

Abstract
Renal tubular epithelial cells (TECs) are one of the main targets of alloreactive T cells during acute rejection. We hypothesize that TECs modulate the outcome of alloimmunity by executing immunosuppressive effects in order to dampen the local inflammation. We studied whether TECs possess immunosuppressive capacities and if indoleamine 2,3-dioxygenase (IDO) might play a role in suppressing T cell alloreactivity. Next, we studied the role of programmed death ligand 1 (PD-L1) and intercellular adhesion molecule-1 (ICAM-1 with regard to TEC-related immunomodulatory effects. CD3/CD28 and alloactivated peripheral blood mononuclear cells were co-cultured with activated TECs. We analysed CD4+ and CD8+ T cell proliferation and apoptosis in the absence or presence of IDO inhibitor 1-methyl-L-tryptophan (1-L-MT), anti-PD-L1 and anti-ICAM-1. Further, we examined whether inhibition of T cell proliferation was cell–cell contact-dependent. We found that TECs dose-dependently inhibited CD4+ and CD8+ T cell proliferation (P < 0·05). Activated TECs showed significantly increased IDO activity and up-regulated PD-L1 and ICAM-1 expression. Suppressed CD4+ and CD8+ T cell proliferation was only partially restored or failed to restore using 1-L-MT. Activated TECs increased early and late apoptosis of proliferating CD4+ and CD8+ T cells; only CD4+ T cell apoptosis was statistically affected by 1-L-MT. Transwell experiments revealed that TEC-mediated immunosuppression is cell–cell contact-dependent. We found that anti-ICAM-1 affected only CD4+ T cell apoptosis and not T cell proliferation. Our data show that TECs suppress both CD4+ and CD8+ T cell proliferation contact dependently. Interestingly, inhibition of proliferation and enhancement of apoptosis of T cell subsets is differentially regulated by indoleamine 2,3-dioxygenase and ICAM-1, with no evidence for the involvement of PD-L1 in our system.
Keywords: human renal tubular epithelial cells (TECs), IDO, immunosuppression, T cells
Introduction
Renal tubular epithelial cells (TECs) represent 75% of the parenchymal cells of the kidney and are one of the main targets of T cells during tubulointerstitial inflammation, such as acute cellular rejection of the kidney graft. The consequences are huge, and will result eventually in structural TEC damage, nephron disruption and graft loss 1,2. The final outcome is determined partially by the local microenvironmental regulation and the interaction between tubular epithelium, T cells and other immune cells 3,4. In our previous work, we reported on the proinflammatory role of the tubular compartment directing selective migration of T helper type 1 (Th1) and Th17 CD4+ T cells in vitro 5. Furthermore, we investigated distinct proliferation of recipient T cell subsets after TEC encounter with variable sensitivity to immunosuppressive drugs. Our data show substantial proliferation of TEC-reactive CD4CD28 null memory T cells, which are resistant to tacrolimus and everolimus 6. Previously, we and others have shown that TECs can resist granzyme B-mediated apoptosis induced by alloreactive cytotoxic T cells through up-regulation of cytosolic serpin peptidase inhibitor, clade B (ovalbumin), member 9 (SERPINB9) during rejection 7,8. Moreover, we showed that TEC-derived SERPINB9 is also high during viral infections such as cytomegalovirus (CMV), Epstein–Barr virus (EBV) or BK virus, pointing towards a general mechanism of self-defence 9,10. Altogether, these findings led to the question of whether other bi-directional pathways of TEC-mediated defence resulting in suppression of T cell mediated allo-immunity exist, and whether TECs are capable of abrogating T-cell activation and/or proliferation in order to limit the burdens of inflammation and the eventual damage.
TECs might possess immune regulatory capacities like other parenchymal cells, with well-established immunomodulatory capacities such as mesenchymal stem cells 11. Indoleamine 2,3-dioxygenase (IDO) is a cell survival-related rate-limiting enzyme which leads to the degradation and depletion of the essential amino acid tryptophan along the kynurenine pathway 12. IDO can be activated directly by a number of proinflammatory cytokines such as interferon (IFN)-γ and tumour necrosis factor (TNF)-α, and is involved in regulating immune responses 13,14. Tryptophan catabolism leads to the production of L-kynurenine derivatives and O2 free radicals, thereby regulating T cell proliferation and survival. Tryptophan starvation inhibits T cell activation and induces cell cycle arrest by blocking their entry into the S phase, eventually leading to T cell apoptosis 15–18.
TECs express IDO upon IFN-γ/TNF-α stimulation resulting in tryptophan depletion, which can be reversed using the IDO inhibitor 1-methyl-L-tryptophan (1-MT) 14. In an experimental glomerulonephritis model, IDO inhibition resulted in acceleration of kidney damage and accumulation of CD4+ T cells, suggesting a protective role for IDO 19. Interestingly, strong immunohistochemical cytoplasmic IDO expression was documented in tubular epithelial cells from kidney transplant recipients with acute rejection compared to non-rejectors. Serum and urine kynurenine/tryptophan ratios of rejecting patients were elevated significantly compared to uncomplicated transplant recipients 20. Even though no functional data are available as yet regarding the immunosuppressive capacities of TECs, we speculate that proliferation of alloreactive T cells within the transplanted organ might also be suppressed via renal TECs with a focus on local TEC-derived IDO activity.
In the present study, we hypothesize that donor-derived TECs possess immunosuppressive capacities and that IDO might play a pivotal role suppressing T cell alloreactivity. Furthermore, we questioned whether inflammatory conditions influence the immunomodulatory functions of TECs via IDO-controlled mechanisms. To these ends, we investigated the differential immunosuppressive effects mediated by TECs on CD4 T cell and CD8 T cell proliferation and apoptosis.
Materials and methods
Isolation of peripheral blood mononuclear cells of healthy volunteers
Peripheral blood mononuclear cells (PBMC) of healthy volunteers (Blood Bank Sanquin, Rotterdam, the Netherlands) were isolated by density gradient centrifugation Ficoll-paque (density 1·077 g/ml; Amersham Pharmacia Biotech, Uppsala, Sweden), frozen in medium containing 10% dimethylsulphoxide (DMSO) and stored at −150°C until analysis.
Culture of primary renal tubular epithelial cells and mesenchymal stem cells used as controls
TECs were cultured from cortical tissue of human kidney nephrectomies due to tumours, as described previously 5. Cells were used between passages 2 and 6 of culture. TEC outgrowth was confirmed by morphological appearance and immunofluorescence staining (CD13+, CD26+ and CD90–). To mimic rejection and proinflammatory intragraft conditions, TECs were stimulated with 50 ng/ml human recombinant IFN-γ (U-Cytech, Utrecht, the Netherlands) and 20 ng/ml human recombinant TNF-α (PeproTech, London, UK) 5. As mesenchymal stem cells (MSC) are well-established parenchymal cells with proven immunomodulatory properties, we used MSCs as the positive control cell line in our experimental set-up. Human MSCs were isolated from adipose tissue fat as described previously 21,22. MSC were activated using 50 ng/ml human recombinant IFN-γ (U-Cytech) and used for experiments between passages 2 and 4. Both TECs and MSCs were kept at 37°C, 5% CO2 and 95% humidity. Unstimulated and IFN-γ/TNF-α-activated TECs were analysed for the surface marker expression of intercellular adhesion molecule-1 (ICAM-1) (CD54; BD Biosciences, San Jose, USA) and PD-L1 (CD274; Biolegend, San Diego, CA, USA).
IDO mRNA expression and IDO activity
To assess the IDO activity in TECs, 75·103 TECs were stimulated in a 24-well culture plate for 24 h with IFN-γ/TNF-α in the absence or presence of the IDO inhibitor; 1-methyl-L-tryptophan (1-L-MT) (Sigma, St Louis, MO, USA) at a concentration of 50 μM. Fresh stock 1-L-MT concentrations of 10 mM were prepared for each experiment. 1-L-MT was dissolved in 0·1 M NaOH and neutralized using an equal volume of 0·1 M HCl. TEC mRNA was stabilized using RNAlater RNA Stabilization Solution (Ambion, Austin, TX, USA). The culture plate was stored for 48 h at 4°C and subsequently at −20°C until analysis. mRNA expression was measured as described previously 5. Briefly, a 500 ng mRNA quantitative real-time reverse transcription–polymerase chain reaction (RT–PCR) containing universal PCR mix (Invitrogen, Carlsbad, CA, USA) was used to quantify the amount of IDO in samples. Assay-on-demand products for the detection and quantification of IDO (Hs00158627.m1) mRNAs were designed by Applied Biosystems (Foster City, CA, USA).
L-Kynurenine accumulation reflecting IDO activity was measured in the supernatants of 24-h cytokine-activated TECs. Briefly, 30% trichloroacetic acid was added to samples at a 1:3 ratio and incubated at 50°C for 30 min. Samples were centrifuged at 12 350 g for 5 min. Supernatants were diluted 1:1 in Ehrlich reagent 200 mg 4-dimethylaminobenzaldehyde (Sigma) in 10 ml of glacial acetic acid. Then, supernatants were measured in duplicate in a 96-well flat-bottomed plate. Absorbance was determined at 490 nm using a multi-label plate reader (VersaMax™; Molecular Devices, Sunnyvale, CA, USA). L-kynurenine (Sigma) diluted in unconditioned medium was used as standard control 23.
Mixed TEC lymphocyte co-culture
PBMC (0·5 × 105) were incubated with irradiated (40 Gy) human leucocyte antigen (HLA)-mismatched (A-B-DR: 2-2-2) PBMC (ratio 1:1) in a mixed lymphocyte reaction (MLR). Both MLR- and anti-CD3/CD28-activated lymphocytes were added to IFN-γ (50 ng/ml)/TNF-α (20 ng/ml)-activated TECs in TEC : PBMC ratios of 120·103:300·103 (1:2·5), 60·103/300·103 (1:5) and 30·103/300·103 (1:10). PBMC proliferation was measured using a [3H]-thymidine incorporation assay (0·5 μCi/well; Amersham Pharmacia Biotech, Roosendaal, the Netherlands) at day 7 for the MLR and at day 3 for the CD3/CD28 stimulation conditions. T cells were activated using 1 μg/ml anti-CD3, 1 μg/ml anti-CD28 and 2 μg/ml polyclonal antibody goat anti-mouse (BD Biosciences). In addition to the above-described experiments, proliferation was measured after 3 days of co-culture using carboxyfluorescein succinimidyl ester (CFSE) dilution assay (Sigma). As positive controls, MSC cell lines were used. MLR- and anti-CD3/CD28-derived activated lymphocytes were added to IFN-γ (50 ng/ml)-activated MSC at MSC : PBMC ratios of 1:2·5, 1:5 and 1:10. Results were analysed as described previously for TEC co-cultures.
To investigate the role of IDO, we performed TEC lymphocyte co-cultures in the presence or absence of IDO inhibitor and measured the T cell proliferation using the CFSE dilution method. TECs (120·103) were seeded in 24-well flat-bottomed culture plates (Corning Costar, Corning, NY, USA) and activated for 3 days with IFN-γ (50 ng/ml)/TNF-α (20 ng/ml) in the absence or presence of 50 μM 1-L-MT (Sigma). CFSE-labelled anti-CD3/CD28 activated PBMC (300·103) were co-cultured with TECs in human culture medium (HCM); RPMI–glutamax (Gibco, Carlsbad, CA, USA) supplemented with 10% heat-inactivated human serum, 100 IU/ml penicillin and 100 μg/ml streptomycin. At day 3, T cells were harvested and proliferation was analysed using flow cytometry. To investigate the role of PD-L1 and ICAM-1, we performed TEC lymphocyte co-cultures in the absence or presence of anti-PD-L1 (1 μg/ml; Biolegend) and anti-ICAM-1 (1 μg/ml; Biolegend) blocking antibodies, and measured the T cell proliferation using the [3H]-thymidine incorporation assay at day 3.
TEC lymphocyte Transwell experiments
IFN-γ/TNF-α-activated TECs (120·103) were seeded in 24-well plates in the absence or presence of 50 μM 1-L-MT. After 24-h IFN-γ/TNF-α stimulation, 0·4 μm pore membranes (ThinCerts; Greiner Bio-One, Frickenhausen, Germany) were placed above the TECs. CFSE-labelled anti-CD3/CD28-activated PBMC (300·103) were placed upon the membrane. As control, anti-CD3/CD28-activated PBMC were placed upon a membrane without TECs. PBMC were harvested at day 3 and analysed for proliferation and subset analysis using CFSE dilution.
Subset analysis of proliferating T cells using flow cytometry
Anti-CD3/CD28-activated T cells were harvested at day 3. Cell surface staining was conducted with the following monoclonal antibodies (mAbs): CD7-eFluor450 (eBioscience), CD4-allophycocyanin (APC)-cyanin 7 (Cy7), CD8-BV510 (Biolegend), CD25-phycoerythrin (PE)-Cy7, CD69 PE, cytotoxic T lymphocyte antigen-4 (CTLA-4) APC, 7-aminoactinomycin D (7-AAD) and annexin V-APC (BD Bioscience). Intracellular forkhead box protein P3 (FoxP3) staining was carried out according to the manufacturer's instructions using the anti-human FoxP3 staining set (eBioscience). Twenty thousand gated lymphocyte events were acquired from each tube by a fluorescence activated cell sorter (FACS)Canto II flow cytometer (BD Biosciences). Fluorescence-minus-one (FMO) controls were used to determine positive or negative boundaries. Data were analysed using FlowJo software (Tree Star, San Carlos, CA, USA). Flow cytometric analysis was performed with at least 100 gated events.
Statistics
Results are expressed as mean ± standard error of the mean. Data were analysed for statistical significance with GraphPad Prism version 5·01 software (Graphpad Software, La Jolla, CA, USA) using the non-parametric Wilcoxon matched-pairs signed-rank test. P-values less than 0·05 were considered significant.
Results
Cytokine-activated renal tubular epithelial cells suppress T cell proliferation
In order to investigate the inhibitory capacity of TECs on T cell proliferation, TECs were preactivated by proinflammatory cytokines IFN-γ and TNF-α to mimic the intragraft inflammatory microenvironment. IFN-γ-activated MSCs were used as the positive control cell line. [3H]-Thymidine incorporation experiments revealed that IFN-γ/TNF-α-activated TECs inhibit T cell proliferation dose-dependently induced by anti-CD3/CD28 stimulation (Fig. 1a, n = 3). In line with this, allogeneic stimulation in an MLR revealed similar immunosuppressive effects of activated TECs. IFN-γ/TNF-α-activated TECs inhibit dose-dependently T cell proliferation induced by MLR (Fig. 1b, n = 3).
Fig 1.

Cytokine-activated renal tubular epithelial cells suppress T cell proliferation. (a,b) Interferon (IFN)-γ/tumour necrosis factor (TNF)-α-stimulated tubular epithelial cells (TECs) (n = 3) inhibit both polyclonal anti-CD3/CD28 and mixed lymphocyte reaction (MLR)-induced antigen-specific T cell proliferation using [3H]-thymidine incorporation. IFN-γ-activated mesenchymal stem cells (MSC) were used as positive controls (n = 3). (c,d) IFN-γ/TNF-α-activated TECs were co-cultured with anti-CD3/CD28-stimulated peripheral blood mononuclear cells (PBMC) according to TEC : PBMC at ratios of 1:2·5, 1:5 and 1:10. CD4+ T cell and CD8+ T cell proliferation was inhibited significantly at the TEC : PBMC ratio 1:2·5.
Subsequently, we analysed proliferation of anti-CD3/CD28-activated CD4 and CD8 T cell subsets by CFSE dilution using flow cytometry. Polyclonal anti-CD3/CD28 stimulation resulted in a proliferation rate of 85 ± 0·8% of CD4+ T cells and 68·4 ± 3·4% of CD8+ T cell pool (n = 6). Both CD4+ T cell and CD8+ T cell proliferation was suppressed dose-dependently by TECs. Activated TECs significantly inhibited T cell proliferation down to a proliferation rate of 34·1 ± 5·9% of CD4+ T cells, and to 31·6 ± 10·8% of CD8+ T cell pool at a TEC : PBMC ratio of 1:2·5, meaning a 60% suppression of CD4+ T cell proliferation and 54% suppression of CD 8 T cell proliferation (P < 0·05; Fig. 1c,d).
Cytokine-activated renal tubular epithelial cells express IDO mRNA
Twenty-four-hour IFN-γ/TNF-α-activated TECs express indoleamine 2,3-dioxygenase (IDO) mRNA in contrast to the unstimulated TECs (Fig. 2a). Harvested supernatants were used to measure the concentration of L-kynurenine as a sign of functional IDO activity showing the catalytic degree of tryptophan. IFN-γ/TNF-α-activated TECs showed abundant IDO enzyme activity after 24 h stimulation as shown by significantly increased L-kynurenine concentration up to 22·2 ± 2·6 μM compared to 0·5 ± 0·2 μM in unstimulated conditions (P < 0·05). In addition, the IDO inhibitor 1-L-MT significantly inhibited IDO enzyme activity of IFN-γ/TNF-α-activated TECs resulting in 12·5 ± 2·0 μM L-kynurenine (P < 0·05) (Fig. 2b).
Fig 2.
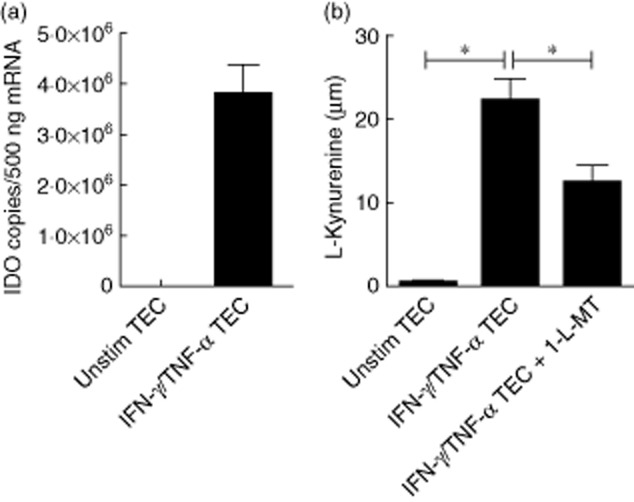
Cytokine-activated renal tubular epithelial cells express indoleamine 2,3-dioxygenase (IDO) mRNA and IDO activity. Tubular epithelial cells (TECs) were cultured for 24 h in the absence or presence of interferon (IFN)-γ/tumour necrosis factor (TNF)-α and in the presence of the IDO inhibitor 1-methyl-L-tryptophan (1-L-MT) (n = 6). (a) IFN-γ/TNF-α activated TECs express IDO mRNA. (b) In order to quantify IDO activity, L-kynurenine was measured in supernatant. IFN-γ/TNF-α stimulation up-regulated L-kynurenine significantly which was, in turn, reduced significantly using 1-L-MT.
Cytokine-activated renal tubular epithelial cells suppress CD4+ T cell proliferation in a cell–cell contact-dependent manner, mediated partially by IDO
In a separate set of six experiments, we examined whether IFN-γ/TNF-α-activated TECs inhibit T cell proliferation via IDO and whether IDO inhibition results in recovery of T cell proliferation. Polyclonal stimulation resulted in a proliferation rate of 79 ± 1·8% of CD4+ T cells and 64·5 ± 3·3% of CD8+ T cells. Cytokine-activated TECs significantly inhibited the proliferation of CD4+ T cells down to 27·5 ± 6·5% and of CD8+ T cells up to 31·2 ± 8·3%, meaning a 65 and 52% suppression at a 1:2·5 ratio. IDO inhibitor 1-L-MT only partly restored CD4+ T cell proliferation by 38%, increasing CD4+ T cell proliferation up to 37·9 ± 8·6% (P < 0·05, Fig. 3a). Surprisingly, we could not detect a significant recovery of CD8+ T cell proliferation using 1-L-MT (34·8 ± 8·6% versus 31·2 ± 8·3%, Fig. 3b). Despite statistical significance, the recovery of CD4+ but also CD8+ T cell proliferation is only partially or not affected by the addition of 1-L-MT.
Fig 3.
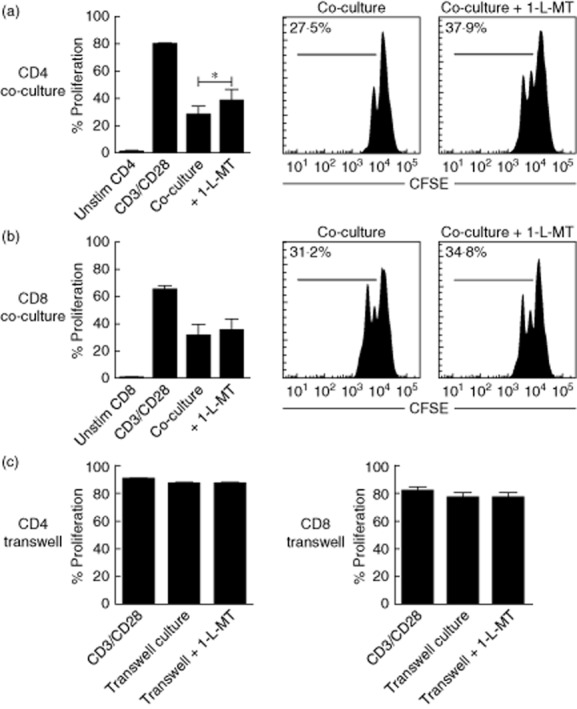
Cytokine-activated renal tubular epithelial cells (TECs) inhibit CD4+ and CD8+ T cell proliferation in a cell–cell contact-dependent manner irrespective of TEC-derived-IDO activity. Interferon (IFN)-γ/tumour necrosis factor (TNF)-α tubular epithelial cells (TEC) were co-cultured with anti-CD3/CD28-activated peripheral blood mononuclear cells (PBMC) (TEC : PBMC ratio of 1:2·5, n = 6). (a) CD4+ T cell proliferation was inhibited significantly by activated TECs. 1-methyl-L-tryptophan (1-L-MT)recovered CD4+ T cell proliferation only partially (*P < 0·05). (b) Proliferating CD8+ T cells were inhibited significantly by activated TECs. 1-L-MT did not recover CD8+ T cell proliferation at all. (c) IFN-γ/TNF-α TECs were cultured with anti-CD3/CD28-activated PBMC using Transwell membranes (TEC : PBMC ratio of 1:2·5, n = 6). Simultaneous Transwell cultures in the absence or presence of 1-L-MT were performed. Activated TECs did not affect CD4+ T cell proliferation and CD8+ T cell proliferation in Transwell experiments. 1-L-MT did not affect CD4+ and CD8+ T cell proliferation.
Interestingly, Transwell experiments revealed that the observed immunomodulatory effects of TECs on proliferating T cell subsets are cell–cell contact-dependent, as separated TECs do not inhibit CD4+ T cell and CD8+ T cell proliferation in a Transwell system. Also 1-L-MT added to the TECs from the beginning did not affect CD4+ T cell or CD8+ T cell proliferation compared to the Transwell experiments without the IDO inhibitor (Fig. 3c). This suggests that TECs need to use cell-surface molecules suppressing CD4+ T cell and CD8+ T cell proliferation.
TEC-induced suppression of T cell proliferation is not mediated by ICAM-1 and PD-L1
Flow cytometric analysis revealed that the cell-surface molecules ICAM-1 and programmed death ligand 1 (PD-L1) are up-regulated significantly on TEC after IFN-γ/TNF-α stimulation and are both known to inhibit T cell proliferation 24,25. Blocking PD-L1 [84 852 ± 17 000 counts per minute (cpm)] and ICAM-1 (77 225 ± 13 539 cpm) did not reverse TEC-inhibited T cell proliferation (87 443 ± 17 273 cpm) (Fig. 4a). To assess other possible regulatory mechanisms in our co-culture system, we analysed the CTLA-4 and FoxP3 expression of both CD4+ and CD8+ T cells. Flow cytometric analysis revealed that antigen encounter during co-culture increased both CTLA-4 and FoxP3 expression by T cells. The percentage of CTLA-4 + CD4+ T cell pool was increased significantly after 3 days co-culture (4·0 ± 0·5% versus 5·5 ± 0·7%, P < 0·05), while CD8+ T cells showed a trend (10·8 ± 0·5% versus 12·3 ± 1·3%) (Fig. 4b). CD4+FoxP3+ T cells (21·3 ± 3·2% versus 31·6 ± 0·1%) and CD8+FoxP3+ T cells (11·3 ± 1·3% versus 15·8 ± 1·7%) were increased significantly after co-culture (Fig. 4c).
Fig 4.
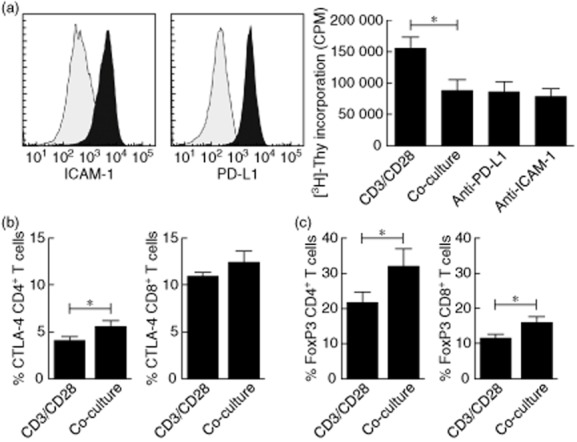
Tubular epithelial cell (TEC)-induced suppression of T cell proliferation is not mediated by intercellular adhesion molecule-1 (ICAM-1) and programmed death ligand-1 (PD-L1). (a) TECs were stained for ICAM-1 and PD-L1 under unstimulated conditions (grey) and after interferon (IFN)-γ/tumour necrosis factor (TNF)-α stimulation (black). Stimulation of TECs resulted in significant up-regulation of ICAM-1 and PD-L1 cell surface expression levels (n = 3). IFN-γ/TNF-α-stimulated TECs were co-cultured with anti-CD3/CD28-activated peripheral blood mononuclear cells (PBMC) (TEC : PBMC ratio of 1:2·5, n = 6) in the absence or presence of blocking PD-L1 and ICAM-1. Blocking PD-L1 and ICAM-1 did not affect the inhibition of T cell proliferation by activated TECs as measured by [3H]-thymidine incorporation (n = 6, *P < 0·05). (b) Co-culture of T cells with activated TECs showed significantly increased percentages of CTLA-4+ CD4+ T cells and an increased trend in the frequency of CD8+ T cell counterparts (n = 6, *P < 0·05). (c) Co-culture of T cells with activated TECs showed significantly increased percentages of forkhead box protein 3 (FoxP3)+CD4+ and FoxP3+CD8+ T cells (n = 6, *P < 0·05).
Cytokine-activated renal tubular epithelial cells induce T cell apoptosis partially mediated via IDO and ICAM-1
Early apoptotic cells are characterized by expression of annexin V+/7-AAD−, and late apoptosis can be designated as annexin V+/7-AAD+ cells. Anti-CD3/CD28-activated T cells showed a low degree of CD4+ T cell early (4·8 ± 0·5%) and late apoptosis (1·2 ± 0·1%) (Fig. 5a) at day 3. However, cytokine-activated TECs significantly increased CD4+ T cell early apoptosis (9·4 ± 2·4%) and late apoptosis (6·3 ± 1·9%) at day 3 co-culture (P < 0·05). IDO inhibition using 50 μM 1-L-MT significantly inhibited CD4+ T cell early apoptosis (6·7 ± 2·0%) and late apoptosis (3·6 ± 0·9%) (P < 0·05). CD8+ T cell early apoptosis (5·4 ± 0·9%) and late apoptosis (0·2 ± 0·04) increased after incubation with activated TECs (8·7 ± 2·0% and 0·5 ± 0·2%). Similarly, 1-L-MT showed the same trend in reduction of CD8+ T cell early apoptosis (6·5 ± 1·6%) and late apoptosis (0·4 ± 0·1%), suggesting that IDO immunosuppressive effects could be mediated via targeting apoptotic pathways (Fig. 5b). Co-culture in the presence of blocking anti-PD-L1 and anti-ICAM-1 (Fig. 5c) revealed that CD4+ T cell apoptosis is inhibited significantly using anti-ICAM-1 antibodies (6·7 ± 0·6% versus 3·3 ± 0·3%, P < 0·05), while anti-PD-L1 did not affect CD4+ T cell apoptosis (6·7 ± 0·6% versus 7·2 ± 0·5%). Also, CD8+ T cell apoptosis was inhibited significantly using anti-ICAM-1 antibodies (8·0 ± 1·1% versus 2·9 ± 0·4%, P < 0·05), and again PD-L1 did not affect CD8+ T cell apoptosis (8·1 ± 0·8%).
Fig 5.

Cytokine-activated renal tubular epithelial cells (TECs) induce T cell apoptosis partially mediated via indoleamine 2,3-dioxygenase (IDO) and intercellular adhesion molecule-1 (ICAM-1). Interferon (IFN)-γ/tumour necrosis factor (TNF)-α-stimulated TECs were co-cultured with anti-CD3/CD28-activated peripheral blood mononuclear cells (PBMC) [tubular epithelial cells (TEC) : PBMC ratio of 1:2·5, n = 6). (a) Proliferating CD4+ T cells and (b) CD8+ T cells were analysed for markers indicating early apoptosis [annexin V+/7-aminoactinomycin D (7-AAD)–] and late apoptosis (annexin V+/7-AAD+). (a) Activated TECs significantly increased CD4+ T cell early and late apoptosis. IDO inhibitor 1-L-MT partly recovered CD4+ T cell early and late apoptosis. (b) The same trends were observed for TEC-induced CD8+ T cell early and late apoptosis. (c) Only blocking ICAM-1 reversed both CD4+ and CD8+ T cell apoptosis, while blocking PD-L1 did not affect T cell apoptosis (n = 6, *P < 0·05).
Discussion
In this study, we investigated whether TECs possess immunosuppressive capacities and if IDO might play a role suppressing T cell alloreactivity. We observed that activated TECs have the capacity to suppress both CD4+ T cell and CD8+ T cell alloreactive proliferation. Transwell experiments showed that TEC-mediated immunosuppression is cell–cell contact-dependent. IFN-γ/TNF-α-activated TECs express high levels of mRNA IDO, and produce abundant amounts of L-kynurenine reflecting a high degree of IDO activity. However, IDO inhibition resulted merely in a partial recovery of CD4+ T cell proliferation and apoptosis, suggesting a role for IDO in the regulation of TEC CD4+ T cell immune interaction. We could not detect any effect of IDO activity on TEC CD8+ T cell immune regulation. The addition of IDO inhibitor to the Transwell system did not result in a significant change in proliferative response of CD4 and CD8 T cells, showing that the immunosuppressive capacities of TECs are cell–cell contact-dependent.
Generally, the role of IDO in inhibition of immune responses has been widely acknowledged 12,15. Immunohistochemical studies have previously shown a significant up-regulation of IDO expression by TECs in rejecting kidney grafts compared to stable grafts 20. Brandacher et al. suggested that IDO activity might offer a novel non-invasive means of immunomonitoring of renal allografts 20. We studied whether IDO expressing TECs also affect lymphocyte responses. IDO is an intracellular rate-limiting enzyme resulting in tryptophan deprivation. The tryptophan catabolite L-kynurenine is involved in T cell apoptosis, as it has been demonstrated that tryptophan deprivation results in blocking entry into the S phase and stagnation of T cell cycle progression at the G0/G1 level. In this way, the T cell is unable to start DNA synthesis 16,26,27. Interestingly, Fallarino et al. found differential susceptibility to cell death by murine Th1 and Th2 clones. T cell exposure to L-kynurenine resulted in a significant degree of Th1 cell apoptosis, as demonstrated by propidium iodide staining, in contrast to Th2 cells 28. Of notable interest, Forouzandeh et al. found a differential sensitivity in cell proliferation between human CD4+ and CD8+ T cells. IDO-expressing fibroblasts were co-cultured with human CD4+ and CD8+ T cells. When grown in the same IDO-induced tryptophan-deficient microenvironment, CD8+ T cells were more sensitive for IDO enzyme activity than CD4+ T cells, showing less CD8+ T cell proliferation by [3H]-thymidine incorporation 29. Remarkably, Sørensen et al. described the effects of IDO-specific cytotoxic CD8+ T cells against IDO enzyme active human cancer cell lines. IDO-specific cytotoxic T cells were capable of killing these cancer cell lines. The authors suggested that IDO-specific T cells might be crucial for an effective immune response during cancer 30. In an IDO active experimental rat lung allograft model, Lui et al. suggested that infiltrating CD8+ T cells remained viable, but the cytotoxic function was affected accompanied by defects in production of granule cytotoxic proteins 31. In line, but not totally comparable with this, we documented a partial recovery of CD4+ T cell proliferation and inhibition of CD4+ T cell apoptosis using the IDO inhibitor 1-L-MT. The question remains as to whether this statistically significant result is biologically relevant. In contrast, neither recovery of CD8+ T cell proliferation nor CD8+ T cell apoptosis could be detected after addition of IDO inhibitor. To our surprise, Transwell experiments revealed the absolute necessity of cell–cell contact in order to suppress the T cell proliferation that was not affected by the addition of IDO inhibitor to the system. This suggests that in a simplified IDO active renal tubular microenvironment model both CD4+ and CD8+ T cells remain fully activated, as not all cells can be in contact with the tubular barrier and will probably mount an effective immune response against alloantigens.
In response to immunostimulation, TECs up-regulate HLA classes I, II 32 and II 33 co-stimulatory molecules ICOS-L 34, CD40 35 and ICAM-1 36, and the inhibitory molecule PD-L1 34,35. Similarly, activated TECs have been shown to execute inhibitory effects on T cell alloreactivity via the PD-L1 pathway. IFN-γ stimulation leads to a strong dose-dependent increase of PD-L1 cell surface expression. In a TEC T cell co-culture system using anti-CD3/CD28 activated T cells, blockade of PD-L1 resulted in a significant increase of CD4+ T cell proliferation and not CD8+ T cell proliferation 24. In an in-vitro model using ovalbumin (OVA)-specific CD8+ T cells and TECs, the PD-L1 pathway significantly inhibited CD8+ T cell effector function 37. We also detected high levels of PD-L1 on IFN-γ/TNF-α-activated TECs. In contrast to others 24,34,37, we could not detect any evidence for the involvement of PD-L1 in TEC-induced inhibition of alloreactive T cell proliferation. We used primary cell cultures activated with IFN-γ and TNF-α, as these two cytokines represent potent proinflammatory cytokines present during renal allograft rejection 38,39. The type of TEC cell lines and the stimuli used for activation of TECs may provide an explanation for the different findings; T cells were not susceptible for PD-L1-mediated immune regulation in our experimental set-up.
Parenchymal cells have been shown to exert their immunosuppressive effects in a cell–cell contact-dependent manner, as supernatant experiments did not reveal any inhibitory effect of IDO 40,41. Similarly, ICAM-1 and vascular cell adhesion protein 1 (VCAM-1) are required for lymphocyte–MSC adhesion. ICAM-1 and VCAM-1 were also critical for MSC-mediated immunosuppression. MSC-mediated immunosuppression was reversed significantly in vitro and in vivo when the adhesion molecules were genetically deleted or functionally blocked 25. We found significant inhibitory properties of activated TECs using cell–cell contact-dependent experiments. We also documented high cell surface expression levels ICAM-1 on IFN-γ/TNF-α-activated TECs in our systems; indeed, ICAM-1 facilitates the cell–cell contact necessary for the suppression of CD4+ T cells, resulting in increased CD4+ T cell apoptosis. This suggests that TECs possess MSC-like immunomodulatory capacities. Interestingly, our co-culture experiments showed higher CTLA-4 + CD8+ T cell frequencies compared with CD4+ T cell counterparts, and higher FoxP3+ CD4+ T cell frequencies compared with CD8+ T cell counterparts. These findings indicate that TECs activate T cells resulting in the expression of CTLA-4 and FoxP3 as activation markers 42,43, and that TEC-reactive CD8+ T cells are more prone for inhibition via CTLA-4-mediated activation-induced cell death 44, which could reasonably underlie why IDO inhibition is not capable of reversing CD8+ T cell proliferation. Moreover, TECs could also induce regulatory CD4+ T cells being able to inhibit the proliferation of other immune cells. We have previously shown similar immune regulatory effects by MSC 45. Supported by our data, it is clear that TECs can act as a double-edged sword, with both immune stimulatory potential and also capacities to inhibit proliferation of antigen-activated T cells. However, the exact mechanisms underlying this intriguing phenomenon need to be unravelled in future studies.
In summary, we show that TECs exert immunosuppressive effects on CD4+ and CD8+ T cell proliferation and lead to enhanced T cell apoptosis. This would mean that, in the renal microenvironment, T cells in contact with the TEC barrier are exposed to more inactivation and death by TECs. Infiltrating T cells in the renal interstitial compartment will still be able to mount effective immune responses against alloantigens. In this light, significant serum and urine kynurenine/tryptophan ratios of rejecting patients might reflect the degree of tubular damage during rejection, rather than an active repair mechanism aiming to dampen the local inflammation. Whether drug strategies aimed to target cell–cell contact and to modulate the expression level of co-stimulatory molecules on TECs will be efficient to prevent overt tubular disease and eventual structural kidney damage remain to be investigated.
Disclosure
The authors of this manuscript have no financial or commercial conflicts of interest to disclose.
Author contributions
M. D. participated in research design, performance of research, data analysis and writing of the paper. C. B. participated in research design, data analysis and writing of the paper. M. R. participated in performance of research. T. B. participated in performance of research. S. K. participated in performance of research and data analysis. M. H. participated in research design. M. B. contributed to the materials. W. W. participated in writing of the paper. A. R. participated in research design, data analysis and writing of the paper.
References
- Bonsib SM, Abul-Ezz SR, Ahmad I, et al. Acute rejection-associated tubular basement membrane defects and chronic allograft nephropathy. Kidney Int. 2000;58:2206–2214. doi: 10.1111/j.1523-1755.2000.00395.x. [DOI] [PubMed] [Google Scholar]
- Jevnikar AM, Mannon RB. Late kidney allograft loss: what we know about it, and what we can do about It. Clin J Am Soc Nephrol. 2008;3(Suppl. 2):S56–S67. doi: 10.2215/CJN.03040707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nankivell BJ, Alexander SI. Rejection of the kidney allograft. N Engl J Med. 2010;363:1451–1462. doi: 10.1056/NEJMra0902927. [DOI] [PubMed] [Google Scholar]
- Halloran PF. T cell-mediated rejection of kidney transplants: a personal viewpoint. Am J Transplant. 2010;10:1126–1134. doi: 10.1111/j.1600-6143.2010.03053.x. [DOI] [PubMed] [Google Scholar]
- Demmers MWHJ, Baan CC, van Beelen E, Ijzermans JNM, Weimar W, Rowshani AT. Differential effects of activated human renal epithelial cells on T-cell migration. PLOS ONE. 2013;8:e64916. doi: 10.1371/journal.pone.0064916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmers MW, Baan CC, Janssen M, et al. Substantial proliferation of human renal tubular epithelial cell-reactive CD4+CD28null memory T cells, which is resistant to tacrolimus and everolimus. Transplantation. 2014;97:47–55. doi: 10.1097/01.TP.0000435697.31148.b2. [DOI] [PubMed] [Google Scholar]
- Stout-Delgado HW, Getachew Y, Rogers TE, Miller BC, Thiele DL. The role of serpinb9/serine protease inhibitor 6 in preventing granzyme B-dependent hepatotoxicity. Hepatology. 2007;46:1530–1540. doi: 10.1002/hep.21820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowshani AT, Florquin S, Bemelman F, Kummer JA, Hack CE, Ten Berge IJ. Hyperexpression of the granzyme B inhibitor PI-9 in human renal allografts: a potential mechanism for stable renal function in patients with subclinical rejection. Kidney Int. 2004;66:1417–1422. doi: 10.1111/j.1523-1755.2004.00903.x. [DOI] [PubMed] [Google Scholar]
- Heutinck KM, Kassies J, Florquin S, Ten Berge IJ, Hamann J, Rowshani AT. SerpinB9 expression in human renal tubular epithelial cells is induced by triggering of the viral dsRNA sensors TLR3, MDA5 and RIG-I. Nephrol Dial Transplant. 2012;27:2746–2754. doi: 10.1093/ndt/gfr690. [DOI] [PubMed] [Google Scholar]
- Rowshani AT, Strik MCM, Molenaar R, et al. The granzyme B inhibitor SERPINB9 (protease inhibitor 9) circulates in blood and increases on primary cytomegalovirus infection after renal transplantation. J Infect Dis. 2005;192:1908–1911. doi: 10.1086/497606. [DOI] [PubMed] [Google Scholar]
- Hoogduijn MJ, Betjes MGH, Baan CC. Mesenchymal stromal cells for organ transplantation: different sources and unique characteristics? Curr Opin Organ Transplant. 2014;19:41–46. doi: 10.1097/MOT.0000000000000036. doi: 10.1097/MOT.0000000000000036. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne GI, Lehmann LK, Kirschbaum JG, Borden EC, Lee CM, Brown RR. Induction of tryptophan degradation in vitro and in vivo: a gamma-interferon-stimulated activity. J Interferon Res. 1986;6:389–396. doi: 10.1089/jir.1986.6.389. [DOI] [PubMed] [Google Scholar]
- Mohib K, Guan Q, Diao H, Du C, Jevnikar AM. Proapoptotic activity of indoleamine 2,3-dioxygenase expressed in renal tubular epithelial cells. Am J Physiol Renal Physiol. 2007;293:F801–812. doi: 10.1152/ajprenal.00044.2007. [DOI] [PubMed] [Google Scholar]
- Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GK, Park HJ, Macleod M, Chandler P, Munn DH, Mellor AL. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology. 2002;107:452–460. doi: 10.1046/j.1365-2567.2002.01526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curti A, Trabanelli S, Salvestrini V, Baccarani M, Lemoli RM. The role of indoleamine 2,3-dioxygenase in the induction of immune tolerance: focus on hematology. Blood. 2009;113:2394–2401. doi: 10.1182/blood-2008-07-144485. [DOI] [PubMed] [Google Scholar]
- Bauer TM, Jiga LP, Chuang J-J, Randazzo M, Opelz G, Terness P. Studying the immunosuppressive role of indoleamine 2,3-dioxygenase: tryptophan metabolites suppress rat allogeneic T-cell responses in vitro and in vivo. Transpl Int. 2005;18:95–100. doi: 10.1111/j.1432-2277.2004.00031.x. [DOI] [PubMed] [Google Scholar]
- Hou W, Li S, Wu Y, Du X, Yuan F. Inhibition of indoleamine 2, 3-dioxygenase-mediated tryptophan catabolism accelerates crescentic glomerulonephritis. Clin Exp Immunol. 2009;156:363–372. doi: 10.1111/j.1365-2249.2009.03902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandacher G, Cakar F, Winkler C, et al. Non-invasive monitoring of kidney allograft rejection through IDO metabolism evaluation. Kidney Int. 2007;71:60–67. doi: 10.1038/sj.ki.5002023. [DOI] [PubMed] [Google Scholar]
- Hoogduijn MJ, Popp F, Verbeek R, et al. The immunomodulatory properties of mesenchymal stem cells and their use for immunotherapy. Int Immunopharmacol. 2010;10:1496–1500. doi: 10.1016/j.intimp.2010.06.019. [DOI] [PubMed] [Google Scholar]
- Crop MJ, Baan CC, Korevaar SS, et al. Donor-derived mesenchymal stem cells suppress alloreactivity of kidney transplant patients. Transplantation. 2009;87:896–906. doi: 10.1097/TP.0b013e31819b3d72. [DOI] [PubMed] [Google Scholar]
- Roemeling-Van Rhijn M, Mensah FK, Korevaar SS, et al. Effects of hypoxia on the immunomodulatory properties of adipose tissue-derived mesenchymal stem cells. Front Immunol. 2013;4:203. doi: 10.3389/fimmu.2013.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke A, Lindenmeyer MT, Segerer S, et al. Renal tubular PD-L1 (CD274) suppresses alloreactive human T-cell responses. Kidney Int. 2010;78:38–47. doi: 10.1038/ki.2010.97. [DOI] [PubMed] [Google Scholar]
- Ren G, Zhao X, Zhang L, et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J Immunol. 2010;184:2321–2328. doi: 10.4049/jimmunol.0902023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196:459–468. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terness P, Bauer TM, Rose L, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196:447–457. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallarino F, Grohmann U, Vacca C, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9:1069–1077. doi: 10.1038/sj.cdd.4401073. [DOI] [PubMed] [Google Scholar]
- Forouzandeh F, Jalili RB, Germain M, Duronio V, Ghahary A. Differential immunosuppressive effect of indoleamine 2,3-dioxygenase (IDO) on primary human CD4+ and CD8+ T cells. Mol Cell Biochem. 2008;309:1–7. doi: 10.1007/s11010-007-9635-y. [DOI] [PubMed] [Google Scholar]
- Sorensen RB, Hadrup SR, Svane IM, Hjortso MC, Thor Straten P, Andersen MH. Indoleamine 2,3-dioxygenase specific, cytotoxic T cells as immune regulators. Blood. 2011;117:2200–2210. doi: 10.1182/blood-2010-06-288498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Liu L, Liu K, Bizargity P, Hancock WW, Visner GA. Reduced cytotoxic function of effector CD8+ T cells is responsible for indoleamine 2,3-dioxygenase-dependent immune suppression. J Immunol. 2009;183:1022–1031. doi: 10.4049/jimmunol.0900408. [DOI] [PubMed] [Google Scholar]
- Lin Y, Proud G, Taylor RM, Kirby JA. Renal allograft rejection: protection of renal epithelium from natural killer cells by cytokine-induced up-regulation of class I major histocompatibility antigens. Immunology. 1993;79:290–297. [PMC free article] [PubMed] [Google Scholar]
- Wuthrich RP, Glimcher LH, Yui MA, Jevnikar AM, Dumas SE, Kelley VE. MHC class II, antigen presentation and tumor necrosis factor in renal tubular epithelial cells. Kidney Int. 1990;37:783–792. doi: 10.1038/ki.1990.46. [DOI] [PubMed] [Google Scholar]
- de Haij S, Woltman AM, Trouw LA, et al. Renal tubular epithelial cells modulate T-cell responses via ICOS-L and B7-H1. Kidney Int. 2005;68:2091–2102. doi: 10.1111/j.1523-1755.2005.00665.x. [DOI] [PubMed] [Google Scholar]
- Waeckerle-Men Y, Starke A, Wahl PR, Wuthrich RP. Limited costimulatory molecule expression on renal tubular epithelial cells impairs T cell activation. Kidney Blood Press Res. 2007;30:421–429. doi: 10.1159/000110578. [DOI] [PubMed] [Google Scholar]
- Wilson JL, Proud G, Forsythe JL, Taylor RM, Kirby JA. Renal allograft rejection. Tubular epithelial cells present alloantigen in the presence of costimulatory CD28 antibody. Transplantation. 1995;59:91–97. [PubMed] [Google Scholar]
- Waeckerle-Men Y, Starke A, Wuthrich RP. PD-L1 partially protects renal tubular epithelial cells from the attack of CD8+ cytotoxic T cells. Nephrol Dial Transplant. 2007;22:1527–1536. doi: 10.1093/ndt/gfl818. [DOI] [PubMed] [Google Scholar]
- Mueller TF, Einecke G, Reeve J, et al. Microarray analysis of rejection in human kidney transplants using pathogenesis-based transcript sets. Am J Transplant. 2007;7:2712–2722. doi: 10.1111/j.1600-6143.2007.02005.x. [DOI] [PubMed] [Google Scholar]
- Pelletier R, Pravica V, Perrey C, et al. Evidence for a genetic predisposition towards acute rejection after kidney and simultaneous kidney–pancreas transplantation. Transplantation. 2000;70:674–680. doi: 10.1097/00007890-200008270-00023. [DOI] [PubMed] [Google Scholar]
- Crop MJ, Baan CC, Korevaar SS, et al. Inflammatory conditions affect gene expression and function of human adipose tissue-derived mesenchymal stem cells. Clin Exp Immunol. 2010;162:474–486. doi: 10.1111/j.1365-2249.2010.04256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krampera M, Glennie S, Dyson J, et al. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood. 2003;101:3722–3729. doi: 10.1182/blood-2002-07-2104. [DOI] [PubMed] [Google Scholar]
- Wang J, Ioan-Facsinay A, van der Voort EIH, Huizinga TWJ, Toes REM. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol. 2007;37:129–138. doi: 10.1002/eji.200636435. [DOI] [PubMed] [Google Scholar]
- Linsley PS, Greene JL, Tan P, et al. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J Exp Med. 1992;176:1595–1604. doi: 10.1084/jem.176.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribben JG, Freeman GJ, Boussiotis VA, et al. CTLA4 mediates antigen-specific apoptosis of human T cells. Proc Natl Acad Sci USA. 1995;92:811–815. doi: 10.1073/pnas.92.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engela AU, Hoogduijn MJ, Boer K, et al. Human adipose-tissue derived mesenchymal stem cells induce functional de-novo regulatory T cells with methylated FOXP3 gene DNA. Clin Exp Immunol. 2013;173:343–354. doi: 10.1111/cei.12120. [DOI] [PMC free article] [PubMed] [Google Scholar]