

Abstract
Background and Purpose
Activation of the α7 nicotinic ACh receptor (nACh receptor) is considered an attractive target for the treatment of cognitive impairment associated with neurological disorders. Here we describe the novel α7-nACh receptor agonist AQW051 as a promising drug candidate for this indication.
Experimental Approach
AQW051 was functionally characterized in vitro and cognitive effects evaluated in rodent behavioural models. Pharmacokinetics and tolerability were evaluated in three phase I placebo-controlled studies in 180 healthy subjects.
Key Results
In vitro, AQW051 bound with high affinity to α7-nACh receptors and stimulated calcium influx in cells recombinantly expressing the human α7-nACh receptor. In vivo, AQW051 demonstrated good oral bioavailability and rapid penetration into the rodent brain. AQW051 administered over a broad dose range facilitated learning/memory performance in the object recognition and social recognition test in mice and the water maze model in aged rats. Clinically, AQW051 was well tolerated in healthy young and elderly subjects, with an adverse event (AE) profile comparable with placebo. No serious AEs were reported and all AEs were either mild or moderate in severity at single oral doses up to 200 mg and multiple daily doses up to 75 mg. Once-daily oral administration of AQW051 resulted in continuous exposure and a two- to threefold accumulation compared with steady state was achieved by 1 week.
Conclusions and Implications
These data support further development of AQW051 as a cognitive-enhancing agent, as a therapeutic, for example, in Alzheimer's disease or schizophrenia.
Tables of Links
| TARGETS |
|---|
| 5-HT3AB receptor |
| α7-nACh receptor |
| nACh receptors |
| LIGANDS | ||
|---|---|---|
| α-bungarotoxin (α-BTX) | Chlordiazepoxide | Nicotine |
| [125I]-α-BTX | Choline | PHA-543613 |
| ACh | Epibatidine | PNU-282987 |
| AQW051 | EVP-6124 | TC-5619 |
| β-amyloid (Aβ) | Methyllycaconitine (MLA) | Rivastigmine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Neuronal nicotinic ACh receptors (nACh receptors) belong to a superfamily of ligand-gated ion channels that includes receptors for GABA, 5–HT and glycine. Two of the most abundant nACh receptor subtypes within the CNS are α7 and α4β2*, formed by the association of five subunits in homopentameric and heteromeric complexes respectively (Gotti and Clementi, 2004; Gotti et al., 2006). α7-nACh receptor is further characterized by its low affinity for the agonist ACh, choline activation, high affinity for the antagonist α-bungarotoxin (α-BTX), rapid desensitization and high permeability to calcium. Within the CNS, the α7-nACh receptor is highly expressed in the hippocampus, thalamus, frontal cingulate and occipital cortices; regions important for learning, memory and sensory gating (Young and Geyer, 2013). Interestingly, outside the CNS, α7-nACh receptor is expressed on tissue-macrophages and synoviocytes, key participants in the cholinergic anti-inflammatory pathway. Choline activation of α7-nACh receptor inhibits NF-κB nuclear translocation and suppresses cytokine release from these cells, supporting resolution of immune responses (Tracey, 2002; 2009,).
Consistent with the CNS expression pattern, several lines of evidence support a functional role for α7-nACh receptor in learning and memory. The first highly selective α7-nACh receptor agonist, AR-R17779, was shown to enhance cognition in a number of animal models (Levin et al., 1999). This evidence has led to the idea that cognitive deficits in certain neuropsychiatric and neurological disorders might be ameliorated by administration of α7-nACh receptor agonists. Indeed, gating deficits experienced by patients with schizophrenia are corrected following α7-nACh receptor agonist administration (Olincy et al., 2006). Furthermore, α7-nACh receptor expression is reduced in post-mortem brain samples of patients with schizophrenia (Freedman et al., 1995), and genetic linkage studies have identified α7-nACh receptor as a predisposing factor for sensory gating deficits (Freedman et al., 1997; Gault et al., 2003); a hallmark of schizophrenia and also associated with Alzheimer's disease (AD) (Jessen et al., 2001). In AD, a number of pathophysiological changes alter the function of the α7-nACh receptor, not least the cholinergic deficit observed because of cholinergic neuronal loss early on in the disease course. Furthermore, β-amyloid (Aβ) binds with high affinity to α7-nACh receptor, inducing a pathway switch that is detrimental to neurons (Wang et al., 2000) and creating a seed for further Aβ accumulation. To this end, a number of selective and potent α7-nACh receptor agonists have been discovered, for example GTS-21 (de Fiebre et al., 1995), JN403 (Feuerbach et al., 2007; 2009,) and PNU-282987 (Hajos et al., 2005), including some that have reached advanced clinical testing (phases II and III) in patients with schizophrenia or AD, for example TC-5619 (Hauser et al., 2009; Mazurov et al., 2012; Lieberman et al., 2013), EVP-6124 (Prickaerts et al., 2012; Preskorn et al., 2014) and ABT-126 (NCT00948909 and NCT01095562) (Bitner et al., 2013). Notably, an α7-nACh receptor positive allosteric modulator, JNJ–39393406, whose activity depends on the cholinergic tone, did not demonstrate efficacy on evoked potentials or EEGs after acute dosing in patients with schizophrenia (Winterer et al., 2013). Here we describe the preclinical characterization and phase I evaluation in healthy volunteers of the potent and selective α7-nACh receptor partial agonist, (R)-3-(6-p-tolyl-pyridin-3-yloxy)-1-aza-bicyclo(2.2.2)octane (AQW051) (Hurth et al., 2014).
Methods
Cell lines
The following cell lines were cultured and maintained as previously described: human rhabdomyosarcoma cell line endogenously expressing α1β1γδ-nACh receptor, TE671 [American Type Culture Collection (ATCC) ]; murine neuroblastoma cell line endogenously expressing the 5-HT receptor 5-HT3AB, N1E-115 (ATCC); HEK cell line, HEK293 (ATCC), transfected with recombinant human α4β2 and α3β4-nACh receptors (HEK293-ha4b2 and HEK293-ha3b4 respectively) (Michelmore et al., 2002); rat pituitary epithelial-like tumour cell line transfected with human neuronal α7-nACh receptors (GH3-ha7-22) (Feuerbach et al., 2005); and human neuroblastoma cell line, transfected with human α7-nACh receptors (SH-SY5Y-ha7) (Charpantier et al., 2005). nACh receptor nomenclature used is according to BJP's Concise Guide to PHARMACOLOGY (Alexander et al., 2013).
In vitro radioligand-binding assays
Radioligand-binding assays were performed as previously described (Feuerbach et al., 2007) using SH-SY5Y cells and 125I-labelled α7-nACh receptor antagonist, α-BTX ( [125I]-α-BTX).
Measurement of intracellular calcium
Calcium influx assays were performed using a fluorescence method as previously described using the full panel of cell lines (Feuerbach et al., 2005).
Electrophysiological studies in Xenopus oocytes
Xenopus oocyte preparation, injection with human α7-nACh receptor mRNA and measurement of response to application of AQW051 were performed as previously described (Feuerbach et al., 2005).
Animals
All experiments were carried out in accordance with the authorization guidelines of the Swiss federal and cantonal veterinary offices for the care and use of laboratory animals. Studies described in this report were approved by the Swiss cantonal veterinary office. Animals were housed in plastic cages in a room with a 12 h light/dark cycle (lights on 07:00 h) and free access to food and water. The following mouse and rat strains were used: OF/1C mice (Charles River, Lille, France), Sprague Dawley rats (RA238, OFA/IC, Iffa Crédo, France) and Listar-hooded rats (Harlan, Venray, The Netherlands). AQW051 was suspended in 0.5% methylcellulose, or dissolved in 0.035 mL 0.1 M hydrochloric acid and diluted in distilled water (pH of about 5.0) to 1 mL. AQW051 (or associated vehicle) was administered p.o. unless otherwise stated. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Concentration of AQW051 in the brain and plasma of mice
Plasma and brain homogenates were extracted and analysed as previously described. (Feuerbach et al., 2009). The limit of detection, defined as the lowest concentration of the extracted standard sample with a signal to noise ratio of about 3, for AQW051 was 1.5 ƒmol per injection (corresponding to 0.15 pmol·mL−1 plasma and 0.75 pmol·g−1 brain).
Ex vivo binding of AQW051
After administration of AQW051, mice were killed at indicated time points by CO2 and brains were removed. Binding of AQW051 was studied in various brain regions. Sagittal sections (10 μm) were thaw-mounted onto silane-coated microscope slides and labelled: 30 min pre-incubation at room temperature (RT) in Krebs-Ringer HEPES buffer (KRH: 20 mM HEPES pH 7.4, 118 mM NaCl, 4.8 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 10 mM NaOH), followed by an incubation for 2 h at RT in KRH buffer supplemented with 0.05 mg·mL−1 BSA and 2.5 nM [125I]-α-BTX (144 Ci·mmol−1, Perkin Elmer, Waltham, MA, USA). Non-specific binding was determined in the presence of 100 μM (−)−nicotine. Sections were rinsed three times for 20 min in ice-cold KRH buffer, then 20 s in ice-cold KRH (diluted 1:10), with a final brief rinse in ice-cold distilled water before drying under a stream of cold air. Labelled sections were exposed to Kodak Biomax MR films (Eastman Kodak Company, Rochester, NY, USA) for 6 days at 4°C and nuclei counterstained with 0.5% cresyl violet and localized (Paxinos and Franklin, 2012). Binding data (total and non-specific) were analysed by optic densitometry using a computerized image analysis system (MCID, Imaging Research, St Catherine's, ON, Canada). Results were compared by one-way anova followed by a Dunnett's multiple comparison test.
Social recognition in mice
Adult female (30–35 g) and young female (18–20 g) mice (OF1/IC strain; n = 10/group) were tested as previously described (Feuerbach et al., 2009). The α7-nACh receptor antagonist, methyllycaconitine (MLA, 2.5 and 10.0 mg·kg−1 or saline) was administered by i.p. injection 30 min after administration of AQW051 (0.03 and 0.3 mg·kg−1; p.o.).
Object recognition in mice
The object recognition test (ORT) records the number of stretched attend posture directed by singly housed mice towards a grey PVC disc (height = 2 cm, diameter = 5 cm) placed in their home cage (22 × 37 × 15 cm) under moderate light (approximately 40 Lux) for 3 min on 3 successive occasions (0 min, 10 min and 24 h). To test for any potential motor disturbances, a novel PVC cone (height = 6 cm, diameter = 5 cm) is presented at 24 h 10 min.
Adult male mice (25–27 g, OF1/IC strain, n = 10 per group) were pretreated with AQW051 (0.03 or 0.3 mg·kg−1 p.o.) or vehicle 1 h before the test, then treated with MLA, (3 mg·kg−1 i.p.) or saline (NaCl 0.9%) 30 min post–dose. The frequency of the stretched attend posture towards the object (SAO) during each exposure was scored from videotape by a trained observer blind to previous treatment of the animals. Intragroup differences (between presentations) were analysed using repeated-measures anova, independently for each treatment group, followed by a post hoc Tukey's test. Intergroup differences (between treatment groups) were analysed per time point using one-way anova, followed by a post hoc Dunnett's test. In addition, this model was used to test whether positive effects of AQW051 (0.3 and 3.0 mg·kg−1 p.o.) were maintained following repeated dosing for 7 consecutive days.
Spatial navigation memory in aged rats
Old male lister-hooded rats (24 months) were tested in the Morris water maze experiment as previously described (Pryce et al., 2003). Rats were assigned to either AQW051 (3 mg·kg−1 p.o.) or vehicle based upon mean-trial platform latency (s). Treatment was administered 1 h pre-test on days 1–4. Rat swimming path, time and speed were recorded by a video activity monitoring system (Ethovision, Noldus, Wageningen, The Netherlands). Parameters measured were distance travelled, swimming speed and latency to find the platform. Intergroup differences (among treatment groups) were analysed using one-way anova, followed by a post hoc Dunnett's test.
Social exploration test in rats
Experiments were performed as previously described (Feuerbach et al., 2009). Adult male rats (350–400 g) were designated residents and male juvenile intruder rats (100–120 g). Intruder rats received AQW051 or vehicle 1, 3, 6 or 16 h before testing. Chlordiazepoxide, an anxiolytic benzodiazepine was used as a positive control.
Human single- and multiple-dose studies
Three randomized, double-blind, placebo-controlled phase I studies evaluated the safety, tolerability and pharmacokinetics (PK) of AQW051 in non-smoking healthy subjects. All doses were given p.o. in fasted state, apart from the fed arm of studies 2 and 3 investigating the effect of food. Each study used an interleaved design and specific details are summarized below:
Study 1 (n = 62): first time in man, single-ascending dose (SAD) study of AQW051 (0.5, 2.5, 7.5, 25 and 75 mg) in two different cohorts of male subjects aged 18–41 years. A third cohort (male subjects aged 20–44) received a dose of 200 mg AQW051 to determine the maximum tolerated dose (MTD). Cohorts 1 (n = 25) and 2 (n = 25) participated in three successive treatment periods of two ascending doses of AQW051 and one placebo, with randomized assignment to placebo. A 2 week washout period (±5 days) was allowed between treatment periods. For determining MTD, subjects were randomized to receive either placebo (n = 4) or AQW051 (n = 8) (planned MTD of 450 mg, actual dose 200 mg).
Study 2 (n = 64): ascending dose study in elderly male and postmenopausal female subjects aged 60–80 years treated with either a single dose followed by once-daily multiple doses of AQW051 over 2 weeks, or 2 weeks of once-daily multiple AQW051 doses. AQW051 was administered at 2.5, 15 or 75 mg.
Study 3 (n = 54): SAD study of AQW051 (2.5, 7.5, 25, 75 and 200 mg) in Japanese male subjects aged 20–45 years. Treatment schedule: cohort 1, 2.5 mg AQW051/placebo then 75 mg AQW051/placebo; cohort 2, 7.5 mg AQW051/placebo then 200 mg AQW051/placebo; cohort 3, 25 mg AQW051/placebo then 25 mg AQW051/placebo (under fasted or fed conditions).
The protocol and amendments were approved by the Independent Ethics Committee and Institutional Review Board at each study centre. All studies were conducted according to the ethical principles of the Declaration of Helsinki and two studies were registered on ClinicalTrials.gov [identifiers NCT00418002 (study 2) and NCT00409500 (study 3) ]. Informed written consent was obtained from all patients.
Safety and PK assessments
Safety assessments included adverse events (AEs) and serious AEs (SAEs) reporting; laboratory tests; vital signs; ECGs; and neurological assessments. For PK assessments, blood samples were collected into EDTA-containing tubes according to a predefined schedule. For single dosing, sampling took place pre-dose and up to 24 h for 0.5 and 2.5 mg dosing or 120 h post-dose for all other dose levels. Urine samples were collected from pre-dose to 48 h. Blood and urine concentrations of AQW051 were determined using liquid chromatography tandem mass spectrometry. The lower limit of quantification was 0.150 ng·mL−1 for blood and 0.500 ng·mL−1 for urine.
Statistical methods
Descriptive statistics were used to summarize safety results. PK parameters were calculated for each dose using non-compartmental analysis. The relationship between dose and PK parameters area under the concentration–time curve (AUC)(0–t), AUC(0–inf) and maximum concentration (Cmax) was explored using the power model PK = α Doseβ. Dose proportionality was assessed by the estimator of β and by exploratively assessing whether the 90% confidence interval (CI) for β is contained in the no effect region of β (1 + ln[0.8]/ln[r], 1 + ln[1.25]/ln[r] ), where r was the dose range tested (highest dose/lowest dose); only doses with reliably estimated PK parameters were included. In studies 2 and 3, food effects were determined using a linear mixed effect model applied to log–transformed PK parameters with food condition (fed/fasted) as fixed effect and subject as random effect. The difference in PK because of food condition was derived and back-transformed into the natural scale to give the estimation for ratio of geometric mean together with corresponding 90% CI.
Reagents
AQW051 (R)-3-(6-p-tolyl-pyridin-3-yloxy)-1-aza-bicyclo(2.2.2)octane (MW 294.40) was synthesized by Novartis Institutes for BioMedical Research (Basel, Switzerland).
Results
AQW051 is a selective partial agonist of the α7-nACh receptor
The chemical structure of AQW051 is shown in Figure 1. AQW051 displayed high affinity for the human α7-nACh receptor with high selectivity towards other nACh receptors. AQW051 had a pKD of 7.56 (SEM ± 0.04, n = 3) at recombinantly expressed human α7-nACh receptor as determined using [125I]-α-BTX radioligand-binding studies. Furthermore, AQW051 showed selectivity over a wide range of neurotransmitter receptors, ion channels, kinases and transporters in radioligand-binding studies; more than 164 targets were tested to a maximal concentration of 10 μM and at 1 μM, there was no significant inhibition of any of the 164 targets (data not shown).
Figure 1.
Structure of AQW051.
Calcium transients, detected after stimulation of human α7-nACh receptors recombinantly expressed in GH3–ha7-22 cells, demonstrated the potent agonist activity of AQW051 (pEC50 7.41 ± 0.09, relative to the positive control epibatidine: 73% ± 4.1, n = 18; Table 2013). This activity was blocked by the α7-nACh receptor antagonist, MLA (100 nM, n = 2). AQW051 showed similar pharmacological properties (pEC50 7.24 ± 0.1, efficacy compared with epibatidine, a full α7-nACh receptor agonist: 68% ± 4, n = 9) when interacting with rat α7-nACh receptor recombinantly expressed in the same cellular background. The functional selectivity of AQW051 over other nACh receptor subtypes and 5-HT3 receptors, using the calcium influx assay, was at least 100-fold (Table 2013). AQW051 did not display agonistic activity up to 100 μM at these receptors.
Table 1.
Effects of AQW051 at human α7-nAChR, α3β4, α4β2, α1β1γδ and murine 5-HT3 receptors using a fluorescence readout (Feuerbach et al., 2005)
| Receptor | n | pEC50 ± SEM | Emax ± SEM | pIC50 ± SEM | Selectivity |
|---|---|---|---|---|---|
| α7 | 18 | 7.41 ± 0.09 | 73 ± 4.1% | ||
| α1β1γδ | 6 | 4.97 ± 0.05 | 274 | ||
| α4β2 | 6 | 5.40 ± 0.07 | 101 | ||
| α3β4 | 5 | 5.28 ± 0.08 | 144 | ||
| 5-HT3 | 6 | 4.72 ± 0.09 | 488 |
In electrophysiological recordings from Xenopus oocytes expressing human α7-nACh receptor, inward currents were generated in response to bath application of AQW051 (30 μM), at a holding potential of −70 mV. The responses were transient with a fast-rising phase followed by a rapid decay. The induced peak current amplitudes ranged between 30 and 170 nA (n = 5). Thus, the signal shape was similar to the response evoked by ACh. The effects of AQW051 were concentration–dependent with a calculated EC50 value of 7.5 μM (95% CI 3.9, 14.6, n = 5). The calculated maximal response amplitude evoked by AQW051 was 75% of the maximally effective ACh response. Thus, AQW051 is best described as a partial agonist at the recombinant human α7-nACh receptor expressed in oocytes. The response evoked by 100 μM AQW051 was fully blocked by MLA.
Levels of AQW051 in the brain and plasma of mice
Levels of AQW051 in mouse brain tissue and plasma were assessed at three time points (0.25, 1 and 4 h) after 10 μmol·kg−1 i.v. and at six time points (0.25, 0.5, 1, 4, 8 and 24 h) after 30 μmol·kg−1 p.o. (Figure 2A and B). AQW051 penetrated rapidly into the brain following p.o. administration with a brain/plasma ratio of 20 after 30 min increasing to 60 after 4 h. Apparent terminal elimination half-lives (t1/2) of approximately 0.6 and 1 h were estimated in plasma following i.v. and p.o. administration respectively. Bioavailability was estimated as 50%.
Figure 2.
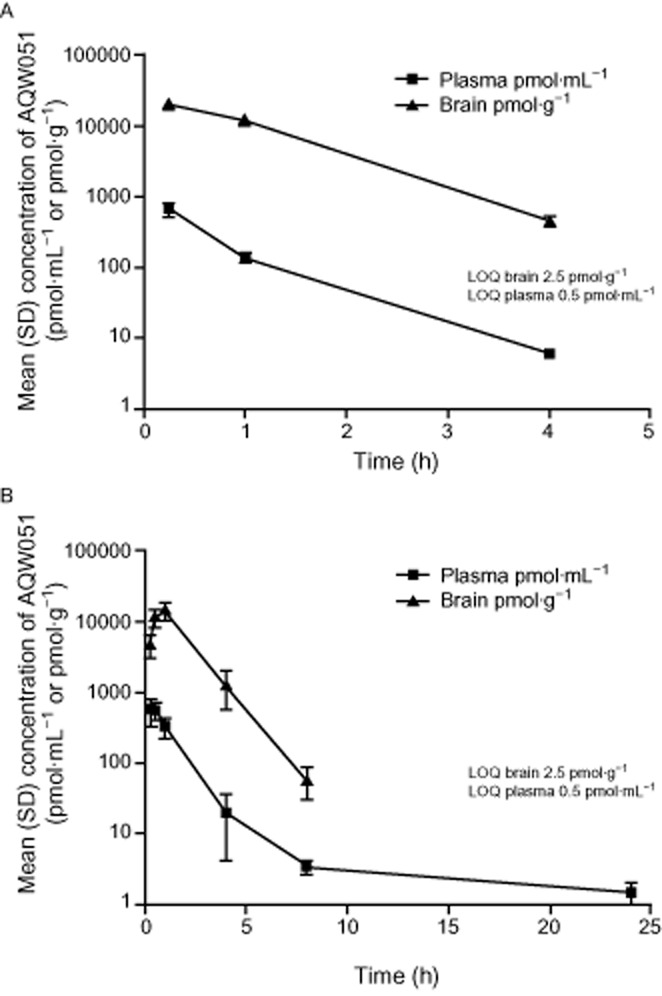
(A) Concentration of AQW051 in mouse brain and plasma following acute i.v. administration of 10 μmol·kg−1. Mean values (±SD) for six mice are given in pmol·mL−1 plasma or pmol·g−1 brain tissue. (B) Concentration of AQW051 in mouse brain and plasma following oral administration of 30 μmol·kg−1. Mean values (±SD) for six mice are given as pmol·mL−1 plasma or pmol·g−1 brain tissue.
Ex vivo AQW051 binding in mouse brain
Using ex vivo autoradiography, the effects of 3 mg·kg−1 i.v. AQW051 on [125I]-α-BTX binding to α7-nACh receptor was determined in nine brain regions: cerebral cortex, caudate putamen, hippocampus, presubiculum, amygdalo-hippocampal area, lateral geniculate nucleus, and more specifically, in the CA1-3 areas of the hippocampus, polymorph layer and granular layer of the dentate gyrus and lacunosum moleculare layer, of which the hippocampus is illustrated in detail in Figure 3. In all regions studied, displacement of bound [125I]α-BTX occurred as early as 5 min following AQW051 administration when the maximal effect was reached. This effect was sustained for at least 4 h in all brain regions and for 7 h in the cortex and hippocampus. By 24 h post-dose, [125I]-α–BTX binding was restored to baseline levels in all brain regions.
Figure 3.
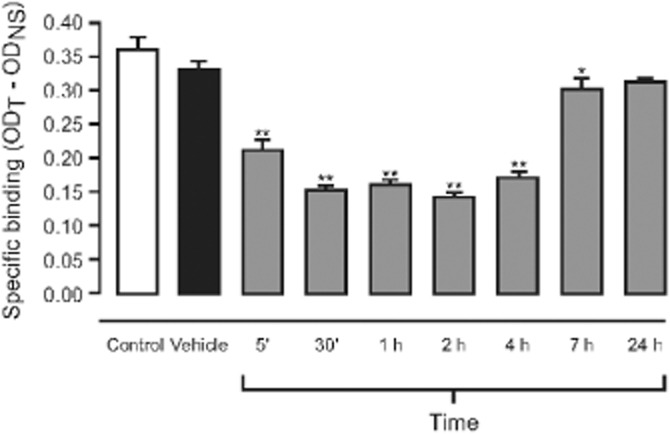
Time-dependent effect of 3 mg·kg−1 i.v. AQW051 on the displacement of [125I]-α-BTX from mouse hippocampus in ex vivo binding studies. Bars represent specific binding at the different time points indicated or control (no injection) or vehicle (NaCl)-treated mice (n = 6). *P < 0.05, **P < 0.01 in comparison with vehicle-treated animals. Error bars indicate SD.
AQW051 treatment enhanced learning/memory in mice in the social recognition test
Mice treated with AQW051 at 0.03 and 0.3 mg·kg−1 p.o. before baseline trial spent significantly more time scrutinizing the novel partner than the familiar partner during the re-test trial at 24 h, suggesting that AQW051 facilitated the learning/memory performance (P < 0.001 and P < 0.01, respectively, Figure 4). Co–administration of MLA (2.5 mg·kg−1or 10 mg·kg−1 i.p.) significantly reversed the positive memory-enhancing effects of 0.3 mg·kg−1 AQW051 p.o. (data not shown).
Figure 4.
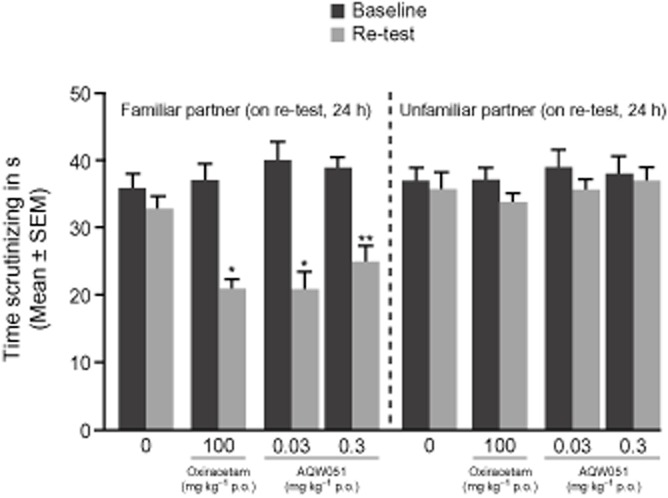
Acute administration of AQW051 improves learning/memory in the social recognition task in mice. Bars represent the mean time (s) within the 3 min trial during which the adult female mice scrutinized the young partner (n = 10 pairs of mice per treatment group). Black bars, time scrutinizing a young mouse during the baseline trial; grey bars, time scrutinizing the familiar (previously encountered) or an unfamiliar partner during the re-test trial performed 24 h after the baseline trial. AQW051 (0.03 or 0.3 mg·kg−1 p.o.) and oxiracetam (100 mg·kg−1 s.c.) was administered 1 h before the baseline trial. Comparisons with baseline trial within each treatment group were made using the Mann–Whitney U-test. Significant values of *P < 0.001 and **P < 0.01 are shown.
MLA reversed the cognitive-enhancing effect of AQW051 in the object recognition test (ORT) in mice
In the ORT, compared with the first presentation to the disc (t = 0 min), the SAO frequency during the second exposure (t = 10 min) was, as expected, decreased in the vehicle-treated group as well as in all drug-treated groups, indicating that the disc was perceived as familiar. (Figure 5A). During the third presentation (t = 24 h), mice treated with 0.03 and 0.3 mg·kg−1 (p.o.) AQW051 displayed less SAO than the vehicle-treated group (P < 0.05), indicating improved recognition memory. MLA-treated mice showed no improvement in retention, even when AQW051 was co administered. No inter-group difference was observed on presentation of a novel object (fourth presentation; t = 24 h 10 min) or change in SAO frequency compared with the first exposure to the original object.
Figure 5.
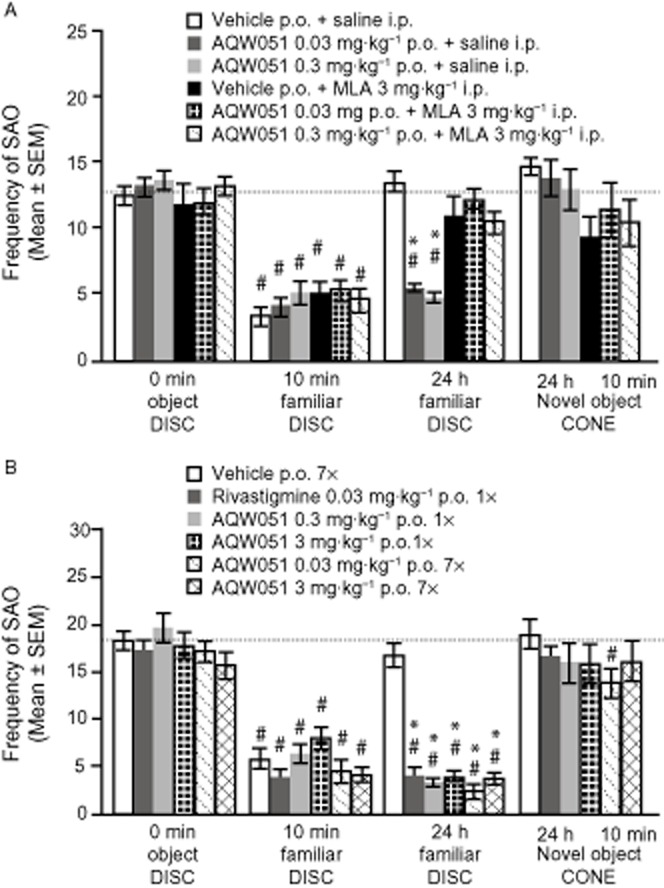
(A) The effects of acute administration of AQW051 and (B) of single (1×) or subchronic [for 7 days (7×), once daily] oral application of AQW051 in the ORT in mice. Mice (n = 10 per group) received vehicle or AQW051 (0.03, 0.3 mg·kg−1) 1 h (p.o.), and saline or MLA (3 mg·kg−1), 30 min (i.p.) before the first disc presentation; (B) vehicle, AQW051 (0.3, 3 mg·kg−1) acutely or after subchronic treatment or rivastigmine (0.03 mg·kg−1; acutely only) was administered, 1 h before the first disc presentation. Each graph represents the mean frequency (±SEM) of stretched attend posture towards the object (SAO) directed by singly housed mice towards a disc (presentations at 0, 10 min and 24 h) or a cone (presentation at 24 h 10 min). Objects were placed for 3 min within the home cage. # P < 0.05 intragroup differences; *P < 0.05 intergroup differences.
As shown in Figure 5B, the cognitive-enhancing effects of AQW051 were maintained with daily dosing for 7 consecutive days and no tolerance developed; the effect was comparable with a single dose of rivastigmine (0.03 mg·kg−1 p.o.).
Effects of AQW051 in the water maze in aged rats
This experiment was conducted to determine whether repeated daily AQW051 enhanced spatial long-term memory between days or working memory within days in aged rats. For acquisition training, anova of swimming distance indicated a significant interaction between drug and day (P < 0.05) (Figure 6). A posteriori re-analysis of single days was conducted: On day 1, there was a drug × trial interaction (P < 0.01) and post hoc analysis indicated that swimming distance was decreased in AQW051-treated compared with vehicle rats in trial 1 (P = 0.01) specifically. Also on day 2, there was a drug × trial interaction (P = 0.01) and swimming distance was again decreased in AQW051-treated compared with vehicle rats in trial 1 (P = 0.01) specifically (Figure 6). By day 3, swimming distance was already at its nadir of around 400 cm at trial 1 in both treatment groups. There was no drug effect in the probe test on day 4; both drug groups spent approximately 40% of the probe trial in the training quadrant (quadrant effect P < 0.001 for AQW051 and vehicle groups). When escape latency was used as the dependent measure, similar findings were obtained (data not shown). AQW051 was without effect on swim speed. The reduced swimming distance to locate the platform on trial 1 on days 1 and 2 is consistent with an enhancing effect of AQW051 on long-term consolidation and/or recall of the context of the task. Aged AQW051 treated rats appeared to have an enhanced memory about the platform per se from day −1 to day 1, and an enhanced memory of its location from day 1 to day 2.
Figure 6.
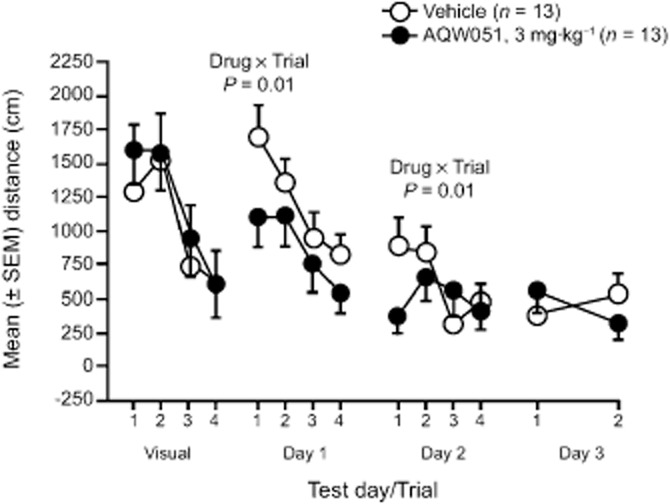
AQW051 enhances spatial navigation in the water maze test. Mice were given four visual cue test trials on day −1 (visual) and counterbalanced to AQW051 and vehicle groups using their trial 4 swimming distance. Compound was administered at day 0 and days 1–3, 1 h before testing. On days 1 and 2, four acquisition trials were run, and to avoid over-training, only two trials were run on day 3. For acquisition training on days 1–3, there was an anova Drug × Day interaction. Single day re-analysis identified anova Drug × Trial interaction, and using post hoc Dunnett's test, decreased swimming distance (i.e. enhanced spatial navigation) on trial 1 specifically in AQW051-treated compared with vehicle-treated rats, on days 1 and 2. Values are expressed as mean (±SEM).
AQW051 treatment increased social exploration in rats with a duration of at least 6 h
Intruder rats pretreated with AQW051 (1 mg·kg−1 p.o.) at 1, 3 and 6 h or the positive control, chlordiazepoxide, an anxiolytic benzodiazepine (1 mg·kg−1 p.o.; except 3 h pretreatment) showed a significant increase in the duration of social contact compared with vehicle-treated rats (P < 0.05), indicating an anxiolytic-like effect (Figure 7). No significant difference versus vehicle group was observed when AQW051 or chlordiazepoxide were administered 16 h prior to testing (P > 0.05). No statistically significant difference was observed between the vehicle-treated animals at different time points.
Figure 7.
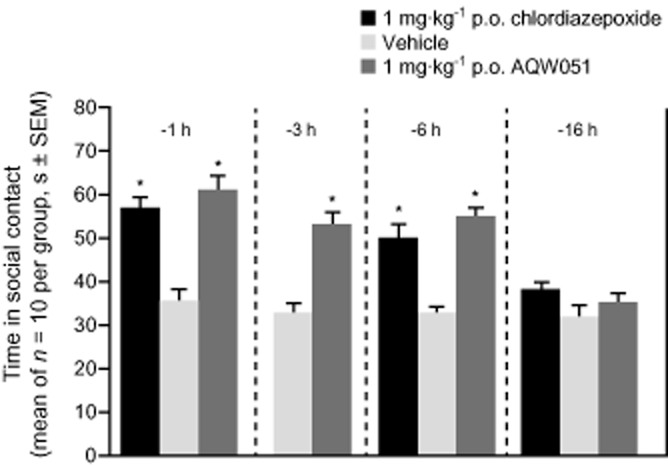
Comparative effects of AQW051 and chlordiazepoxide administered to rats at different time points before the social exploration test. Bars represent the mean time (s ± SEM per 5 min trial) spent by the ‘intruder’ juvenile male rat actively exploring the ‘resident’ adult male rat. Intruder rats received either vehicle or single doses of AQW051 (1 mg·kg−1 p.o.) at 1, 3, 6 or 16 h before the test. Three additional groups of intruder rats received single doses of the positive control, chlordiazepoxide (1 mg·kg−1 p.o.), at 1, 6 or 16 h before the test. For each group, 10 rats were used. Data were analysed by anova followed by a post hoc Dunnett's test. *P < 0.05 versus vehicle-treated rats.
In humans, AQW051 has a favourable safety profile
A total of 180 healthy volunteers were randomized to receive AQW051. In the first two cohorts of study 1, 50 healthy volunteers (48 + 2 replacements) were assigned to sequences of three successive treatment periods consisting of two ascending doses of AQW051 and one of placebo, with randomized assignment to placebo; the remaining 12 were assigned to the MTD determination (200 mg) and randomized to active : placebo parallel groups in a 2:1 ratio. In study 2 (n = 64), 24 volunteers participated in the sequential single-dose phase, followed by multiple-dose phase; 40 volunteers were assigned to the multiple-dose phase only. In study 3, 54 healthy Japanese volunteers were assigned to three cohorts (n = 18 per cohort) and randomized to receive ascending doses of AQW051 or placebo.
AQW051 was generally well tolerated in all three studies at dose levels up to 200 mg, the highest single dose tested. MTD was not established, as dose escalation was stopped based on a predefined ceiling exposure. There were no SAEs reported. All AEs were transient and mild or moderate in severity; 72/180 subjects experienced at least one AE. AEs reported under fasting conditions are summarized in Tables ,,. Headache was the most frequently reported AE for subjects receiving either single (seven subjects) or multiple (six subjects) doses of AQW051. The incidence of AEs under fed conditions was only available for study 3, where one subject receiving AQW051 experienced haematuria. In study 1, treatment-related AEs were reported in cohort 2 (headache, chest pain and muscle twitching under active treatment), but not cohort 1. Three subjects treated with AQW051 200 mg reported CNS-related AEs (dizziness, headache and/or nervousness) within 30 min of receiving AQW051 that were considered by the investigator to be treatment-related; no effects on vital signs were reported. In study 2, the most commonly reported treatment-related AEs were dizziness (five subjects), headache (five subjects), nausea (four subjects), pharyngolaryngeal pain (three subjects) and somnolence (three subjects). Subjects receiving 2.5 mg AQW051 experienced more treatment-related AEs (20 subjects) than subjects receiving 15 (four subjects) and 75 mg AQW051 (eight subjects), and placebo (three subjects), but this was not thought to be clinically significant. In study 3, nine AEs were considered to be treatment-related, six were reported by subjects receiving low doses of AQW051 [2.5, 7.5, and 25 mg (fasted) ] and three by subjects receiving placebo (fed and fasted). No treatment-related AEs were reported for subjects treated with AQW051 75 or 200 mg (fasted) or 25 mg (fed). Postural dizziness (three events in three subjects) and headache (two events in two subjects) were the most frequent treatment-related AEs; two of three and one of two subjects, respectively, experienced the AE while receiving AQW051. There were no clinically significant abnormalities in ECG readings (including no evidence of treatment-related QT prolongation), vital signs or clinical laboratory parameters reported for any of the studies.
Table 2.
AEs reported by ≥2 healthy subjects in study 1
| AE, n (%) | AQW051, mg | Placebo | |||||
|---|---|---|---|---|---|---|---|
| 0.5 (N = 17) | 2.5 (N = 17) | 7.5 (N = 32) | 25 (N = 15) | 75 (N = 16) | 200 (N = 8) | (N = 50) | |
| Any | 5 (29.4) | 3 (17.6) | 5 (15.6) | 1 (6.7) | 0 | 3 (37.5) | 10 (20.0) |
| Cough | 1 (5.9) | 0 | 1 (3.1) | 0 | 0 | 0 | 0 |
| Dizziness | 1 (5.9) | 0 | 0 | 0 | 0 | 2 (25.0) | 0 |
| Eye injury | 1 (5.9) | 0 | 1 (3.1) | 0 | 0 | 0 | 0 |
| Headache | 1 (5.9) | 0 | 1 (3.1) | 1 (6.7) | 0 | 1 (12.5) | 4 (8.0) |
| Musculoskeletal chest pain | 0 | 1 (5.9) | 1 (3.1) | 0 | 0 | 0 | 0 |
| Pharyngolaryngeal pain | 0 | 0 | 1 (3.1) | 0 | 0 | 0 | 1 (2.0) |
Table 3.
AEs reported by ≥2 healthy subjects in study 2
| AE, n (%) | Single-dose phase | Multiple-dose phase | ||||||
|---|---|---|---|---|---|---|---|---|
| AQW051, mg | Placebo | AQW051, mg | Placebo | |||||
| 2.5 (N = 6) | 15 (N = 6) | 75 (N = 6) | (N = 6) | 2.5 (N = 18) | 15 (N = 18) | 75 (N = 12) | (N = 16) | |
| Any | 3 (50.0) | 2 (33.3) | 2 (33.3) | 1 (16.7) | 12 (66.7) | 5 (27.8) | 8 (66.7) | 4 (25.0) |
| Abnormal dreams | 0 | 0 | 0 | 0 | 2 (11.1) | 0 | 0 | 0 |
| Constipation | 1 (16.7) | 0 | 0 | 0 | 2 (11.1) | 0 | 0 | 0 |
| Diarrhoea | 1 (16.7) | 0 | 0 | 1 (16.7) | 2 (11.1) | 0 | 0 | 0 |
| Dizziness | 0 | 0 | 1 (16.7) | 0 | 3 (16.7) | 1 (5.6) | 2 (16.7) | 0 |
| Dizziness postural | 0 | 0 | 0 | 0 | 2 (11.1) | 0 | 0 | 0 |
| Headache | 0 | 2 (33.3) | 0 | 0 | 2 (11.1) | 2 (11.1) | 2 (16.7) | 1 (6.3) |
| Nausea | 0 | 0 | 0 | 0 | 4 (22.2) | 0 | 1 (8.3) | 0 |
| Pharyngolaryngeal pain | 0 | 0 | 0 | 0 | 2 (11.1) | 0 | 1 (8.3) | 1 (6.3) |
| Somnolence | 0 | 0 | 0 | 0 | 2 (11.1) | 0 | 1 (8.3) | 1 (6.3) |
Table 4.
AEs reported by ≥2 healthy subjects in study 3
| AE, n (%) | AQW051 (mg) | Placebo | ||||
|---|---|---|---|---|---|---|
| 2.5 (N = 14) | 7.5 (N = 14) | 25 (N = 14) | 75 (N = 13) | 200 (N = 12) | (N = 19) | |
| Any | 2 (14.3) | 2 (14.3) | 1 (7.1) | 2 (15.4) | 2 (16.7) | 3 (15.8) |
| Blood glucose increased | 0 | 0 | 0 | 0 | 1 (8.3) | 1 (5.3) |
| Dizziness postural | 1 (7.1) | 1 (7.1) | 0 | 0 | 0 | 1 (5.3) |
| Gastrointestinal infection | 0 | 0 | 0 | 2 (15.4) | 0 | 0 |
| Headache | 0 | 0 | 1 (7.1) | 0 | 0 | 1 (5.3) |
AQW051 provides continuous systemic exposure with limited fluctuation at once-daily p.o. administration
PK data for AQW051 across all three studies are summarized in Tables ,,. There was a negligible lag time in the absorption of AQW051, with median time to maximum concentration (Tmax) ranging from 4 to 8 h post-dose in all three studies under fasting conditions (Tables ,,). The geometric mean elimination t1/2 following single-dosing ranged from 18.8 to 22.7 h in young male Caucasian subjects (study 1; Table 2007), 35.3–43.2 h in elderly subjects (study 2; Table 1997) and 22.9–26.4 h in young male Japanese subjects (study 3; Table 1998a). In all three studies, urinary excretion of unchanged AQW051 was <1.5% of the dose over 24 or 48 h. When adjusted for body weight, there was little difference in exposure per dose between Caucasian and Japanese subjects (data not shown). Cmax and all AUC values showed no relevant deviation from dose proportionality (Table ,,). When normalized for dose, AUC(0–24h) and AUC(0–inf) values were approximately twofold higher in the elderly (study 2; Table 1997) than in young subjects (study 1; Table 2007).
Table 5.
Pharmacokinetic profile of AQW051 in Caucasian healthy male subjects following single-dose administration (study 1)
| PK parameter (Gmean, %Gmean CV) | AQW051, mg | |||||
|---|---|---|---|---|---|---|
| 0.5 (N = 17) | 2.5 (N = 17) | 7.5 (N = 32) | 25 (N = 15) | 75 (N = 16) | 200 (N = 8) | |
| Cmax, ng·mL−1 | 0.266 (28.3) | 1.20 (44.0) | 4.34 (37.0) | 20.6 (30.0) | 47.0 (30.8) | 162 (39.2) |
| Tmax, h | 8 (4–12) | 8 (3.98–10) | 7.98 (3.98–8.13) | 8 (3.98–8) | 4.04 (4–7.98) | 4.77 (3.98–5.62) |
| AUC(0–24), ng·h·mL−1 | – | 18.5 (51.5) | 67.3 (41.1) | 277 (26.8) | 614 (31.7) | 2240 (46.4) |
| AUC(0–24)/dose, ng·h·mL−1·mg−1 | – | 7.40 (51.5) | 8.97 (41.1) | 11.1 (26.8) | 8.19 (31.7) | 11.2 (46.4) |
| AUC(0–inf), ng·h·mL−1 | – | – | 136 (56.8) | 533 (42.7) | 1030 (41.9) | 3680 (63.0) |
| AUC(0–inf)/dose, ng·h·mL−1·mg−1 | – | – | 18.1 (56.8) | 21.3 (42.7) | 13.8 (41.9) | 18.4 (63.0) |
| t1/2, h | – | – | 22.0 (29.1) | 22.7 (31.7) | 18.8 (20.0) | 20.0 (30.6) |
| Ae(0–24), % of dose | – | 0.207 (63.5) | 0.311 (63.6) | 0.401 (57.5) | 0.210 (43.8) | 0.360 (42.0) |
| Ae(0–48), % of dose | – | 0.291 (67.7) | 0.439 (62.9) | 0.537 (63.4) | 0.276 (42.3) | 0.489 (50.5) |
Ae, amount of drug excreted unchanged in urine; CV, coefficient of variation; Gmean, geometric mean.
Table 6.
Pharmacokinetic profile of AQW051 in Japanese healthy male subjects following single-dose administration (study 2)
| PK parameter (Gmean, %Gmean CV) | AQW051, mg | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Fed | Fasted | ||||||||
| 2.5 (N = 6) | 15 (N = 6) | 75 (N = 6) | 2.5 (N = 18) | 15 (N = 18) | 75 (N = 12) | ||||
| SD | SS | SD | SS | SD | SS | ||||
| Cmax , ng·mL−1 | 1.37 (30) | 13.4 (19) | 60.7 (23) | 1.9 (27) | 5.6 (26) | 11.6 (20) | 31.0 (39) | 74.2 (20) | 163.0 (39) |
| Tmax, h | 8.0 (6.0–10.0) | 6.02 (6.0–8.02) | 6.01 (4.0–8.02) | 8.0 (4.0–12.0) | 8.01 (4.0–12.0) | 6.01 (4.0–10.0) | 6.0 (4.0–8.13) | 6.0 (4.03–8.0) | 6.0 (2.0–8.0) |
| AUC(0–24), ng·h·mL−1 | 21.3 (41) | 219 (15) | 988 (26) | 31.2 (28) | 114 (25) | 191 (25) | 568 (48) | 1190 (23) | 2980 (417) |
| AUC(0–24)/dose, ng·h·mL−1·mg−1 | 8.52 (41) | 14.6 (15) | 13.2 (26) | 12.5 (28) | 45.5 (25) | 12.7 (25) | 37.9 (48) | 15.9 (23) | 39.7 (47) |
| AUC(0–inf), ng·h·mL−1 | 76.5 (19) | 715 (12) | 2700 (28) | – | – | – | – | – | – |
| AUC(0–inf)/dose, ng·h·mL−1·mg−1 | 30.6 (19) | 47.7 (12) | 36.0 (28) | – | – | – | – | – | – |
| t1/2, h | 35.3 (27) | 43.2 (12) | 38.1 (12) | – | 45.6 (19) | – | 44.1 (17) | – | 36.4 (23) |
| Ae(0–24), % of dose | 0.32 (91) | 0.48 (81) | 1.01 (69) | 0.36 (45) | 1.37 (62) | 0.45 (67) | 1.46 (73) | 0.52 (74) | 1.28 (83) |
Ae, amount of drug excreted unchanged in urine; CV, coefficient of variation; Gmean, geometric mean; SS, steady state.
Table 7.
Pharmacokinetic profile of AQW051 in elderly healthy subjects after single- and multiple-dose administration (study 3)
| Pharmacokinetic parameter (Gmean, %Gmean CV) | AQW051, mg | |||||
|---|---|---|---|---|---|---|
| Fasted | Fed | |||||
| 2.5 (N = 14) | 7.5 (N = 14) | 25 (N = 14) | 75 (N = 13) | 200 (N = 12) | 25 (N = 11) | |
| Cmax, ng·mL−1 | 1.92 (16.1) | 6.07 (18.5) | 21.3 (18.9) | 71.8 (29.6) | 237 (25.1) | 26.2 (19.9) |
| Tmax, h | 6 (6–8) | 6 (4–6) | 6 (4–6) | 4 (1–6) | 4 (2–6) | 6 (6–6) |
| AUC(0–24), ng·h·mL−1 | 28.7 (19.9) | 91.7 (21.0) | 315 (19.8) | 1030 (26.0) | 3240 (30.1) | 344 (16.5) |
| AUC(0–inf), ng·h·mL−1 | 64.1 (30.0) | 185 (27.2) | 594 (22.5) | 1850 (29.1) | 5310 (38.4) | 662 (19.9) |
| t1/2, h | 26.4 (21.4) | 23.0 (18.0) | 24.9 (19.7) | 23.4 (14.0) | 22.9 (22.7) | 25.3 (24.5) |
| Ae(0–24), % of dose | 0.42 (0.27) | 0.52 (0.25) | 0.36 (0.25) | 0.50 (0.35) | 0.43 (0.17) | 0.84 (0.63) |
| Ae(0–48), % of dosea | 0.71 (0.53) | 0.79 (0.42) | 0.56 (0.43) | 0.66 (0.41) | 0.68 (0.30) | 1.09 (0.79) |
Mean (SEM).
After multiple dosing, steady state was achieved by day 7 in elderly subjects. In this population AUC(0–24h) was 2.49- to 3.64-fold higher after 2 weeks compared with single-dose values, showing that overall accumulation was reflecting the observed t1/2 and single-dose PK. In studies 2 and 3, a light breakfast had no major effect on the PK of AQW051. In study 2, five subjects per dose group received a single dose of AQW051 in both the fasting and fed state. An average increase of <20% in Cmax and AUC(0–24h) was observed in the fed state compared with the fasting state. In study 3, the mean Tmax for fed and fasted states were identical and t1/2, Cmax as well as AUC values were similar (Table 1998a).
Discussion
In recent years, a number of novel synthetic α7-nACh receptor agonists have been discovered and characterized preclinically, some of which have undergone clinical evaluation. One of the first molecules identified was the prodrug GTS-21 (Briggs et al., 1997), which demonstrated limited selectivity towards other nACh receptor subtypes (Michelmore et al., 2002). Consistent with preclinical findings, the compound displayed efficacy on cognitive functions in healthy male volunteers (Kitagawa et al., 2003). In subsequent studies, GTS-21 was shown to improve P50 inhibition in patients with schizophrenia (Olincy et al., 2006), but a phase II study where cognitive parameters after chronic dosing were assessed was negative (Freedman et al., 2008). Clinical findings for three highly potent and selective α7-nACh receptor agonists (PHA-543613, PHA-568487, CP-810123) were recently reported (Rogers et al., 2011). In contrast to the anabaseine-derived GTS-21, these compounds belong to the quinuclidine (PHA-543613 and PHA-568487) or 4-azabenzoxazole (CP-810123) chemotype. After single- and multiple-dose finding studies, development of all three compounds was terminated for cardiovascular safety reasons (Rogers et al., 2011). The aim of the current treatise was the identification of a selective α7-nACh receptor agonist that demonstrates efficacy in preclinical animal models and has a favourable PK and safety profile in humans.
We characterized AQW051 as a potent and selective agonist at the human α7-nACh receptor. Importantly, AQW051 demonstrated similar pharmacological properties at the rat α7-nACh receptor, distinguishing it from other α7-nACh receptor agonists that have been reported to manifest marked species-dependent differences in potency, for example GTS-21. Along the same lines, AQW051 does not display any interaction at the 5-HT3 receptor, a common feature reported for some of the recently characterized α7-nACh receptor agonists, for example EVP-6124 (Prickaerts et al., 2012) and ABT-126 (Bitner et al., 2013). Although 5-HT3 receptor antagonists have been shown to enhance cholinergic function in animal models clinical results have been variable with both encouraging (Broocks et al., 1998a) and disappointing results (Broocks et al., 1998b). In addition, patients with irritable bowel syndrome report ischaemic colitis and constipation as the most common side effects when treated with long-acting 5-HT3 receptor antagonists, for example alosetron (Camilleri, 2000). Therefore, a α7-nACh receptor agonist devoid of this activity might offer a more favourable safety profile and improve compliance.
Functional assessment of AQW051 revealed that the compound acted as a partial agonist of α7-nACh receptor in calcium influx assays and evoked currents that reached 75% of the ACh response in electrophysiological studies on Xenopus oocytes. Under similar electrophysiological conditions two nACh receptors α7 agonists, EVP-6124 (Prickaerts et al., 2012) and TC-5619 (Mazurov et al., 2012), acted as partial or full agonists demonstrating 82 or 100% of the activity of ACh respectively. AQW051 demonstrated excellent brain penetration, good oral bioavailability and a short t1/2 following p.o. administration. The favourable mouse PK profile permitted in vivo testing of the compound using p.o. delivery. In ex vivo binding experiments, direct interaction of the compound with the α7-nACh receptor was shown in nine brain regions. To elucidate the effect of AQW051 on learning and memory, mice treated with the compound were evaluated in the social recognition test. Time spent scrutinizing a familiar partner was significantly reduced 24 h after p.o. administration of AQW051 at 0.03 and 0.3 mg·kg−1, an effect that was blocked by co-administration of the selective α7-nACh receptor antagonist MLA. Similar results have been reported for other α7-nACh receptor agonists, for example AR-R17779 (Levin et al., 1999), EVP-6124 (Prickaerts et al., 2012) and JN403 (Feuerbach et al., 2007). Furthermore, subchronic p.o. dosing of AQW051 did not affect the memory retention efficacy of AQW051, indicating that under these experimental conditions, tachyphylaxis does not play a role.
To investigate the duration of action of AQW051, the social exploration model was applied in rats. In this test, nicotine has been shown to exert anxiolytic effects at low doses, and anxiogenic effects at high doses (File et al., 1998). Our data indicate that AQW051 has an anxiolytic-like effect (disinhibition of social approaches/contacts) that is preserved up to 6 h after treatment. Preclinical learning/memory experiments suggest that α7-nACh receptor agonists are notably active in experimental models involving long-term memory. Data presented here from the ORT, social recognition test and spatial navigation test further support this feature. Similar findings have been presented for a variety of different α7-nACh receptor agonists (Arendash et al., 1995). Furthermore, facilitation of long term potentiation has been demonstrated by other α7-nACh receptor agonists (e.g. SSR180711) (Biton et al., 2007). In support of this notion, ABT-107 when tested in the delayed matching to sample in monkeys showed improved accuracy only at long, but not the immediate, short or medium delay (Bitner et al., 2010). Clinically, this might translate into efficacy of delayed recall and memory consolidation rather than attentional tasks.
Single (0.5–200.0 mg) and multiple doses (2.5, 15.0 and 75.0 mg·day−1) of AQW051 were well tolerated in young healthy male Caucasian and Japanese subjects, and elderly healthy subjects of both sexes. MTD was not determined, as a predefined exposure cap was applied limiting dose escalation. AEs were either mild or moderate in severity, and no AEs led to study discontinuation. The most frequently reported AE across all three studies was headache, irrespective of dosing schedule. The absence of any cardiovascular AEs or clinically significant changes in ECG parameters, suggests AQW051 does not produce cardiovascular side effects that have been reported for compounds with similar modes of action [e.g. PHA-543613 and PHA-568487 (Rogers et al., 2011) ]. Treatment-related AEs were more frequent in the cohorts receiving AQW051 at the lower dose ranges in studies 3 (2.5, 7.5 and 25.0 mg) and 2 (2.5 mg). In study 1, only cohorts 2 and 3 reported treatment-related AEs. The PK profile for AQW051 showed that the compound provided a continuous systemic exposure with limited fluctuation at once-daily p.o. administration and was not affected by food intake. This contrasts with the rodent PK profile of a fast PK with a high peak exposure. The exposure and elimination t1/2 were approximately twofold higher in elderly subjects compared with younger subjects.
In conclusion, AQW051 demonstrates high affinity for the α7-nACh receptor, acting as a potent and selective partial agonist for the receptor. In vivo, AQW051 rapidly crossed the blood–brain barrier and enhanced memory function. Clinical data showed that AQW051 was generally well tolerated and provided a continuous systemic exposure upon once-daily administration. Together, these data support further development of AQW051 as a cognitive-enhancing agent in AD and/or schizophrenia.
Acknowledgments
Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals. This research is sponsored by Novartis Pharmaceuticals, Basel, Switzerland. We thank Louise Prince, PhD, of iMed Comms (funded by Novartis Pharmaceuticals, Basel, Switzerland) for medical editorial assistance with this manuscript.
Glossary
Abbreviations
- α-BTX
α-bungarotoxin
- AD
Alzheimer's disease
- AE
adverse event
- ATCC
American Type Culture Collection
- AUC
area under the concentration–time curve
- Aβ
β-amyloid
- CI
confidence interval
- Cmax
maximum concentration
- KRH
Krebs-Ringer HEPES buffer
- MLA
methyllycaconitine
- MTD
maximum tolerated dose
- nACh receptor
nicotinic ACh receptor
- ORT
object recognition test
- p.o
orally
- PK
pharmacokinetic
- RT
room temperature
- SAD
single-ascending dose
- SAE
serious adverse event
- SAO
stretched attend posture towards the object
- t1/2
terminal elimination half-life
- Tmax
time to maximum concentration
Author contributions
D. F., K. L., D. H., K. H., G. B., C. R. P., K. M., F. C. (preclinical studies) and D. F., N. P., M. W., K. S.-N., K. K., D. J., T. B. and C. L. L. (clinical studies) designed and performed the studies, and analysed the data. D. F., N. P., M. W., C. R. P., F. C. and C. L. L. wrote the paper.
Conflict of interest
D. F., N. P., M. W., K. S-N., K. L., K. H., K. M., F. C., K. K., D. J. and C. L. L. are employees of, and hold shares in, Novartis Pharma AG. T. B. is an employee of, and holds shares in, Roche Pharma AG. Otherwise, no conflicts of interest exist.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ligand-gated ion channels. Br J Pharmacol. 2013;170:1582–1606. doi: 10.1111/bph.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendash GW, Sengstock GJ, Sanberg PR, Kem WR. Improved learning and memory in aged rats with chronic administration of the nicotinic receptor agonist GTS-21. Brain Res. 1995;674:252–259. doi: 10.1016/0006-8993(94)01449-r. [DOI] [PubMed] [Google Scholar]
- Bitner R, Anderson D, Drescher K, Kohlhaas K, Gronlien H, Hu M, et al. Preclinical characterization of a selective alpha-7 neuronal nicotinic acetylcholine receptor agonist ABT-126: a novel therapeutic agent for the treatment of cognitive impairment in Alzheimer's disease and schizophrenia. Alzheimers Dement. 2013;9:P817–P818. [Google Scholar]
- Bitner RS, Bunnelle WH, Decker MW, Drescher KU, Kohlhaas KL, Markosyan S, et al. In vivo pharmacological characterization of a novel selective alpha7 neuronal nicotinic acetylcholine receptor agonist ABT-107: preclinical considerations in Alzheimer's disease. J Pharmacol Exp Ther. 2010;334:875–886. doi: 10.1124/jpet.110.167213. [DOI] [PubMed] [Google Scholar]
- Biton B, Bergis OE, Galli F, Nedelec A, Lochead AW, Jegham S, et al. SSR180711, a novel selective alpha7 nicotinic receptor partial agonist: (1) binding and functional profile. Neuropsychopharmacology. 2007;32:1–16. doi: 10.1038/sj.npp.1301189. [DOI] [PubMed] [Google Scholar]
- Briggs CA, Anderson DJ, Brioni JD, Buccafusco JJ, Buckley MJ, Campbell JE, et al. Functional characterization of the novel neuronal nicotinic acetylcholine receptor ligand GTS-21 in vitro and in vivo. Pharmacol Biochem Behav. 1997;57:231–241. doi: 10.1016/s0091-3057(96)00354-1. [DOI] [PubMed] [Google Scholar]
- Broocks A, Little JT, Martin A, Minichiello MD, Dubbert B, Mack C, et al. The influence of ondansetron and m-chlorophenylpiperazine on scopolamine-induced cognitive, behavioral, and physiological responses in young healthy controls. Biol Psychiatry. 1998a;43:408–416. doi: 10.1016/s0006-3223(97)00388-0. [DOI] [PubMed] [Google Scholar]
- Broocks A, Pigott TA, Hill JL, Canter S, Grady TA, L'Heureux F, et al. Acute intravenous administration of ondansetron and m-CPP, alone and in combination, in patients with obsessive-compulsive disorder (OCD): behavioral and biological results. Psychiatry Res. 1998b;79:11–20. doi: 10.1016/s0165-1781(98)00029-8. [DOI] [PubMed] [Google Scholar]
- Camilleri M. Pharmacology and clinical experience with alosetron. Expert Opin Investig Drugs. 2000;9:147–159. doi: 10.1517/13543784.9.1.147. [DOI] [PubMed] [Google Scholar]
- Charpantier E, Wiesner A, Huh KH, Ogier R, Hoda JC, Allaman G, et al. Alpha7 neuronal nicotinic acetylcholine receptors are negatively regulated by tyrosine phosphorylation and Src-family kinases. J Neurosci. 2005;25:9836–9849. doi: 10.1523/JNEUROSCI.3497-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerbach D, Lingenhohl K, Dobbins P, Mosbacher J, Corbett N, Nozulak J, et al. Coupling of human nicotinic acetylcholine receptors alpha 7 to calcium channels in GH3 cells. Neuropharmacology. 2005;48:215–227. doi: 10.1016/j.neuropharm.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Feuerbach D, Nozulak J, Lingenhoehl K, McAllister K, Hoyer D. JN403, in vitro characterization of a novel nicotinic acetylcholine receptor alpha7 selective agonist. Neurosci Lett. 2007;416:61–65. doi: 10.1016/j.neulet.2007.01.045. [DOI] [PubMed] [Google Scholar]
- Feuerbach D, Lingenhoehl K, Olpe HR, Vassout A, Gentsch C, Chaperon F, et al. The selective nicotinic acetylcholine receptor alpha7 agonist JN403 is active in animal models of cognition, sensory gating, epilepsy and pain. Neuropharmacology. 2009;56:254–263. doi: 10.1016/j.neuropharm.2008.08.025. [DOI] [PubMed] [Google Scholar]
- de Fiebre CM, Meyer EM, Henry JC, Muraskin SI, Kem WR, Papke RL. Characterization of a series of anabaseine-derived compounds reveals that the 3-(4)-dimethylaminocinnamylidine derivative is a selective agonist at neuronal nicotinic alpha 7/125I-alpha-bungarotoxin receptor subtypes. Mol Pharmacol. 1995;47:164–171. [PubMed] [Google Scholar]
- File SE, Kenny PJ, Ouagazzal AM. Bimodal modulation by nicotine of anxiety in the social interaction test: role of the dorsal hippocampus. Behav Neurosci. 1998;112:1423–1429. doi: 10.1037//0735-7044.112.6.1423. [DOI] [PubMed] [Google Scholar]
- Freedman R, Hall M, Adler LE, Leonard S. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol Psychiatry. 1995;38:22–33. doi: 10.1016/0006-3223(94)00252-X. [DOI] [PubMed] [Google Scholar]
- Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci U S A. 1997;94:587–592. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Olincy A, Buchanan RW, Harris JG, Gold JM, Johnson L, et al. Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am J Psychiatry. 2008;165:1040–1047. doi: 10.1176/appi.ajp.2008.07071135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault J, Hopkins J, Berger R, Drebing C, Logel J, Walton C, et al. Comparison of polymorphisms in the alpha7 nicotinic receptor gene and its partial duplication in schizophrenic and control subjects. Am J Med Genet B Neuropsychiatr Genet. 2003;123B:39–49. doi: 10.1002/ajmg.b.20061. [DOI] [PubMed] [Google Scholar]
- Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol. 2004;74:363–396. doi: 10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Gotti C, Riganti L, Vailati S, Clementi F. Brain neuronal nicotinic receptors as new targets for drug discovery. Curr Pharm Des. 2006;12:407–428. doi: 10.2174/138161206775474486. [DOI] [PubMed] [Google Scholar]
- Hajos M, Hurst RS, Hoffmann WE, Krause M, Wall TM, Higdon NR, et al. The selective alpha7 nicotinic acetylcholine receptor agonist PNU-282987 [N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride] enhances GABAergic synaptic activity in brain slices and restores auditory gating deficits in anesthetized rats. J Pharmacol Exp Ther. 2005;312:1213–1222. doi: 10.1124/jpet.104.076968. [DOI] [PubMed] [Google Scholar]
- Hauser TA, Kucinski A, Jordan KG, Gatto GJ, Wersinger SR, Hesse RA, et al. TC-5619: an alpha7 neuronal nicotinic receptor-selective agonist that demonstrates efficacy in animal models of the positive and negative symptoms and cognitive dysfunction of schizophrenia. Biochem Pharmacol. 2009;78:803–812. doi: 10.1016/j.bcp.2009.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurth K, Gomez-Mancilla B, Hoyer D, Johns D, Lopez-Lopez C, Nozulak J, et al. 2014. Abstract MEDI 222 at the Division of Medicinal Chemistry , 247th American Chemical Society (ACS) National Meeting, Dallas, Texas, USA, March 16–20, 2014.
- Jessen F, Kucharski C, Fries T, Papassotiropoulos A, Hoenig K, Maier W, et al. Sensory gating deficit expressed by a disturbed suppression of the P50 event-related potential in patients with Alzheimer's disease. Am J Psychiatry. 2001;158:1319–1321. doi: 10.1176/appi.ajp.158.8.1319. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa H, Takenouchi T, Azuma R, Wesnes KA, Kramer WG, Clody DE, et al. Safety, pharmacokinetics, and effects on cognitive function of multiple doses of GTS-21 in healthy, male volunteers. Neuropsychopharmacology. 2003;28:542–551. doi: 10.1038/sj.npp.1300028. [DOI] [PubMed] [Google Scholar]
- Levin ED, Bettegowda C, Blosser J, Gordon J. AR-R17779, and alpha7 nicotinic agonist, improves learning and memory in rats. Behav Pharmacol. 1999;10:675–680. doi: 10.1097/00008877-199911000-00014. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Dunbar G, Segreti AC, Girgis RR, Seoane F, Beaver JS, et al. A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacology. 2013;38:968–975. doi: 10.1038/npp.2012.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurov AA, Kombo DC, Hauser TA, Miao L, Dull G, Genus JF, et al. Discovery of (2S,3R)-N-[2-(pyridin-3-ylmethyl)-1-azabicyclo[2.2.2]oct-3-yl]benzo[b]furan-2-car boxamide (TC-5619), a selective alpha7 nicotinic acetylcholine receptor agonist, for the treatment of cognitive disorders. J Med Chem. 2012;55:9793–9809. doi: 10.1021/jm301048a. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelmore S, Croskery K, Nozulak J, Hoyer D, Longato R, Weber A, et al. Study of the calcium dynamics of the human alpha4beta2, alpha3beta4 and alpha1beta1gammadelta nicotinic acetylcholine receptors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:235–245. doi: 10.1007/s00210-002-0589-z. [DOI] [PubMed] [Google Scholar]
- Olincy A, Harris JG, Johnson LL, Pender V, Kongs S, Allensworth D, et al. Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry. 2006;63:630–638. doi: 10.1001/archpsyc.63.6.630. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. Paxinos and Franklin's the Mouse Brain in Stereotaxic Coordinates. Waltham, MA: Academic Press; 2012. [Google Scholar]
- Preskorn SH, Gawryl M, Dgetluck N, Palfreyman M, Bauer LO, Hilt DC. Normalizing effects of EVP-6124, an alpha-7 nicotinic partial agonist, on event-related potentials and cognition: a proof of concept, randomized trial in patients with schizophrenia. J Psychiatr Pract. 2014;20:12–24. doi: 10.1097/01.pra.0000442935.15833.c5. [DOI] [PubMed] [Google Scholar]
- Prickaerts J, van Goethem NP, Chesworth R, Shapiro G, Boess FG, Methfessel C, et al. EVP-6124, a novel and selective alpha7 nicotinic acetylcholine receptor partial agonist, improves memory performance by potentiating the acetylcholine response of alpha7 nicotinic acetylcholine receptors. Neuropharmacology. 2012;62:1099–1110. doi: 10.1016/j.neuropharm.2011.10.024. [DOI] [PubMed] [Google Scholar]
- Pryce CR, Bettschen D, Nanz-Bahr NI, Feldon J. Comparison of the effects of early handling and early deprivation on conditioned stimulus, context, and spatial learning and memory in adult rats. Behav Neurosci. 2003;117:883–893. doi: 10.1037/0735-7044.117.5.883. [DOI] [PubMed] [Google Scholar]
- Rogers BN, Jacobsen EJ, O'Donnell CJ, Shaffer CL, Walker DP, Wishka DG. Identification of a7 nicotinic acetylcholine receptor agonists for their assessment in improving cognition in schizophrenia. In: Barrish JC, Carter PH, Cheng PT, Zahler R, editors. Accounts in Drug Discovery: Case Studies in Medicinal Chemistry. London: Royal Society of Chemistry; 2011. pp. 332–362. [Google Scholar]
- Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009;9:418–428. doi: 10.1038/nri2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. Beta-amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem. 2000;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- Winterer G, Gallinat J, Brinkmeyer J, Musso F, Kornhuber J, Thuerauf N, et al. Allosteric alpha-7 nicotinic receptor modulation and P50 sensory gating in schizophrenia: a proof-of-mechanism study. Neuropharmacology. 2013;64:197–204. doi: 10.1016/j.neuropharm.2012.06.040. [DOI] [PubMed] [Google Scholar]
- Young JW, Geyer MA. Evaluating the role of the alpha-7 nicotinic acetylcholine receptor in the pathophysiology and treatmen tof schizophrenia. Biochem Pharmacol. 2013;15:1122–1132. doi: 10.1016/j.bcp.2013.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]