

Abstract
Background and Purpose
MLN4924 prevents the formation of active cullin-RING ubiquitin ligase complexes and thus inhibits NF-κB signalling. Here, we evaluated the effects of this compound on monocytes and dendritic cells (DCs).
Experimental Approach
Monocytes and DCs were challenged with TNF or LPS in the presence and absence of MLN4924. The effects of MLN4924 on cellular viability, pro-inflammatory gene induction and DC maturation were investigated using the MTT assay, elisa and FACS analysis. Mechanisms of cell death induction were evaluated by using inhibitors of caspases, RIPK1 and MLKL.
Key Results
MLN4924 inhibited NF-κB activation and sensitized monocytes and immature DCs (iDCs) for TNFR1-induced cell death. Neither the caspase inhibitor zVAD-fmk, the RIPK1 inhibitor necrostatin-1 (nec-1) nor the MLKL inhibitor necrosulfonamide (NSA) alone prevented TNF-induced cell death. A combination of zVAD-fmk and nec-1 or NSA, however, rescued monocytes and iDCs from MLN4924/TNF-induced cell death indicating that MLN4924 affects anti-apoptotic and anti-necrotic activities in TNFR1 signalling. MLN4924 also converted the response of iDCs to LPS from maturation to cell death. LPS-induced cell death in MLN4924-treated iDCs was again only effectively blocked by cotreatment with zVAD-fmk and nec-1 or NSA. Noteworthy, MLN4924/LPS-induced cell death was almost completely independent of endogenous TNF. MLN4924 also strongly inhibited maturation and activation of iDCs that were rescued from cell death by zVAD-fmk and nec-1.
Conclusions and Implications
Our data reveal a strong dual suppressive effect of MLN4924 on DC activity. The targeting of NAE by MLN4924 could be a new way to treat inflammatory diseases.
Tables of Links
| TARGETS | |
|---|---|
| Catalytic receptorsa | Enzymesb |
| Fas (CD95/TNFRSF5) | Caspase-3 |
| TNFR1 (TNFRSF1A) | Caspase-8 |
| TNFR2 (TNFRSF1B) | Caspase-9 |
| Toll-like receptor 3 (TLR3) | IKK2 |
| Other protein targets | MLKL |
| CD86 | RIPK1 |
| cIAP2 | RIPK3 |
| Tubulin |
| LIGANDS | |
|---|---|
| A20 | IL-4 |
| β-catenin | IL-6 |
| Bortezomib | IL-12 |
| CD40L | LPS |
| CD95L | Lymphotoxin-α |
| Enbrel | PGE2 |
| FLIP | TNF |
| GM-CSF | TPCA-1 |
| Humira | TWEAK |
| IL-1β |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Introduction
The success of proteasome inhibitors such as Bortezomib in cancer therapy has prompted the development of other therapeutic compounds targeting proteins of the ubiquitin proteasome system. One such compound is the neural precursor cell expressed, developmentally down-regulated 8 (NEDD8) activating enzyme (NAE) inhibitor MLN4924 (Soucy et al., 2009). NEDD8 is a ubiquitin-like protein that is conjugated to the cullin subunits of E3 ligases of the cullin-RING ubiquitin ligase (CRL) family in a ubiquitination-related process by the sequential action of the NEDD8-activating E1 enzyme, the E2 enzyme UbcH12 and an E3 ligase (Watson et al., 2011; Tanaka et al., 2013). Modification of cullins with NEDD8 allows assembly of the active CRL complexes, and an intact neddylation pathway is thus of crucial relevance for the activity of CRL E3 ligases. CRLs mark a variety of proteins for proteasomal degradation including the cell cycle inhibitors p21 and p27, various cyclins and the DNA replication licensing protein CTD1 (Watson et al., 2011; Tanaka et al., 2013). Accumulation of these CRL substrates in MLN4924-treated cells triggers S-phase defects and DNA damage and eventually p53 activation, cell cycle arrest and apoptosis. The CRL family member β-transducin repeat containing protein (βTrCP) furthermore triggers proteasomal degradation of IκBα and p100 processing, key events in signalling pathways that mediate the activation of transcription factors of the NF-κB family (Read et al., 2000; Amir et al., 2004). MLN4924 thus also acts as a potent inhibitor of NF-κB activation (Milhollen et al., 2010; Swords et al., 2010; Chang et al., 2012; Mathewson et al., 2013; Rauert-Wunderlich et al., 2013; Godbersen et al., 2014). The NF-κB system regulates the transcription of a considerable number of anti-apoptotic proteins and pro-inflammatory cytokines. The inhibitory effect of MLN4924 on NF-κB signalling not only contributes to the induction of apoptosis by down-regulating survival factors but also has an anti-inflammatory effect. Indeed, recent publications revealed that MLN4924 inhibits the NF-κB-mediated induction of pro-inflammatory cytokines in macrophages and dendritic cells (DCs; Chang et al., 2012; Li et al., 2013; Mathewson et al., 2013). From preclinical models, there is broad evidence that MLN4924 has anti-tumoural activity. However, it is noteworthy that MLN4924 exerts its anti-tumoural activity not only by triggering cell death and cell cycle arrest but also by suppressing tumour growth and metastasis as a results of its inhibition of angiogenesis (Tan et al., 2014; Yao et al., 2014). Currently, MLN4924 is under consideration in multiple phase I studies to treat solid tumours, metastatic melanoma and various haematological malignancies (http://clinicaltrials.gov, identifiers: NCT02122770, NCT01862328, NCT01011530, NCT01814826, NCT00677170, NCT00911066 and NCT00722488). Substrates of CRLs regulate a diverse spectrum of cellular processes. MLN4924 might therefore also have therapeutic potential beyond tumour therapy. This is indicated not only by the aforementioned studies reporting anti-inflammatory effects of MLN4924, but also by the recent findings showing that MLN4924 restricts retroviral infection of myeloid cells by preventing virus-induced degradation of the deoxynucleotide triphosphohydrolase SAMHD1, which limits retroviral replication (Hofmann et al., 2013; Nekorchuk et al., 2013; Wei et al., 2014).
Here, we reveal a dual suppressive effect of MLN4924 on monocytes and DCs. Our experiments showed that MLN4924 not only effectively inhibits the NF-κB-dependent induction of chemokines/cytokines and the maturation of DCs but also sensitizes monocytes and immature DCs (iDCs) to TNF- and LPS-induced necroptosis. NAE targeting with MLN4924 could, therefore, be a new way to treat inflammatory diseases.
Methods
Preparation of monocytes, iDCs and mature dendritic cells (mDCs)
Blood buffy coats of totally anonymous donors, for which no special written informed consent is required, were obtained from the Institute of Clinical Transfusion Medicine and Hemotherapy of the University Hospital Würzburg. Peripheral blood mononuclear cells were isolated from blood buffy coats using density gradient centrifugation with lymphocyte separation medium (PAA Laboratories, Pasching, Germany). Pure monocytes were isolated using anti-CD14-coated beads and magnetic bead separation (Miltenyi Biotec, Bergisch Gladbach, Germany). The purity of the monocytes was controlled by FACS analysis of CD14 expression. To obtain iDCs, monocytes were cultivated immediately in 10 cm Petri dishes containing RPMI 1640, 10% FCS and 1% penicillin to induce the differentiation of monocytes into iDCs by adding 30 ng·mL−1 of IL-4 (Miltenyi Biotec) and 50 ng·mL−1 of granulocyte-macrophage colony-stimulating factor (GM-CSF) (Miltenyi Biotec) every second day for 1 week. Differentiation to iDCs was controlled by FACS evaluation for the absence of CD14 expression. mDCs were furthermore obtained by stimulating iDCs for 2 days with Fc-CD40L (1 μg·mL−1) in the presence of IL-4 (30 ng·mL−1) and GM-CSF (50 ng·mL−1). Successful stimulation of DC maturation was controlled by FACS evaluation of the up-regulation of CD83 and CD86.
Cell death assay
Freshly isolated monocytes were seeded on 96-well plates at a density of 200 × 103 cells per well and were stimulated the same day. iDCs were seeded in 96-well plates at a density of 50 × 103 cells per well in the presence of IL-4 and GM-CSF. Monocytes, iDCs and mDCs were then challenged with the following reagents as indicated in the corresponding figure legends: MLN4924 (5 μM; Active Biochemicals Co., Hong Kong, China), necrostatin-1 (nec-1; 90 μM; Enzo Life Sciences, Lörrach, Germany), necrosulfonamide (NSA; 2.5 μM, Merck Calbiochem, San Diego, CA, USA), benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (zVAD-fmk; 50 μM; Bachem AG, Weil am Rhein, Germany), Humira (10 μg·mL−1; Abbott Biotechnology, Maplewood, NJ, USA) or Enbrel (10 μg·mL−1; Wyeth Pharma GmbH, Münster, Germany), TNF (kind gift of Prof. Daniela Männel, University of Regensburg), LPS (Sigma, Deisenhofen, Germany), poly IC (Sigma), Fc-CD40L (Wyzgol et al., 2009), a mixture of 10 ng·mL−1 IL-1β (R&D Systems, Wiesbaden-Nordenstadt, Germany) and 1 μg·mL−1 of PGE2 (Biomol, Hamburg, Germany), TNF (32W/86T) (TNFR1-specific TNF mutant), TNC-scTNF(143N/145R) (TNFR2-specific nonameric TNF mutant) (Rauert et al., 2010) and Fc-CD95L. With exception of TNF, all TNF ligand variants were produced in HEK293 cells and purified using anti-Flag M2 agarose affinity purification. LPS contents were verified using the Pierce LAL chromogenic endotoxin quantification kit (Thermo Fischer Scientific, Waltham, MA, USA) and if necessary LPS contaminations were removed by help of the Pierce high-capacity endotoxin removal resin (Thermo Fischer Scientific). In all experiments evaluating the protective effect of zVAD-fmk/nec-1, zVAD-fmk/NSA, Humira and Enbrel, four randomly chosen wells of a 96-well plate per group were stimulated. DMSO was used as a negative control for the vehicle of MLN4924. Nec-1 alone caused a slight increase in the optical density that may interfere with the interpretation of its protective effect against cell death. Therefore, a group of cells treated only with Nec-1 was included in all cell viability experiments using Nec-1. MLN4924 was added 30 min before challenging the cells with the indicated stimuli. Cellular viability was finally assayed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
Western blotting
For Western blot analysis, cells were harvested in ice-cold PBS, washed twice in PBS and pelleted for 4 min at 1280× g (4°C). To prepare total cell lysates, cell pellets were lysed in 4× Laemmli buffer (0.2 M Tris, 10% β-mercaptoethanol, 8% SDS, 40% glycerol, pH 8.0) containing phosphatase inhibitor mixture II (Sigma) and protease inhibitor (Roche Diagnostics, Basel, Switzerland), sonicated for 20 s, boiled for 5 min at 96°C and finally cleared by centrifugation for 10 min at 20 800× g. Cell lysates were subjected to fractionation by vertical SDS-PAGE, and subsequently proteins were transferred to nitrocellulose membranes by wet blotting. Western blot analyses were performed with primary antibodies specific for caspase-8 (clone C15; Enzo Life Sciences), caspase-3 (#9662; Cell Signaling Technology, USA), caspase-9 (#9502; Cell Signaling Technology, Frankfurt, Germany), cellular inhibitor of apoptosis protein 2 (cIAP2) (#3130; Cell Signaling Technology), tubulin (Dunn Labortechnik, Asbach, Germany) and NEDD8 (Cell Signaling Technology), β-catenin (clone 6B3; Cell Signaling Technology), IκBα (clone L35A5; Cell Signaling Technology), pIκBα (clone 14D4; Cell Signaling Technology) and TNF receptor-associated factor 1/2 (TRAF1; H-132, Santa Cruz Biotechnology, Heidelberg, Germany). For detection of antigen primary antibody complexes, anti-mouse-HRP (Dako-Cytomation, Hamburg, Germany) and anti-rabbit-HRP (Dako-Cytomation or Cell Signaling Technology) were used. To finally visualize the antigen–antibody complexes, the ECL Western blotting detection system (Thermo Fischer Scientific) was used according to the protocol of the manufacturer.
FACS analyses
Monocytes, iDCs and mDCs were treated as indicated in the corresponding figure legends, harvested, washed twice in ice-cold PBS and prepared for FACS analysis as follows: cells were incubated with specific antibodies or corresponding isotype control antibodies for 30 min at 4°C. After three washes with PBS, cells were resuspended in 150 μL PBS and analysed using a FACSCalibur. Directly labelled PE-conjugated antibodies were used to detect CD14 (clone M5E2; BD Biosciences, Heidelberg, Germany), CD83 (clone HB15e; R&D Systems), CD86 (clone 37301; R&D Systems), membrane TNF (clone MAb11; eBioscience, Germany), TNFR1 (clone 16803; R&D Systems) and TNFR2 (clone 22235; R&D Systems). The corresponding PE-conjugated isotype control antibodies were from BD Biosciences (mouse IgG2aκ), eBioscience (mouse IgG1κ) and R&D Systems (mouse IgG1, clone 11711 and mouse IgG2a). To detect CD95 (Fas) expression, cells were incubated with anti-Fas (clone DX2, R&D Systems) and with anti-Flag mAb M2 (mouse IgG1 monoclonal, Sigma) as a negative control group. After three washes with ice-cold PBS, cells were incubated for 30 min with anti-mouse IgG-PE (Sigma) and processed as described above for directly labelled primary antibodies.
elisa assay
iDCs were seeded (50 × 103 cells per well) on 96-well plates and stimulated as indicated for 24 h. Next day, the plates were centrifuged for 2 min at 1200× g, and the supernatants were collected for further analysis of their content of IL-6 (human IL-6 elisa set, BD Biosciences), IL-12 (human IL-12 elisa DuoSet, R&D Systems) and TNF (human TNF elisa set, BD Biosciences).
qPCR
Total RNAs were purified from monocytes and iDCs (RNeasy Mini kit, Qiagen, Hilden, Germany) and 1 μg of each sample was subjected to reverse transcription PCR (QuantiTect Reverse Transcription kit, Qiagen). Quantitative real-time PCR reactions were then performed with 2% of the reverse transcription PCR reactions using the Cycler CT1000 (BioRad) with the CFX96 real-time system and the QuantiTect SYBR Green PCR Kit (Qiagen). The amplification programme consisted of the following three steps: (i) 95°C, hot start; (ii) 40 cycles of 15 s at 95°C; and (iii) 30 s at 52°C and 30 s at 72°C. The primers used for amplification (FLIP: HS_CFLAR_1_SG; A20: HS_TNFAIP3_1_SG; β-actin: HS_ACTB_2_SG) were from Qiagen. To calculate fold induction values for A20 and FLIP, the CT values of the β-actin PCR reactions of the treated samples were set to the CT value of the β-actin PCR reaction derived from cDNA of untreated cells. Then the differences of the latter and the CT values of the β-actin PCR reactions of the treated samples were used to correct the CT values of the A20- and FLIP-specific PCR reactions. To finally obtain fold induction values (fi), the differences between the corrected CT values derived from the untreated control samples and the various treatment groups (ΔCT) were transformed using the equation fi = 2ΔCT.
Statistical analyses
All statistical analyses were performed using the GraphPad Prism 5.0 program (GraphPad Software, Inc., La Jolla, CA, USA). P-values were calculated using Student's t-test (*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.0001). All results shown are representative experiments from different independent donors. Cell viability experiments were independently performed with monocytes and DCs prepared from more than four donors. FACS and elisa analyses were independently performed with cells from at least three donors. Western blotting analysis was performed with cells from two donors.
Results
MLN4924 sensitizes monocytes and iDCs for TNF-induced cell death
Human monocytes as well as iDCs and mDCs were challenged with increasing concentrations of TNF in the presence and absence of MLN4924. In all three cases, the treatment with MLN4924 strongly reduced the modification of cullins with NEDD-8 (Figure 1A). Accordingly, there was an accumulation of the established CRL substrates pIκBα and β-catenin (Figure 1A). MLN4924 had no, or only a very moderate, cytotoxic effect in the time frame of the experiment (Figure 1B). Likewise, treatment with soluble TNF alone elicited no major cytotoxicity. More intriguing, soluble TNF induced considerable cell death at moderate concentrations in monocytes and iDCs treated with MLN4924 (Figure 1C). In contrast, mDCs remained largely resistance against TNF also in the presence of MLN4924 (Figure 1C).
Figure 1.
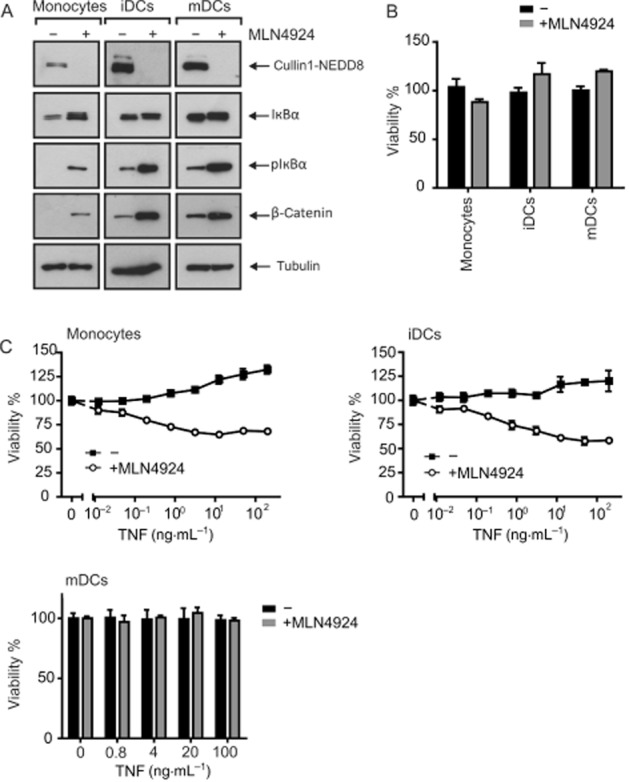
MLN4924 sensitizes monocytes and iDCs for TNF-induced cell death. (A) Monocytes, iDCs and mDCs were challenged overnight with 5 μM MLN4924, and total lysates were analysed by Western blotting for the presence of the indicated proteins. (B) Cells were stimulated in triplicate with MLN4924 (5 μM), and next day cellular viability was determined using the MTT assay. (C) Cells were stimulated in triplicate in 96-well plates with the indicated concentrations of TNF in the presence and absence of 5 μM MLN4924. MLN4924 was added 30 min before stimulation with TNF. Next day, cellular viability was again quantified with MTT and normalized against samples treated with a mixture of cytotoxic reagents.
MLN4924/TNF-induced cell death is mediated by TNFR1
TNF typically induces cell death via TNFR1 while the pro-apoptotic effects of TNFR2 are indirect and base on induction of death ligands, for example, TNF itself, and/or the depletion of anti-apoptotic TRAF2–cIAP complexes (Wajant et al., 2003). Although FACS analysis of monocytes revealed the co-expression of TNFR1 and TNFR2, iDCs and mDCs showed no significant TNFR1 staining but revealed robust TNFR2 expression (Figure 2A). However, the expression of TNFR1 is notoriously low on many cell types and thus negative FACS data do not necessarily rule out the expression of functionally relevant amounts of TNFR1. Thus, to clarify the relevance of the two TNF receptors for the cytotoxic effects of TNF in the presence of MLN4924, we performed experiments with TNFR1- [TNF(32W/86T)] and TNFR2-specific [TNF(143N/145R)] mutants of soluble TNF that effectively discriminate between human TNFR1 and human TNFR2 binding (Loetscher et al., 1993). We previously found that oligomeric TNF variants have a significantly higher ability to stimulate TNFR2 than TNF trimers while ligand oligomerization has no effect on TNFR1 signalling (Rauert et al., 2010). In the case of TNFR2 stimulation, we therefore used a nonameric variant of the human TNFR2-specific TNF mutant, in which three protomers of this TNF mutant were connected by peptide linkers and fused to the trimerization domain of tenascin-C [TNC-scTNF(143N/145R)] (Rauert et al., 2010). The TNFR1-specific TNF mutant again triggered significant cell death in MLN4924-sensitized cells while the TNFR2-stimulating variant showed no cytotoxic activity indicating that MLN4924 sensitizes for TNFR1-mediated cell death (Figure 2B). iDCs showed significant expression of the TNFR1-related death receptor CD95 (Figure 2A). Therefore, we tested the effect of MLN4924 on the cytotoxic activity of Fc-CD95L. Noteworthy, treatment with MLN4924 showed no enhancing effect on CD95L-induced cell death in iDCs (Figure 2C), indicating that MLN4924 interferes with a TNFR1-specific survival mechanism that is irrelevant (or at least less relevant) for other death receptors.
Figure 2.
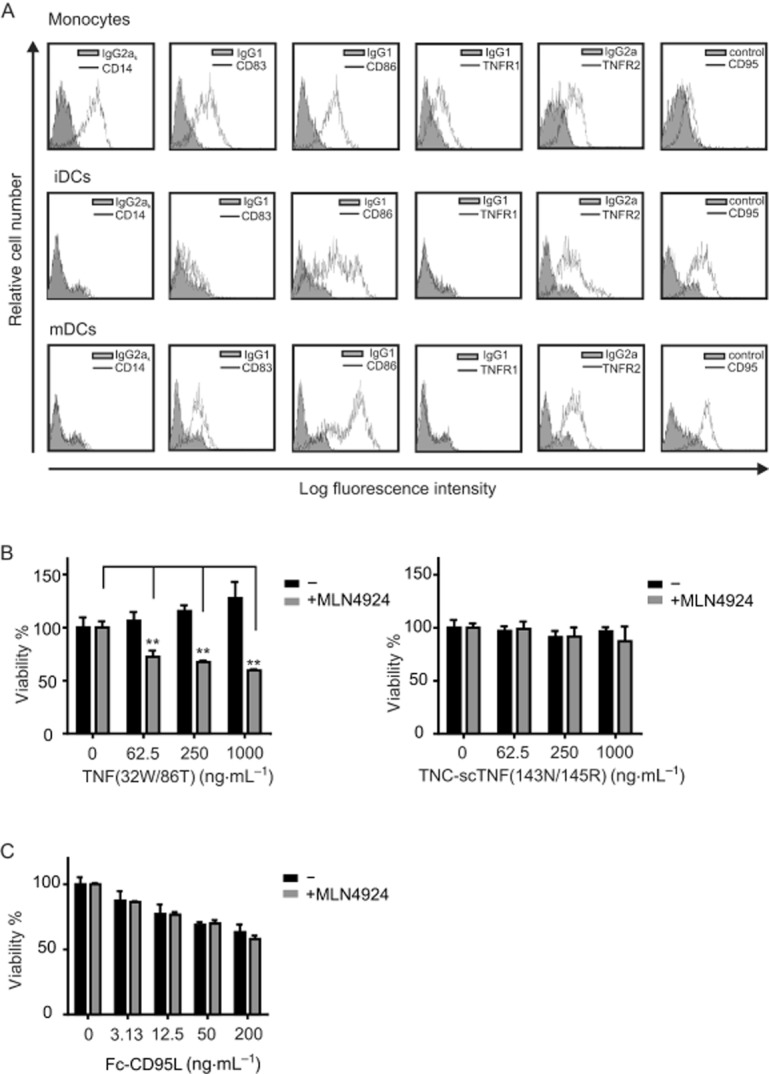
MLN4924/TNF-induced cell death is mediated by TNFR1. (A) Human monocytes, iDCs and CD40L-matured DCs (mDCs) were analysed by FACS evaluation for the cell surface expression of the indicated proteins. (B) iDCs were stimulated in triplicate in 96-well plates with the indicated concentrations of the TNFR1-specific TNF mutant TNF(32W/86T) or of the selectively TNFR2-activating TNF variant TNC-scTNF(143N/145R) in the presence and absence of 5 μM MLN4924. Next day, cellular viability was quantified and normalized against samples treated with a mixture of cytotoxic reagents. (C) Untreated and MLN4924-treated iDCs were challenged with the indicated concentrations of Fc-CD95L and assayed the next day for viability using the MTT assay.
MLN4924 sensitizes monocytes and iDCs for TNF-induced apoptosis and necroptosis
TNFR1-induced cell death can occur by two not mutually exclusive pathways: the caspase-8-dependent apoptotic pathway and the RIP1-RIP3-MLKL-mediated necroptotic pathway, whereby the activity of the latter pathway is antagonized by caspase-8 (Salvesen and Walsh, 2014; Vanden Berghe et al., 2014). We therefore evaluated the effect of well-established inhibitors of apoptosis and necroptosis on TNF/MLN4924-induced cell death in monocytes and iDCs. The pan-caspase inhibitor zVAD-fmk only partly rescued iDCs treated with MLN4924 and even strongly sensitized monocytes for the cytotoxic activity of TNF/MLN4924 (Figure 3A). Treatment with MLN4924 alone showed a very slight reduction in the viability of monocytes but even seemed to slightly improve the viability of iDCs (Figure 3A). As the MTT assay used for evaluation of cell viability quantifies metabolic activity, the MLN4924-induced increase in MTT staining does not necessarily reflect DC proliferation but could simply reflect changes in metabolic activity triggered by MLN4924. The RIPK1 inhibitor nec-1 as well as the MLKL inhibitor NSA showed no or only a mild protective effect when given alone to monocytes, but each of the two inhibitors almost completely inhibited TNF/MLN4924-induced death of monocytes and iDCs when applied in combination with zVAD-fmk (Figure 3A). In the case of monocytes, there was already significant processing of pro-caspase-8 to the p43/41 intermediate, but the combination of MLN4924 and TNF not only resulted in enhanced processing of the proform of caspase-8 but also allowed detection of the less stable p18 subunit of heterotetrameric mature caspase-8 (Figure 3B). Neither MLN4924 nor TNF alone induced significant caspase-8 processing in iDCs after 8 h of stimulation; however, the combination of MLN4924 with TNF triggered the processing not only of caspase-8 but also of the downstream caspases: caspase-3 and caspase-9 (Figure 3C). Furthermore, the processing of caspases was inhibited in the presence of zVAD-fmk (Figure 3C). In summary, these data suggest that MLN4924 inhibits the expression/activity of one or more factors that antagonize both TNFR1-induced apoptosis and necroptosis.
Figure 3.
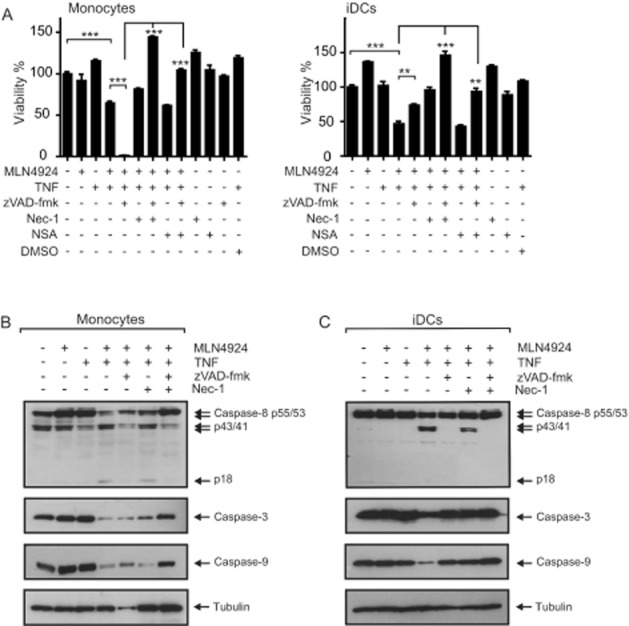
MLN4924 sensitizes monocytes and iDCs for TNF-induced apoptosis and necroptosis. (A) Monocytes and iDCs were challenged with TNF (100 ng·mL−1) and MLN4924 (5 μM) in the presence of the indicated mixtures of nec-1 (90 μM), NSA (2.5 μM) and zVAD-fmk (50 μM). Next day, cells were assayed for cellular viability using the MTT assay. (B and C) Monocytes and iDCs were treated with TNF (200 ng·mL−1) and MLN4924 (5 μM) in the presence of the indicated mixtures of nec-1 (90 μM) and zVAD-fmk (50 μM) for 8 h, and total cell lysates were subjected to Western blot analysis for processing of the indicated proteins.
MLN4924 sensitizes monocytes for endogenous TNF-induced necroptosis in the presence of zVAD-fmk
zVAD-fmk strongly enhanced the cytotoxic effect of MLN4924/TNF mixtures on monocytes (Figure 3A) and the latter express significant amounts of endogenous TNF (Figure 4A). Therefore, we wondered whether in the absence of exogenous TNF or in the presence of zVAD-fmk, MLN4924 has the capacity to trigger monocyte cell death in the presence of only endogenous TNF. Indeed, in the presence of zVAD-fmk, MLN4924 triggered dose-dependent cell death in monocytes but not in iDCs that showed no membrane TNF expression (Figure 4A and B). The cytotoxic effect of MLN4924 in the presence of zVAD-fmk in monocytes was completely abolished by treatment with nec-1 or NSA (Figure 4C). Moreover, blockade of TNF completely prevented the cell death induced by MLN4924/zVAD-fmk (Figure 4C). It is thus tempting to speculate that the modest cytotoxic effect of MLN4924 on monocytes that was observed in Figure 1B is also due to endogenous TNF-mediated activation of TNFR1. In any case, our data suggest that in monocytes endogenous TNF-induced autocrine/paracrine TNFR1-mediated necroptosis is prevented by caspase-8 and a factor that is inhibited by MLN4924.
Figure 4.
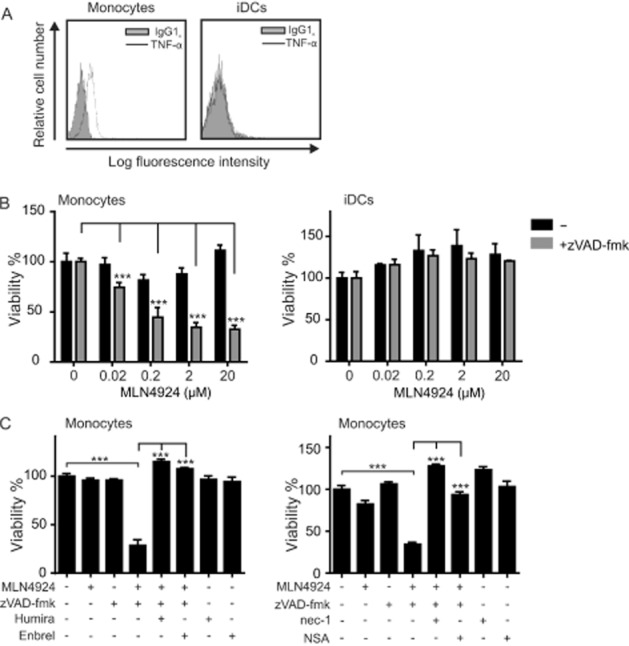
In the presence of zVAD-fmk, MLN4924 sensitizes monocytes for endogenous TNF-induced necroptosis. (A) Monocytes and iDCs were analysed by FACS for their cell surface expression of TNF. (B) Monocytes and iDCs were challenged with the indicated concentrations of MLN4924 in the presence and absence of zVAD-fmk (50 μM). The next day, cellular viability was assayed by MTT assay. (C) Monocytes were seeded on 96-well plates in the presence and absence of zVAD-fmk (50 μM), Humira (10 μg·mL−1), Enbrel (10 μg·mL−1), nec-1 (90 μM) or NSA (2.5 μM). Thirty minutes after adding the various reagents, cells were treated with MLN4924 (5 μM) and the next day cellular viability was assayed using the MTT assay.
MLN4924 inhibits activation and maturation of DCs
In view of the inhibitory effect of MLN4924 on the two NF-κB signalling pathways and the relevance of these pathways for several aspects of DC biology, we next investigated the effect of MLN4924 on the maturation of DCs and cytokine production of maturing DCs. It should be noted that TNF alone is able to trigger DC maturation. To clarify whether MLN4924 affects TNF-induced DC maturation not only by sensitizing the cells to factors that induce their death, but also by modulating the non-apoptotic signalling abilities of TNFR1, we evaluated the inhibitory effect of MLN4924 on TNF-induced DC maturation in the presence of a mixture of zVAD-fmk and nec-1 (Figure 5A and B). zVAD-fmk and nec-1 alone showed no major effect on TNF-induced expression of the DC maturation markers CD83 and CD86 (Figure 5A). More importantly, however, treatment with MLN4924 completely abolished TNF-induced maturation of iDCs rescued from cell death induction by a mixture of zVAD-fmk and nec-1 (Figure 5A). Likewise, MLN4924 inhibited TNF-induced IL-6 and IL-12 production in zVAD-fmk/nec-1-protected DCs (Figure 5B). In accordance with the idea that the observed inhibition of TNF-induced hallmarks of DC maturation by MLN4924 is due to inhibition of the classical NF-κB pathway, MLN4924 efficiently antagonized TNF-induced stimulation of phosphorylation and degradation of IκBα (Figure 5C). In the same scenario (in the presence of zVAD-fmk and nec-1), we evaluated the ability of MLN4924 to inhibit DC maturation triggered by LPS, CD40L or a mixture of IL-1β and PGE2 (Figure 6A and B). Again MLN4924 completely inhibited the up-regulation of the DC markers CD83 and CD86 (Figure 6A) as well as markedly inhibiting the secretion of IL-6 and IL-12 induced by LPS, CD40L or the mixture of IL-1β and PGE2 (Figure 6B). Thus, MLN4924, possibly, has at least a dual inhibitory effect on the pro-inflammatory activities of TNF on DCs. It not only limits the availability of mature DCs by sensitizing monocytes and iDCs for the cytotoxic activities of TNFR1, but also prevents the maturation and activation of DCs, most likely by inhibiting the classical NF-κB pathway. Importantly, the latter is not only crucial for the pro-inflammatory activities of TNFR1 but also positively regulates the expression of various factors that render cells resistant to TNF-induced cell death, such as A20, cIAP2, TRAF1 and FLIP (Krikos et al., 1992; Chu et al., 1997; Wang et al., 1998; Schwenzer et al., 1999; Kreuz et al., 2001; Micheau et al., 2001). Therefore, we analysed whether the ability of MLN4924 to sensitize cells for TNF-induced death correlates with its inhibitory effect on TNF-induced expression of the aforementioned survival factors. Western blot experiments showed that TNF strongly induces the expression of TRAF1 and cIAP2 in monocytes and iDCs and of cIAP2 in iDCs (Figure 7A). Furthermore, quantitative PCR revealed an up-regulation of A20 and FLIP (Figure 7B). Although the induction of cIAP2, A20 and TRAF1 was severely reduced in the presence of MLN4924, the expression of FLIP was slightly increased by MLN4924; however, this latter effect appeared not to be additive with that of TNF (Figure 7B). Thus, the ability of MLN4924 to enhance TNF-induced cell death largely correlates with its capacity to inhibit TNF-induced expression of NF-κB-regulated survival proteins.
Figure 5.
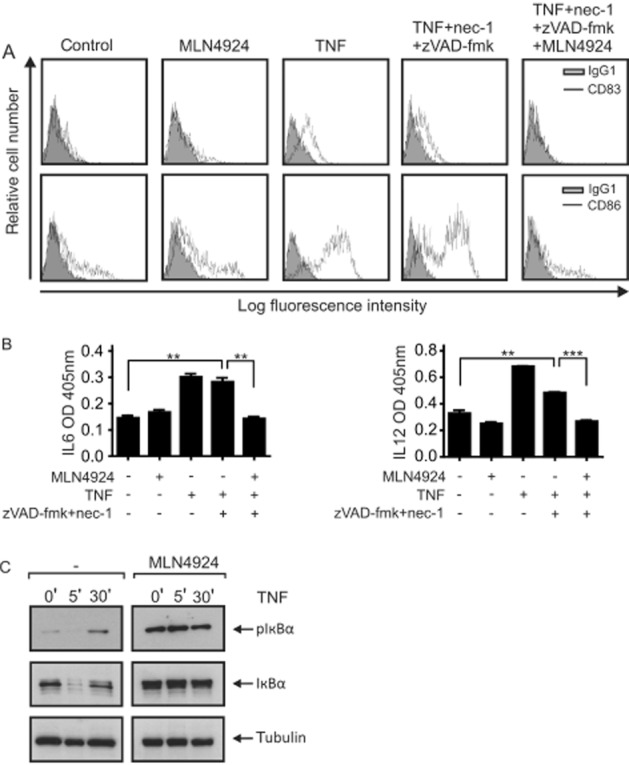
MLN4924 inhibits TNF-induced maturation of DCs independently of cell death induction. (A and B) iDCs were seeded in medium supplemented with nec-1 (90 μM) and zVAD-fmk (50 μM). Cells were then stimulated with TNF (500 ng·mL−1) and MLN4924 (5 μM) and were investigated after 48 h for DC maturation by analysing cell surface expression of CD83 and CD86 by FACS (A) and after 24 h for the secretion of IL-12 and IL-6 by elisa (B). (C) iDCs were stimulated with 500 ng·mL−1 TNF for the indicated time in the presence and absence of MLN4924 (5 μM). Total cell lysates were finally analysed by Western blotting for phosphorylation and degradation of IκBα.
Figure 6.
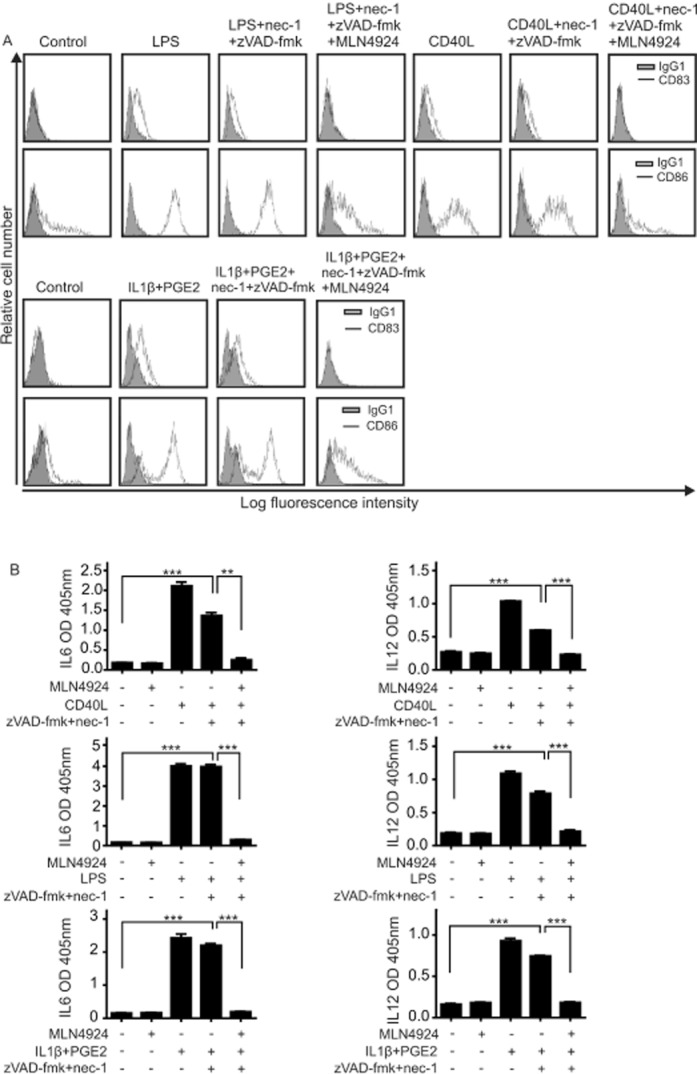
MLN4924 inhibits LPS-, CD40L- and IL-1β/PGE2-induced maturation of DCs. (A) iDC maturation was triggered with Fc-CD40L (1 μg·mL−1), LPS (10 ng·mL−1) and a mixture of IL-1β (10 ng·mL−1) and PGE2 (1 μg·mL−1) in the presence and absence of the indicated mixtures of nec-1 (90 μM), zVAD-fmk (50 μM) and MLN4924 (5 μM). After 48 h, cells were evaluated by FACS for the cell surface expression of CD83 and CD86. (B) iDCs were seeded in 96-well plates with the indicated mixtures of nec-1 (90 μM), zVAD-fmk (50 μM) and MLN4924 (5 μM) and were then stimulated with Fc-CD40L (1 μg·mL−1), LPS (1 ng·mL−1) or a mixture of IL-1β (10 ng·mL−1) and PGE2 (1 μg·mL−1). Next day, supernatants were analysed by elisa for their IL-6 and IL-12 content.
Figure 7.
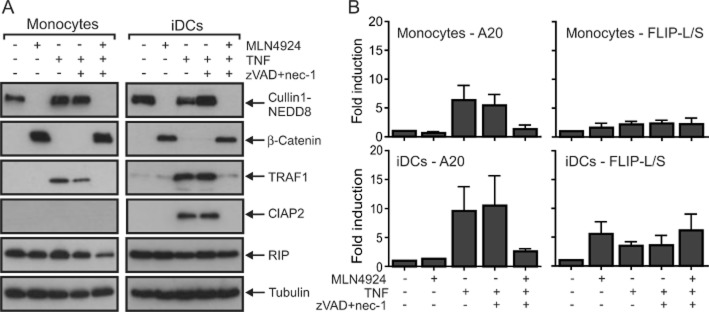
MLN4924 inhibits TNF-induced expression of cIAP2, TRAF1 and A20. (A and B) Monocytes and iDCs were incubated in medium in the presence of the indicated mixtures of nec-1 (90 μM), zVAD-fmk (50 μM), TNF (200 ng·mL−1) and MLN4924 (5 μM). Total cell lysates were then subjected to Western blot analysis to evaluate the expression of the indicated proteins. (A). Alternatively, total RNA was isolated and subjected to qPCR evaluation of expression of A20, TRAF1 and FLIP. (B) Data show the average of three experiments with three independent donors.
A mixture of MLN4924 and LPS induces necroptotic cell death in monocytes and iDCs independently of endogenous TNF
Next, we tested whether ML4924 only triggers cell death in maturing DCs in concert with exogenous TNF or also in combination with other reagents inducing DC maturation, such as LPS, poly IC (a synthetic toll-like receptor 3 agonist), Fc-CD40L or a mixture of IL-1β and PGE2. We observed robust cell death only in iDCs upon treatment with a combination of MLN4924 and LPS (Figure 8A). Moreover, treatment with MLN4924/LPS also induced cell death in monocytes (Figure 8B). It has been shown, previously, that zVAD-fmk sensitizes microglia to LPS-induced cell death (Kim and Li, 2013). Similarly, we observed that monocytes were strongly sensitized for LPS- as well as MLN4924/LPS-induced cell death in the presence of zVAD-fmk (Figure 8B). As in the case of MLN4924/TNF-induced cell death, cotreatment with zVAD-fmk/nec-1 or zVAD-fmk/NSA was required to prevent the death of monocytes and iDCs induced by MLN4924/LPS (Figure 8C). This suggests that MLN4924/LPS-induced cell death is due to the triggering of the apoptosis–necroptosis signalling network. As shown in Figure 4, MLN4924 was able to sensitize monocytes to endogenous TNF-induced necroptosis, and LPS also increased TNF production in DCs. Therefore, we evaluated whether MLN4924/LPS-induced cell death was related to the activity of endogenously produced TNF. Neither anti-human TNF (Humira) nor soluble human TNFR2-Fc, which neutralizes TNF and lymphotoxin α, a second TNF-related cytokine with TNFR1-stimulating activity, was able to antagonize cell death induced by MLN4924/LPS in monocytes and iDCs (Figure 8D). Moreover, in accordance with recent publications showing that inhibition of NF-κB signalling blocks LPS-induced TNF production (Li et al., 2013; Mathewson et al., 2013) and the strong inhibitory effect of MLN4924 on NF-κB activation (Figures 5 and 6), we found that MLN4924 even inhibited LPS-induced TNF production (Figure 8E). Thus, MLN4924/LPS-induced cell death in monocytes and DCs is largely independent of TNF/TNFR1.
Figure 8.
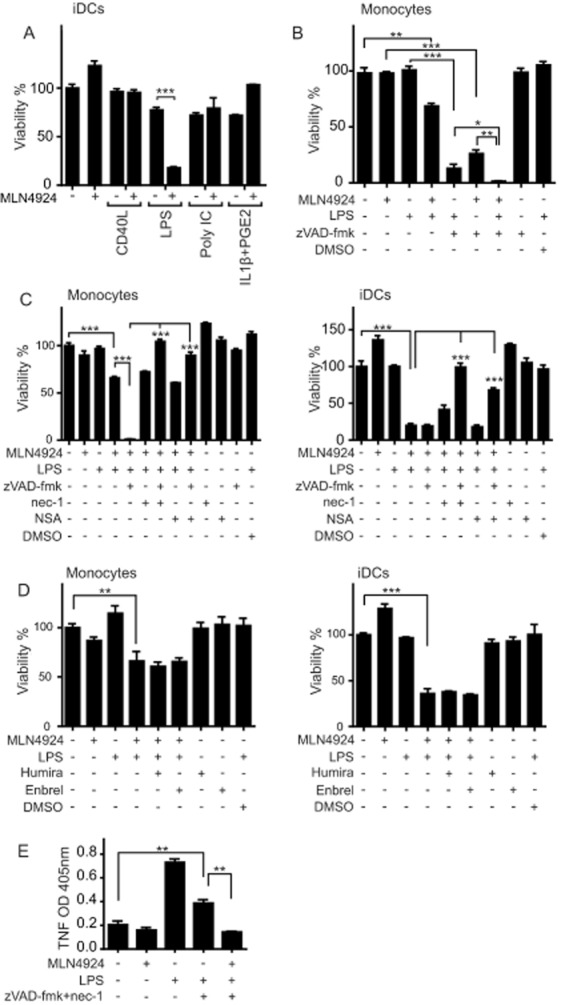
MLN4924 triggers necroptosis and apoptosis in LPS-maturing DCs. (A) DC maturation was triggered (triplicates, 96-well) with Fc-CD40L (1 μg·mL−1), LPS (10 ng·mL−1), poly IC (20 μg·mL−1) or a mixture of IL-1β (10 ng·mL−1) and PGE2 (1 μg·mL−1) in the presence and absence of 5 μM MLN4924. After 24 h, cell viability was evaluated using the MTT assay and normalized according to untreated cells and cells treated with a cocktail of cytotoxic substances. (B) Monocytes were treated with the indicated mixtures of LPS (1 ng·mL−1), zVAD-fmk (50 μM) and MLN4924 (5 μM). Next day, cellular viability was determined using the MTT assay. (C) Monocytes and iDCs were treated with the indicated mixtures of LPS (1 ng·mL−1), MLN4924 (5 μM), nec-1 (90 μM), NSA (2.5 μM) and zVAD-fmk (50 μM). After 24 h, cell viability was again determined by help of the MTT assay. (D) Monocytes and iDCs were challenged with LPS (0.1 ng·mL−1) and MLN4924 (5 μM) in the presence and absence of 10 μg·mL−1 Humira or 10 μg·mL−1 Enbrel and the next day, cell viability was evaluated by help of the MTT assay. (E) iDCs were stimulated in triplicate with 1 ng·mL−1 of LPS and the indicated mixtures of MLN4924 (5 μM), zVAD (50 μM) and nec-1 (90 μM). After 24 h, supernatants were collected and analysed for TNF production by elisa.
Discussion
In this study, we investigated the effects of MLN4924 on monocytes and DCs. Although the compound alone showed no major effect on cellular viability, it significantly sensitized monocytes as well as iDCs for TNFR1-mediated caspase activation and cell death. Intriguingly, cells were only protected from TNF/MLN4924-induced cell death when caspase inhibition was combined with inhibitors of the necroptotic RIP1-RIPK3-MLKL pathway. Thus, MLN4924 has to inhibit the expression/activity of one or more factors that antagonize both TNFR1-induced apoptosis and necroptosis or it has to trigger the accumulation of a TNFR1-dependent cell death-promoting factor. Cell death induced by CD95L, which stimulates the TNFR1-related death receptor CD95, remained unaffected by MLN4924 (Figure 2C). The TNFR1-specific sensitizing effect of MLN4924 argues against an accumulation of an apoptosis/necroptosis-promoting factor because TNFR1 and CD95 share the ability to trigger caspase-8-mediated apoptosis and RIPK1-mediated necroptosis (Holler et al., 2000). Although it cannot be fully ruled out that MLN4924 targets different proteins to sensitize for TNFR1-induced apoptosis and TNFR1-induced necroptosis, the simplest explanation would be that MLN4924 affects a single TNFR1-specific step that controls both apoptotic and necroptotic TNFR1 signalling.
In this respect, TNFR1-induced apoptosis and necroptosis bifurcate at the level of RIPK1 (Vanden Berghe et al., 2014). Upon TRAF2–cIAP1/2 complex-mediated K63 ubiquitination of RIPK1 in the TNFR1 signalling complex, the modified RIPK1 species contributes to activation of the classical NF-κB pathway and, after its release from the receptor, is hindered to trigger RIPK1-mediated caspase-8 activation and RIPK-induced necroptosis. The cell death-stimulating activity of the TNF-induced caspase-8/RIP complex is controlled by several factors that are regulated by the classical NF-κB pathway, especially by A20, cIAP2, TRAF1 and FLIP (Krikos et al., 1992; Chu et al., 1997; Wang et al., 1998; Schwenzer et al., 1999; Kreuz et al., 2001; Micheau et al., 2001). So, FLIP-L, the long isoform of the FLIP protein, inhibits not only caspase-8 activation by TNFR1 but also the induction of necroptosis (Chan et al., 2003; Geserick et al., 2009; Feoktistova et al., 2011; Oberst et al., 2011; He and He, 2013). However, a full-blown TNF-induced cell death response requires not only inhibition of FLIP-L but also formation of complexes of caspase-8 and de-ubiquitinated RIPK1. The levels of the latter are mainly controlled by the ubiquitination editing enzyme A20, which stimulates the degradation of RIPK1 and prevents necroptosis, the constitutively expressed deubiquitinase CYLD, the TRAF2–cIAP1/2 complex and presumably complexes of cIAP1 or cIAP2 with TRAF1–TRAF2 heteromers (Wertz et al., 2004; Zheng et al., 2010; Vanlangenakker et al., 2011; Moquin et al., 2013; Vanden Berghe et al., 2014). A straight forward explanation for the apoptotic and necroptosis-sensitizing effect of MLN4924 in TNFR1-signalling is, therefore, the inhibition of the induction of one or more of the protective proteins regulated by the classical NF-κB pathway. Indeed, we observed a marked upregulation of cIAP2 and/or A20 in monocytes and iDCs treated with TNF (Figure 7A and B). Moreover, the induction of both molecules was abolished by MLN4924 (Figure 7A and B). The effects of MLN4924 on FLIP expression were rather weak but more complex. Treatment with MLN4924 alone resulted, as TNF, in a moderate induction of FLIP mRNA in monocytes and iDCs but showed no additive effect. In both cell types, TNF also strongly induced the expression of TRAF1, and this was again inhibited in the presence of MLN4924 (Figure 7A and B). Noteworthy, TRAF1 not only forms heterotrimeric complexes with TRAF2 with a much higher affinity for cIAPs than TRAF2 homotrimers (Zheng et al., 2010) but has also been found to be protective against TNF-induced cell death in concert with TRAF2 and the cIAPs (Wang et al., 1998). Future knockout/knockdown studies must now prove the causal relevance of MLN4924-mediated inhibition of TNF-induced expression of cIAP2, TRAF1 and A20 for the cytotoxic TNF/MLN4924 crosstalk.
The idea that the sensitizing effect of MLN4924 on cytotoxic TNFR1 signalling is due to inhibition of NF-κB-dependent pro-survival activities nicely matches with the fact that phosphorylated IκBα and phosphorylated p100 represent two major substrates of the NEDD-8 activating enzyme-regulated E3 ligase βTrCP (Read et al., 2000; Amir et al., 2004). Although βTrCP-mediated K48 ubiquitination of phospho-IκBα results in proteasomal IκBα degradation and activation of the classical NF-κB pathway, βTrCP-induced K48 ubiquitination of p100 triggers limited proteasomal proteolysis of this precursor protein and activation of the so-called alternative NF-κB pathway (Read et al., 2000; Amir et al., 2004). As p100 can also act as an IκBα-like inhibitor, its processing can result in a cell type-specific activation of the classical NF-κB pathway, too (Basak et al., 2007). Accordingly, we observed that MLN4924 inhibits activation of the classical NF-κB pathway by TNF and also p100 processing induced by the TNF-related cytokine TWEAK (Rauert-Wunderlich et al., 2013). In particular, this was also evident in zVAD-fmk/nec-1-protected monocytes and iDCs in which TNF-induced production of the NF-κB targets IL-6 and IL-12 were completely abolished (Figure 5B). Thus, the ability of MLN4924 to target the TNF-induced classical NF-κB pathway and inhibit the induction of caspase-8/RIP1 antagonists, such as cIAP2, TRAF1 and A20, might straightforwardly explain why MLN4924 sensitizes for both TNFR1-induced apoptosis and necroptosis.
The idea that MLN4924 sensitizes for TNF-induced cell death by interfering with TNF-induced NF-κB-mediated up-regulation of survival factors is also in good accordance with a recent report showing that various NF-κB inhibitors, including the proteasome inhibitor Mg-132 and the IKK2-specific inhibitor TPCA-1, triggered TNF-dependent reactive oxygen species-mediated cell death in murine macrophages and DC lines and primary macrophages (Tilstra et al., 2014). Future studies that focus on the evaluation of RIPK1 modifications and the composition of TNF-induced RIPK1- and/or caspase-8-containing complexes in MLN4924-treated cells must now finally verify the molecular basis of the cell death sensitizing effect of MLN4924 in the context of TNFR1 signalling.
Strikingly, MLN4924 alone showed no major toxic effects on monocytes although these cells express endogenous TNF and are significantly killed by mixtures of MLN4924 and exogenous TNF (Figure 3A). A possible explanation for this observation could be that a certain, comparably high threshold of TNFR1 stimulation is required to induce cell death in the presence of MLN4924. As yet, the amounts of soluble TNF endogenously produced are almost undetectable in the supernatants of monocytes, and the considerable amounts of membrane-bound TNF might substantially be diverted by binding to TNFR2. Noteworthy, the threshold for TNFR1-induced necroptosis in the presence of zVAD-fmk appears to be significantly lower because these conditions not only result in strongly enhanced TNF/MLN4924-induced cell death of monocytes but also enable MLN4924 to kill the cells in concert with endogenous TNF (Figures 3A and 4C).
We also observed that MLN4924 sensitized cells to the apoptosis and necroptosis-effect of LPS (Figure 8). As before in the case of TNF, the cell death sensitizing effect of MLN4924 in monocytes was especially evident in the presence of zVAD-fmk (Figure 8B and C). Importantly, the LPS/MLN4924-induced cell death in monocytes and iDCs is not an indirect effect of LPS-induced TNF production and subsequent TNFR1 stimulation. Firstly, LPS-induced TNF production is inhibited by MLN4924 as we have shown in zVAD-fmk/nec-1-protected cells (Figure 8E) and secondly, LPS-induced cell death remained largely unaffected in the presence of high concentrations of TNF- and TNF/LTα-neutralizing reagents (Figure 8D) that were able to antagonize endogenous TNF in other experiments (Figure 4C). In accordance with a role of the caspase-8–RIPK1 dyad in LPS-induced cell death of MLN4924-sensitized monocytes and iDCs, both molecules have already previously been implicated in LPS signalling, especially in the context of LPS-induced NF-κB activation (Vivarelli et al., 2004; Su et al., 2005; Lehner et al., 2007; Lemmers et al., 2007; Kang et al., 2008; Maelfait et al., 2008; McComb et al., 2014). In particular, enhanced LPS-induced cell death has also been reported in LPS-stimulated caspase-8 knockout B-cells (Lemmers et al., 2007). Furthermore, RIPK3-mediated TNF-independent necroptosis has been reported in caspase inhibitor-treated LPS-stimulated microglia and macrophages challenged with a SMAC mimetic (He et al., 2011; Kim and Li, 2013).
It is tempting to speculate that the sensitizing effect of MLN4924 for TNF- and LPS-induced apoptosis can be exploited to suppress bacterial infection or other inflammatory syndromes. In particular, the cell death sensitizing activity of MLN4924 on immune cells might cooperate with its ability to inhibit the pro-inflammatory NF-κB pathways. In this regard, a recent publication showed that MLN4924 inhibits the production of pro-inflammatory cytokines and stimulation of allogeneic T cells by bone marrow-derived dendritic cells stimulated with LPS or other NF-κB inducers (Mathewson et al., 2013). Likewise, an inhibitory effect on anti-CD3/anti-CD28-stimulated T cells has been shown in this study. Noteworthy, in contrast to the results of our experiments with iDCs, Mathewson et al. (2013) found that bone marrow-derived dendritic cell viability was not affected by MLN4924. This could reflect the use of lower concentrations of MLN4924, species differences or differences in the state of DC maturation. Indeed, we found no MLN4924-associated toxicity in iDCS and mDCs (Figure 1B) and FLIP-L is up-regulated during DC maturation (Leverkus et al., 2000). To boost the immune inhibitory effects of MLN4924 related to cell death induction, one might consider co-treatment with a caspase inhibitor. The feasibility of this idea will have to be clarified in future in vivo studies. Cotreatment with MLN4924 and caspase inhibitors leads to a switch from apoptotic to necroptotic cell death. In contrast to apoptosis, the latter form of cell death is typically stimulated by immunogenic factors. It is tempting to speculate that the in vivo net effect of a mixture of MLN4924 and a caspase inhibitor can be steered not only in an immune inhibitory but also in an immune-stimulant direction by use of appropriate protocols. This might allow it to be applied not only for the treatment of immune disease but also for the immune therapy of cancer.
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft (grant KFO216/TP8) and Deutsche Krebshilfe (grant 109922). M. E.-M. is a German Egyptian Research Long Term Scholarship (GERLS) holder funded by DAAD.
Glossary
Abbreviations
- cIAP
cellular inhibitor of apoptosis protein
- CRL
cullin-RING ubiquitin ligase
- FLIP-L
long form of FLICE inhibitory protein
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- iDCs
immature dendritic cells
- mDCs
mature dendritic cells
- MLKL
mixed lineage kinase domain-like protein
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAE
NEDD-8 activating enzyme; nec-1, necrostatin-1
- NEDD-8
neural precursor cell expressed, developmentally down-regulated 8
- NSA
necrosulfonamide
- RIPK
receptor-interacting protein
- TRAF1/2
TNF receptor associated factor 1/2
- zVAD-fmk
benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone
- βTrCP
β-transducin repeat containing protein
Author contributions
M. E.-M., T. S. and D. S. performed experiments and analysed the data. M. E.-M. and H. W. designed experiments, analysed data and wrote the paper.
Conflict of interest
The authors declare no competing financial interests.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic receptors. Br J Pharmacol. 2013a;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Haecker H, Karin M, Ciechanover A. Mechanism of processing of the NF-kappa B2 p100 precursor: identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of NEDD8-modification on the SCF(beta-TrCP) ubiquitin ligase. Oncogene. 2004;23:2540–2547. doi: 10.1038/sj.onc.1207366. [DOI] [PubMed] [Google Scholar]
- Basak S, Kim H, Kearns JD, Tergaonkar V, O'Dea E, Werner SL, et al. A fourth IkappaB protein within the NF-kappaB signaling module. Cell. 2007;128:369–381. doi: 10.1016/j.cell.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278:51613–51621. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- Chang FM, Reyna SM, Granados JC, Wei SJ, Innis-Whitehouse W, Maffi SK, et al. Inhibition of neddylation represses lipopolysaccharide-induced proinflammatory cytokine production in macrophage cells. J Biol Chem. 2012;287:35756–35767. doi: 10.1074/jbc.M112.397703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc Natl Acad Sci U S A. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, et al. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43:449–463. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geserick P, Hupe M, Moulin M, Wong WW, Feoktistova M, Kellert B, et al. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J Cell Biol. 2009;187:1037–1054. doi: 10.1083/jcb.200904158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbersen JC, Humphries LA, Danilova OV, Kebbekus PE, Brown JR, Eastman A, et al. The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NF-kappaB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin Cancer Res. 2014;20:1576–1589. doi: 10.1158/1078-0432.CCR-13-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He MX, He YW. A role for c-FLIP(L) in the regulation of apoptosis, autophagy, and necroptosis in T lymphocytes. Cell Death Differ. 2013;20:188–197. doi: 10.1038/cdd.2012.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108:20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H, Norton TD, Schultz ML, Polsky SB, Sunseri N, Landau NR. Inhibition of CUL4A neddylation causes a reversible block to SAMHD1-mediated restriction of HIV-1. J Virol. 2013;87:11741–11750. doi: 10.1128/JVI.02002-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- Kang TB, Oh GS, Scandella E, Bolinger B, Ludewig B, Kovalenko A, et al. Mutation of a self-processing site in caspase-8 compromises its apoptotic but not its nonapoptotic functions in bacterial artificial chromosome-transgenic mice. J Immunol. 2008;181:2522–2532. doi: 10.4049/jimmunol.181.4.2522. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Li J. Caspase blockade induces RIP3-mediated programmed necrosis in toll-like receptor-activated microglia. Cell Death Dis. 2013;4:e716. doi: 10.1038/cddis.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuz S, Siegmund D, Scheurich P, Wajant H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol. 2001;21:3964–39673. doi: 10.1128/MCB.21.12.3964-3973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J Biol Chem. 1992;267:17971–17976. [PubMed] [Google Scholar]
- Lehner M, Bailo M, Stachel D, Roesler W, Parolini O, Holter W. Caspase-8 dependent apoptosis induction in malignant myeloid cells by TLR stimulation in the presence of IFN-alpha. Leuk Res. 2007;31:1729–1735. doi: 10.1016/j.leukres.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Lemmers B, Salmena L, Bidere N, Su H, Matysiak-Zablocki E, Murakami K, et al. Essential role for caspase-8 in toll-like receptors and NFkappaB signaling. J Biol Chem. 2007;282:7416–7423. doi: 10.1074/jbc.M606721200. [DOI] [PubMed] [Google Scholar]
- Leverkus M, Walczak H, McLellan A, Fries HW, Terbeck G, Brocker EB, et al. Maturation of dendritic cells leads to up-regulation of cellular FLICE-inhibitory protein and concomitant down-regulation of death ligand-mediated apoptosis. Blood. 2000;96:2628–2631. [PubMed] [Google Scholar]
- Li L, Liu B, Dong T, Lee HW, Yu J, Zheng Y, et al. Neddylation pathway regulates the proliferation and survival of macrophages. Biochem Biophys Res Commun. 2013;432:494–498. doi: 10.1016/j.bbrc.2013.02.028. [DOI] [PubMed] [Google Scholar]
- Loetscher H, Stueber D, Banner D, Mackay F, Lesslauer W. Human tumor necrosis factor alpha (TNF alpha) mutants with exclusive specificity for the 55-kDa or 75-kDa TNF receptors. J Biol Chem. 1993;268:26350–26357. [PubMed] [Google Scholar]
- Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, et al. Stimulation of toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J Exp Med. 2008;205:1967–1973. doi: 10.1084/jem.20071632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathewson N, Toubai T, Kapeles S, Sun Y, Oravecz-Wilson K, Tamaki H, et al. Neddylation plays an important role in the regulation of murine and human dendritic cell function. Blood. 2013;122:2062–2073. doi: 10.1182/blood-2013-02-486373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McComb S, Shutinoski B, Thurston S, Cessford E, Kumar K, Sad S. Cathepsins limit macrophage necroptosis through cleavage of Rip1 kinase. J Immunol. 2014;192:5671–5678. doi: 10.4049/jimmunol.1303380. [DOI] [PubMed] [Google Scholar]
- Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21:5299–5305. doi: 10.1128/MCB.21.16.5299-5305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116:1515–1523. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- Moquin DM, McQuade T, Chan FK. CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE. 2013;8:e76841. doi: 10.1371/journal.pone.0076841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekorchuk MD, Sharifi HJ, Furuya AK, Jellinger R, de Noronha CM. HIV relies on neddylation for ubiquitin ligase-mediated functions. Retrovirology. 2013;10:138. doi: 10.1186/1742-4690-10-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauert H, Wicovsky A, Muller N, Siegmund D, Spindler V, Waschke J, et al. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2) J Biol Chem. 2010;285:7394–7404. doi: 10.1074/jbc.M109.037341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauert-Wunderlich H, Siegmund D, Maier E, Giner T, Bargou RC, Wajant H, et al. The IKK inhibitor Bay 11-7082 induces cell death independent from inhibition of activation of NFkappaB transcription factors. PLoS ONE. 2013;8:e59292. doi: 10.1371/journal.pone.0059292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read MA, Brownell JE, Gladysheva TB, Hottelet M, Parent LA, Coggins MB, et al. Nedd8 modification of cul-1 activates SCF(beta(TrCP))-dependent ubiquitination of IkappaBalpha. Mol Cell Biol. 2000;20:2326–2333. doi: 10.1128/mcb.20.7.2326-2333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvesen GS, Walsh CM. Functions of caspase 8: the identified and the mysterious. Semin Immunol. 2014;26:246–252. doi: 10.1016/j.smim.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenzer R, Siemienski K, Liptay S, Schubert G, Peters N, Scheurich P, et al. The human tumor necrosis factor (TNF) receptor-associated factor 1 gene (TRAF1) is up-regulated by cytokines of the TNF ligand family and modulates TNF-induced activation of NF-kappaB and c-Jun N-terminal kinase. J Biol Chem. 1999;274:19368–19374. doi: 10.1074/jbc.274.27.19368. [DOI] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- Su H, Bidere N, Zheng L, Cubre A, Sakai K, Dale J, et al. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005;307:1465–1468. doi: 10.1126/science.1104765. [DOI] [PubMed] [Google Scholar]
- Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115:3796–3800. doi: 10.1182/blood-2009-11-254862. [DOI] [PubMed] [Google Scholar]
- Tan M, Li H, Sun Y. Endothelial deletion of Sag/Rbx2/Roc2 E3 ubiquitin ligase causes embryonic lethality and blocks tumor angiogenesis. Oncogene. 2014;33:5211–5220. doi: 10.1038/onc.2013.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Nakatani T, Kamitani T. Negative regulation of NEDD8 conjugation pathway by novel molecules and agents for anticancer therapy. Curr Pharm Des. 2013;19:4131–4139. doi: 10.2174/1381612811319220017. [DOI] [PubMed] [Google Scholar]
- Tilstra JS, Gaddy DF, Zhao J, Dave SH, Niedernhofer LJ, Plevy SE, et al. Pharmacologic IKK/NF-kappaB inhibition causes antigen presenting cells to undergo TNFalpha dependent ROS-mediated programmed cell death. Sci Rep. 2014;4:3631. doi: 10.1038/srep03631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Vanden Berghe T. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2011;17:e230. doi: 10.1038/cddis.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivarelli MS, McDonald D, Miller M, Cusson N, Kelliher M, Geha RS. RIP links TLR4 to Akt and is essential for cell survival in response to LPS stimulation. J Exp Med. 2004;200:399–404. doi: 10.1084/jem.20040446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Watson IR, Irwin MS, Ohh M. NEDD8 pathways in cancer, Sine Quibus Non. Cancer Cell. 2011;19:168–176. doi: 10.1016/j.ccr.2011.01.002. [DOI] [PubMed] [Google Scholar]
- Wei W, Guo H, Liu X, Zhang H, Qian L, Luo K, et al. A first-in-class NAE inhibitor, MLN4924, blocks lentiviral infection in myeloid cells by disrupting neddylation-dependent Vpx-mediated SAMHD1 degradation. J Virol. 2014;88:745–751. doi: 10.1128/JVI.02568-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- Wyzgol A, Muller N, Fick A, Munkel S, Grigoleit GU, Pfizenmaier K, et al. Trimer stabilization, oligomerization, and antibody-mediated cell surface immobilization improve the activity of soluble trimers of CD27L, CD40L, 41BBL, and glucocorticoid-induced TNF receptor ligand. J Immunol. 2009;183:1851–1861. doi: 10.4049/jimmunol.0802597. [DOI] [PubMed] [Google Scholar]
- Yao WT, Wu JF, Yu GY, Wang R, Wang K, Li LH, et al. Suppression of tumor angiogenesis by targeting the protein neddylation pathway. Cell Death Dis. 2014;5:e1059. doi: 10.1038/cddis.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Kabaleeswaran V, Wang Y, Cheng G, Wu H. Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: affinity, specificity, and regulation. Mol Cell. 2010;38:101–113. doi: 10.1016/j.molcel.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]