

Abstract
The role of complement in inflammatory bowel disease (IBD) has been studied primarily using acute models, and it is unclear how complement affects processes in more relevant chronic models of IBD in which modulation of adaptive immunity and development of fibrosis have pathogenic roles. Using mice deficient in C1q/mannose-binding lectin (MBL) or C3, we demonstrated an important role for these opsonins and/or the classical pathway C3 convertase in providing protection against mucosal injury and infection in a model of chronic dextran sulphate sodium (DSS)-induced colitis. In contrast, deficiency of the alternative pathway (fB–/– mice) had significantly less impact on injury profiles. Consequently, the effect of a targeted inhibitor of the alternative pathway was investigated in a therapeutic protocol. Following the establishment of colitis, mice were treated with CR2-fH during subsequent periods of DSS treatment and acute injury (modelling relapse). CR2-fH significantly reduced complement activation, inflammation and injury in the colon, and additionally reduced fibrosis. Alternative pathway inhibition also altered the immune response in the chronic state in terms of reducing numbers of B cells, macrophages and mature dendritic cells in the lamina propria. This study indicates an important role for the alternative pathway of complement in the pathogenesis and the shaping of an immune response in chronic DSS-induced colitis, and supports further investigation into the use of targeted alternative pathway inhibition for the treatment of IBD.
Keywords: colon, complement, inflammation, inflammatory bowel disease
Introduction
Inflammatory bowel disease (IBD) is a chronic inflammatory condition that comprises both ulcerative colitis and Crohn's disease in humans, and is characterized by periods of relapse and remission. The exact etiology of IBD is unclear, but inflammation and injury is generally believed to be the result of a dysregulated mucosal immune response to mucosal antigens.
The complement system is involved in the pathology of several inflammatory diseases and disease states, and there is mounting clinical and evidence that complement plays an important role in IBD 1–9. The vast majority of the experimental evidence comes from work done in rodent models of acute dextran sodium sulfate (DSS)-induced colitis 10–15, and only a single study has investigated the role of complement in a chronic model 16. Thus, while it seems clear that complement activation in the gut leads to acute mucosal injury, it is less clear how complement may impact disease as it progresses to chronicity when other factors known to be modulated by complement may come into play. These other factors include host defense (important if mucosal barrier is compromised), wound healing and tissue repair, and the potential modulation of an adaptive immune response. Indeed, even in an acute model of DSS-induced colitis, we have shown that complement can play a dual role in pathogenesis, with the classical and/or lectin pathway of complement providing an important protective role in terms of host defense 12.
Complement can be activated by the classical, lectin or alternative pathways, all of which culminate in the cleavage of C3 and the subsequent generation of C3 opsonins and the complement activation products C3a, C5a and the membrane attack complex (MAC). Under certain conditions, C5a and the MAC can also be generated by proteases independent of C3 activation 17,18. Complement is implicated in the pathogenesis of IBD by studies showing that C3 deficient mice are protected from acute DSS-induced colitis 15,19, and that complement inhibitor (decay accelerating factor) deficient mice have increased susceptibility to acute DSS-induced colitis 13. Several studies using either C5/C5aR deficient mice, anti-C5 mAb or C5a receptor antagonists have reported a pathogenic role for C5a in the pathogenesis of IBD 3,20,21, although one study reported that C5 deficiency increased susceptibility to DSS-induced colitis 14. The C3aR has also been shown to play a role in the pathogenesis of acute DSS-induced colitis in BALB/c mice, although not C57BL/6 mice 10. In a single study that investigated the role of complement in a chronic model of IBD, C5aR deficiency was not protective, although C5aR deficiency provided protection against acute DSS-induced colitis 16.
Thus, there is not a clearly delineated role for complement in murine IBD, and considering data indicates there exists a balance between pathogenic and protective roles for complement, it is not clear if complement inhibition would be therapeutic option in a clinical setting of chronic disease with periods of relapse and remission. In this study we investigated the effect of complement deficiency and complement inhibition in a 68 day chronic model of DSS-induced colitis. The complement inhibitor used in these studies was complement receptor 2 (CR2)-fH, a previously described inhibitor of the alternative pathway that specifically targets to sites of complement activation 22. We have shown previously that CR2-mediated targeting of complement inhibitors obviates the need for systemic complement inhibition and does not increase host susceptibility to infection 22,23, a potentially important characteristic in a disease model where the integrity of the intestinal mucosal barrier is compromised.
Materials and methods
Mice
Wild type C57BL/6 mice were purchased from Jackson Labs (Bar Harbor, ME). C3–/– and fB–/– mice on C57BL/6 background were obtained from an in house breeding colony. A breeding pair of double deficient C1qα–/– MBL-A/C–/– mice on C57BL/6 background [referred to as C1q/mannose-binding lecton (MBL–/–)] were kindly provided by Dr. Kazue Takahashi (Massachusetts General Hospital for Children, Boston, MA) and bred in house. All animals used were female between 8–10 weeks old. Animals were maintained under standard laboratory conditions, and all animal procedures were approved by the Medical University of South Carolina (MUSC) Institutional Animal Care and Use Committee, in accordance with the guidelines of the National Institutes of Health Guide for Care and Use of Laboratory Animals.
DSS-induced colitis and CR2-fH treatment protocol
Chronic colitis was induced by 4 cycles of oral administration of 3% (w/v) dextran sodium sulfate (DSS, MP Biomedical, Solon, OH) for 7 days followed by normal drinking water for 10 days. Sham control mice received normal drinking water throughout. During cycles 2–4, mice were treated with 0·25 mg of CR2-fH i.p. on day 1 of 3% DSS water administration and every 48 h thereafter for the duration of DSS treatment. Mice were monitored every other day for weight loss. At the end of cycle 4 DSS water or cycle 4 rest, mice were sacrificed, colons removed and colon length measured. Colitis was assessed by percent weight loss, colon length and histological damage. The fusion protein CR2-fH was prepared and purified as described previously 22. The dose of CR2-fH was determined by previously published dose response data in intestinal ischemia reperfusion injury (IRI) 22 and acute colitis 12.
Histology
Formalin fixed colon sections were stained with H&E. H&E stained sections were scored according to a previously described scoring system 12 by a blinded observer. A cumulative scale with a maximum score of 10 was used. Three parameters were assessed: (i) severity of inflammation (0, none; 1, slight; 2, moderate; and 3, severe); (ii) depth of injury (0, none; 1, mucosal; 2, mucosal and submucosal; and 3, transmural); and (iii) crypt damage (0, none; 1, basal one-third damaged; 2, basal two-thirds damaged; 3, only surface epithelium intact; and 4, complete loss of crypt and epithelium).
Collagen
The collagen content in colons following induction of colitis was assessed using a Picrosirius red stain kit (Polysciences, Inc, Warrington, PA) on formalin fixed colon sections. The percentage of positive red staining was assessed by ImageJ software (NIH, Bethesda, MD) and calculated by summation of 5 high power random fields per section. Analyses were performed by an observer blinded to experimental groups.
Complement activation and cytokine analysis
Complement activation in the colon was assessed by C5a levels in colon homogenates using a mouse C5a ELISA (R&D Systems, Minneapolis, MN, and BD biosciences). Cytokine levels in colon homogenates were analyzed by IL-6, IL-10, IFNγ (BD biosciences) and IL-17 (R&D systems) specific ELISAs according to the manufacturer's protocols.
Tissue isolation and single-cell preparations
The lamina propria was isolated from colons by using a collagenase based digestion and separation protocol. Briefly, the colon was removed, washed and cut into pieces. The colon pieces were subsequently digested with collagenase type VIII. The resulting digest was washed and filtered through a 100 micron cell strainer followed by a 40 micron cell strainer. Single cell preparations from lymph nodes and splenocytes were prepared by mechanical disruption of the respective tissue, followed by red cell lysis. Cell preparations were washed and filtered through a 40 micron cell strainer. Single cell populations were counted and resuspended at 1 × 107 cells/ml for flow cytometry analysis.
Flow cytometry analysis of cell populations
Immune cell populations within the lamina propria, mesenteric lymph nodes and spleen were analyzed by flow cytometry. Staining for specific cell types were performed using the following antibodies; for B cells, anti-CD19; for mature DC, anti-CD11c, anti-CD80 and anti-CD86; for M1/M2 macrophages, anti-F4/80 (total), anti-CD86 (M1) and anti-CD206 (M2); for CD8+ T cells, anti-CD3e and anti-CD8 and CD4+ T cells, anti-CD3e and anti-CD4. Antibodies were purchased from BD biosciences, except CD206 (Biolegend, San Diego, CA). Samples were analyzed on a BD LSRFortessa flow cytometer (BD biosciences) and analyzed with FlowJo 9·3.3 software (TreeStar, Inc.).
Statistical analysis
All data were subjected to statistical analysis using Prism Software version 5 (GraphPad, San Diego, CA). Comparison between multiple groups was done using one-way ANOVA with Bonferroni's multiple comparison test (parametrical) or Dunn's multiple comparison test (non-parametrical). Comparisons between two groups were performed by Students t test (parametric) or Mann-Whitney U test (non-parametric). A P value of <0·05 was considered significant.
Results
The effect of complement deficiencies on chronic DSS-induced colitis
We demonstrated previously that C3 or fB deficiency is protective in a 5 day model of acute DSS-induced colitis 12. However, injury and immune profiles change with progression to chronic disease in this model, and we investigated the effect of various complement deficiencies on chronic DSS-induced colitis. To induce chronic colitis, mice were given 4 × 7 day cycles of DSS (3%) with a 10 day rest period between DSS treatments. Mice deficient in C1q/MBL (blocked in classical and lectin pathways of activation) all died at the beginning of the second DSS cycle, and by the end of the fourth and last rest period, survival of C3 deficient mice (blocked in all pathways at C3 activation step) was approximately 45%. Survival of fB–/– mice (blocked in alternative pathway activation) was approximately 85% and was not statistically different to that of wt mice (Fig. 1). Analysis of surviving DSS treated mice at the end of the last cycle revealed that C3–/– mice had a significantly worse outcome compared to wt and fB–/– mice in terms of weight change, colon length and stool consistency (Table 1). In addition, histopathological damage (characterized by crypt loss, epithelial ulceration and infiltration of inflammatory cells into the bowel wall) was significantly worse in C3–/– mice with chronic DSS-induced colitis compared with either wt or fB–/– mice. C3–/– mice also had increased neutrophil infiltration as measured by MPO content in bowel samples compared to wt and fB–/– mice (Fig. 1).
Fig 1.
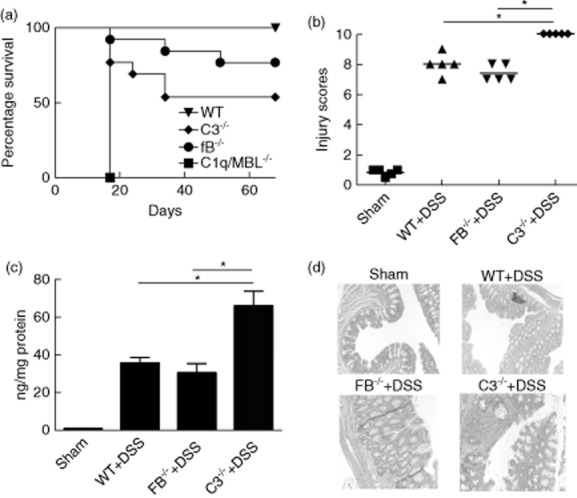
Survival of and histological assessment of injury and neutrophil infiltration in complement deficient mice during induction of chronic dextran sulphate sodium (DSS)-induced colitis. (a) Wild-type (WT) or complement-deficient mice were subjected to the DSS protocol [without complement receptor 2 (CR2)-fH treatment], and survival was monitored; n = 13, *P < 0·05 versus WT. (b) Sections of colons isolated from mice after fourth cycle of DSS and the rest were stained with haematoxylin and eosin (H&E) and injury and inflammation quantified (see Materials and methods). Horizontal bar indicates mean, n = 5, *P < 0·05. (c) Myeloperoxidase (MPO) content in colon samples isolated from mice after fourth cycle and normalized for protein content. Mean ± standard error of the mean (s.e.m.); n = 5, *P < 0·05. (d) Representative H&E-stained sections.
Table 1.
Clinical effects and colon length at end of last cycle in complement-deficient mice with chronic dextran sulphate sodium (DSS)-induced colitis
| Group | Weight change | Stool | Colon length (cm) |
|---|---|---|---|
| WT | 22·73% gain | Normal | 6·8 |
| WT+ DSS | 11·8% gain | Soft | 5·46 |
| FB–/– | 18% gain | Normal | 6·5 |
| FB–/– + DSS | 13% gain | Soft | 5·6 |
| C3–/– | 21% gain | Normal | 6·6 |
| C3–/– + DSS | 6% gain* | Bloody diarrhoea | 2·25# |
P < 0·01 versus wild-type (WT) + DSS;
P < 0·01 versus WT + DSS and fB–/– + DSS. Results expressed as mean, n = 8–12.
We further assessed colon inflammation by measuring the levels of 3 inflammatory cytokines associated with DSS-induced colitis. Levels of IFNγ in surviving DSS treated mice were not significantly different between any group, but compared to wt mice, levels of TNFα and IL-12 were significantly increased in C3–/– mice (markedly so for TNFα). In fB–/– mice, TNFα levels were similar to wt mice, and IL-12 levels were significantly lower compared to C3–/– mice (Fig. 2).
Fig 2.
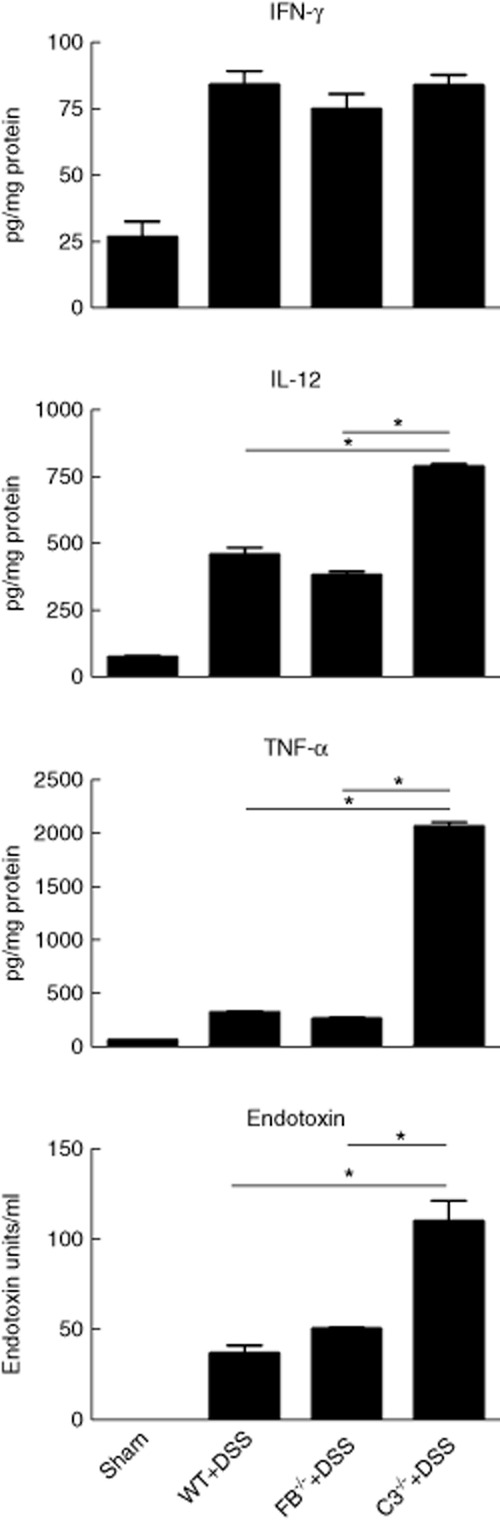
Cytokine levels in the colon and serum endotoxin levels in complement-deficient mice with chronic dextran sulphate sodium (DSS)-induced colitis. Cytokines were measured in homogenates of colons isolated after fourth cycle of DSS and rest. Mean ± standard error of the mean (s.e.m.); n = 5, *P < 0·05. Endotoxin was measured in serum prepared from blood collected after fourth cycle of DSS and rest. Mean ± s.e.m.; n = 5, P < 0·05.
Our previous data demonstrated that in a high dose and extended 12 day model of acute (5%) DSS-induced colitis, there was 100% mortality of C3–/– and fB–/– mice during the recovery period 12. The cause of mortality was linked to impaired intestinal epithelial barrier function in the complement deficient mice, since antibiotic treatment prevented the lethal effects of DSS treatment and reduced serum endotoxin levels. We therefore investigated whether this may also be the cause of mortality of C3–/– mice during progression to a chronic condition. In support of this contention, at the end of the fourth cycle, endotoxin levels were significantly increased in surviving DSS treated C3–/– mice, but not in fB–/– mice, compared to wt (Fig. 2).
The worse outcome in C3–/– mice compared to wt mice demonstrate that C3 activation is important for protection against chronic DSS-induced colitis. Factor B–/– mice fared similarly to wt mice, whereas C1q/MBL–/– mice rapidly succumb to the disease with 100% mortality. Together, these data indicate that the classical pathway C3 convertase, generated via the classical and/or lectin pathway, directly or indirectly provides an important protective mechanism against infection and mucosal injury in chronic DSS-induced colitis. Deficiency of the alternative pathway of complement activation, which generates the alternative pathway C3 convertase and simultaneously acts as an amplification loop and drives injury in many models of inflammation, had minimal impact on injury profiles in this model.
Targeted pharmacological inhibition of the alternative pathway is protective against chronic DSS-induced colitis
Based on the above data, we investigated whether targeted and temporary inhibition of the alternative pathway would be protective in a therapeutic protocol. Mice were treated with CR2-fH during DSS treatment cycles 2–4, i.e. after the first full cycle of DSS treatment and rest and the establishment of colitis. These DSS treatment periods model acute inflammatory episodes. We evaluated the effect of CR2-fH treatment at two different time points; at the end of cycle 4 DSS treatment and at the end of cycle 4 rest to determine therapeutic effect on both the relapse and remission phases of the disease model. CR2-fH treatment significantly improved outcome in terms of both colon length and histological score at both time points of analysis (Fig. 3). Note that at sacrifice, colon length from all mice was recorded and colons were then either fixed for histological analysis or digested to analyze cell infiltrates, resulting in lower sample numbers for histological score compared to colon length.
Fig 3.
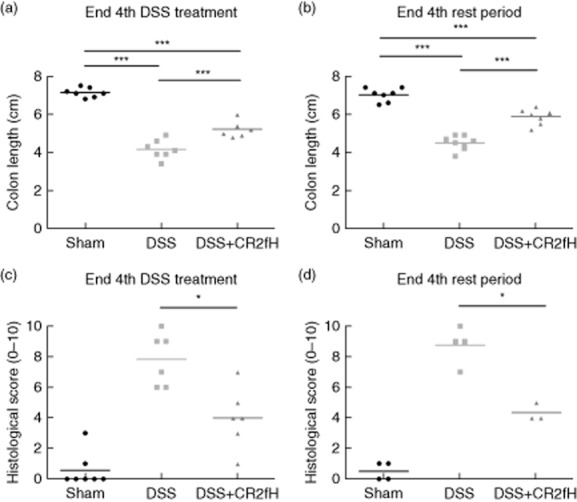
Colon length and histological damage after complement receptor 2 (CR2)-fH treatment of chronic dextran sulphate sodium (DSS-induced colitis. Colon length measured at the end of fourth cycle of DSS treatment (a) or at end of fourth cycle rest period (b). Histological injury score (see Materials and methods) for sections of colons isolated from mice at end of fourth cycle of DSS treatment (c) or at end of fourth cycle rest period (d). Horizontal bar indicates mean, n = 4–8 (representative of two independent experiments). *P < 0·05; ***P < 0·001.
Fibrosis is a hallmark of injury in chronic colitis, and is a major cause of the need for surgical intervention. Collagen, as a marker of fibrosis, is increased in colons of patients and experimental animals with chronic colitis. We therefore also assessed the effect of CR2-fH on development of fibrosis by staining colon sections for collagen. Collagen content in colons from CR2-fH treated mice was significantly reduced compared to DSS treated mice, and was not significantly different to that in sham controls (Fig. 4).
Fig 4.
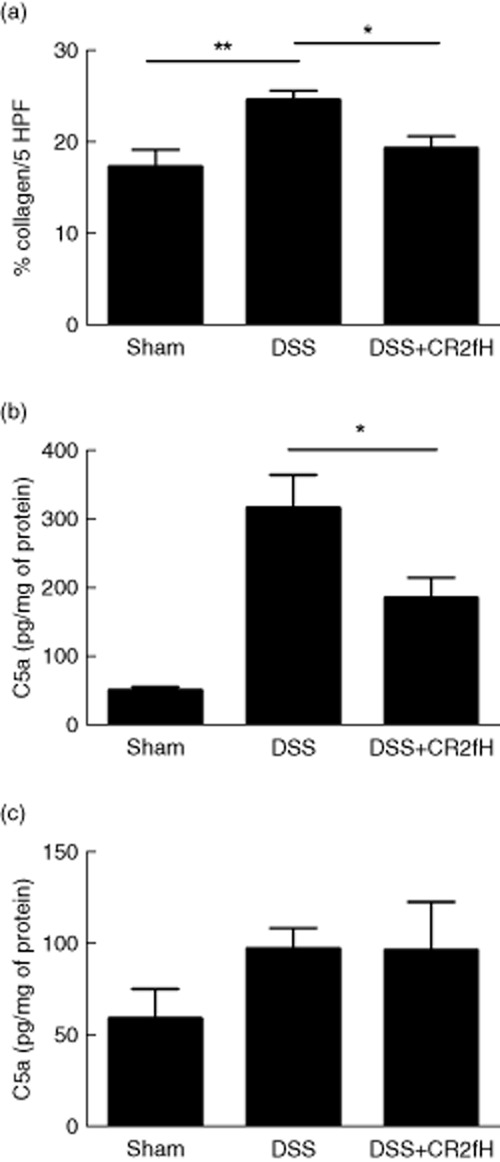
Effect of complement receptor 2 (CR2)-fH on development of fibrosis and local complement activation in mice with chronic dextran sulphate sodium (DSS)-induced colitis. (a) Picrosirius red staining of colon sections isolated from mice at end of fourth cycle rest period was used to quantify percent collagen as a marker of fibrosis development. Data expressed as percentage of collagen per five high-powered fields (HPF). Mean ± standard error of the mean (s.e.m.); n = 7–10, *P < 0·05; **P < 0·01. (b,c) C5a levels were assessed in colon homogenates at the end of fourth cycle of DSS treatment (b) or at end of fourth cycle rest period (c). Mean ± s.e.m.; a, n = 5–6; b, n = 4 (representative of two independent experiments). *P < 0·05.
CR2-fH decreases complement activation in the colon during treatment
We have previously shown that the complement activation product C3d is deposited on the colon mucosa of DSS treated mice, and we have demonstrated that systemically administered CR2-fH targets to the acutely inflamed intestine and reduces C3 activation 12,22. To further relate the protective effect of CR2-fH to complement inhibition, we determined the effect of CR2-fH on the generation of C5a, a complement activation product directly associated with the pathogenesis of DSS-induced colitis, and which can also be generated by proteases independent of C3/C5 convertase under certain conditions 17. At the end of the last DSS treatment cycle (active disease), C5a levels in colon homogenates were significantly increased in DSS vs. sham treated mice, but CR2-fH treatment significantly reduced levels of C5a. At the end of the last rest period, however, 10 days after DSS and CR2-fH treatment (remission), C5a levels were similar in DSS compared to DSS + CR2-fH treated mice, and were reduced compared to C5a levels immediately following the DSS treatment cycle (Fig. 4). These data demonstrate that CR2-fH is only inhibiting complement activation during treatment in the acute phase of injury, but not during remission.
CR2-fH alters the immune response
Since there are reported differences in innate and adaptive immune cell populations and cytokine profiles in acute vs. chronic DSS-induced colitis 24–27, we investigated the effect of targeted inhibition of the alternative pathway on the shaping of an immune response. At the end of the final rest period, we analyzed immune cell populations and cytokines. Cell populations from the spleen, mesenteric lymph nodes (MLNs) and lamina propria (LP) were analyzed to investigate systemic and local mucosal immune responses, but differences were seen only in the LP, and only these data are shown. We found significantly lower numbers of B cells, M1 macrophages and M2 macrophages in the LP of mice treated with DSS + CR2-fH compared to DSS alone (Fig. 5). Compared to sham, we also found a significant increase in the number of mature dendritic cells in DSS treated mice, but not DSS + CR2-fH treated mice. We detected no differences in the numbers of CD4+ or CD8+ T cells between DSS and DSS + CR2-fH and treated groups. At the end of the final rest period, modeling remission after progression of disease to chronicity, only mature DC's and CD4+ T cells were present in higher numbers in colons from DSS treated vs. sham animals. Overall, these data indicate a decreased inflammatory/immunostimulatory environment in CR2-fH treated mice.
Fig 5.
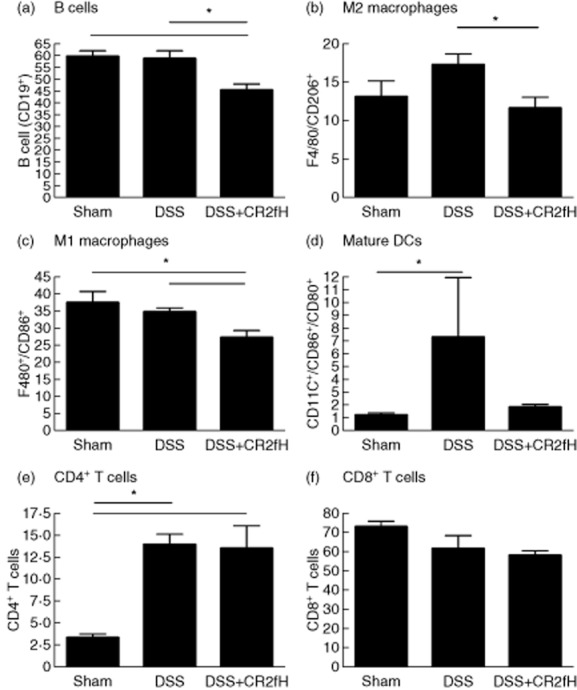
Modulation of immune cell populations in the lamina propria (LP) at the end of fourth cycle rest. The percentage of CD19+ B cells (a), F4/80+/CD206+ macrophages (b), F4/80+/CD86+ macrophages (c), CDllc+/CD80+/CD86+ dendritic cells (d), CD4+ T cells (e) and CD8+ T cells (f) were analysed in the LP of mice at the end of fourth cycle rest; n = 5, *P < 0·05.
We also analyzed an array of cytokines in colon homogenates at the end of the 4th rest period. We found strong trends towards a decrease in IL-6 and IL-17 in colons from CR2-fH treated vs. untreated mice, but the differences did not reach statistical significance (P = 0·14 and 0·11, respectively). There was no difference between IFNγ or TNFα levels in colons from CR2-fH treated vs. untreated mice (data not shown).
Discussion
Most experimental studies investigating the role of complement in IBD have used a model of DSS-induced colitis, and previous studies have shown that complement inhibition is protective in acute models (see introduction). However, these models provide limited information on the effects of complement or complement inhibition on wound healing, the development of fibrosis, or the modulation of adaptive immunity. Here, we investigated the effect of different complement deficiencies on outcomes in a chronic model of DSS-induced colitis, and further investigated the effect of a site-targeted inhibitor of the alternative pathway in a more clinically relevant paradigm. We demonstrated that specific inhibition of the alternative pathway with CR2-fH during DSS treatment periods (corresponding to clinical relapse) improved clinical outcome, reduced tissue inflammation and injury, reduced the level of fibrosis, and altered the local immune response relative to basal levels and DSS treated mice.
Unlike complement deficiency, CR2-fH did not increase animal mortality, and neither fB deficiency nor CR2-fH treatment increased serum endotoxin levels, unlike C3 deficiency that impacts all complement pathways. In this regard, we have previously shown that CR2-mediated targeting of complement inhibition to sites of complement activation obviates the need for systemic inhibition and doses not increase host susceptibility to infection 23. On the other hand, many current treatments for human IBD involve suppression of inflammation and the host immune response; for example, anti-TNFα therapy increases the risk of opportunistic infections in IBD patients 28, and additionally does not appear to reduce the overall need for surgery for Crohn's disease patients due to fibrosis 29. These considerations, together with our current and published findings, suggest that human CR2-fH may be an effective and safer alternative to current biological treatments for colitis. The reason we see protection from DSS-induced colitis when the alternative pathway is temporarily inhibited, but not when it is permanently absent, is likely due to a dual role of complement in the pathogenesis of this disease. Mice received CR2-fH only during periods of DSS treatment, which models acute disease flare-ups, and the absence of complement inhibition during remission will allow alternative pathway complement activity that may function in tissue repair/regeneration and host defense.
In the only previously published study investigating the role of complement in a chronic DSS-induced colitis model, C5aR–/– mice fared worse than wt mice 12. Here, we found that C5a is reduced in the colons of CR2-fH treated mice at the end of the fourth cycle of DSS, and these mice fared better than untreated controls. There are several potential explanations for this apparent contradiction on the role of C5a. First, colitis was induced with 3% DSS in our model compared with only 1·5% in the previously published data with C5aR–/– mice. The amount of DSS affects the extent of injury and may impact inflammatory mediators and the induced immune response. Additionally, C5aR deficiency was shown to affect other mediators that may alter disease outcome such as expression of C3aR and C5L2. Finally, it is likely that C5a has opposing roles during active disease and repair/regeneration. C5aR–/– mice are protected from acute disease, demonstrating that the absence of C5a signaling during active disease protects from tissue damage. On the other hand, C5a signaling has documented roles in tissue repair and regeneration. In the current study, we blocked complement activation only during DSS administration, while allowing complement activation during the regeneration phase. In line with this treatment protocol, we found that C5a was reduced in CR2-fH treated animals at the end the fourth cycle of DSS, but had returned to levels seen in untreated mice at the end of rest.
Since human colitis is a disease characterized by acute flare-ups followed by periods of remission, analysis of both the mucosal immune response and the systemic response is warranted when investigating potential therapeutics. We therefore assessed whether CR2-fH altered immune cell populations in the spleen, MLN and colon mucosa (LP) at the end of rest cycle 4. Alteration of immune cell populations in the mucosa is important for understanding acute disease, but modulation of cell populations in the spleen and MLN are likely to be important in preventing relapse. Colons from mice with chronic DSS-induced colitis have been shown to contain increased B cells and CD11c+ cells 24. Similarly, colitis patients have increased accumulation of B cells 30 and dendritic cells in the LP, and their DC's have increased activation markers 31. Dendritic cells in the LP are thought to be essential for maintaining tolerance and inducing appropriate adaptive immune responses to ingested antigens 31. In the DSS-induced model of colitis, depletion of CD11c+ cells resulted in less severe disease 32, highlighting the importance of these cells in disease progression. In agreement with these results, we detected a significant increase in mature CD11c+ cells following DSS-induced colitis, and we demonstrated that this increase is prevented by CR2-fH treatment. We also found that CR2-fH treatment significantly reduced B cells in the colons of mice with chronic DSS-induced colitis, although we did not see any difference in B cell numbers between mice treated with DSS alone compared to sham controls. The finding that there was no difference between B cells in LP from sham vs. DSS treated mice in chronic DSS-induced colitis is consistent with a previous report that showed differences only in the MLN and spleen 24. B cells have also been implicated in the pathogenesis of human IBD by a study showing that B cells from patients have heightened activation status that correlates with disease severity 33. Thus, the protective effect of CR2-fH against chronic DSS-induced colitis correlates with the local modulation of immunity and immune cells associated with human disease.
M1 macrophages have a pro-inflammatory phenotype that can cause tissue damage, while M2 macrophages are generally anti-inflammatory and can play an important role in wound healing. In a setting of chronic inflammation, excessive wound healing can lead to the development of fibrosis, which is the leading cause of surgery in human IBD patients 34. In our model, we detect a decrease in both M1 and M2 type macrophages in the LP after treatment with CR2-fH. We also detected a decrease in fibrosis in colons from CR2-fH treated mice, consistent with less damage to the colon, and therefore requiring less healing/wound repair. This indicates that CR2-fH may not only decrease inflammation in the colon, but also decrease the need for surgery due to fibrosis, which is currently common even after successful treatment with anti-inflammatory drugs.
In summary, we investigated the use of a targeted complement inhibitor, CR2-fH, in a chronic DSS-induced model of colitis. First and foremost, CR2-fH was shown to be therapeutically beneficial, and this targeted inhibitor may potentially have an improved safety profile over current therapies for IBD. Additionally, we show that CR2-fH alters the local immune environment in the chronic phase of modeled IBD. This study supports the use of targeted complement inhibition as a potential treatment for IBD.
Acknowledgments
This work was supported by a grant from the Crohn's and Colitis Foundation of America (to ST) and National Institutes of Health Training Grant T32 AR050958 (to J. S. B.). We thank Emily Paulling for histology work and expert technical assistance.
Disclosure
The authors declare no competing financial interests.
References
- Ueki T, Mizuno M, Uesu T, et al. Distribution of activated complement, C3b, and its degraded fragments, iC3b/C3dg, in the colonic mucosa of ulcerative colitis (UC) Clin Exp Immunol. 1996;104:286–292. doi: 10.1046/j.1365-2249.1996.17721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstensen TS, Mollnes TE, Fausa O, Brandtzaeg P. Deposits of terminal complement complex (TCC) in muscularis mucosae and submucosal vessels in ulcerative colitis and Crohn's disease of the colon. Gut. 1989;30:361–366. doi: 10.1136/gut.30.3.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff TM, Arumugam TV, Shiels IA, Reid RC, Fairlie DP, Taylor SM. A potent human C5a receptor antagonist protects against disease pathology in a rat model of inflammatory bowel disease. J Immunol. 2003;171:5514–5520. doi: 10.4049/jimmunol.171.10.5514. [DOI] [PubMed] [Google Scholar]
- Halstensen TS, Mollnes TE, Garred P, Fausa O, Brandtzaeg P. Epithelial deposition of immunoglobulin G1 and activated complement (C3b and terminal complement complex) in ulcerative colitis. Gastroenterology. 1990;98:1264–1271. doi: 10.1016/0016-5085(90)90343-y. [DOI] [PubMed] [Google Scholar]
- Halstensen TS, Brandtzaeg P. Local complement activation in inflammatory bowel disease. Immunol Res. 1991;10:485–492. doi: 10.1007/BF02919746. [DOI] [PubMed] [Google Scholar]
- Ahrenstedt O, Knutson L, Nilsson B, Nilsson-Ekdahl K, Odlind B, Hallgren R. Enhanced local production of complement components in the small intestines of patients with Crohn's disease. N Engl J Med. 1990;322:1345–1349. doi: 10.1056/NEJM199005103221903. [DOI] [PubMed] [Google Scholar]
- Scheinin T, Bohling T, Halme L, Kontiainen S, Bjorge L, Meri S. Decreased expression of protectin (CD59) in gut epithelium in ulcerative colitis and Crohn's disease. Hum Pathol. 1999;30:1427–1430. doi: 10.1016/s0046-8177(99)90163-6. [DOI] [PubMed] [Google Scholar]
- Berstad AE, Brandtzaeg P. Expression of cell membrane complement regulatory glycoproteins along the normal and diseased human gastrointestinal tract. Gut. 1998;42:522–529. doi: 10.1136/gut.42.4.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uesu T, Mizuno M, Inoue H, Tomoda J, Tsuji T. Enhanced expression of decay accelerating factor and CD59/homologous restriction factor 20 on the colonic epithelium of ulcerative colitis. Lab Invest. 1995;72:587–591. [PubMed] [Google Scholar]
- Wende E, Laudeley R, Bleich A, et al. The complement anaphylatoxin C3a receptor (C3aR) contributes to the inflammatory response in dextran sulfate sodium (DSS)-induced colitis in mice. PLOS ONE. 2013;8:e62257. doi: 10.1371/journal.pone.0062257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain U, Woodruff TM, Stadnyk AW. The C5a receptor antagonist PMX205 ameliorates experimentally induced colitis associated with increased IL-4 and IL-10. Br J Pharmacol. 2013;168:488–501. doi: 10.1111/j.1476-5381.2012.02183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepp-Berglind J, Atkinson C, Elvington M, Qiao F, Mannon P, Tomlinson S. Complement-dependent injury and protection in a murine model of acute dextran sulfate sodium-induced colitis. J Immunol. 2012;188:6309–6318. doi: 10.4049/jimmunol.1200553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F, Spencer D, Hatala DA, Levine AD, Medof ME. Decay-accelerating factor deficiency increases susceptibility to dextran sulfate sodium-induced colitis: role for complement in inflammatory bowel disease. J Immunol. 2004;172:3836–3841. doi: 10.4049/jimmunol.172.6.3836. [DOI] [PubMed] [Google Scholar]
- Deguchi Y, Andoh A, Inatomi O, et al. Development of dextran sulfate sodium-induced colitis is aggravated in mice genetically deficient for complement C5. Int J Mol Med. 2005;16:605–608. [PubMed] [Google Scholar]
- Lu F, Fernandes SM, Davis AE., III The role of the complement and contact systems in the dextran sulfate sodium-induced colitis model: the effect of C1 inhibitor in inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2010;298:G878–883. doi: 10.1152/ajpgi.00400.2009. [DOI] [PubMed] [Google Scholar]
- Johswich K, Martin M, Bleich A, et al. Role of the C5a receptor (C5aR) in acute and chronic dextran sulfate-induced models of inflammatory bowel disease. Inflamm Bowel Dis. 2009;15:1812–1823. doi: 10.1002/ibd.21012. [DOI] [PubMed] [Google Scholar]
- Huber-Lang M, Sarma JV, Zetoune FS, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- Krisinger MJ, Goebeler V, Lu Z, et al. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood. 2012;120:1717–1725. doi: 10.1182/blood-2012-02-412080. [DOI] [PubMed] [Google Scholar]
- Muller S, Schaffer T, Flogerzi B, et al. Mannan-binding lectin deficiency results in unusual antibody production and excessive experimental colitis in response to mannose-expressing mild gut pathogens. Gut. 2010;59:1493–1500. doi: 10.1136/gut.2010.208348. [DOI] [PubMed] [Google Scholar]
- Aomatsu T, Imaeda H, Takahashi K, et al. Neutralization of complement component C5 ameliorates the development of dextran sulfate sodium (DSS)-colitis in mice. J Clin Biochem Nutr. 2013;52:72–75. doi: 10.3164/jcbn.12-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Yang Y, Gao X, et al. Blockade of complement activation product C5a activity using specific antibody attenuates intestinal damage in trinitrobenzene sulfonic acid induced model of colitis. Lab Invest. 2011;91:472–483. doi: 10.1038/labinvest.2010.183. [DOI] [PubMed] [Google Scholar]
- Huang Y, Qiao F, Atkinson C, Holers VM, Tomlinson S. A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. J Immunol. 2008;181:8068–8076. doi: 10.4049/jimmunol.181.11.8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson C, Song H, Lu B, et al. Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection. J Clin Invest. 2005;115:2444–2453. doi: 10.1172/JCI25208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall LJ, Faivre E, Quinlan A, Shanahan F, Nally K, Melgar S. Induction and activation of adaptive immune populations during acute and chronic phases of a murine model of experimental colitis. Dig Dis Sci. 2011;56:79–89. doi: 10.1007/s10620-010-1240-3. [DOI] [PubMed] [Google Scholar]
- Bento AF, Leite DF, Marcon R, et al. Evaluation of chemical mediators and cellular response during acute and chronic gut inflammatory response induced by dextran sodium sulfate in mice. Biochem Pharmacol. 2012;84:1459–1469. doi: 10.1016/j.bcp.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Melgar S, Karlsson A, Michaelsson E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am J Physiol. 2005;288:G1328–1338. doi: 10.1152/ajpgi.00467.2004. [DOI] [PubMed] [Google Scholar]
- Alex P, Zachos NC, Nguyen T, et al. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2008;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford AC, Peyrin-Biroulet L. Opportunistic infections with anti-tumor necrosis factor-alpha therapy in inflammatory bowel disease: meta-analysis of randomized controlled trials. Am J Gastroenterol. 2013;108:1268–1276. doi: 10.1038/ajg.2013.138. [DOI] [PubMed] [Google Scholar]
- de Buck van Overstraeten A, Wolthuis A, D'Hoore A. Surgery for Crohn's disease in the era of biologicals: a reduced need or delayed verdict? World J Gastroenterol. 2012;18:3828–3832. doi: 10.3748/wjg.v18.i29.3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn I, Schlenke P, Mascher B, Stange EF, Seyfarth M. Lamina propria plasma cells in inflammatory bowel disease: intracellular detection of immunoglobulins using flow cytometry. Immunobiology. 2002;206:546–557. doi: 10.1078/0171-2985-00203. [DOI] [PubMed] [Google Scholar]
- Niess JH. Role of mucosal dendritic cells in inflammatory bowel disease. World J Gastroenterol. 2008;14:5138–5148. doi: 10.3748/wjg.14.5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe K, Nguyen KP, Fine SD, et al. Conventional dendritic cells regulate the outcome of colonic inflammation independently of T cells. Proc Natl Acad Sci USA. 2007;104:17022–17027. doi: 10.1073/pnas.0708469104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noronha AM, Liang Y, Hetzel JT, et al. Hyperactivated B cells in human inflammatory bowel disease. J Leukoc Biol. 2009;86:1007–1016. doi: 10.1189/jlb.0309203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder F, Brenmoehl J, Leeb S, Scholmerich J, Rogler G. Wound healing and fibrosis in intestinal disease. Gut. 2007;56:130–139. doi: 10.1136/gut.2006.090456. [DOI] [PMC free article] [PubMed] [Google Scholar]