

ABSTRACT
Neisseria gonorrhoeae (the gonococcus) causes gonorrhea and is uniquely adapted to survive within the human reproductive tract. Gonococci evade host immune surveillance in part by varying their pili and opacity-associated proteins. These variable surface antigens influence interactions with host epithelial and immune cells. A potent polymorphonuclear leukocyte (PMN) response is a hallmark of symptomatic gonococcal infection, with vast numbers of PMNs recruited to the site of infection. A large body of literature describes gonococcus-PMN interactions, but the factors driving the outcome of infection are not fully understood. Gonococci have been described to both induce and suppress the PMN oxidative burst, but we determined that gonococci differentially affect induction of the PMN oxidative burst depending on the multiplicity of infection (MOI). Infecting PMN at an MOI of <20 gonococci elicits an oxidative burst, while an MOI of >20 suppresses the burst. Oxidative burst in response to gonococci is enhanced by, but does not require, expression of pili or opacity proteins. Neutrophil extracellular traps (NETs) were observed in gonococcus-infected PMNs, a process which requires an oxidative burst, yet gonococci induced NETs under suppressing conditions. The NETs were unable to kill gonococci despite killing the common vaginal bacterium Lactobacillus crispatus. Thus, gonococci influence PMN biology to promote their own survival by suppressing the oxidative burst of PMNs and stimulating the formation of NETs, which do not effectively kill gonococci, illustrating how N. gonorrhoeae has evolved to modulate PMN responses to promote infection.
IMPORTANCE
Neisseria gonorrhoeae, the gonococcus, is the only causative agent of gonorrhea and is exclusively found within the human host. Gonococci stochastically vary the composition of antigens on their surface to evade immune surveillance. We used gonococcal mutants which stably express different surface antigens to dissect interactions between gonococci and primary human polymorphonuclear leukocytes (PMNs). We found that gonococci, depending on the number of bacteria present, either induce or suppress the oxidative burst of PMNs regardless of other stimuli. Gonococci also cause PMNs to release DNA, forming neutrophil extracellular traps (NETs) independently of the oxidative burst. The NETs were unable to kill gonococci but were able to kill commensal bacteria, suggesting that NET production can help gonococci outcompete other bacterial species. We propose that gonococci have evolved to manipulate PMN responses to promote their own survival during infection.
INTRODUCTION
Neisseria gonorrhoeae (the gonococcus) is an obligate human pathogen and the sole causative agent of the sexually transmitted infection gonorrhea. Infection typically results in urethritis or cervicitis, but more-severe disseminating infections can also occur (1). The prevalence of gonococcal infections, with 820,000 estimated cases per year in the United States, is made more troubling because an increasing number of gonococcal isolates are multidrug resistant, which led the CDC to label the gonococcus as an urgent antibiotic resistance threat in 2013 (2–4). Symptomatic gonorrheal disease is characterized by a potent inflammatory immune response and infiltration of polymorphonuclear leukocytes (PMNs) to the site of infection (reviewed in reference 5). Despite the presence of large numbers of PMNs, the purulent exudates of infected individuals contain live gonococci (6). Interactions between PMNs and gonococci have been studied extensively, and this prior work indicates that the bacteria have evolved many mechanisms to resist PMN killing mechanisms (5, 7, 8).
Gonococcal surface proteins, including type IV pili and opacity-associated (Opa) proteins, mediate interactions with host cells. The type IV pili are protein polymers that extend from the gonococcal surface and mediate attachment of gonococci to host cells (9). The primary amino acid sequence of the type IV pili varies during the course of infection (10, 11). N. gonorrhoeae FA1090 possesses 11 distinct, phase-variable, opa genes (opaA to -K) that encode structural proteins expressed on the surface of gonococci (12). Each of the Opa proteins is stochastically expressed due to a slipped-strand mispairing mechanism that allows for the translation of any combination of Opa proteins or none at all (13, 14). Opa switching occurs frequently enough (1/1,000 cells/generation) to produce heterogeneity in an infecting population of gonococci (14–16). The processes of antigenic and phase variation are thought to contribute to gonococcal immune evasion (17).
N. gonorrhoeae interacts with epithelial cells and innate immune cells at the site of infection through several cell surface receptors. Toll-like receptors (TLRs) on host cells recognize gonococcal surface structures, including the lipooligosaccharides (LOSs) and porin (18, 19), while NOD-like receptors (NLRs) recognize peptidoglycan components (20, 21). TLR or NLR binding results in the production of inflammatory cytokines (e.g., interleukin 8 [IL-8] and IL-17), which recruit PMNs to the site of infection. IL-8 has been shown to be produced in response to gonococcal infection both in vivo (22) and in cell culture (23, 24). In addition to TLR and NLR recognition, Opas on the gonococcal outer membrane engage carcinoembryonic antigen cell adhesion molecule (CEACAM) receptors expressed on the surface of epithelial cells, endothelial cells, and PMNs (25–27). Specifically, it has been suggested that Opa binding to CEACAM3 promotes phagocytic uptake of gonococci, resulting in PMN degranulation and oxidative burst—the production of microbicidal, reactive oxygen species (ROS) (27–30). Despite these intimate interactions with host cells, some gonococci resist PMN-mediated killing (6–8, 31).
N. gonorrhoeae has multiple mechanisms to evade PMN-mediated killing during host-pathogen interactions. Phase variation of Opa expression on the cell surface allows for populations of gonococci to be less efficiently phagocytosed (25). If gonococci are taken up by PMNs, they can delay phagosomal maturation by inhibiting granule fusion with phagosomes (32), potentially impairing the ability of PMNs to destroy phagocytosed material. Similarly, gonococcal Opa expression influences the assembly of NADPH oxidase in PMNs (33). It has also been shown that PMN apoptosis is inhibited when the cells are infected with gonococci (24, 34), which could assist in the dissemination of viable gonococci that persist within PMNs. Finally, gonococci proteolytically inactivate antimicrobial proteins and peptides and detoxify reactive oxygen species (35). In fact, gonococci have been shown to directly suppress the PMN oxidative burst resulting from stimulation by phorbol myristate acetate (PMA) or nonviable gonococci (24, 36). Here, we present evidence that gonococcal induction and suppression of the PMN oxidative burst are more universal and highly dependent on the multiplicity of infection (MOI). Gonococci induce an oxidative burst at low MOI and suppress the burst at high MOI without requiring the expression of pili or opacity proteins.
Neutrophil extracellular traps (NETs) are formed when PMNs undergo a specialized cell death program (37) that is thought to be dependent on the PMN oxidative burst activating myeloperoxidase (MPO) (38–40). NETs are proposed to be a last line of defense against infection because the PMN chromatin, imbued with PMN granule components, is released into the extracellular matrix to trap microbes (37, 41). In addition to preventing the dissemination of pathogens, NETs have been shown to directly kill a wide variety of bacteria and fungi, including Staphylococcus aureus, Shigella flexneri, and Candida albicans (37, 41). The role of NETs in gonococcal infection is investigated here, and the data show that NETs are produced in response to infection with gonococci. This is true whether or not the gonococci express a specific Opa or pili on their surface. Contrary to our expectations, we found that gonococcal stimulation of NETs occurs through both oxidative and oxidative burst-independent mechanisms. Gonococci resist NET-mediated killing, while Lactobacillus crispatus, the predominant member of the normal flora of the female reproductive tract, is killed. The data support a model where gonococci have evolved mechanisms in an MOI-dependent fashion, to stimulate PMNs to form NETs despite suppressing the oxidative burst. Thus, gonococci directly modulate PMN biology, thereby facilitating their survival within the human reproductive tract at the expense of resident flora.
RESULTS
Gonococci stimulate PMNs to form NETs containing histones and granule components.
4′,6-Diamidino-2-phenylindole (DAPI)-stained networks of extracellular DNA were observed in PMNs infected with gonococci (Fig. 1A). To test whether these structures were neutrophil extracellular traps (NETs), immunofluorescence (IF) staining was utilized to determine the molecular composition of the DNA structures. The DNA contains histone H2A (Fig. 1B), suggesting that the DNA is indeed chromatin and of eukaryotic origin rather than DNA from lysed gonococci. Further IF microscopy revealed that the granule components cathelicidin (LL-37), cathepsin G, and neutrophil elastase (Fig. 1D, E, and F, respectively) are localized to the extracellular chromatin, in addition to cytoplasmic granules. Thus, the extracellular DNA structures observed in gonococcal infections are consistent with published reports of NET composition.
FIG 1 .

Gonococcal infection stimulates PMNs to form neutrophil extracellular traps (NETs). (A) Gonococci that express the mCherry protein are trapped in DAPI-stained DNA (NET) produced by primary human PMNs. (B) OpaD+ P+ gonococcus-infected PMN stained for histone H2A. Gonococci with a grape cluster appearance are present in the NETs and are more easily distinguished in the insets, which are different planes of focus through the three-dimensional NET structure. (C to F) OpaD+ P+ gonococcus-infected PMNs were stained with secondary antibody alone as a negative control or using antibodies specific to the granule components, as indicated in each panel. Arrows indicate NETs.
Gonococci with stably expressed pili and OpaD stimulate PMN to form NETs.
We next sought to define the molecular interactions between gonococci and PMNs that are required for the formation of NETs. Interactions between gonococci and PMNs are heavily influenced by the expression of Opa and pili, and these proteins are stochastically expressed in wild-type gonococci. To minimize the effects of phase and antigenic variation in our model infections, we utilized a set of four gonococcal strains that are genetically locked and more stably maintain their phenotypes: (i) a strain that is locked in a piliated state (42) and stably expresses a single opacity protein (43) on its surface (OpaD+ P+ strain), (ii) a strain that stably expresses OpaD but is nonpiliated (OpaD+ P− strain), (iii) a strain that has been made genetically Opaless by deletion of all its opa genes (43) but is locked in a piliated state (Opaless P+ strain), and (iv) an Opaless strain that is also nonpiliated (Opaless P− strain). OpaD was chosen from the 11 gonococcal opacity proteins because it is known to enhance gonococcus-PMN interactions (25–27, 43). All of the four strains were similar in their abilities to stimulate PMNs to form NETs (see Fig. S1 in the supplemental material), as were heat-killed gonococci (see Fig. S2), suggesting that neither OpaD, pili, nor viable gonococci are required to stimulate NETs from PMNs. We also observed that lactate dehydrogenase (LDH) release does not increase during the same time as NET production or degranulation (data not shown); thus, the release of PMN DNA is specific and not simply the result of PMN lysis.
Gonococcal surface antigens enhance, but are not required for, the stimulation of NETs.
To more accurately assess the ability of gonococci to stimulate NETs, we measured integrated H2A staining area, as a fraction of the total DAPI staining area. The H2A/DAPI ratio serves as a proxy for NET formation because the cells were not permeabilized prior to antibody staining, and only extracellular DNA (NETs) will stain with the H2A antibody. A higher H2A/DAPI staining ratio reflects a greater proportion of NETs. Treating PMN with phorbol myristate acetate (PMA), a protein kinase C (PKC) activator, stimulated NET formation and resulted in an H2A/DAPI ratio of 0.9, meaning that 90% of DAPI-stained DNA was in NETs. Using the same measurement, uninfected PMNs had a ratio of 0.05 (5% of DAPI-stained DNA was in NETs). PMNs were infected with gonococci, and the H2A/DAPI ratio was plotted against the multiplicity of infection (MOI, Fig. 2). OpaD+ P+ strain-infected PMNs showed the largest increase in NETs as MOI increased. The PMNs infected with Opaless P− gonococci showed the smallest increase, with OpaD+ P− and Opaless P+ strain-infected PMNs falling in between. This suggests that both OpaD and pili can contribute to NET formation by PMNs but that neither is required.
FIG 2 .
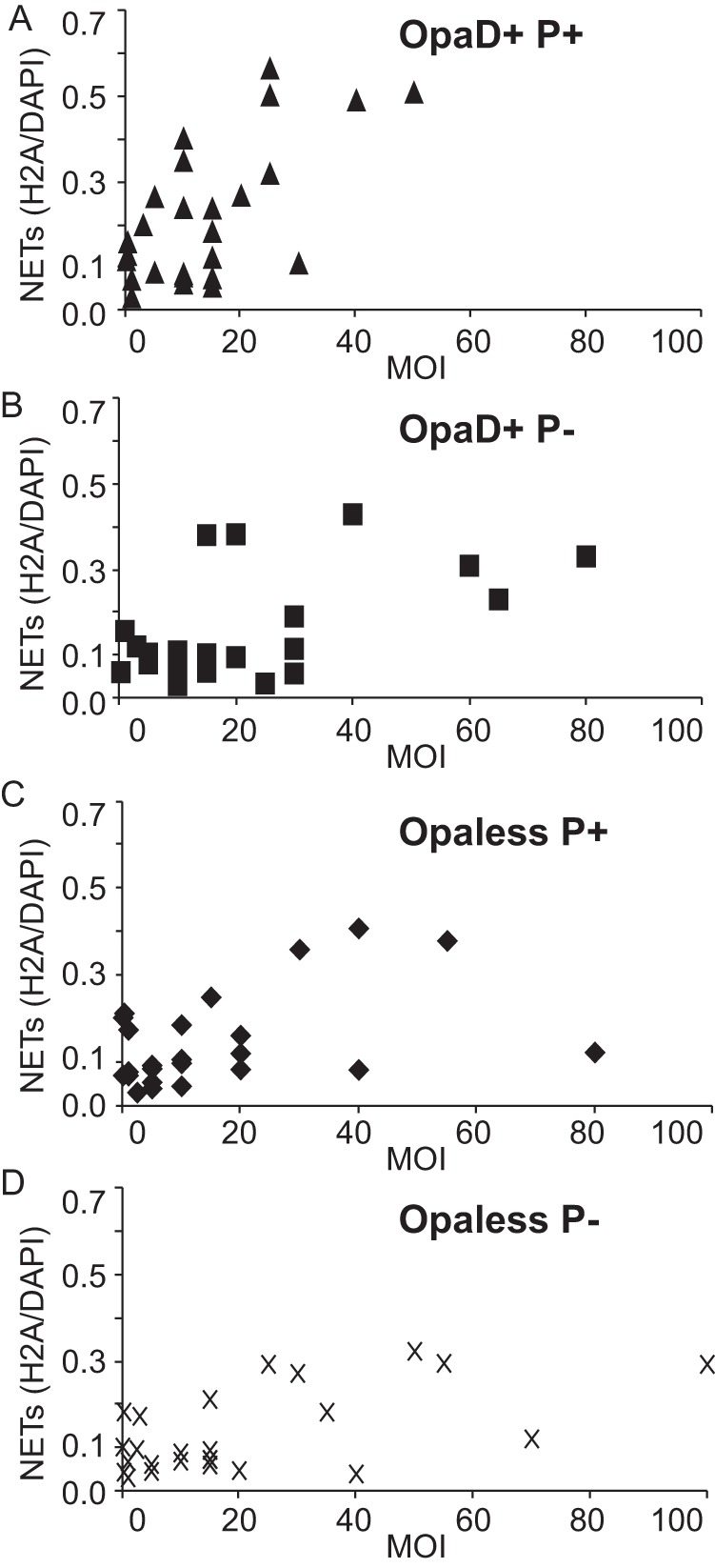
OpaD-expressing gonococci robustly stimulate PMNs to form NETs. NET formation was quantified by taking a ratio of the integrated H2A and DAPI staining area density measurements. NET measurements were plotted against the infection MOI, for PMNs infected with OpaD+ P+, OpaD+ P−, Opaless P+, and Opaless P− gonococci (A to D, respectively). Data are cumulative from 11 independent experiments using PMNs from one of six different donors. Due to the intrinsic variability of both the primary PMNs and the gonococcus, it is impossible to average these data.
Gonococcal NETs are produced via an oxidative signaling-independent pathway.
There is an extensive body of literature showing that stimulation of the oxidative burst is required for the production of NETs (38–40). More specifically, it has been shown previously that myeloperoxidase is required for PMNs to produce NETs (39). To determine whether the oxidative burst was required for NETs produced in response to gonococcal infection, the oxidative burst (via luminol-dependent chemiluminescence [LDCL]) and NET formation (H2A/DAPI ratio) were measured (Fig. 3). We observed that NET formation did not correlate with oxidative burst. Piliated (triangles) and nonpiliated (squares) gonococci that express OpaD show no correlation between oxidative burst and NET production (Fig. 3A). Separating the data by MOI (filled symbols, MOI of <15; open symbols, MOI of >15) does not reveal a stronger correlation. Similarly, NET formation in response to infection by Opaless gonococci also failed to correlate with oxidative burst (Fig. 3B). Comparing the data between OpaD+ and Opaless strain gonococcal infections (Fig. 3A versus B), the extent of oxidative burst was smaller in response to Opaless gonococci than in the OpaD+ strain gonococcal infections, and yet the extents of NET formation were very similar. To test whether gonococci stimulate myeloperoxidase (MPO) without increasing the luminol signal and to confirm that this was inducing NETs, PMNs were treated with the myeloperoxidase inhibitor (MPOI) 4-aminobenzhydrazide and then PMA, Opaless P+ gonococci, or OpaD+ P+ gonococci (Fig. 3C). The MPOI had no effect on NET production in uninfected or gonococcus-infected PMNs but inhibited NET formation after PMA treatment. These results confirm that the formation of NETs by PMNs in response to gonococcal infection is by a pathway independent of the oxidative burst and MPO activation.
FIG 3 .
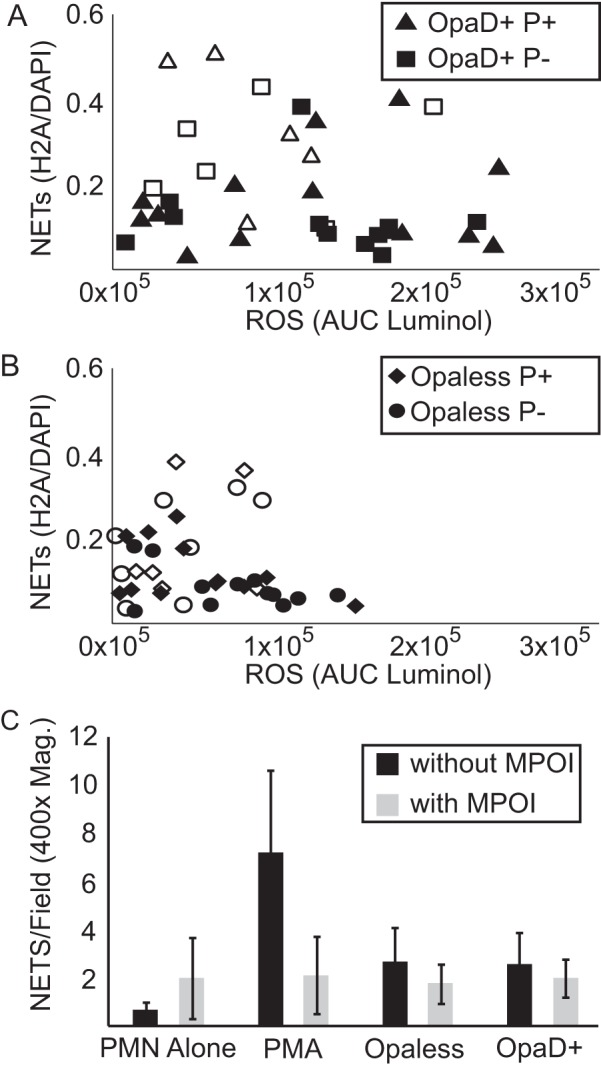
Gonococcus-stimulated NET production does not correlate with oxidative burst. (A and B) NET production quantified in Fig. 2 was plotted against reactive oxygen species burst (AUC of luminol). (A and B) Data from PMNs infected with OpaD+ (A) and Opaless (B) gonococci. Filled symbols represent infections where the MOI was less than 15, and open symbols represent infections with MOIs greater than 15. Data are cumulative from 7 independent experiments using PMNs from 5 different donors. (C) PMNs were left untreated, treated with PMA, or infected with Opaless P+ or OpaD+ P+ gonococci. These treatments were performed alone or in combination with an inhibitor of myeloperoxidase (MPOI) (gray bars). The number of NETs in each ×400 magnification field was counted (see Materials and Methods). All data are a representative experiment from at least three independent experiments using PMNs from at least 3 individual donors.
Gonococci both activate and suppress the oxidative burst.
The lack of a correlation between NET production and oxidative burst prompted a detailed study on the reactive oxygen species (ROS) burst elicited from PMNs by the four defined strains at different MOIs. All four strains of gonococci elicit a peak oxidative response from PMNs at an MOI of 10 to 20 but differ in the extents of the oxidative burst induced at that peak (i.e., Fig. 4A versus D). Whether or not the infecting gonococci express pili or OpaD, the oxidative burst from the PMNs drops to background levels when MOIs are greater than 50. The extent of the oxidative burst activation and suppression and how these phenotypes change with MOI were not appreciated in previous reports (7, 24, 36). It was possible that the suppression phenotype at high MOIs was an artifact of high levels of catalase produced by the gonococci (44). However, ROS burst induction and suppression in OpaD+ gonococci had identical burst kinetics, and this was the case at both inducing and suppressing MOIs independent of katA function (Fig. 4E), showing that catalase is not responsible for the suppression phenotype.
FIG 4 .
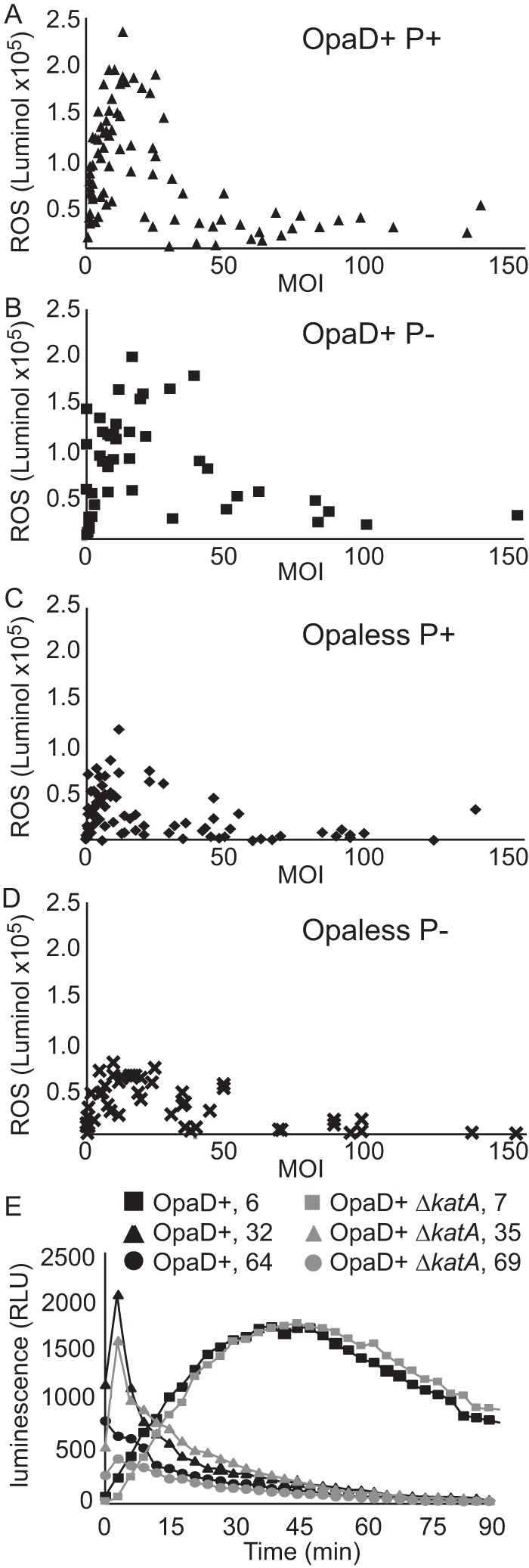
Gonococcal induction and suppression of the PMN oxidative burst are MOI dependent. Primary human PMNs were infected with gonococci in the presence of luminol, and luminescence increases were monitored over the course of 90 min. The area under the curve was calculated, and the value was plotted against the MOI. (A to D) PMNs were infected with OpaD+ P+ (A), OpaD+ P− (B), Opaless P+ (C), and Opaless P− (D) gonococci, at MOIs ranging from 1 to 150. Data are cumulative from 9 to 12 different experiments using PMNs isolated from one of seven different donors. (E) Gonococcal suppression of PMN oxidative burst is not due to catalase activity. The curves show the accumulation of LDCL by PMNs infected with OpaD+ P+ gonococci (black curves) compared to infections with a ΔkatA mutant (gray curves). Data are one representative set of data from at least 3 independent experiments, with PMNs from 3 individual donors.
Gonococcal suppression of the oxidative burst is downstream of PKC activation.
To better understand the mechanism of the suppression phenotype, LDCL assays were performed by infecting PMNs with gonococci in combination with PMA, diphenyleneiodonium (DPI), or formyl-Met-Leu-Phe peptide (fMLF). As expected, DPI, an inhibitor of NADPH oxidase, completely inhibited the oxidative burst of PMNs in the presence of gonococci (Fig. 5A, black bars). Gonococci were capable of suppressing the ROS burst caused by direct stimulation of protein kinase C (PKC) by PMA (Fig. 5A, gray bars). Together, these data suggest that gonococcal suppression of ROS burst occurs downstream of PKC activation but before NADPH oxidase activation. We hypothesized that if suppression is downstream of PKC, then stimulation or inhibition of the upstream pathways of neutrophil activation should not affect gonococcus-mediated activation and suppression of the oxidative burst. We observed an additive effect in the ROS burst elicited from PMNs treated with the formyl peptide receptor (FPR) ligand, fMLF, and infected with gonococci (Fig. 5B, black bars). This result suggests that activation of PMN by fMLF or gonococci is occurring through independent pathways. The suppression phenotype was unchanged in the presence of fMLF, suggesting that formyl peptide receptor signaling is not involved. Next, FPR, CXCR2, and CXCR4 antagonists (cyclosporine H, SB225002, and plerixafor, respectively) were assessed for their ability to interfere with gonococcus-PMN interactions. LDCL assays revealed that none of the three inhibitors had a large effect on the induction or suppression of the oxidative burst caused by Opaless P+ gonococcal infection (see Fig. S3A in the supplemental material). Similar results were observed for infections using the OpaD+ P+ strain (data not shown). We thought that, due to significant cross talk between PMN signaling pathways, combining the treatments might result in a more distinct phenotype. The combination treatment did not result in greater reduction in oxidative burst (see Fig. S3B). We tested peptide rD-7, which antagonizes CEACAM receptors (45, 46). The oxidative burst was inhibited, and this was true not only for the OpaD+ gonococci but also for Opaless gonococci (Fig. 5C). Together, these results suggest that the CEACAM-dependent signaling is important for mounting an oxidative burst against gonococci, whether the bacteria are expressing a CEACAM-engaging opacity protein or not. The rD-7 peptide had no effect on the formation of NETs in response to gonococcal infection (Fig. 5D). Thus, oxidative signaling is dependent on interactions with CEACAMs, but the stimulation of NETs is not.
FIG 5 .
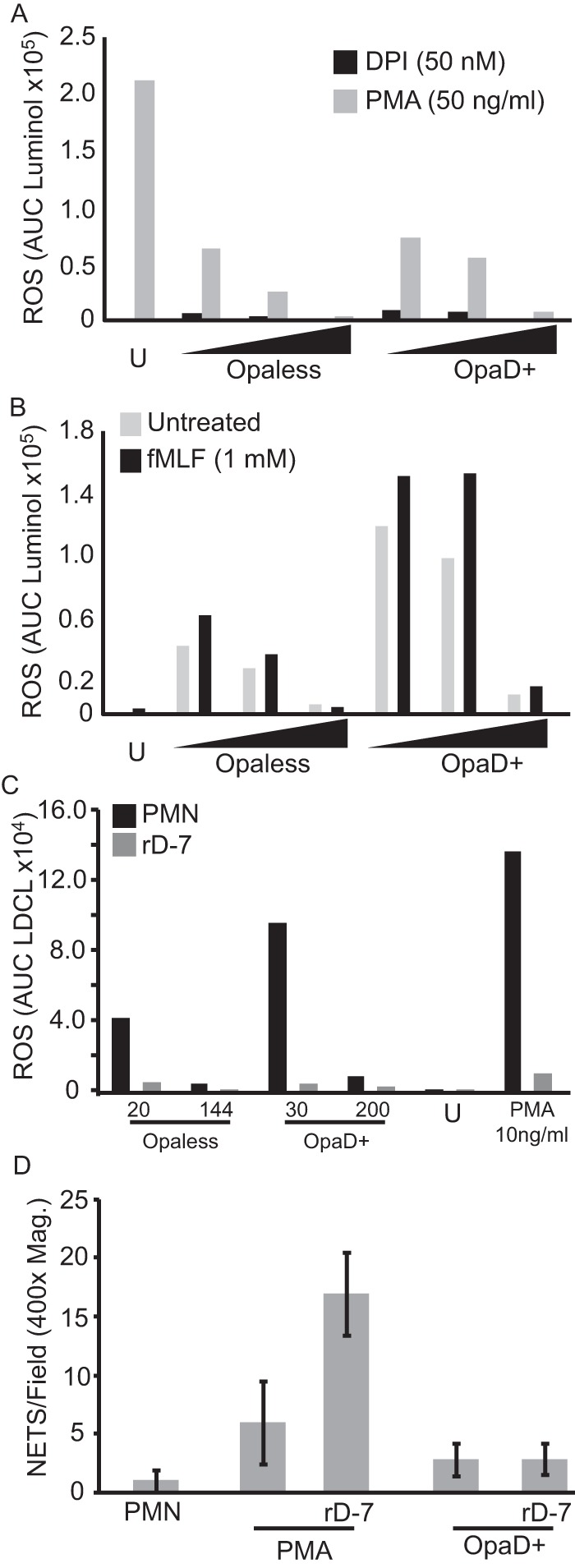
Gonococcal suppression of the oxidative burst occurs downstream of protein kinase C activation. PMNs were infected with gonococci in the presence of luminol, and LDCL increase was monitored over the course of 90 min. (A) PMNs treated with DPI (black bars) or PMA (gray bars). Opaless P+ or OpaD+ gonococci were titrated over a range of MOIs (Opaless, 1, 14, and 67; OpaD+ strain, 5, 9, and 47), moving left to right on the x axis, as indicated by the triangles underneath the axis. (B) PMNs were treated with fMLF, in combination with gonococcal infection. Opaless P+ or OpaD+ gonococci were titrated over a range of MOIs (Opaless, 1, 10, 50; OpaD+ strain, 2, 16, 80), moving left to right on the x axis, represented by the triangles underneath the axis. (C and D) The effect of CEACAM-binding peptide rD-7 on oxidative burst and NET formation was assessed by LDCL (C) and counting the number of NETs in each ×400 magnification field (D) (see Materials and Methods). All data are a representative experiment from at least three independent experiments using PMNs from at least 3 individual donors.
Gonococci are resistant to NET-mediated killing in vitro.
We hypothesized that NETs might influence gonococcal infection through localized killing of gonococci. After several attempts to quantify NET-mediated killing by quantitative enumeration of colonies after nuclease treatment with ambiguous results, a LIVE/DEAD staining protocol was utilized. Coupled with microscopy, the LIVE/DEAD staining allowed us to better resolve localized killing of the bacteria. PMNs were stimulated with PMA to ensure uniform NET induction and were then infected with gonococci and stained. For each image, the viability of each gonococcus and its location relative to the PMN were determined (see Materials and Methods). PMN- and NET-mediated killing of gonococci was tested in parallel with Staphylococcus aureus and Lactobacillus crispatus (Table 1). Gonococci in NETs were 92% to 97% viable, which is similar to the values obtained for gonococci in the absence of PMNs (93% to 97% viable). Over 97% of S. aureus bacteria were viable, but those staphylococci in NETs were only 63% viable (P < 0.05), suggesting that a portion of staphylococci are killed in NETs. Similarly, NET-bound L. crispatus bacteria were 43% viable compared to the input bacteria (91% viable). Extracellular (but not NET-bound) bacteria showed similar phenotypes; gonococci were not killed, but S. aureus and L. crispatus were. Killing of gonococci was observed only with OpaD+ P− bacteria and then only when they had been internalized by PMNs. This result is consistent with OpaD+ gonococci being more readily taken up by PMNs (27–30) and with pili being involved in resisting oxidative killing (47). The presence of DPI in the infected cells abolished killing of intracellular OpaD+ P− gonococci and staphylococci, suggesting that the killing after phagocytosis is partially dependent on the oxidative burst.
TABLE 1 .
Gonococci resist PMA-activated PMN NET-mediated killing
| Localization | % viable bacteria (SD) for straina,b: |
|||||
|---|---|---|---|---|---|---|
| Opaless P+ strain | Opaless P− strain | OpaD+ P+ strain | OpaD+ P− strain | S. aureus | L. crispatus | |
| Bacteria alone | 95.8 (2.9) | 97.5 (0.5) | 93.7 (5.4) | 96.0 (1.3) | 97.4 (1.7) | 91.6 (3.1) |
| Extracellular | 98.2 (0.05) | 98.2 (0.6) | 91.7 (1.3) | 95.1 (2.2) | 62.5 (28.1) | 39.5 (4.8)** |
| NET associated | 97.0 (0.4) | 96.2 (1.6) | 95.9 (2.0) | 92.3 (3.2) | 63.6 (19.8)* | 43.5 (7.3)* |
| PMN associated | 94.9 (1.7) | 92.9 (0.5) | 88.1 (6.9) | 75.8 (8.2)** | 50.0 (26.9)* | NTc |
| PMN associated (DPI) | NT | NT | NT | 93.8 (0.02) | 76.1 (0.1) | NT |
Bacterial survival was determined by LIVE/DEAD staining. Opaless P+, Opaless P−, and OpaD+ P+ strain and DPI treatments, n = 2, from 2 donors (a total of 143 to 1,857 gonococci were counted in each location across a minimum of three fields in each experiment); OpaD+ P− strain, n = 5, from 5 donors (a total of 50 to 1,697 gonococci were counted in each location across a minimum of three fields in each experiment); S. aureus and L. crispatus, n = 4, from 4 donors (a total of 93 to 311 bacteria were counted in each location across a minimum of three fields in each experiment).
Significance: *, significant (P < 0.05), and **, highly significant (P < 0.001), compared to bacteria alone (two-tailed Student’s t test).
NT, not tested.
DISCUSSION
We have shown that N. gonorrhoeae stimulates PMNs to form NETs but resists NET-mediated killing. Neisseria meningitidis has also been shown to be resistant to NET-mediated killing (48); however, it is not known whether or not N. meningitidis also suppresses the oxidative burst of PMNs in a similar manner. The production of NETs by PMNs in response to gonococcal infection is influenced by the presence of pili and opacity proteins on the surface of the bacteria, but expression of these virulence factors modulates but does not change the response. Despite its being described as an oxidative burst-dependent phenomenon, we observed that the extents of PMN oxidative burst and of NET induction in response to gonococcal infection do not correlate. This discrepancy is a result of gonococci suppressing the oxidative burst of PMNs at high MOIs. That more NETs are observed as oxidative burst is more strongly suppressed shows that gonococci are activating NET formation independently from oxidative signaling. This conclusion is supported by the observation that an MPO inhibitor has no effect on the production of gonococcus-stimulated NETs. These gonococcus-stimulated NETs are more consistent with the rapid NETs reported by the Kubes group in response to staphylococcal infection (49, 50). NETs induced during gonococcal infection are similar to the rapid NETs in at least four ways: (i) they are produced in less than 90 min, (ii) they are produced under conditions where the oxidative burst is suppressed, (iii) they are released prior to lysis of the PMNs, and (iv) PMN nuclei decondense during the process. Thus, NETs produced by human PMNs in response to gonococcal infection might arise through a mechanism similar to that of the rapid NETs that form in response to S. aureus.
The ability of gonococci to induce and suppress the oxidative burst of PMNs has been previously described (24, 36, 43). The lack of correlation between the oxidative burst and NET formation prompted a closer look at ROS burst induction and suppression during PMN infection. We have shown that the ROS burst is elicited in response to infection by gonococci independently of opacity protein or pilus expression. The extent of the induction, however, is increased by the presence of a CEACAM-engaging opacity protein. Because the strains included in this study suppress the oxidative burst at similar MOIs, this result suggests that ROS burst suppression is independent of the presence of pili or OpaD. For the first time, we show that either a PMA or an Opa-induced burst can be suppressed by Opaless, nonpiliated gonococci. Thus, N. gonorrhoeae is influencing PMN behavior by modulating the oxidative burst. These phenotypes will be all the more complex during human infections, where multiple subpopulations of gonococci with different pilus and opacity protein composition on their surfaces are present.
The mechanism of gonococcal suppression of the PMN oxidative burst is not dependent on formyl peptide receptor or chemokine receptor signaling and occurs downstream of PKC activation. The rD-7 peptide (45, 46) abolishes the PMN oxidative burst in response to gonococci, suggesting that CEACAM binding is important to the activation of the oxidative burst but has no effect on suppression. Recent work from the Gray-Owen lab revealed that when human opacity protein-binding CEACAM3 receptors were expressed in mice, the researchers saw that CEACAM receptors were responsible for oxidative burst and PMN migration to the site of infection in response to Opa-expressing gonococci (51). Interestingly, we observed oxidative burst elicited from PMNs, albeit at lower levels, when cells were infected with Opaless gonococci, suggesting that there may CEACAM-independent mechanisms of ROS burst induction with human PMNs or that Opaless gonococci are capable of binding to CEACAM receptors to some extent. Recent evidence has shown that gonococci delay the assembly of NADPH oxidase by interfering with granule fusion with phagosomes (32). This phenomenon has been shown to be influenced by the expression of opacity proteins on the surface of gonococci (33). It is possible that these phenotypes are the mechanism behind the suppression of the oxidative burst. However, our data that show Opaless and OpaD-expressing gonococci suppressing the ROS burst argue against this as a possibility. Despite these inconsistencies, interference with phagosomal maturation will be considered a possible mechanism for oxidative burst suppression in future studies.
Our data suggest a model where the local abundance (MOI) of gonococci has a large influence on the outcome of their interactions with PMNs. While it is possible to try to observe NETs in patient purulent exudates, the background lysis and death of PMNs in patient samples would create a high level of background that would be impossible to control for. Suppressing levels of gonococci (MOIs of >20) are likely to be achieved within microenvironments during infection, where subpopulations of PMNs are subject to a higher relative MOI due to localized gonococcal growth. When gonococci are present in low abundance relative to the PMNs, there will be a preferential uptake of gonococci that express opacity proteins that engage CEACAM receptors (Fig. 6). This would result in an oxidative burst and killing of the intracellular bacteria, as well as the release of cytokines that will recruit more PMNs to the site of infection (51). As PMNs infiltrate, subpopulations of gonococci that are resistant to oxidative killing mechanisms will preferentially multiply. As the relative numbers of gonococci increase, they will suppress the oxidative burst of PMNs and also stimulate NET formation. In addition, while the NETs are unable to kill gonococci, they are still capable of killing L. crispatus and other genital commensal bacteria, potentially opening the niche to further colonization by gonococci due to reduced competition. These PMN-suppressing activities of gonococci may also inhibit PMN surveillance to open the niche for other pathogens. In addition, it may be possible that gonococcal extracellular nuclease (52) will degrade NETs, reducing their ability to prevent dissemination of the infection, but this hypothesis remains to be tested. Thus, gonococci are influencing PMN antimicrobial activities to promote infection, creating a better environment for gonococcal replication, with reduced ROS and less competition from normal flora. Future studies will aim to more accurately define the mechanism of gonococcal suppression of the PMN oxidative burst and oxidative burst-independent NET induction.
FIG 6 .

Model for how gonococci may be driving the outcome of infection through differential effects caused by the localized MOI. The outcome of infection is dictated, in part, by both the MOI and the pilus/Opa composition of the infecting population. See the text for a discussion.
MATERIALS AND METHODS
Growth and maintenance of bacterial strains.
The N. gonorrhoeae strains used in this study (Table 2) are derived from strain FA1090 and are genetically “locked” to stably express or not express pili and opacity proteins (42, 43). The stably piliated strains carry a mutation in a G-quadruplex structure that is required for antigenic variation of the pili (42). The opacity proteins are stochastically expressed due to slipped-strand mispairing at pentameric repeats at the 5′ end of their genes. The stable opacity protein strain was initially deleted of all 11 known opa genes, rendering it Opaless (43). The coding sequence for opaD was then reintroduced into the Opaless background but was mutated to remove the repeats without changing the protein coding (43). The key strains used in this study include piliated and nonpiliated derivatives of a strain that stably expresses only OpaD, “OpaD+ P+” and “OpaD+ P−” strains, and piliated and nonpiliated derivatives of a strain that is genetically Opaless, “Opaless P+” and “Opaless P−” strains. The strains were freshly back-crossed into the parental background to minimize the effect of other phase-variable proteins or LOS variation on the phenotypes. Strains were grown from −80°C freezer stocks for each experiment and never passaged for longer than 36 h prior to infections. Gonococci were maintained on Difco gonococcus base (GCB) agar or gonococcus base liquid (GCBL) medium (Becton Dickinson, Sparks, MD) with Kellogg’s supplements (53). N. gonorrhoeae bacteria used for infection of PMNs were grown in liquid culture to achieve a viable, rapidly dividing population, as described previously (47). Prior to infection, cultures were spun down and washed in RPMI 1640 containing 10% fetal bovine serum (FBS). All strains were incubated at 37°C in an atmosphere containing 5% CO2, unless otherwise stated. S. aureus was maintained on LB medium, and overnight cultures of S. aureus were washed in RPMI prior to infection of PMNs. Lactobacilli were grown on Lactobacillus MRS agar plates (Becton Dickinson, Sparks, MD) or in Lactobacillus MRS broth. Infection MOIs for all bacteria were estimated by the optical density of the input culture and confirmed by serial dilution and plate counts.
TABLE 2 .
Bacterial strains used in this study
| Strain background and designation | Description | Source (references) |
|---|---|---|
| N. gonorrhoeae FA1090 | ||
| Opaless P+ | Opaless phase-locked, locked pilE | A. Criss (42, 43) |
| Opaless P− | Opaless phase-locked, ΔpilE | This study: ΔpilE allele transformed into Opaless P+ strain, above |
| OpaD+ P+ | OpaD+ phase-locked, locked pilE | A. Criss (42, 43) |
| OpaD+ P− | OpaD+ phase-locked, ΔpilE | This study: ΔpilE allele transformed into OpaD+ P+ strain, above |
| Opaless P+ ΔkatA | Opaless phase-locked, locked pilE, katA::eph | katA allele, transformed into Opaless P+ strain |
| OpaD+ P+ ΔkatA | OpaD+ phase-locked, locked pilE, katA::eph | katA allele, transformed into OpaD+ P+ strain |
| mCherry | PkatA-mCherry | This study; strain had to be freshly constructed for each use |
| S. aureus | ATCC 25923 | ATCC |
| L. crispatus | ATCC 33820, VPI 3199 | ATCC |
Ethics statement.
All protocols involving human subjects were approved by the Institutional Review Board at Northwestern University Feinberg School of Medicine, All subjects included in this study were adults and gave written informed consent prior to enrollment in the study.
Isolation of primary human PMNs.
The PMN isolation protocol has been described previously (47). Briefly, blood from healthy volunteers was obtained via venipuncture of the forearm using the BD Vacutainer Safety-Lok blood collection set (Becton Dickinson, Sparks, MD), collecting venous blood in Vacutainers containing 158 USP units of sodium heparin (Becton Dickinson, Sparks, MD). PMNs were isolated using density centrifugation and ultimately resuspended in Dulbecco’s phosphate-buffered saline supplemented with 0.1% glucose (DPBS+G; Gibco, Life Technologies, Carlsbad, CA). PMNs were verified for purity and enumerated by manual counting using a hemocytometer. PMNs were seeded in RPMI with glutamine without phenol red (Gibco, Life Technologies, Carlsbad, CA) and supplemented with 5% heat-inactivated fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA). Cells were incubated at 37°C with 5% atmospheric CO2.
Luminol assays.
Luminol-dependent chemiluminescence assays were performed as described previously (24, 36). PMNs were seeded at a concentration of 106 in 200 µl of RPMI plus FBS containing 100 µM luminol. Infecting bacteria and chemical treatments were added as described in the figure legends. Phorbol myristate acetate (PMA; Sigma, St. Louis, MO) was used as a positive control at a concentration of 10 µg/ml, and diphenyleneiodonium (DPI; an inhibitor of NADPH oxidase) was used to verify that the oxidative burst was NADPH oxidase dependent. Luminescence was read in a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA) with settings of: 1,000-ms integration, reading every 3 min for 2 h at 37°C. Luminol data are largely presented as area under the curve (AUC) luminol measurements for ease of comparison.
Immunofluorescence microscopy.
PMNs were seeded at a concentration of 106/ml on poly-l-lysine (Sigma, St. Louis, MO)-coated coverslips in 12-well tissue culture plates and infected with liquid-grown gonococci. Infections were centrifuged at a relative centrifugal force (RCF) of 130 for 5 min to facilitate contact between gonococci and PMNs and synchronize the infection. Infected cells were incubated at 37°C for 90 min, rinsed with 1× PBS, and fixed with 4% paraformaldehyde for 5 min, followed by two washes with PBS. Slides were blocked with PBS containing 10% normal goat serum overnight at 4°C. Antibody staining and subsequent washes were performed with coverslips upside down on 100-µl drops of reagent. Rabbit IgG primary antibodies (Abcam, Cambridge, MA) were diluted in PBS containing 1% goat serum: anti-histone H2A (ab18255; 1:200), anticathelicidin (LL-37, ab64892; 1:400), anti-cathepsin G (ab64891; 1:100), and anti-neutrophil elastase (ab68672; 1:100). Primary antibody staining was performed at room temperature for 1 h. Coverslips were washed with PBS three times and then stained with a goat anti-rabbit, fluorescein isothiocyanate (FITC) conjugate (Jackson ImmunoResearch, West Grove, PA; catalog no. 111-096-144; 1:250) for 1 h in the dark. After staining, coverslips were washed three times and mounted on slides with 5 µl Vectashield with DAPI (Vector Labs, Burlingame, CA). Coverslips were sealed with nail polish and stored at 4°C or on ice until viewed in the microscope.
Microscopy.
Microscopy was performed on a Nikon 90i upright fluorescence microscope equipped with a CoolSNAP HQ2 charge-coupled device (CCD) camera (Photometrics, Tucson, AZ) and an X-Cite Series 120 PCQ fluorescence light source (EXFO/Lumen Dynamics, Mississauga, ON, Canada). Image acquisition was performed using NIS Elements software (Nikon Instruments, Melville, NY), and image analysis was performed using NIS Elements and ImageJ (NIH.gov). For most experiments, image stacks ranging from 10 to 30 half-micrometer slices were imaged to better visualize the infected PMNs. The quantification of NETs using anti-H2A immunofluorescence staining was done by measuring the integrated density (a measurement that takes into account both area and intensity of staining) of the anti-H2A staining using ImageJ software (NIH). The integrated density of anti-H2A staining was normalized to the integrated density of DAPI staining as described in the text (Fig. 2). NET/field observations were made under ×400 magnification, and the number of NETs in each field was counted by hand. The NET/field observations allowed for higher-throughput analysis (Fig. 3C and 5D). The myeloperoxidase inhibitor (MPOI) is 4-aminobenzhydrazide and was used at a final concentration of 100 µM. In microscopy experiments, PMA was used at a 50-ng/ml concentration.
LIVE/DEAD staining.
Bacterial viability was determined using the Molecular Probes LIVE/DEAD BacLight bacterial viability kit (L7012; Life Technologies, Carlsbad, CA). PMNs were seeded on glass coverslips at a concentration of 5 × 105/ml in a 1-milliliter volume. PMA (Sigma, St. Louis, MO) was added at a final concentration of 2 µg/ml at the time of PMN seeding to stimulate uniform NET formation. In control experiments, 2 µM DPI was also added at the time of seeding to prevent the oxidative burst. Two hours of treatment with PMA and/or DPI at 37°C was followed by infection at high MOIs (100 to 200) with gonococci, S. aureus, or L. crispatus. Infected cells were incubated at 37°C for 60 min, followed by removal of infected cell supernatant. The infected cells were washed with 1 milliliter of fresh 37°C RPMI. The wash medium was removed, and stain mix was added to the wells. Stain mixture was prepared in PBS and contained dye components A and B at a ratio of 1:2, which was empirically determined to achieve superior staining of gonococci in the presence of PMNs. Infected cells were stained at 37°C for 30 min, washed twice, fixed with 4% paraformaldehyde (PFA), washed again, and then mounted using ProLong Gold mounting medium (Life Technologies, Carlsbad, CA). The mounting medium was allowed to set overnight at room temperature and then sealed with nail polish. Microscopy was performed as described above, and the image stacks were hand scored by visually counting live and dead bacteria and noting their localization relative to the PMNs: NET associated (in contact with NETs but not directly with PMNs), PMN associated (in contact with or inside PMNs but not in contact with NETs), or extracellular (in contact with neither NETs or cells).
SUPPLEMENTAL MATERIAL
Gonococcal pili and Opa variants elicit NETs from PMNs. The OpaD+ P+, OpaD+ P−, Opaless P+, or Opaless P− strain was used to infect primary human PMNs, and the infected cells were stained using an antibody against histone H2A and visualized with an FITC-conjugated secondary antibody. (A) Gonococcus-infected PMNs were stained with only secondary antibody as a negative control. (B) Uninfected PMNs. (C to F) PMNs infected with OpaD+ P+ (C), OpaD+ P− (D), Opaless P+ (E), and Opaless P− (F) gonococci. Images are a representative from one of 14 independent experiments using PMNs from 7 different donors. Arrows indicate NETs. Bar, 25 µm. All images are at the same scale. Download
Heat-killed (HK) gonococci stimulate PMNs to form NETs. Gonococci were heat killed for 30 min at 65°C, washed, resuspended in RPMI, and used to infect PMNs. After 120 min, the infected cells were fixed with PFA and mounted with Vectashield with DAPI. Micrographs are labeled according to treatment condition and indicate that uninfected PMNs produce minimal NETs, while HK gonococci elicit NET production from PMNs. HK gonococcal infections are representative of two independent experiments with PMNs from two individual donors. Black-and-white images of the DAPI channel were inverted for ease of visualization and comparison. Bars, 25 µm. Arrows indicate NETs. Download
Receptor antagonists do not affect gonococcus-stimulated oxidative burst. (A) PMNs were infected with Opaless P+ gonococci alone or in the presence of cyclosporine H SB225002 and plerixafor and assessed for LDCL accumulation (ROS burst). (B) Cyclosporine H SB225002 and plerixafor were combined into a single treatment (light gray bars). Data are a representative experiment from at least three independent experiments using PMNs from at least 3 individual donors. Download
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 AI044239 and R37 AI033493 to H.S.S. C.W.G. was partially supported by NIH institutional training grant T32 AI747614.
rD-7 peptide was generously provided by D. Hill and M. Virji (University of Bristol, United Kingdom). Alison Criss (University of Virginia) provided the Opaless and OpaD+ strains. Paul Schumacker (Northwestern University) gave advice regarding neutrophil signaling pathways. Teng-Leong Chew of the Cell Imaging Facility and Nikon Imaging Center at Northwestern University provided helpful discussion on quantifying fluorescence image data. Thanks go to M. Anderson, A. Chen, and members of the Seifert group for critical reading of the manuscript.
Footnotes
Citation Gunderson CW, Seifert HS. 2015. Neisseria gonorrhoeae elicits extracellular traps in primary neutrophil culture while suppressing the oxidative burst. mBio 6(1):e02452-14. doi:10.1128/mBio.02452-14.
REFERENCES
- 1.Centers for Disease Control and Prevention 2009. Sexually transmitted disease surveillance, 2008. US Department of Health and Human Services, Atlanta, GA. [Google Scholar]
- 2.Akasaka S, Muratani T, Yamada Y, Inatomi H, Takahashi K, Matsumoto T. 2001. Emergence of cephem- and aztreonam-high-resistant Neisseria gonorrhoeae that does not produce beta-lactamase. J Infect Chemother 7:49–50. doi: 10.1007/s1015610070049. [DOI] [PubMed] [Google Scholar]
- 3.Bolan GA, Sparling PF, Wasserheit JN. 2012. The emerging threat of untreatable gonococcal infection. N Engl J Med 366:485–487. doi: 10.1056/NEJMp1112456. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention 2013. Antibiotic resistance threats in the United States, 2013. US Department of Health and Human Services, Atlanta, GA. [Google Scholar]
- 5.Criss AK, Seifert HS. 2012. A bacterial siren song: intimate interactions between Neisseria and neutrophils. Nat Rev Microbiol 10:178–190. doi: 10.1038/nrmicro2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hook EW III, Holmes KK. 1985. Gonococcal infections. Ann Intern Med 102:229–243. doi: 10.7326/0003-4819-102-2-229. [DOI] [PubMed] [Google Scholar]
- 7.Criss AK, Katz BZ, Seifert HS. 2009. Resistance of Neisseria gonorrhoeae to non-oxidative killing by adherent human polymorphonuclear leucocytes. Cell Microbiol 11:1074–1087. doi: 10.1111/j.1462-5822.2009.01308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seib KL, Wu HJ, Kidd SP, Apicella MA, Jennings MP, McEwan AG. 2006. Defenses against oxidative stress in Neisseria gonorrhoeae: a system tailored for a challenging environment. Microbiol Mol Biol Rev 70:344–361. doi: 10.1128/MMBR.00044-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards JL, Brown EJ, Uk-Nham S, Cannon JG, Blake MS, Apicella MA. 2002. A co-operative interaction between Neisseria gonorrhoeae and complement receptor 3 mediates infection of primary cervical epithelial cells. Cell Microbiol 4:571–584. doi: 10.1046/j.1462-5822.2002.t01-1-00215.x. [DOI] [PubMed] [Google Scholar]
- 10.Swanson J, Robbins K, Barrera O, Corwin D, Boslego J, Ciak J, Blake M, Koomey JM. 1987. Gonococcal pilin variants in experimental gonorrhea. J Exp Med 165:1344–1357. doi: 10.1084/jem.165.5.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seifert HS, Wright CJ, Jerse AE, Cohen MS, Cannon JG. 1994. Multiple gonococcal pilin antigenic variants are produced during experimental human infections. J Clin Invest 93:2744–2749. doi: 10.1172/JCI117290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dempsey JA, Litaker W, Madhure A, Snodgrass TL, Cannon JG. 1991. Physical map of the chromosome of Neisseria gonorrhoeae FA1090 with locations of genetic markers, including opa and pil genes. J Bacteriol 173:5476–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stern A, Brown M, Nickel P, Meyer TF. 1986. Opacity genes in Neisseria gonorrhoeae: control of phase and antigenic variation. Cell 47:61–71. doi: 10.1016/0092-8674(86)90366-1. [DOI] [PubMed] [Google Scholar]
- 14.Murphy GL, Connell TD, Barritt DS, Koomey M, Cannon JG. 1989. Phase variation of gonococcal protein II: regulation of gene expression by slipped-strand mispairing of a repetitive DNA sequence. Cell 56:539–547. doi: 10.1016/0092-8674(89)90577-1. [DOI] [PubMed] [Google Scholar]
- 15.James JF, Swanson J. 1978. Studies on gonococcus infection. XIII. Occurrence of color/opacity colonial variants in clinical cultures. Infect Immun 19:332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jerse AE, Cohen MS, Drown PM, Whicker LG, Isbey SF, Seifert HS, Cannon JG. 1994. Multiple gonococcal opacity proteins are expressed during experimental urethral infection in the male. J Exp Med 179:911–920. doi: 10.1084/jem.179.3.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Virji M. 2009. Pathogenic neisseriae: surface modulation, pathogenesis and infection control. Nat Rev Microbiol 7:274–286. doi: 10.1038/nrmicro2097. [DOI] [PubMed] [Google Scholar]
- 18.Massari P, Henneke P, Ho Y, Latz E, Golenbock DT, Wetzler LM. 2002. Cutting edge: immune stimulation by neisserial porins is Toll-like receptor 2 and MyD88 dependent. J Immunol 168:1533–1537. doi: 10.4049/jimmunol.168.4.1533. [DOI] [PubMed] [Google Scholar]
- 19.Fisette PL, Ram S, Andersen JM, Guo W, Ingalls RR. 2003. The Lip lipoprotein from Neisseria gonorrhoeae stimulates cytokine release and NF-kappaB activation in epithelial cells in a Toll-like receptor 2-dependent manner. J Biol Chem 278:46252–46260. doi: 10.1074/jbc.M306587200. [DOI] [PubMed] [Google Scholar]
- 20.Kaparakis M, Turnbull L, Carneiro L, Firth S, Coleman HA, Parkington HC, Le Bourhis L, Karrar A, Viala J, Mak J, Hutton ML, Davies JK, Crack PJ, Hertzog PJ, Philpott DJ, Girardin SE, Whitchurch CB, Ferrero RL. 2010. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol 12:372–385. doi: 10.1111/j.1462-5822.2009.01404.x. [DOI] [PubMed] [Google Scholar]
- 21.Mavrogiorgos N, Mekasha S, Yang Y, Kelliher MA, Ingalls RR. 2014. Activation of NOD receptors by Neisseria gonorrhoeae modulates the innate immune response. Innate Immun 20:377–389. doi: 10.1177/1753425913493453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramsey KH, Schneider H, Cross AS, Boslego JW, Hoover DL, Staley TL, Kuschner RA, Deal CD. 1995. Inflammatory cytokines produced in response to experimental human gonorrhea. J Infect Dis 172:186–191. doi: 10.1093/infdis/172.1.186. [DOI] [PubMed] [Google Scholar]
- 23.Fichorova RN, Desai PJ, Gibson FC III, Genco CA. 2001. Distinct proinflammatory host responses to Neisseria gonorrhoeae infection in immortalized human cervical and vaginal epithelial cells. Infect Immun 69:5840–5848. doi: 10.1128/IAI.69.9.5840-5848.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen A, Seifert HS. 2011. Neisseria gonorrhoeae-mediated inhibition of apoptotic signaling in polymorphonuclear leukocytes. Infect Immun 79:4447–4458. doi: 10.1128/IAI.01267-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray-Owen SD, Lorenzen DR, Haude A, Meyer TF, Dehio C. 1997. Differential Opa specificities for CD66 receptors influence tissue interactions and cellular response to Neisseria gonorrhoeae. Mol Microbiol 26:971–980. doi: 10.1046/j.1365-2958.1997.6342006.x. [DOI] [PubMed] [Google Scholar]
- 26.Sadarangani M, Pollard AJ, Gray-Owen SD. 2011. Opa proteins and CEACAMs: pathways of immune engagement for pathogenic Neisseria. FEMS Microbiol Rev 35:498–514. doi: 10.1111/j.1574-6976.2010.00260.x. [DOI] [PubMed] [Google Scholar]
- 27.Sarantis H, Gray-Owen SD. 2012. Defining the roles of human carcinoembryonic antigen-related cellular adhesion molecules during neutrophil responses to Neisseria gonorrhoeae. Infect Immun 80:345–358. doi: 10.1128/IAI.05702-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hauck CR, Meyer TF, Lang F, Gulbins E. 1998. CD66-mediated phagocytosis of Opa52 Neisseria gonorrhoeae requires a Src-like tyrosine kinase- and Rac1-dependent signalling pathway. EMBO J 17:443–454. doi: 10.1093/emboj/17.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Booth JW, Telio D, Liao EH, McCaw SE, Matsuo T, Grinstein S, Gray-Owen SD. 2003. Phosphatidylinositol 3-kinases in carcinoembryonic antigen-related cellular adhesion molecule-mediated internalization of Neisseria gonorrhoeae. J Biol Chem 278:14037–14045. doi: 10.1074/jbc.M211879200. [DOI] [PubMed] [Google Scholar]
- 30.Sarantis H, Gray-Owen SD. 2007. The specific innate immune receptor CEACAM3 triggers neutrophil bactericidal activities via a Syk kinase-dependent pathway. Cell Microbiol 9:2167–2180. doi: 10.1111/j.1462-5822.2007.00947.x. [DOI] [PubMed] [Google Scholar]
- 31.Johnson MB, Criss AK. 2011. Resistance of Neisseria gonorrhoeae to neutrophils. Front Microbiol 2:77. doi: 10.3389/fmicb.2011.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson MB, Criss AK. 2013. Neisseria gonorrhoeae phagosomes delay fusion with primary granules to enhance bacterial survival inside human neutrophils. Cell Microbiol 15:1323–1340. doi: 10.1111/cmi.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smirnov A, Daily KP, Criss AK. 2014. Assembly of NADPH oxidase in human neutrophils is modulated by the opacity-associated protein expression state of Neisseria gonorrhoeae. Infect Immun 82:1036–1044. doi: 10.1128/IAI.00881-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simons MP, Nauseef WM, Griffith TS, Apicella MA. 2006. Neisseria gonorrhoeae delays the onset of apoptosis in polymorphonuclear leukocytes. Cell Microbiol 8:1780–1790. doi: 10.1111/j.1462-5822.2006.00748.x. [DOI] [PubMed] [Google Scholar]
- 35.Johnson SR, Steiner BM, Cruce DD, Perkins GH, Arko RJ. 1993. Characterization of a catalase-deficient strain of Neisseria gonorrhoeae: evidence for the significance of catalase in the biology of N. gonorrhoeae. Infect Immun 61:1232–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Criss AK, Seifert HS. 2008. Neisseria gonorrhoeae suppresses the oxidative burst of human polymorphonuclear leukocytes. Cell Microbiol 10:2257–2270. doi: 10.1111/j.1462-5822.2008.01205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. 2004. Neutrophil extracellular traps kill bacteria. Science 303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 38.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. 2007. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, Wahn V, Papayannopoulos V, Zychlinsky A. 2011. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood 117:953–959. doi: 10.1182/blood-2010-06-290171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, Noppen S, Delforge M, Willems J, Vandenabeele P. 2011. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 21:290–304. doi: 10.1038/cr.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A. 2009. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog 5:e1000639. doi: 10.1371/journal.ppat.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cahoon LA, Seifert HS. 2009. An alternative DNA structure is necessary for pilin antigenic variation in Neisseria gonorrhoeae. Science 325:764–767. doi: 10.1126/science.1175653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ball LM, Criss AK. 2013. Constitutively Opa-expressing and Opa-deficient Neisseria gonorrhoeae differentially stimulate and survive exposure to human neutrophils. J Bacteriol 195:2982–2990. doi: 10.1128/JB.00171-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson SR, Steiner BM, Perkins GH. 1996. Cloning and characterization of the catalase gene of Neisseria gonorrhoeae: use of the gonococcus as a host organism for recombinant DNA. Infect Immun 64:2627–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hill DJ, Edwards AM, Rowe HA, Virji M. 2005. Carcinoembryonic antigen-related cell adhesion molecule (CEACAM)-binding recombinant polypeptide confers protection against infection by respiratory and urogenital pathogens. Mol Microbiol 55:1515–1527. doi: 10.1111/j.1365-2958.2005.04487.x. [DOI] [PubMed] [Google Scholar]
- 46.Hill DJ, Virji M. 2003. A novel cell-binding mechanism of Moraxella catarrhalis ubiquitous surface protein UspA: specific targeting of the N-domain of carcinoembryonic antigen-related cell adhesion molecules by UspA1. Mol Microbiol 48:117–129. doi: 10.1046/j.1365-2958.2003.03433.x. [DOI] [PubMed] [Google Scholar]
- 47.Stohl EA, Criss AK, Seifert HS. 2005. The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol Microbiol 58:520–532. doi: 10.1111/j.1365-2958.2005.04839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lappann M, Danhof S, Guenther F, Olivares-Florez S, Mordhorst IL, Vogel U. 2013. In vitro resistance mechanisms of Neisseria meningitidis against neutrophil extracellular traps. Mol Microbiol 89:433–449. doi: 10.1111/mmi.12288. [DOI] [PubMed] [Google Scholar]
- 49.Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FH, Surette MG, Sugai M, Bowden MG, Hussain M, Zhang K, Kubes P. 2010. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol 185:7413–7425. doi: 10.4049/jimmunol.1000675. [DOI] [PubMed] [Google Scholar]
- 50.Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, Malawista SE, de Boisfleury Chevance A, Zhang K, Conly J, Kubes P. 2012. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med 18:1386–1393. doi: 10.1038/nm.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sintsova A, Sarantis H, Islam EA, Sun CX, Amin M, Chan CH, Stanners CP, Glogauer M, Gray-Owen SD. 2014. Global analysis of neutrophil responses to Neisseria gonorrhoeae reveals a self-propagating inflammatory program. PLoS Pathog 10:e1004341. doi: 10.1371/journal.ppat.1004341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steichen CT, Cho C, Shao JQ, Apicella MA. 2011. The Neisseria gonorrhoeae biofilm matrix contains DNA, and an endogenous nuclease controls its incorporation. Infect Immun 79:1504–1511. doi: 10.1128/IAI.01162-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kellogg DS Jr, Peacock WL Jr, Deacon WE, Brown L, Pirkle DI. 1963. Neisseria gonorrhoeae. I. Virulence genetically linked to clonal variation. J Bacteriol 85:1274–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gonococcal pili and Opa variants elicit NETs from PMNs. The OpaD+ P+, OpaD+ P−, Opaless P+, or Opaless P− strain was used to infect primary human PMNs, and the infected cells were stained using an antibody against histone H2A and visualized with an FITC-conjugated secondary antibody. (A) Gonococcus-infected PMNs were stained with only secondary antibody as a negative control. (B) Uninfected PMNs. (C to F) PMNs infected with OpaD+ P+ (C), OpaD+ P− (D), Opaless P+ (E), and Opaless P− (F) gonococci. Images are a representative from one of 14 independent experiments using PMNs from 7 different donors. Arrows indicate NETs. Bar, 25 µm. All images are at the same scale. Download
Heat-killed (HK) gonococci stimulate PMNs to form NETs. Gonococci were heat killed for 30 min at 65°C, washed, resuspended in RPMI, and used to infect PMNs. After 120 min, the infected cells were fixed with PFA and mounted with Vectashield with DAPI. Micrographs are labeled according to treatment condition and indicate that uninfected PMNs produce minimal NETs, while HK gonococci elicit NET production from PMNs. HK gonococcal infections are representative of two independent experiments with PMNs from two individual donors. Black-and-white images of the DAPI channel were inverted for ease of visualization and comparison. Bars, 25 µm. Arrows indicate NETs. Download
Receptor antagonists do not affect gonococcus-stimulated oxidative burst. (A) PMNs were infected with Opaless P+ gonococci alone or in the presence of cyclosporine H SB225002 and plerixafor and assessed for LDCL accumulation (ROS burst). (B) Cyclosporine H SB225002 and plerixafor were combined into a single treatment (light gray bars). Data are a representative experiment from at least three independent experiments using PMNs from at least 3 individual donors. Download