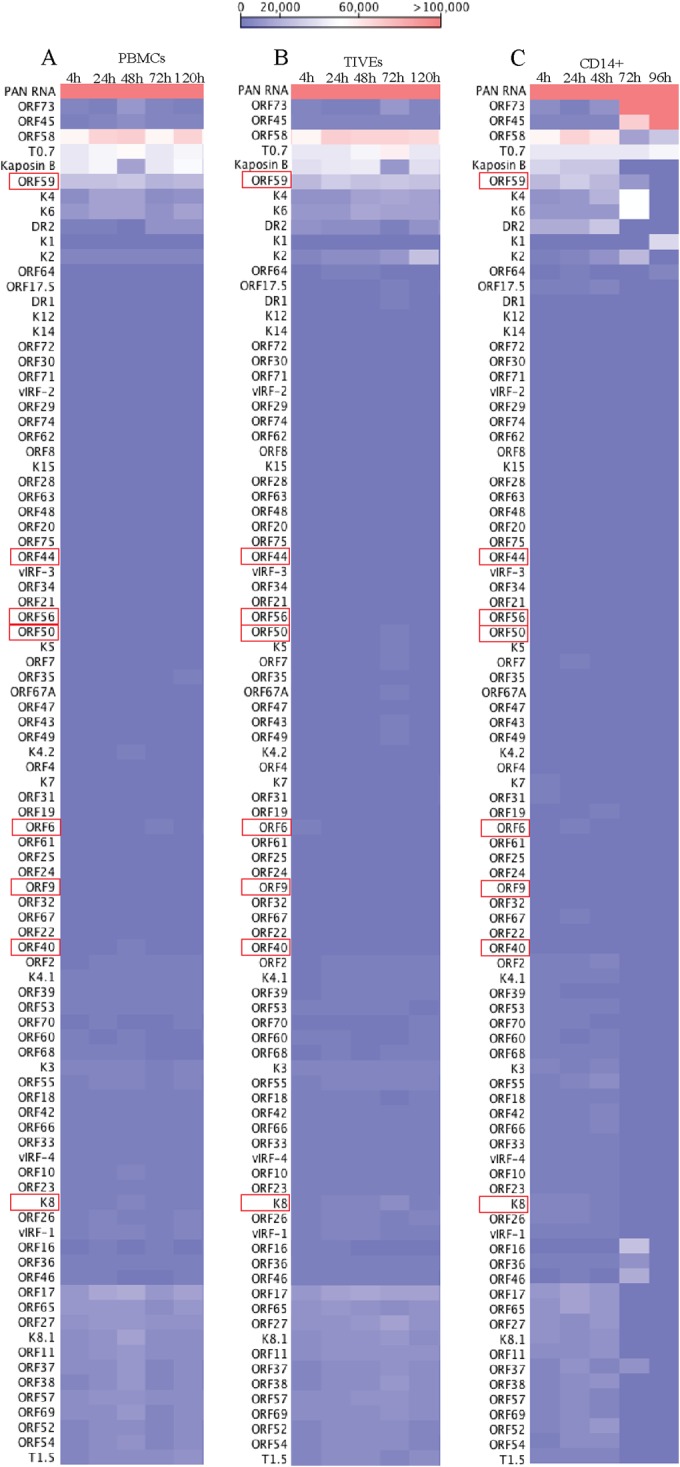
Transcriptome analysis of KSHV in de novo-infected PBMCs, TIVE cells, and CD14+ cells. RPKM values, calculated based the number of reads for each gene, were used for analyzing relative expression of KSHV genes as heat maps. Hierarchal clustering of genes was performed using CLC Workbench 7.0. Increasing RPKM values are represented from blue to red. (A) KSHV transcriptome from de novo-infected human PBMCs. (B) KSHV transcriptome from de novo-infected human TIVE cells. (C) KSHV transcriptome from de novo-infected human CD14+ cells.
Earlier studies reported that immediately after de novo infection, the herpesviruses express genes involved in lytic DNA replication (21, 28, 37). Our RNA-seq analysis detected the transcripts of proteins involved in viral DNA replication as early as 4 hpi, and these included ORF59 (processivity factor), ORF9 (DNA polymerase), ORF6 (single-stranded DNA binding protein), ORF56 (DNA replication protein), ORF40 (primase-associated factor), and ORF54 (dUTPase), along with ORF60 and -61 (ribonucleoprotein reductase), ORF70 (thymidylate synthase), and ORF37 (alkaline exonuclease). Apart from these transcripts, several other transcripts, encoding the viral structural proteins ORF8 (glycoprotein B), K8.1 (glycoprotein), and ORF64 and -75 (tegument protein), were also detected in our transcription profiling data. Moreover, many of the noncoding RNAs, including PAN RNA, T0.7, and T1.5 (OriLyt transcript), were abundantly detected in the RNA-seq analysis. RNA-seq profiling of the virion encapsidated RNA, as relative abundance, showed packaging of a large number of viral transcripts, which are brought into the target cells by the KSHV virion during the de novo infection (Fig. 2 and Table 1).
TABLE 1.
mRNAs detected in purified TRExBCBL1-RTA virions by RNA-seq analysisa
| No. | mRNA | RPKM value | Total gene reads | Function | Transcription 4 h after de novo infection of human PBMCs | Source or reference |
|---|---|---|---|---|---|---|
| 1 | ORF4 | 2,633.55 | 101 | Complement control protein (KCP), regulates complement activation | This study | |
| 2 | ORF6 | 3,804.2 | 300 | Single-stranded DNA binding protein essential for lytic DNA replication | This study | |
| 3 | ORF7 | 2,332.60 | 113 | Probably DNA packaging | This study | |
| 4 | ORF8 | 4,432.43 | 261 | Virion envelope-associated glycoprotein | This study | |
| 5 | ORF9 | 5,318.55 | 375 | KSHV DNA polymerase | This study | |
| 6 | ORF10 | 6,446.37 | 188 | Specific suppressor of interferon signaling | This study | |
| 7 | ORF11 | 6,275.82 | 191 | Viral lytic protein | Yes | This study |
| 8 | K2 | 7,218.64 | 103 | K2 (viral interleukin-6), regulates cellular proliferation | Yes | 27 |
| 9 | K3 | 6,004.86 | 135 | E3 ubiquitin ligases that reduce surface expression in infected cells | This study | |
| 10 | ORF70 | 5,568.35 | 131 | Homolog of viral thymidylate synthase | This study | |
| 11 | K4.2 | 4,632.04 | 59 | This study | ||
| 12 | T1.5 | 10,245.93 | 358 | This study | ||
| 13 | K6 | 10,027.11 | 67 | vMIP-IA | 27 | |
| 14 | PAN RNA | 159,519.90 | 3,986 | Late gene expression, lytic DNA replication, immune modulation | 27 | |
| 15 | ORF17 | 13,534.71 | 504 | Yes | 27 | |
| 16 | ORF18 | 2,840.02 | 51 | Late gene regulation | This study | |
| 17 | ORF19 | 3,134.66 | 120 | This study | ||
| 18 | ORF21 | 3,140.50 | 127 | Thymidine kinase | This study | |
| 19 | ORF22 | 1,729.56 | 88 | Glycoprotein H | This study | |
| 20 | ORF23 | 2,305.84 | 65 | Predicted glycoprotein | This study | |
| 21 | ORF24 | 2,823.83 | 148 | Essential for DNA replication | This study | |
| 22 | ORF25 | 3,338.78 | 320 | Major capsid protein | This study | |
| 23 | ORF26 | 3,239.66 | 69 | Minor capsid protein | This study | |
| 24 | ORF27 | 6,270.22 | 127 | Glycoprotein | Yes | This study |
| 25 | ORF29 | 3,163.27 | 390 | Packaging protein | This study | |
| 26 | ORF34 | 2,233.92 | 51 | This study | ||
| 27 | ORF35 | 5,613.67 | 59 | This study | ||
| 28 | ORF36 | 4,907.44 | 152 | Serine protein kinase | Yes | This study |
| 29 | ORF37 | 2,625.62 | 89 | Alkaline exonuclease Sox protein | Yes | This study |
| 30 | ORF39 | 2,149.70 | 60 | Glycoprotein M | This study | |
| 31 | ORF40 | 2,904.36 | 144 | Helicase/primase | This study | |
| 32 | ORF44 | 2,676.778 | 147 | Helicase | This study | |
| 33 | ORF45 | 6,972.31 | 198 | Virion-associated tegument protein regulating IFN function | Yes | This study |
| 34 | ORF46 | 7,969.30 | 142 | Uracil deglycosylase | This study | |
| 35 | ORF48 | 3,101.60 | 87 | Activates JNK/p38 | This study | |
| 36 | ORF50 | 4,389.71 | 309 | Replication transcriptional activator protein | This study | |
| 37 | K8 | 4,072.23 | 89 | bZIP | Yes | 27 |
| 38 | K8.1 | 3,918.32 | 71 | Glycoprotein | Yes | This study |
| 39 | ORF52 | 6,095.17 | 56 | Tegument protein | Yes | This study |
| 40 | ORF54 | 9,659.02 | 199 | dUTPase/immune modulation | 27 | |
| 41 | ORF55 | 6,364.41 | 101 | Tegument protein | This study | |
| 42 | ORF56 | 3,830.11 | 225 | DNA replication | This study | |
| 43 | ORF57 | 3,533.39 | 121 | Lytic replication protein homologous to EBV SM protein, mRNA splicing | Yes | This study |
| 44 | vIRF-1 | 7,407.08 | 232 | Immune modulation | Yes | This study |
| 45 | vIRF-4 | 2,324.47 | 153 | Facilitates lytic replication by targeting cellular IRF4 and Myc gene | Yes | This study |
| 46 | vIRF-3 | 2,302.58 | 96 | Immune modulation | This study | |
| 47 | vIRF-2 | 2,429.94 | 122 | Type I interferon signaling | This study | |
| 48 | ORF58 | 16,775.11 | 418 | Yes | 27 | |
| 49 | ORF59 | 26,201.13 | 724 | Processivity factor | Yes | 27 |
| 50 | ORF60 | 4,601.25 | 98 | Ribonucleoprotein reductase | This study | |
| 51 | ORF61 | 3,659.74 | 202 | Ribonucleoprotein reductase | This study | |
| 52 | ORF63 | 2,891.99 | 187 | NLR homolog | This study | |
| 53 | ORF64 | 3,897.02 | 715 | Deubiquitinase | Yes | This study |
| 54 | ORF65 | 5,629.25 | 67 | Capsid protein | This study | |
| 55 | ORF66 | 5,646.64 | 169 | Capsid protein | This study | |
| 56 | ORF67 | 4,753.85 | 90 | Nuclear egress complex | This study | |
| 57 | ORF68 | 5,372.34 | 175 | Glycoprotein | This study | |
| 58 | ORF69 | 6,353.81 | 134 | BRLF2 nuclear egress | Yes | This study |
| 59 | T0.7 | 9,310.93 | 151 | This study | ||
| 60 | ORF72 | 3,118.46 | 56 | vCyclin | This study | |
| 61 | ORF73 | 23,623.23 | 1,858 | Latency-associated nuclear antigen | This study | |
| 62 | K14 | 3,116.41 | 59 | vOX2 | This study | |
| 63 | ORF74 | 4,481.89 | 107 | vGPCR | This study | |
| 64 | ORF75 | 3,810.57 | 344 | Tegument protein vFGARAT | Yes | This study |
Total gene reads above 50 and a qPCR relative fold change above 10 were considered significant.
In agreement with a previous report, our results showed the concurrent expression of ORF50 and ORF73 transcripts during early primary infection (Fig. 2A to C and 3). However, the expression of ORF50 decreased after 24 h, whereas the expression of ORF73 increased exponentially (Fig. 3). The KSHV latency-associated protein ORF73 is the predominant protein expressed during latency and is responsible for immune evasion, latent DNA replication, and genome maintenance, whereas the ORF50-encoded protein RTA activates lytic cycle-specific KSHV genes in a cascaded manner to facilitate the lytic replication process (38–42). In a cell culture system, the overexpression of RTA is capable of triggering the lytic DNA replication (38, 42). Our data detecting the transcripts of ORF50 and other lytic genes along with ORF73 during early infection suggest that KSHV may enter into a DNA replicative phase before establishing latent infection.
Real-time qPCR validation of the RNA sequencing analysis.
To further confirm the results of the transcriptome analysis by an independent method, we performed a real-time qPCR analysis on de novo-infected PBMCs harvested at different time points (4, 24, 48, 96, and 120 hpi). A custom qPCR array of the KSHV genes in a 96-well format (Bar Harbor Biotechnology) was used for the quantification of the viral mRNA levels. The fold changes for the individual KSHV genes were calculated for the various times postinfection using the ΔΔCT method and plotted separately (Fig. 4). The viral genes showed expression kinetics similar to those seen with the RNA-seq analysis. As first shown by the RNA-seq analysis, even at 4 hpi, several lytic transcripts (ORF58/59, ORF11, ORF8, K8, K8.1, ORF17, ORF22, ORF57, ORF 45, ORF27, ORF 37, and ORF64) were detected at high levels, and the expression of these transcripts was gradually depleted after 24 hpi (Fig. 4). In addition, latent transcripts (ORF73, K12, and ORF72) were consistently detected at significant levels during early infection (Fig. 4). As reported previously (9), the real-time qPCR analysis showed the concurrent expression of ORF50 and ORF73 at 4 hpi, and ORF50 showed a much higher fold increase during early infection than ORF 73. However, the expression of the ORF73 transcript increased exponentially until 96 hpi. Additionally, the noncoding PAN RNA and recently identified T0.7 transcripts were also detected abundantly at 4 hpi (Fig. 4). The qPCR validation of the RNA-seq analysis suggested that the limited lytic gene accumulation observed during primary infection might have been influenced by the RTA-mediated gene expression as well as by the transduction of viral transcripts with the virions during the de novo infection.
FIG 4.

Real-time qPCR validation of de novo-infected PBMCs. Real-time qPCR analysis was performed on the cDNA prepared from total RNA harvested at 4 h, 24 h, 48 h, 96 h, and 120 h postinfection of human PBMCs. A 96-well format qPCR plate representing each of the KSHV-specific ORF primers was used for the qPCR analysis. Fold changes of individual KSHV transcripts at various time points are plotted. The lytic genes are shown in red.
Detection of actively transcribing genes during de novo infection.
The transcriptome analysis of de novo-infected cells during early infection revealed a significantly higher expression of several genes which are expressed during the lytic replication cycle, including ORF50, ORF6, ORF9, ORF8, ORF7, ORF10, ORF11, ORF22, ORF27, ORF31, ORF37, ORF40/41, ORF45, ORF54, ORF55, ORF56, ORF58, ORF59, and ORF69 (Fig. 2A to C and 3). In addition, many of the immediate early unique KSHV genes, such as K7, K8, K3, K5, K9, and K12, as well as late K8.1, were also detected. Many of these transcripts have crucial regulatory roles in immune evasion and nucleic acid processing and synthesis, which are associated with lytic DNA replication. The real-time qPCR analysis of the viral transcripts fully corroborated the RNA-seq analysis (Fig. 4).
To further identify the actively transcribing genes during de novo infection, we used an approach to capture the nascent transcribed RNA from the KSHV-infected PBMCs by labeling them with EdU and isolating them by the Click-It approach. The sequencing of newly synthesized RNA captured from the KSHV-infected PBMCs showed peaks of many viral genes at 4 hpi, and these included ORF2, K3, T1.5, PAN RNA, ORF26, ORF29, ORF36, ORF37, ORF40, ORF50, K8.1, ORF55, vIRF-2, ORF58/59, ORF60, ORF63, ORF64, ORF67, T0.7, ORF72, ORF73, and ORF74 (Fig. 5). Importantly, the majority of these were showed increased expression in the graphs of relative abundance as well as in the heat maps (Fig. 2, 3, and 5). Interestingly, the majority of the active transcription was limited to ORF73, detected by the sequence reads, at 24 hpi as suggested for the beginning of latency establishment (Fig. 5). We also confirmed the transcription during de novo infection by treating the cells with actinomycin D to block active transcription. Comparison of the transcript levels at 4 hpi in the untreated actinomycin D-treated cells revealed profiles similar to those for the data for nascent RNA capture of gene transcription at 4 hpi (data not shown). As expected, the ORF50 gene showed a moderate level of de novo transcription within 4 hpi (Fig. 5). Interestingly, not all the genes required for lytic DNA replication showed active transcription during de novo infection, but they were detected in RNA-seq analysis, suggesting that those genes were transduced with the virions. Not surprisingly, the ORF50 gene transcripts and those of genes regulated by ORF50 decreased after 24 hpi. Also, a gradual increase in ORF73 transcripts by 24 hpi may aid in suppressing the expression of other viral genes to promote the establishment of latency.
FIG 5.
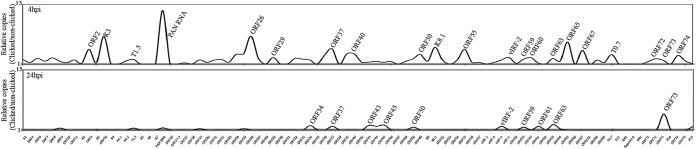
Viral genes transcribed at 4 hpi and 24 hpi, identified by a nascent RNA capture approach. Human PBMCs infected with KSHV virions were incubated with EdU (alkyne) at 4 hpi and 24 hpi to label the newly transcribing RNA. Total RNA extracted from these cells was subjected to click reaction with biotin-azide (Invitrogen, Inc.). DMSO instead of biotin-azide was used in control click reactions. Newly synthesized, biotin-labeled RNA was enriched by streptavidin magnetic beads as per the manufacturer's instructions. These enriched RNAs were subjected to RNA-seq analysis by HiSeq, and the sequences were mapped to the KSHV reference genome (NC_009333) using CLC Bio Workbench 7.5. The relative numbers of copies of the newly synthesized viral RNA were determined by ratios of specific (biotin-labeled) to nonspecific (DMSO) sequence reads.
KSHV genome copy numbers increase exponentially after de novo infection.
To investigate whether the detection of lytic reactivation genes leads to lytic DNA replication and genome amplification, we analyzed the KSHV genome copy number during the de novo infection of PBMCs. Total DNAs from different time points (0, 4, 24, 48, 72, 96, and 120 hpi) were subjected to a real-time qPCR analysis to analyze the relative numbers of viral copies in the infected cells. We used ORF73-specific primers and ORF73 plasmid standards to obtain the copy numbers of the KSHV genome. The relative number of KSHV genome copies was calculated in the real-time qPCR assay by amplifying the ORF73 gene and normalizing it against GAPDH. KSHV DNA was detected as early as 4 hpi, increased exponentially up to 48 hpi, and then slightly decreased over time after establishing latency (Fig. 6A). This exponential increase in the genome copies confirms genome replication during early infection until the establishment of latency. The punctate immunolocalization of LANA in the de novo-infected PBMCs (120 hpi) clearly demonstrated the establishment of latency in these cells (Fig. 6B). A flow cytometry analysis of de novo-infected PBMCs using anti-LANA and B- and T-lymphocyte markers gated for LANA expression showed that the populations of both the B (∼55.94%) and T (∼24.75%) lymphocytes were successfully infected with KSHV (Fig. 6C and D).
FIG 6.

KSHV genome copies exponentially increase after infection. (A) Approximately 8 × 107 human PBMCs were infected with KSHV isolated from reactivated TRExBCBL1-RTA, with a multiplicity of infection (MOI) of 10. De novo-infected PBMCs were harvested at 4 h, 24 h, 48 h, 72 h, 96 h, and 120 h postinfection, followed by extraction of total DNA using a modified Hirt lysis method. This DNA was used to analyze the genome copy number by real-time qPCR using ORF73-specific primers and ORF73 plasmid as a standard. (B) Indirect immunofluorescence assay (IFA) for LANA on de novo-infected PBMCs. KSHV-infected PBMCs at 120 h postinfection were subjected to IFA with rat anti-LANA antibody followed by detection with Alexa Fluor 488 secondary antibody. Nuclear stain TO-PRO-3 was used for staining the nuclei. LANA is shown in green, and nuclear stain TO-PRO 3 is shown in blue. (C and D) Flow cytometry analysis of de novo-infected PBMCs. KSHV-infected PBMCs at 120 h postinfection were subjected to flow cytometry analysis to detect the percentages of B and T lymphocytes infected with KSHV. Mouse anti-CD19 was used for the detection of B lymphocytes, and mouse anti-CD3 was used for the detection of T lymphocytes. Rat anti-LANA was used to gate KSHV-positive cells. Data were acquired on a FACSCalibur equipped with CellQuest Pro software and analyzed using FlowJo software. (E) PAA treatment reduces the expression of late genes. Untreated or PAA-pretreated human PBMCs were infected with KSHV virions (MOI of 10) for 4 h or 24 h. Total RNAs extracted at 4 hpi and 24 hpi were subjected to detection of selected viral transcripts by real-time qPCR, which showed significant reduction in ORF65 (late gene) expression with PAA treatment. (F) PAA treatment reduces viral genome copies in KSHV-infected human PBMCs. Untreated or PAA-treated human PBMCs infected with KSHV virions (MOI of 10) were subjected to genome copy analysis. Viral genome copies at different time postinfection were determined relative to the copies at 4 hpi.
Since the number of viral genome copies increased about 6-fold at 48 hpi compared to 4 hpi (the noninternalized virions were removed at 2 hpi by treating the cells with trypsin), we wanted to determine whether the virus had undergone latent or lytic modes of DNA synthesis to amplify the genome copies. To this end, we treated the cells with a viral DNA polymerase inhibitor, phosphonoacetic acid (PAA), to block the lytic DNA replication. Cells pretreated with PAA or untreated were infected with KSHV virions for 4 h and 24 h for the transcriptome analysis. Analysis of selected latent, immediate early, and early genes showed almost no effect of PAA on the transcription of latent (ORF73) and immediate early (ORF50) genes at 4 hpi (Fig. 6E). However, the expression of a late gene (ORF65) was significantly reduced with PAA at both 4 and 24 hpi (Fig. 6D). We also determined the number of genome copies during de novo infection of PBMCs in the presence of PAA. The relative number of genome copies showed a significant reduction in viral genome amplification in cells treated with PAA (Fig. 6F). Since there was still a slight increase in the viral genome copies at 24 hpi, we speculate that another mechanism besides the lytic DNA replication takes place during de novo infection of PBMCs.
KSHV virions package the viral transcripts.
To analyze the RNA composition in the encapsidated virions, total RNA extracted from the purified C-type KSHV virions was used for the RNA-seq analysis. After confirming the purity of the RNA, the total RNA from two independent virus purifications was used to construct cDNA libraries. After quantifying, 10 pM concentrations of the mature library samples were used for the RNA-seq analysis. In comparison with previous reports (26, 27), the transcriptome analysis with the CLC Genomic Workbench 7 software using KSHV as the reference genome detected a larger number of KSHV mRNAs in the virions. A previous DNA microarray study identified 11 KSHV mRNAs in the virions (27), but in this study, we identified more than 60 KSHV-specific transcripts, including mRNAs for latent genes (Table 1). The mRNAs with 50 or more specific gene reads and a 10-fold higher abundance in the qPCR assays were considered significant (Fig. 7A and Table 1). The most abundant transcripts detected in the virions included PAN RNA, K7, K14, K12, ORF58/59, and the recently identified T0.7, with PAN RNA being the most abundant (Fig. 7A). In addition, many lytic-specific mRNAs were also present in the virions, such as ORF50, ORF8, ORF9, ORF21, ORF22, ORF31, ORF40/41, ORF45, ORF54, ORF55, ORF69, and ORF75. Moreover, many of the immediate early and early unique KSHV K genes (K7, K8, K3, K5, and K9) and the late K8.1 gene were also detected. Interestingly, in contrast to the previous report (27), a significant amount of latent-specific ORF73 mRNA was detected in the virions (Fig. 7A). Both this and the previous studies suggest that virion transcripts are released into the target cells immediately after de novo infection (26, 27). This may be the reason for the immediate concurrent expression of ORF50 and ORF73 in the target cells (9). It has been shown that proteins encoded by these transcripts have significant regulatory roles in various cellular pathways, including cell signaling, immune modulation, and apoptosis, that could critically affect the phenotype of the newly infected cell and its microenvironment (14, 15, 43, 44) to provide the necessary factors required for a successful infection.
FIG 7.

RNA-seq and real-time qPCR validation of the KSHV virion transcriptome. (A) RNA-seq analysis of virions purified from induced TRExBCBL1-RTA. (B) RNA-seq analysis of induced TRExBCBL1-RTA. Arrows indicate the most abundantly expressed KSHV genes. (C) Specificity of RNA encapsidation by KSHV virion. Graphs show the ratio of virion-encapsidated mRNA to that in induced TRExBCBL1-RTA cells. The ratio was calculated by dividing the number of virion-packaged transcripts copies by the number of copies expressed during lytic reactivation.
Virion-packaged transcript specificity.
To further clarify the RNA-seq results, we performed a real-time qPCR analysis on cDNA prepared from the same source of total RNA (extracted from purified C-type KSHV virions) used in the sequencing analysis. A custom qPCR array representing each of the KSHV-specific ORF primers was used for the analysis. The uninfected PBMCs used as a reference control did not amplify, and a no-RT control was used to ensure the quality of the RNA. The qPCR data showed results similar to those of the RNA-seq analysis, confirming the presence of viral transcripts in the virions.
To determine whether there is a specific mechanism of RNA encapsidation during virion assembly, we compared the abundance of viral transcripts in the virions with the amounts present during the lytic reactivation of the cells. An earlier study indicated that RNA encapsidation by the KSHV virion could be a specific event (27), and a recent report on KSHV virion miRNA also suggested that transcript encapsidation might be a specific process (26). To confirm this, we performed transcriptome (Fig. 7B) and real-time qPCR analyses of the viral genes in the cDNA extracted from induced BCBL1 cells for comparison with the transcripts present in the purified virions (Fig. 7A and B). Many of the transcripts present in the virions were expressed in abundance during lytic reactivation in the TRExBCBL1-RTA cells. To understand the specificity of RNA encapsidation in the virion, we calculated the ratio of virion transcripts to the transcript expression levels present during reactivation in the induced TRExBCBL1-RTA cells. Several of the transcripts, including PAN RNA, T0.7, DR1, ORF58, and ORF59, that were detected in high abundance in the virions were highly expressed during reactivation, and thus the ratios of encapsidated to mRNA transcripts in the induced cells were lower. We therefore concluded that they were packaged simply due to their abundance and not because of any specificity. However, some of the transcripts, including ORF73, ORF31, ORF40, ORF50, ORF56, ORF49, and ORF64, showed significantly higher ratios (≥0.5) of virion transcripts to their abundance during reactivation, suggesting the involvement of a specific mechanism in their encapsidation. The viral genes with a ratio of virions packaged to induced cells above 0.5 were considered significant and are marked by asterisks in Fig. 7C. Many of the virion-encapsidated latent- and lytic-specific transcripts that were detected early during de novo infection showed a significantly higher ratio; the ratio for the latent transcript ORF73 was 1.0843, whereas the ratio for the lytic transcript ORF50 was 2.6103. Taken together, these data suggest that KSHV may selectively encapsidate the latent and lytic transcripts required during early infection for priming the cells to successfully establish latency.
Early ORF59 expression is required for KSHV de novo infection and genome amplification.
Our transcriptome data showed a high expression of ORF59 during the primary early infection of both B and endothelial cells, which led us to believe that ORF59 may be required during the early events of KSHV infection and latency establishment. To examine this, we used a recombinant KSHV with a stop codon in the ORF59 gene (BAC36ΔORF59) (45) for de novo infection. Knowing that the ORF59-deleted BAC36 cannot produce virion particles, we generated a 293L cell line stably expressing the ORF59 gene fused with DsRed to complement the ORF59 in BAC36ΔORF59. These bacterial artificial chromosomes (BACs), WT in 293L and BAC36ΔORF59 in ORF59-DsRed-complemented 293L, were transfected and selected with hygromycin to obtain pure cell populations maintaining the BACs. These stable cells were induced with sodium butyrate (NaB) and tetradecanoyl phorbol acetate to produce virion particles, and the C-type virions were purified by sucrose density gradient centrifugation for DNA extraction and infection studies.
To determine the composition of the KSHV virion-encapsidated RNA in BAC36WT and BAC36ΔORF59, we performed an RNA-seq analysis on the virions from these two cell lines. Unsurprisingly, the patterns of the viral mRNA transcripts were similar (Fig. 8A and Table 2). We also compared the virion mRNA patterns from the BACs with those of the TRExBCBL1-RTA virions, which showed a slightly different packaging of the viral mRNA but similar levels of PAN RNA (Fig. 8A). Interestingly, the virions produced in the 293L cells complemented with ORF59-DsRed and containing BAC36ΔORF59 also encapsidated ORF59, which was confirmed by mapping the sequence reads with ORF59-DsRed (Fig. 8B). The sequence reads from BAC36WT mapped only to the ORF59 regions, confirming that BAC36WT packaged the parental ORF59 gene (Fig. 8B), as expected. Interestingly, the total number of virions produced by BAC36ΔORF59 was three logs lower than that produced by BAC36WT (Fig. 9B), even with the ORF59-DsRed complementation, which is reflected in the C-type band intensity in the BAC36ΔORF59 tube (Fig. 9A).
FIG 8.

Comparative RNA-seq analysis of virions purified from induced TRExBCBL1-RTA, BAC36WT, and BAC36ΔORF59 transcomplemented with ORF59-DsRed. Total RNA extracted from micrococcal nuclease-treated virions was subjected to DNase treatment before preparing cDNA libraries using a TrueSeq RNA-seq library kit. The libraries were sequenced using HiSeq, and the sequences were mapped to the reference sequence using the RNA-seq analysis tool of CLC Genomic Workbench 7.0. Arrows indicate the location of viral gene peaks. (B) Read mappings of the ORF59 gene from sequences of BAC36WT virions and BAC36ΔORF59 virions complemented with ORF59-DsRed using pLVxDsRed-ORF59 as a reference. ORF59-DsRed-complemented virions showed read mapping to the ORF59-DsRed fusion protein.
TABLE 2.
RPKM and gene reads of viral genes packaged in BAC36WT and ΔORF59-BAC36 virions
| No. | mRNA | BAC36WT virion |
BAC36Δ59 virion |
Function | ||
|---|---|---|---|---|---|---|
| RPKM | Gene reads | RPKM | Gene reads | |||
| 1 | K1 | 4,106.397 | 65 | 3,545.13 | 36 | Glycoprotein |
| 2 | ORF11 | 9,104.682 | 210 | 5,136.19 | 76 | Viral lytic protein |
| 3 | K2 | 264,559.9 | 3,066 | 1800,100.6 | 1,339 | K2 (viral interleukin-6), regulates cellular proliferation |
| 4 | K3 | 13,800.78 | 252 | 1,621.957 | 19 | E3 ubiquitin ligases that reduce surface expression in infected cells |
| 5 | K4 | 14,151.28 | 76 | 17,124.46 | 59 | vMIP-II |
| 6 | T1.5 | 2,572.319 | 73 | 3,735.02 | 68 | |
| 7 | K5 | 9,016.62 | 131 | 4,291.56 | 40 | E3 ubiquitin ligase |
| 8 | K6 | 10,134.38 | 55 | 2,010.55 | 7 | vMIP-IA |
| 9 | PAN RNA | 456,221 | 9,259 | 554,384.1 | 7,218 | Late gene expression, lytic DNA replication, immune modulation |
| 10 | ORF16 | 5,125.818 | 51 | 2,506.66 | 16 | Bcl2 homolog |
| 11 | ORF17 | 61,895.31 | 1,872 | 40,406.45 | 784 | Protease |
| 12 | ORF18 | 12,752.6 | 186 | 13,786.64 | 129 | Late gene regulation |
| 13 | ORF45 | 5,332.74 | 123 | 4,798.29 | 71 | Virion-associated tegument protein regulating interferon function |
| 14 | ORF54 | 4,601.55 | 77 | 94.15 | 1 | dUTPase/immune modulation |
| 15 | ORF57 | 16,862.17 | 469 | 4,203.24 | 75 | Lytic replication protein homologous to EBV SM protein, mRNA splicing |
| 16 | vIRF-4 | 6,565.60 | 351 | 10,321.76 | 354 | Facilitates lytic replication by targeting cellular IRF4 and Myc gene |
| 17 | vIRF-3 | 8,888.84 | 301 | 11,738.21 | 255 | Immune modulation |
| 18 | vIRF-2 | 2,305.14 | 94 | 2,493.35 | 77 | Type I interferon signaling |
| 19 | ORF58 | 167,550.29 | 339 | 17,252.55 | 224 | |
| 20 | ORF59 | 9,980.75 | 224 | 14,863.18 | 214 | Processivity factor |
| 21 | ORF60 | 3,005.99 | 52 | 2,793.37 | 31 | Ribonucleoprotein reductase |
| 22 | ORF61 | 1,672.91 | 75 | 1,599.45 | 46 | Ribonucleoprotein reductase |
| 23 | ORF64 | 899.21 | 134 | 1,234.312 | 118 | Deubiquitinase |
| 24 | ORF65 | 8,585.93 | 83 | 8,384.85 | 52 | Capsid protein |
| 25 | ORF66 | 3,085.30 | 75 | 3,077.94 | 48 | Capsid protein |
| 26 | T0.7 | 10,097.21 | 125 | 18,224.4 | 154 | |
| 27 | ORF73 | 2,081.99 | 133 | 1,445.63 | 68 | Latency-associated nuclear antigen |
| 28 | ORF50 | 192.39 | 11 | 545.28 | 20 | Replication and transcription activator |
FIG 9.
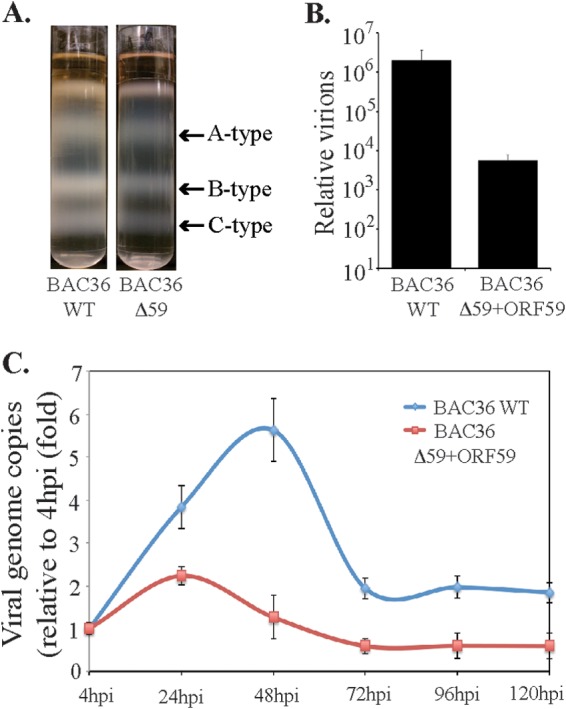
De novo infection of PBMCs with BAC36WT and BAC36ΔORF59 virions. (A) Virions were purified from culture supernatant of approximately 9 × 108 reactivated 293L cells harboring either BAC36WT or BAC36ΔORF59 on a 20 to 50% sucrose gradient. The three white bands as indicated represent A-type (empty), B-type (intermediate), and C-type (mature) KSHV virus particles, respectively. (B) Supernatants from 293L cells containing BAC36WT or BAC36ΔORF59 were used for real-time qPCR quantification of viral genome copies using ORF73-specific primers along with ORF73 plasmid standards. (C) Human PBMCs were infected with KSHV isolated from reactivated 293L cells containing BAC36WT or BAC36ΔORF59, with a multiplicity of infection (MOI) of 10. De novo-infected PBMCs were harvested at 4 h, 24 h, 48 h, 72 h, 96 h, and 120 h postinfection, followed by extraction of total DNA using a modified Hirt lysis method. This DNA was used for analyzing the genome copy number by real-time qPCR using ORF73-specific primers and ORF73 plasmid as a standard.
We normalized the number of virions used for the infection of human PBMCs by extracting the virion DNA and quantifying it in a real-time qPCR assay. Cells were collected at 4, 24, 48, 72, 96, and 120 hpi and analyzed for viral gene expression and the number of KSHV genome copies during early infection. Our results showed that both BAC36WT and BAC36ΔORF59 transcomplemented with ORF59-DsRed produced infectious virions and infected PBMCs (Fig. 9C). However, the KSHV genome copy analysis of the PBMCs infected with the transcomplemented BAC36ΔORF59 showed a reduced number of KSHV genome copies in comparison to BAC36WT (Fig. 9C). This suggested that the lytic cycle gene ORF59 is required for amplifying the viral genome during primary infection, probably by allowing DNA replication through lytic origins.
The total RNAs extracted from the de novo-infected PBMCs at different time points (4, 24, and 48 hpi) were used for a transcriptome analysis to determine whether there were differences in the viral gene expression profiles in the ORF59-deleted virus. The expression profiles of the viral genes were determined based on RPKM values and compared with those for the wild-type virus at each of the time points. The heat map generated based on the RPKM values showed subtle differences in the viral gene expression profiles (Fig. 10). The genes required for lytic DNA replication (ORF9, ORF6, ORF40/41, ORF44, ORF56, ORF50, and K8), encircled by red boxes, showed almost no difference in their expression profiles (Fig. 10). ORF59 showed a gradual decrease in virion-released ORF59 transcripts for the ORF59-deleted virus. However, BAC36WT showed a slight increase in the number of ORF59 transcripts. The PAN RNA levels were comparable in the wild-type and ORF59-deleted viruses at the time points analyzed. The expression of K2 and ORF17 was distinctly different in the ORF59-deleted virus, occurring earlier than in the PBMCs with the BAC36WT virus (Fig. 10). Interestingly, the ORF59-deleted virus showed a higher expression of the ORF73 gene (LANA) at earlier time points than did BAC36WT, suggesting an early onset of latency in the virus lacking ORF59. Overall, these data suggest that ORF59 is required for replicating the DNA and amplifying the viral genome during primary infection of human PBMCs.
FIG 10.
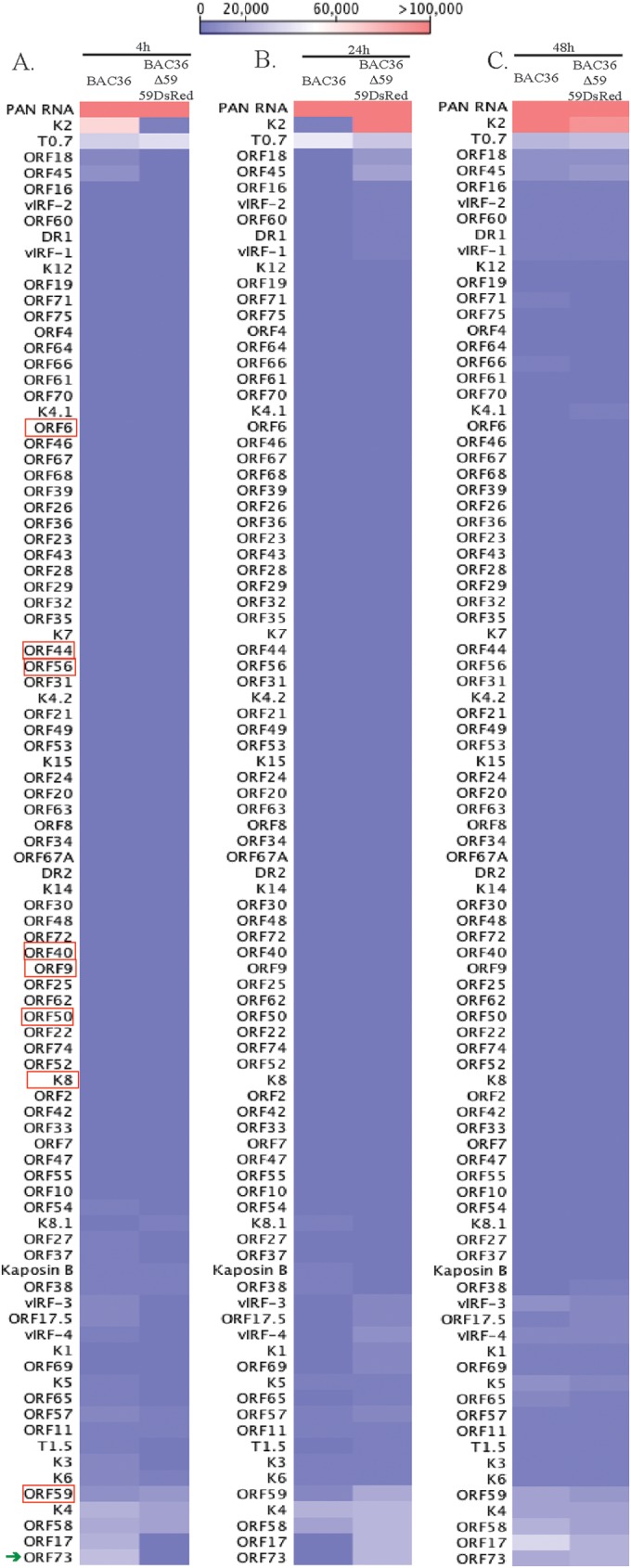
Expression profiles of viral genes in BAC36WT and BAC36ΔORF59 at different times postinfection. Normalized expression (RPKM values) of viral genes in BAC36WT and BAC36ΔORF59 at different times postinfection was used for generating hierarchal clustering. (A) Relative expression of KSHV genes at 4 h postinfection of PBMCs with BAC36WT and BAC36ΔORF59 virions. (B) Relative expression of KSHV genes at 24 h postinfection of PBMCs with BAC36WT and BAC36ΔORF59 virions. (C) Relative expression of KSHV genes at 48 h postinfection of PBMCs with BAC36WT and BAC36ΔORF59 virions. The red boxes encircle KSHV genes required for lytic DNA replication. The latency-associated gene (LANA), which showed early expression in cells infected with BAC36ΔORF59, is indicated with a green arrow.
DISCUSSION
KSHV infection is a complex multistep process involving the regulation of various cellular pathways. KSHV infects a variety of cellular targets and establishes a latent infection, generally by 24 hpi (14, 16–20). To understand the differential expression of viral transcripts early during infection, PBMCs, CD14+ cells, and TIVE cells were de novo infected with KSHV for different lengths of time (0, 4, 24, 48, 72, and 120 h). The transcriptome and real-time qPCR analyses of the de novo-infected PBMCs revealed that several lytic- and latent-specific mRNAs were concurrently expressed very early following the de novo infections. Previous reports have shown that immediately after de novo infection with herpesviruses such as EBV and herpes simplex virus, the virus undergoes a short lytic DNA replication phase (21, 37). Our data also showed the presence of the lytic DNA replication-associated transcripts, suggesting the activation of a lytic cycle. Many of the lytic cycle replication-associated transcripts were detected as early as 4 hpi, including ORF59 (processivity factor), ORF9 (DNA polymerase), ORF6 (single-stranded DNA binding protein), ORF56 (DNA replication protein), ORF40 (primase associated factor), ORF54 (dUTPase), ORF60 and -61 (ribonucleoprotein reductase), ORF70 (thymidylate synthase), and ORF37 (alkaline exonuclease). In addition, the ORF50, ORF2, K3, PAN RNA, ORF26, ORF29, ORF37, ORF55, ORF60 ORF63, ORF73, and ORF74 transcripts were found to be actively transcribing at 4 hpi, as detected by capturing the nascent RNA and actinomycin D treatment.
Apart from these lytic transcripts, the expression of several other transcripts encoding structural proteins, such as K8.1 (glycoprotein) and ORFs 65 (tegument protein), were also detected early during infection. Moreover, many of the noncoding RNAs, including PAN RNA, T0.7, and T1.5 (OriLyt transcript), were also expressed during early infection. PAN RNA has been shown to modulate viral and cellular transcription as well as to activate the KSHV lytic cycle by interacting with the ORF50 promoter and viral genome (46, 47). Furthermore, there were a large number of viral transcripts whose expression did not increase from their levels at 4 hpi, suggesting that those were transduced with the virions.
The detection of PAN RNA in the virion and during infection suggests that PAN RNA could be playing major role in triggering the lytic cycle cascade. The KSHV ORF50-encoded immediate early protein RTA is known to activate the cascade of lytic genes for facilitating the lytic replication process (38–42). Studies on the primary infection of B cells by EBV showed an accumulation of lytic transcripts during infection, indicating the requirement of a short lytic replication prior to the establishment of latency (21, 23). Earlier studies have shown that the KSHV virion also contains several KSHV lytic proteins, including ORF50, ORF8, ORF25, ORF26, K12, ORF62, ORF63, and ORF75, that are brought into the target cells during de novo infection (24). The ORF50 protein accompanying the virion may also be important for orchestrating the activation of RTA-responsive genes early during infection to induce a short lytic cycle. A significant amount of the concurrent expression of ORF50 and the KSHV latent protein ORF73 was detected as early as 4 hpi. Similar observations were reported in a previous study based on microarray and real-time qPCR analyses, but the possibility of lytic DNA replication was not proposed due to the lack of detection of all of the proteins required for lytic DNA replication (9).
A recent study on histone modifications of the KSHV genome during de novo infection demonstrated that there is a distinct pattern of activating the histone H3K4me3 mark across the KSHV genome, which is gradually replaced by the repressive H3K27me3 mark by the LANA-mediated displacement of soluble sp100 (48). Additionally, in comparison to the repressive H3K27me3 mark and the rest of the genome, a LANA ChIP-seq analysis of TRExBCBL1-RTA and TIVE-LTC showed a clear association of H3K4me3 mark and RNA polymerase II activation on the latent gene promoters (49). Our results showed that immediately after the de novo infection of PBMCs, the viral genome increased exponentially up to 48 hpi, followed by a slight decrease at 72 hpi. The accumulation of ORF50 and other RTA-regulated lytic cycle genes during early infection is suggestive of the involvement of lytic DNA replication in genome amplification. Treating the cells with a late gene expression inhibitor, PAA, led to a significantly reduced number of viral genome copies, further substantiating the involvement of lytic DNA replication in genome copy amplification in human PBMCs.
Discriminating whether the transcripts detected at 4 hpi were due to the virion-transduced mRNA or active transcription, RNA-seq analysis of the newly synthesized transcripts at 4 hpi of human PBMCs showed transcription of only a limited number of genes during primary infection. However, a large number of viral transcripts are detected at 4 hpi as well as in the virion particles, which confirms that those transcripts are introduced into the infected cells along with the virions. Detection of actively synthesizing mRNA in KSHV-infected PBMCs at 24 hpi showed transcription of primarily the ORF73 gene, which confirmed that the viral genome is chromatinized and epigenetically modified for restricted gene expression by 24 hpi. An earlier study reported the viral gene transcription of ORF59 during primary infection (27), and here we provide a comprehensive list of the genes transcribed during a de novo infection. The transcription of the viral genes as early as 4 hpi suggests that the viral DNAs entering the target cells are capable of transcribing genes before the assembly of the epigenetic histone marks. Not surprisingly, the latent protein ORF73 gene showed active transcription at 4 hpi, suggesting that LANA begins transcription as early as 4 hpi and accumulates over time to help establish latency. The immediate early protein ORF50 undergoes a moderate level of transcription immediately after infection, which may contribute in triggering the transcription of the lytic cascade to complete the DNA replication. The KSHV virions have also been shown to encapsidate a small quantity of the RTA protein, which may also be important in triggering the lytic gene expression cascade (24, 27).
It has been shown that both KSHV and EBV encapsidate biologically functional virion transcripts that are transported into the target cells during de novo infections (22, 26, 27). A previous study using a DNA microarray and real-time qPCR identified 11 of the virus transcripts packaged into the virion (27). In this study, using RNA-seq and transcriptome analyses, we detected additional KSHV latent and lytic cycle transcripts encapsidated in the virions that were also transported into the targets cells. These data were validated by a real-time qPCR analysis using KSHV gene-specific primer arrays, which showed consistent results. The transcriptome analysis identified a variety of virion transcripts in high abundance, including PAN RNA, K7, K14, K12, ORF 58/59, and the recently identified T1.5 and T0.7. Additionally, many lytic-specific mRNAs (ORF50, ORF8, ORF9, ORF21, ORF22, ORF31, ORF 40/41, ORF45, ORF54, ORF55, ORF69, and ORF75) were present in the virion in moderate amounts. A number of immediate early and early unique KSHV K genes (K7, K8, K3, K5, K9, and late K8.1) were also detected in the virion, in addition to a significant amount of latent-specific mRNAs (ORF73, ORF72, and K2).
The mechanism of RNA encapsidation by KSHV is currently unknown. Reports show that, similarly to the herpes simplex virus, the RNA packaging and encapsidation by KSHV could be specific events (26, 27). In addition, the specific encapsidation of miRNA by the KSHV virion has recently been demonstrated (26). However, both the previous study and this study determined that the majority of mRNAs detected in the KSHV virions were present at very high levels during lytic reactivation (9, 27). This suggests that these transcripts might have been randomly packaged into the virions simply due to their higher abundance. Nonetheless, the qPCR analysis to determine the specificity of the virion versus the reactivated TRExBCBL1-RTA showed that the ORF31, ORF40, ORF50, ORF56, ORF49, ORF64, and ORF73 transcripts had significantly higher proportions of encapsidated mRNA than did the PAN RNA, ORF58, ORF59, T0.7, and DR1 transcripts, suggesting a specific packaging mechanism for those transcripts.
Several of these highly expressed KSHV lytic genes have been shown to have regulatory roles in immune modulation, antiapoptosis, and lytic DNA replication, indicating that their expression may prime the host cell for retaining the incoming viral DNA during the initial infection. This is evident from the de novo infection of PBMCs with virions produced from the BAC36ΔORF59 virus. Although significantly fewer virions were produced from BAC36ΔORF59 than from BAC36WT, a similar number of virions were used to compare the viral genome copy numbers and expression profiles. The lower number of virion copies in BAC36ΔORF59 complemented with ORF59-DsRed may have been due to a comparably lower expression of ORF59 in those cells. The transcriptome analysis of the de novo-infected PBMCs showed the differential expression of only a few latent- and lytic-specific genes (PAN RNA, T0.7, K2, K4, ORF17, ORF18, ORF58, ORF45, and ORF73), confirming that the incoming DNA is transcription competent.
The virions from both the BAC36WT- and ORF59-complemented cells showed the packaging of ORF59 transcripts. However, the BAC36ΔORF59 virus genome in de novo-infected PBMCs failed to show an appreciable increase of copy number in comparison to BAC36WT. This indicates that the extended expression of ORF59 during early infection is probably required for synthesizing viral DNA through lytic DNA replication to increase the copy number. The exact mechanism requiring the initial transient accumulation of lytic-specific transcripts for the establishment of latency currently remains elusive. To fully understand the early events of KSHV infection, i.e., latency establishment and genome amplification, further experiments identifying the viral proteins expressed and the mechanism of viral DNA replication during primary infection in various cells are ongoing.
ACKNOWLEDGMENTS
We thank Erle S. Robertson at the University of Pennsylvania for providing the cell lines and LANA expression plasmids.
This work was supported by public health grants from the NIH (CA174459 and AI105000) to S.C.V. S.T. was supported by the Mick Hitchcock Fellowship at the University of Nevada, Reno.
REFERENCES
- 1.Moore PS, Chang Y. 2010. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat Rev Cancer 10:878–889. doi: 10.1038/nrc2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore PS, Chang Y. 2003. Kaposi's sarcoma-associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu Rev Microbiol 57:609–639. doi: 10.1146/annurev.micro.57.030502.090824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verma SC, Robertson ES. 2003. Molecular biology and pathogenesis of Kaposi sarcoma-associated herpesvirus. FEMS Microbiol Lett 222:155–163. doi: 10.1016/S0378-1097(03)00261-1. [DOI] [PubMed] [Google Scholar]
- 4.Fakhari FD, Dittmer DP. 2002. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J Virol 76:6213–6223. doi: 10.1128/JVI.76.12.6213-6223.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenner RG, Alba MM, Boshoff C, Kellam P. 2001. Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J Virol 75:891–902. doi: 10.1128/JVI.75.2.891-902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verma SC, Lan K, Robertson E. 2007. Structure and function of latency-associated nuclear antigen. Curr Top Microbiol Immunol 312:101–136. doi: 10.1007/978-3-540-34344-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong W, Wang H, Herndier B, Ganem D. 1996. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc Natl Acad Sci U S A 93:6641–6646. doi: 10.1073/pnas.93.13.6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ballestas ME, Chatis PA, Kaye KM. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641–644. doi: 10.1126/science.284.5414.641. [DOI] [PubMed] [Google Scholar]
- 9.Krishnan HH, Naranatt PP, Smith MS, Zeng L, Bloomer C, Chandran B. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J Virol 78:3601–3620. doi: 10.1128/JVI.78.7.3601-3620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannon JS, Ciufo D, Hawkins AL, Griffin CA, Borowitz MJ, Hayward GS, Ambinder RF. 2000. A new primary effusion lymphoma-derived cell line yields a highly infectious Kaposi's sarcoma herpesvirus-containing supernatant. J Virol 74:10187–10193. doi: 10.1128/JVI.74.21.10187-10193.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grundhoff A, Ganem D. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J Clin Invest 113:124–136. doi: 10.1172/JCI200417803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med 2:342–346. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- 13.Wang CY, Sugden B. 2004. New viruses shake old paradigms. J Clin Invest 113:21–23. doi: 10.1172/JCI20662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandran B. 2010. Early events in Kaposi's sarcoma-associated herpesvirus infection of target cells. J Virol 84:2188–2199. doi: 10.1128/JVI.01334-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chakraborty S, Veettil MV, Chandran B. 2012. Kaposi's sarcoma associated herpesvirus entry into target cells. Front Microbiol 3:6. doi: 10.3389/fmicb.2012.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akula SM, Naranatt PP, Walia NS, Wang FZ, Fegley B, Chandran B. 2003. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) infection of human fibroblast cells occurs through endocytosis. J Virol 77:7978–7990. doi: 10.1128/JVI.77.14.7978-7990.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akula SM, Pramod NP, Wang FZ, Chandran B. 2002. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108:407–419. doi: 10.1016/S0092-8674(02)00628-1. [DOI] [PubMed] [Google Scholar]
- 18.Bechtel JT, Liang Y, Hvidding J, Ganem D. 2003. Host range of Kaposi's sarcoma-associated herpesvirus in cultured cells. J Virol 77:6474–6481. doi: 10.1128/JVI.77.11.6474-6481.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lagunoff M, Bechtel J, Venetsanakos E, Roy AM, Abbey N, Herndier B, McMahon M, Ganem D. 2002. De novo infection and serial transmission of Kaposi's sarcoma-associated herpesvirus in cultured endothelial cells. J Virol 76:2440–2448. doi: 10.1128/jvi.76.5.2440-2448.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakraborty S, Veettil MV, Bottero V, Chandran B. 2012. Kaposi's sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc Natl Acad Sci U S A 109:E1163–E1172. doi: 10.1073/pnas.1119592109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halder SMM, Verma SC, Kumar P, Yi F, Robertson ES. 2009. Early events associated with infection of Epstein-Barr virus infection of primary B-cells. PLoS One 4:e7214. doi: 10.1371/journal.pone.0007214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jochum SRR, Moosmann A, Hammerschmidt W, Zeidler R. 2012. RNAs in Epstein-Barr virions control early steps of infection. Proc Natl Acad Sci U S A 109:E1396–E1404. doi: 10.1073/pnas.1115906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wen W, Iwakiri D, Yamamoto K, Maruo S, Kanda T, Takada K. 2007. Epstein-Barr virus BZLF1 gene, a switch from latency to lytic infection, is expressed as an immediate-early gene after primary infection of B lymphocytes. J Virol 81:1037–1042. doi: 10.1128/JVI.01416-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bechtel JT, Winant RC, Ganem D. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J Virol 79:4952–4964. doi: 10.1128/JVI.79.8.4952-4964.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu FX, Chong JM, Wu L, Yuan Y. 2005. Virion proteins of Kaposi's sarcoma-associated herpesvirus. J Virol 79:800–811. doi: 10.1128/JVI.79.2.800-811.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin X, Li X, Liang D, Lan K. 2012. MicroRNAs and unusual small RNAs discovered in Kaposi's sarcoma-associated herpesvirus virions. J Virol 86:12717–12730. doi: 10.1128/JVI.01473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bechtel J, Grundhoff A, Ganem D. 2005. RNAs in the virion of Kaposi's sarcoma-associated herpesvirus. J Virol 79:10138–10146. doi: 10.1128/JVI.79.16.10138-10146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toth Z, Brulois K, Lee HR, Izumiya Y, Tepper C, Kung HJ, Jung JU. 2013. Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog 9:e1003813. doi: 10.1371/journal.ppat.1003813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verma SC, Bajaj BG, Cai Q, Si H, Seelhammer T, Robertson ES. 2006. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus recruits uracil DNA glycosylase 2 at the terminal repeats and is important for latent persistence of the virus. J Virol 80:11178–11190. doi: 10.1128/JVI.01334-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verma SC, Choudhuri T, Kaul R, Robertson ES. 2006. Latency-associated nuclear antigen (LANA) of Kaposi's sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J Virol 80:2243–2256. doi: 10.1128/JVI.80.5.2243-2256.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 32.Lallemand F, Desire N, Rozenbaum W, Nicolas JC, Marechal V. 2000. Quantitative analysis of human herpesvirus 8 viral load using a real-time PCR assay. J Clin Microbiol 38:1404–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bates I, Bedu-Addo G, Jarrett RF, Schulz T, Wallace S, Armstrong A, Sheldon J, Rutherford T. 2001. B-lymphotropic viruses in a novel tropical splenic lymphoma. Br J Haematol 112:161–166. doi: 10.1046/j.1365-2141.2001.02517.x. [DOI] [PubMed] [Google Scholar]
- 34.Medveczky MM, Horvath E, Lund T, Medveczky PG. 1997. In vitro antiviral drug sensitivity of the Kaposi's sarcoma-associated herpesvirus. AIDS 11:1327–1332. doi: 10.1097/00002030-199711000-00006. [DOI] [PubMed] [Google Scholar]
- 35.Chang PJ, Boonsiri J, Wang SS, Chen LY, Miller G. 2010. Binding of RBP-Jkappa (CSL) protein to the promoter of the Kaposi's sarcoma-associated herpesvirus ORF47 (gL) gene is a critical but not sufficient determinant of transactivation by ORF50 protein. Virology 398:38–48. doi: 10.1016/j.virol.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nealon K, Newcomb WW, Pray TR, Craik CS, Brown JC, Kedes DH. 2001. Lytic replication of Kaposi's sarcoma-associated herpesvirus results in the formation of multiple capsid species: isolation and molecular characterization of A, B, and C capsids from a gammaherpesvirus. J Virol 75:2866–2878. doi: 10.1128/JVI.75.6.2866-2878.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roizman B, Knipe DM. 2001. Herpes simplex viruses and their replication, p 2399–2459 InKnipe DM, Howley PM, et al. (ed), Fields virology, 4th ed Lippincott, Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 38.Guito J, Lukac DM. 2012. KSHV Rta promoter specification and viral reactivation. Front Microbiol 3:30. doi: 10.3389/fmicb.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dourmishev LA, Dourmishev AL, Palmeri D, Schwartz RA, Lukac DM. 2003. Molecular genetics of Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol Mol Biol Rev 67:175–212. doi: 10.1128/MMBR.67.2.175-212.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. 1999. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J Virol 73:2232–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.West JT, Wood C. 2003. The role of Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression. Oncogene 22:5150–5163. doi: 10.1038/sj.onc.1206555. [DOI] [PubMed] [Google Scholar]
- 42.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol 77:4205–4220. doi: 10.1128/JVI.77.7.4205-4220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan SR, Bloomer C, Chandran B. 1998. Identification and characterization of human herpesvirus-8 lytic cycle-associated ORF 59 protein and the encoding cDNA by monoclonal antibody. Virology 240:118–126. doi: 10.1006/viro.1997.8911. [DOI] [PubMed] [Google Scholar]
- 44.Unal A, Pray TR, Lagunoff M, Pennington MW, Ganem D, Craik CS. 1997. The protease and the assembly protein of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8). J Virol 71:7030–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McDowell ME, Purushothaman P, Rossetto CC, Pari GS, Verma SC. 2013. Phosphorylation of Kaposi's sarcoma-associated herpesvirus processivity factor ORF59 by a viral kinase modulates its ability to associate with RTA and oriLyt. J Virol 87:8038–8052. doi: 10.1128/JVI.03460-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rossetto CC, Pari G. 2012. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog 8:e1002680. doi: 10.1371/journal.ppat.1002680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossetto CC, Pari GS. 2011. Kaposi's sarcoma-associated herpesvirus noncoding polyadenylated nuclear RNA interacts with virus- and host cell-encoded proteins and suppresses expression of genes involved in immune modulation. J Virol 85:13290–13297. doi: 10.1128/JVI.05886-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gunther T, Schreiner S, Dobner T, Tessmer U, Grundhoff A. 2014. Influence of ND10 components on epigenetic determinants of early KSHV latency establishment. PLoS Pathog 10:e1004274. doi: 10.1371/journal.ppat.1004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu J, Yang Y, Turner PC, Jain V, McIntyre LM, Renne R. 2014. LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex. PLoS Pathog 10:e1004240. doi: 10.1371/journal.ppat.1004240. [DOI] [PMC free article] [PubMed] [Google Scholar]