

Abstract
Background and Purpose
Memantine and ketamine are clinically used, open-channel blockers of NMDA receptors exhibiting remarkable pharmacodynamic similarities despite strikingly different clinical profiles. Although NMDA channel gating constitutes an important difference between memantine and ketamine, it is unclear how positive allosteric modulators (PAMs) might affect the pharmacodynamics of these NMDA blockers.
Experimental Approach
We used two different PAMs: SGE-201, an analogue of an endogenous oxysterol, 24S-hydroxycholesterol, along with pregnenolone sulphate (PS), to test on memantine and ketamine responses in single cells (oocytes and cultured neurons) and networks (hippocampal slices), using standard electrophysiological techniques.
Key Results
SGE-201 and PS had no effect on steady-state block or voltage dependence of a channel blocker. However, both PAMs increased the actions of memantine and ketamine on phasic excitatory post-synaptic currents, but neither revealed underlying pharmacodynamic differences. SGE-201 accelerated the re-equilibration of blockers during voltage jumps. SGE-201 also unmasked differences among the blockers in neuronal networks – measured either by suppression of activity in multi-electrode arrays or by neuroprotection against a mild excitotoxic insult. Either potentiating NMDA receptors while maintaining the basal activity level or increasing activity/depolarization without potentiating NMDA receptor function is sufficient to expose pharmacodynamic blocker differences in suppressing network function and in neuroprotection.
Conclusions and Implications
Positive modulation revealed no pharmacodynamic differences between NMDA receptor blockers at a constant voltage, but did expose differences during spontaneous network activity. Endogenous modulator tone of NMDA receptors in different brain regions may underlie differences in the effects of NMDA receptor blockers on behaviour.
Tables of Links
| TARGETS |
|---|
| Ligand-gated ion channels |
| AMPA receptor |
| NMDA receptor |
| GluN2A |
| GluN1a |
| LIGANDS |
|---|
| CYZ, cyclothiazide |
| Ketamine |
| Memantine |
| MK-801, dizocilpine |
| NMDA |
| PS, pregnenolone sulphate |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Memantine and ketamine are two clinically important NMDA receptor open-channel blockers. Despite prominent clinical differences, the drugs' pharmacodynamic properties are strikingly similar (Gilling et al., 2009; Emnett et al., 2013). We recently showed that strong depolarization reveals biophysical differences between the blockers, but transient depolarization during physiological activity is not sufficient to unveil this difference (Emnett et al., 2013). This, in part, results from the low open probability (Popen) of the fully liganded NMDA receptor channel (Rosenmund et al., 1995; Emnett et al., 2013). It remains unclear whether positive modulators of NMDA receptor function, which increase channel Popen, might alter blocker behaviour and expose pharmacodynamic differences among the channel blockers during physiological or pathophysiological activity. If so, areas of the nervous system affected by positive modulation may be sites relevant to the clinical differences between drugs.
Both ketamine and memantine are trapping-type channel blockers (Chen et al., 1992; Chen and Lipton, 1997; Kotermanski et al., 2009b), but have largely non-overlapping clinical applications. Channel opening is required for blocker association. Channel gating can occur with blocker bound (Blanpied et al., 1997; Chen and Lipton, 1997), and blocker dissociates mainly from the open state of the channel. Dissociation is dramatically accelerated by depolarization. However, the low Popen of the channel is rate limiting for exit from the channel. Upon strong, prolonged depolarization, memantine re-equilibrates faster than ketamine, but both drugs remain trapped during brief depolarizations associated with physiological activity (Emnett et al., 2013). Memantine is neuroprotective during ischaemic injury and is modestly effective at stemming the cognitive decline of Alzheimer's disease (Chen et al., 1998; Culmsee et al., 2004; Tariot et al., 2004; Babu and Ramanathan, 2009). Ketamine is both an anaesthetic and analgesic, and has recently generated excitement in psychiatry as a rapidly acting antidepressant (Aan Het Rot et al., 2012; Marland et al., 2013; Murrough et al., 2013). Although pharmacokinetic differences between the drugs are likely to contribute to the different clinical actions (Gilling et al., 2009; but see Kotermanski et al., 2013), pharmacodynamic differences might also contribute.
The brain contains many endogenous positive allosteric modulators (PAMs) of NMDA channel function, including pregnenolone sulphate (PS) and 24S-hydroxycholesterol. PS is a neurosteroid that potentiates NMDA receptor function, but also negatively modulates GABAA receptors (Wu et al., 1991; Horak et al., 2004). 24S-hydroxycholesterol is the major metabolite of brain cholesterol and accounts for nearly 50% of cholesterol's turnover and clearance from the brain (Lund et al., 2003; Karu et al., 2007). It is a potent PAM of NMDA receptors at concentrations found in vivo. Its synthetic analogue, analogue of 24S-hydroxycholesterol (SGE-201), which we have used in the present study, has experimental advantages, including greater water solubility and reversibility (Paul et al., 2013). 24S-hydroxycholesterol and SGE-201 selectively potentiate NMDA receptor-, but not AMPA receptor)- or GABAA receptor-mediated synaptic currents (Paul et al., 2013; Linsenbardt et al., 2014).
Here we test the hypothesis that PAMs may expose biophysical differences among channel blockers. In the first part of our work, we evaluated the effects of two PAMs, PS and SGE-201, on trapping channel block. We then evaluated the more specific modulator, SGE-201, on the pharmacodynamic differences between ketamine and memantine, two blockers that exhibit very subtle pharmacodynamic differences at baseline. We hypothesized that positive modulation would increase the probability of blocker dissociation during depolarization, thereby allowing physiological activity to expose biophysical difference between blockers. We found that PS and SGE-201 did not alter steady-state block but altered blocker actions during phasic agonist presentation. Despite accelerating kinetics of block, increasing channel Popen did not disclose blocker differences when voltage was constant. However, positive modulation did accelerate voltage-dependent blocker re-equilibration into a time domain potentially relevant for synaptic responses. Exploiting the selectivity of SGE-201 for NMDA receptors, we found that SGE-201 exposes differences between the actions of memantine and ketamine on network activity and on cell survival following excitotoxic insult. Either NMDA receptor potentiation during basal network activity or strongly increased network activity without NMDA receptor potentiation was sufficient to expose these pharmacodynamic differences. Thus, we concluded that unusually low-channel Popen masks pharmacodynamic differences between ketamine and memantine, but PAMs expose this difference. We propose that differences in endogenous modulator tone in different areas of the nervous system could lead to differences between effects of memantine and ketamine on behaviour.
Methods
Cell cultures
All animal care and experimental procedures were consistent with National Institutes of Health guidelines and were approved by the Washington University Animal Studies Committee. Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 120 animals were used in the experiments described here.
Hippocampal cultures were prepared as either mass cultures or microcultures (as indicated in figure legends) from postnatal day 1 to 3, female rat pups anaesthetized with isoflurane. Methods were adapted from earlier descriptions (Huettner and Baughman, 1986; Bekkers et al., 1990; Tong and Jahr, 1994; Mennerick et al., 1995). Hippocampal slices (500 μm thickness) were digested with 1 mg·mL−1 papain in oxygenated Leibovitz L-15 medium (Life Technologies, Gaithersburg, MD, USA). Tissue was mechanically triturated in modified Eagle's medium (Life Technologies) containing 5% horse serum, 5% FCS, 17 mM D-glucose, 400 μM glutamine, 50 U·mL−1 penicillin and 50 μg·mL−1 streptomycin. Cells were seeded in modified Eagle's medium at a density of ∼650 cells per mm2 as mass cultures (onto 25 mm cover glasses coated with 5 mg·mL−1 collagen or 0.1 mg·mL−1 poly-D-lysine with 1 mg·mL−1 laminin) or 100 cells per mm2 as ‘microisland’ cultures (onto 35 mm plastic culture dishes coated with collagen microdroplets on a layer of 0.15% agarose). Cultures were incubated at 37°C in a humidified chamber with 5%CO2/95% air. Cytosine arabinoside (6.7 μM) was added 3–4 days after plating to inhibit glial proliferation. The following day, half of the culture medium was replaced with Neurobasal medium (Life Technologies) plus B27 supplement (Life Technologies).
Xenopus oocyte expression
cRNA encoding rat NMDA receptor subunits were injected into stages V–VI oocytes harvested from sexually mature female Xenopus laevis frogs (Xenopus 1, Northland, MI, USA). Frogs were anaesthetized with 0.1% tricane (3-aminobenzoic acid ethyl ester) during a partial ovariectomy. Oocytes were defolliculated by shaking for 20 min at 37°C in 2 mg·mL−1 collagenase dissolved in a calcium-free solution containing (in mM): 96 NaCl, 2 KCl, 1 MgCl2, and 5 HEPES, pH 7.4. Capped RNA for GluN1a and GluN2A subunits was prepared in vitro (mMESSAGE mMachine kit, Ambion, Austin, TX, USA) from linearized pBluescript vectors. Subunit RNA was injected in equal parts (50 ng of total RNA). After injection, oocytes were incubated at 18°C in ND96 solution containing (in mM): 96 NaCl, 2 KCl, 2 CaCl2, 1 MgCl2, and 5 HEPES, pH 7.4. ND96 was supplemented with pyruvate (5 mM), penicillin (100 U·mL−1), streptomycin (100 μg·mL−1) and gentamycin (50 μg·mL−1).
Electrophysiology
Whole-cell recordings were performed at room temperature from neurons cultured for 4–10 days (depending on the experiment) using a Multiclamp 700B amplifier, Digidata 1440A converter and Clampex 10.1 software (Molecular Devices, Sunnyvale, CA, USA). Young cells were favoured for biophysical experiments to minimize voltage-clamp errors, and older, synaptically mature cells were used for excitatory post-synaptic current (EPSC) measurements. For recordings, cells were transferred to an extracellular (bath) solution containing (in mM): 138 NaCl, 4 KCl, 2 CaCl2, 10 glucose, 0.01 μM glycine and 10 HEPES, pH 7.25 adjusted with NaOH, 25–50 μM D-2-amino-5-phosphonovalerate (D-APV) was included until seal formation to prevent excitotoxicity. Solutions with adjusted composition described later were perfused using a gravity-driven local perfusion system from a common tip. The estimated solution exchange times were <100 ms (10–90% rise), estimated from junction current rises at the tip of an open patch pipette. For synaptic recordings, these solutions contained 1 μM 2,3-dihydroxy-6-nitro-7-sulphonyl-benzo[f]quinoxaline (NBQX) and 25 μM bicuculline methobromide. For exogenous NMDA application, 0.25 mM CaCl2 was used (in APV-free perfusion solutions) to minimize Ca2+-dependent NMDA receptor desensitization (Zorumski et al., 1989), and 250 nM tetrodotoxin (TTX) was added to prevent network activity. Unless otherwise noted, exogenous NMDA concentration was 30 μM.
The tip resistance of patch pipettes was 3–6 MΩ when filled with an internal solution containing (in mM): 130 potassium gluconate, 0.5 CaCl2, 5 EGTA, 4 NaCl, and 10 HEPES at pH 7.25, adjusted with KOH. Potassium was used as the main intracellular cation for autaptic stimulation to preserve action potential waveform, but in experiments examining current responses to exogenous agonists, cesium methanesulphonate replaced potassium gluconate to block potassium channels and improve spatial voltage-clamp quality. Holding voltage was typically −70 mV unless otherwise noted. Access resistance (8–10 MΩ was compensated 80–100% for EPSC measurements. For evoked EPSCs, cells were stimulated with 1.5 ms pulses to 0 mV from −70 mV to evoke autaptic transmitter release (Mennerick et al., 1995). EPSC decays were fit with a biexponential function in Clampfit using Levenberg Marquart or variable metric fit parameters. Weighted time constants (τw) were calculated for data analysis.
Oocyte responses to NMDA were recorded 2–5 days following RNA injection using a two-electrode voltage clamp (OC725C amplifier; Warner Instruments, Hamden, CT, USA) at a membrane potential of −70 mV. The bath solution was ND96 solution with the following changes: Mg2+ was omitted, Ba2+ replaced Ca2+ to avoid activation of endogenous Ca2+-dependent currents, and 10 μM glycine was added. Glass recording pipettes (∼1 MΩ resistance) were filled with 3 M KCl. Compounds were applied by gravity to the oocytes using a multi-barrel pipette with a common output tip. Data acquisition and analysis were performed with pCLAMP software (Molecular Devices).
Multi-electrode arrays (MEAs)
Mass cultures were seeded on MEAs (Multichannel Systems, Reutlingen, Germany) for network analysis (Mennerick et al., 2010). Recordings were performed 14–15 days in vitro (DIV) at 37°C. For figures and statistics, effects of drugs over a 30 min recording period were compared with the average of activity (30 min) before drug administration and following drug washout. If a PAM [such as cyclothiazide (CYZ) or SGE-201] was present, the baseline recordings contained the PAM. Array-wide spike detection rate (ASDR) was measured as the total number of spikes across the entire array in each second of recording, averaged across the entire recording (Wagenaar et al., 2006; Mennerick et al., 2010). Average ASDRs are given in the figure legends. Because of the large differences in absolute baseline ASDR among cultures (Wagenaar et al., 2006), statistics were performed on drug effects normalized to baseline ASDR.
To reduce activity to baseline levels in the presence of SGE-201, we titrated NBQX or TTX concentration in the presence of the PAM. We employed 100 nM NBQX (n = 5) or 10 nM TTX (n = 6). Experiments in which activity was not reduced below twofold from baseline were excluded from analysis. Trends with both activity blockers were similar, so results were pooled for analysis.
Hypoxia
Mass cultures (13–14 DIV) were exposed to hypoxia in a commercially available chamber (Billups-Rothenberg, Del Mar, CA, USA), humidified and saturated with 95% nitrogen and 5% CO2 at 37°C, for 2.5 h. The gas exchange followed the specifications of the chamber manufacturer (flow of 20 L·min−1 for 4 min to achieve 100% gas exchange). Original medium was exchanged for conditioned media containing the specified drugs immediately prior to hypoxia exposure. Following hypoxia, cells were returned to their original medium and incubated under standard culture conditions until the cell death assay (24 h later). We used Hoechst 33342 (5 μM) to identify all nuclei and propidium iodide (PI, 3 μM) for 30 min to stain nuclei of cells with compromised membranes. Five 10× microscope fields were quantified per condition per experiment, yielding >100 total neurons for each condition. Ratios of healthy neurons were quantified as the fraction of PI-negative neuronal nuclei to total neuronal nuclei. Automated cell counting algorithms (ImageJ software, National Institutes of Health, Bethesda, MD, USA) were used for cell counts. Toxicity experiments were treated as a dependent sample design (Zar, 1999) in which sibling cultures plated in identical media and exposed to hypoxia at the same time were compared by repeated measures statistics.
Data analysis
Results are shown as means ± SEM. Comparative statistics were performed with Student's t-test or with two-way anova where indicated.
Materials
SGE-201 was synthesized by previously published methods (Paul et al., 2013). Memantine, D-APV, NBQX and TTX were purchased from Tocris (Bristol, UK). (R,S) ketamine, bicuculline methobromide, PS and CYZ were purchased from Sigma (St. Louis, MO, USA).
Results
Positive allosteric modulation does not alter steady-state inhibition by memantine
Previous studies are equivocal on whether channel Popen, usually manipulated with agonist concentration, influences steady-state inhibition by trapping channel blockers. Here we used memantine to test the principle. Inhibition by trapping blockers increases with agonist concentration in vitro and in simulations (Chen et al., 1992; Chen and Lipton, 1997), but other studies found that agonist concentration did not influence IC50 values for memantine and, like our study, found that kinetics are altered by Popen (Gilling et al., 2007). To test whether PAMs that increase channel Popen influence the steady-state potency of trapping-type blockers, we generated concentration-inhibition plots for memantine in the presence and absence of SGE-201 or PS in Xenopus oocytes expressing a homogeneous receptor population of GluN1a/GluN2A subunits. PS (50 μM) increased peak responses to 30 μM NMDA 3.0 ± 0.3-fold (n = 7), but failed to significantly influence memantine IC50 (Figure 1A–B). Similarly, SGE-201 (1 μM) potentiated NMDA responses 3.4 ± 0.5-fold (n = 11), but did not affect steady-state inhibition by memantine (Figure 1C–D). We also tested the comparator channel blocker ketamine, which we and others have shown to be pharmacodynamically very similar to memantine (Gilling et al., 2009; Emnett et al., 2013). Figure 1E–F shows that positive modulation by SGE-201 also did not affect the IC50 for ketamine block. This demonstrates that conclusions about the effect of positive modulation extend to other similar trapping channel blockers.
Figure 1.
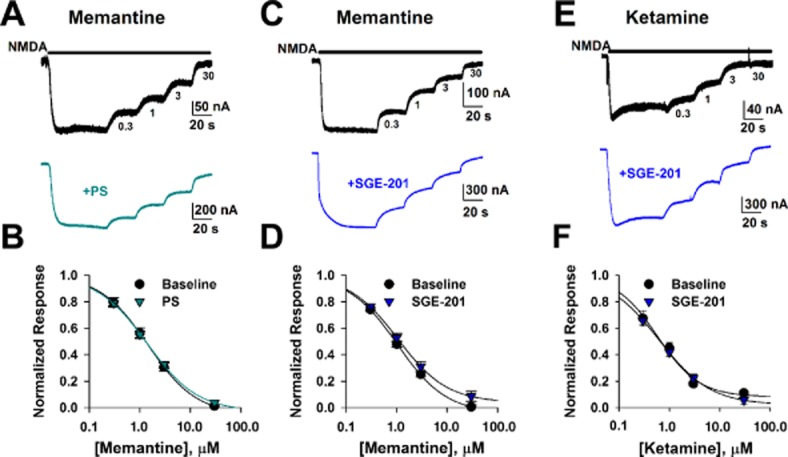
Positive allosteric modulation does not affect steady-state memantine IC50. A. NMDA (30 μM) responses in Xenopus oocytes injected with GluN1a-GluN2A receptor RNA were recorded in the absence and then presence of increasing concentrations of memantine (0.3, 1, 3, 30 μM) in saline or in the presence of PS (50 μM). Memantine concentrations are indicated below the top trace. B. Concentration–response curves in control conditions and in the presence of PS (n = 11). Solid lines represents fits to the Hill equation of the form I = Imax * Cn/(IC50n + Cn), where Imax is maximum current in the absence of agonist, C is antagonist concentration, n is the Hill coefficient. Memantine IC50 was calculated to be 1.4 μM in the presence or absence of PS. C and D. Representative traces and summary of memantine effects in the absence and presence of SGE-201 (1 μM; n = 7). Solid lines represent fits to the Hill equation. Memantine IC50 values are 1.0 μM in the absence of SGE-201 and 1.1 μM in the presence of SGE-201. Neither potentiator significantly altered IC50 concentration of memantine (P > 0.05, unpaired t-tests). E and F. Similar experiment as C and D except ketamine was employed as channel blocker. IC50 values were 0.5 μM (absence of SGE-201; n = 11) and 0.6 μM (presence of SGE-201; n = 5). There was no significant difference in calculated IC50s between control and SGE-201 treated cells (P > 0.05, unpaired t-test). Although ketamine IC50 values trended lower than those for memantine, fits of individual cells produced average IC50 values that did not significantly differ from those of memantine (Emnett et al., 2013).
This result suggests that increasing channel Popen does not significantly affect the potency of trapping channel blockers. To extend these results, we examined steady-state memantine inhibition at both positive and negative membrane potentials to test whether PAMs interact with the voltage dependence of inhibition (Figure 2A–B). For these studies we used cultured neurons, where kinetics of block could be more readily examined in whole-cell recordings. We found that neither PS nor SGE-201 influenced steady-state inhibition observed with a fixed concentration of memantine (Figure 2C–D). Both the onset and offset time constant of block at −70 mV, significantly decreased with SGE-201 (Figure 2E). The alteration in kinetics is consistent with a mechanism of allosteric modulation that increases channel Popen, enhancing blocker access to its channel binding site. The effect of PS on blocker onset/offset time constants was much weaker than that of SGE-201, and neither rate constant was significantly altered (Figure 2F). This is consistent with the observation that although PS potentiated peak NMDA current, it had less effect on steady-state current. This is likely explained by the mixed potentiating/inhibiting actions of PS (Horak et al., 2004). These results suggest that SGE-201 may be a purer potentiator and/or have a different mechanism of action than PS (Paul et al., 2013).
Figure 2.
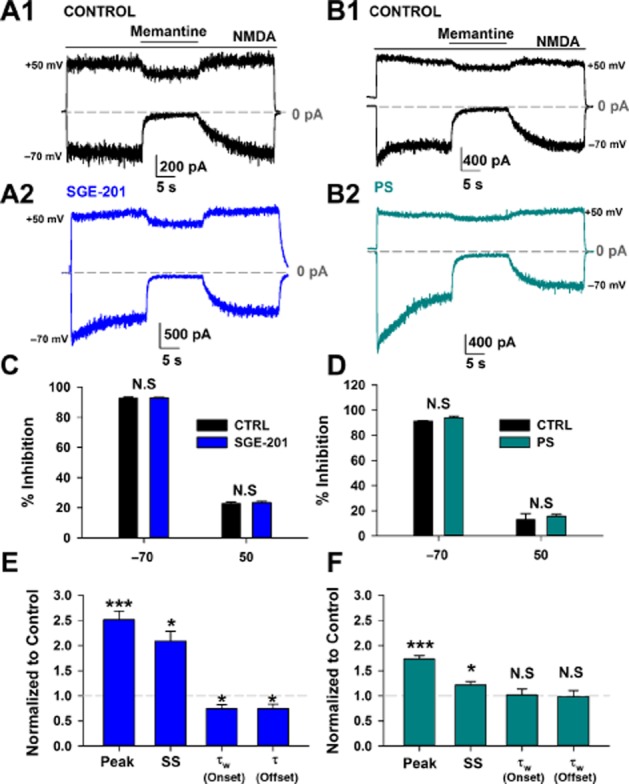
Positive allosteric modulation does not affect the voltage dependence of memantine at steady-state (SS). A–B. Representative traces depicting memantine (10 μM) block of a baseline NMDA (30 μM) response (A1, B1,) and then from the same cell in the presence of SGE-201 (1 μM, A2) or PS (50 μM, B2) at holding potentials of −70 and +50 mV. SGE-201 and PS were pre-applied for 1min and subsequently co-applied with NMDA. C–D. Average block achieved at −70 or +50 mV in the absence or presence of SGE-201 (C) or PS (D). Neither SGE-201 nor PS altered steady-state memantine block at either holding potential (N.S. = not significant; P > 0.05, unpaired t-test). E. SGE-201 significantly potentiated both peak and SS currents and significantly decreased the weighted time constant (τw) of onset and the single time constant of offset of memantine block at −70 mV. ***P < 0.001, *P < 0.05; unpaired single-sample t-test, n = 9. F. PS also significantly potentiated the response at the peak and steady state, but did not significantly alter onset or offset time constants of memantine block. ***P < 0.001, *P < 0.05, unpaired single-sample t-test, n = 10.
Positive allosteric modulation enhances EPSC suppression by memantine
The actions of trapping channel blockers are strongly affected by temporal features of agonist presentation and ensuing NMDA receptor activation (Xia et al., 2010; Wroge et al., 2012; Emnett et al., 2013). Despite the lack of change in steady-state inhibition, perhaps transient, synaptic glutamate presentation to NMDA receptors supports a PAM-induced change in blocker effect because of faster blocker equilibration kinetics. We tested this principle on evoked, recurrent EPSCs stimulated in hippocampal autapses challenged with PS and with PS plus blocker ( Figure 3). PS (50 μM) had prominent effects on the NMDA receptor EPSC decay time constant and also increased peak NMDA receptor EPSC amplitude (Figure 3A–B), resulting in a strong potentiation of post-synaptic charge (Figure 3C). From the potentiated baseline, PS exaggerated the effect of memantine on EPSC decay kinetics and on peak EPSC amplitude (Figure 3D–F). Thus, in contrast to steady-state effects of memantine, which were unaffected by PAMs, synaptic effects of the channel blocker were augmented by PAMs.
Figure 3.
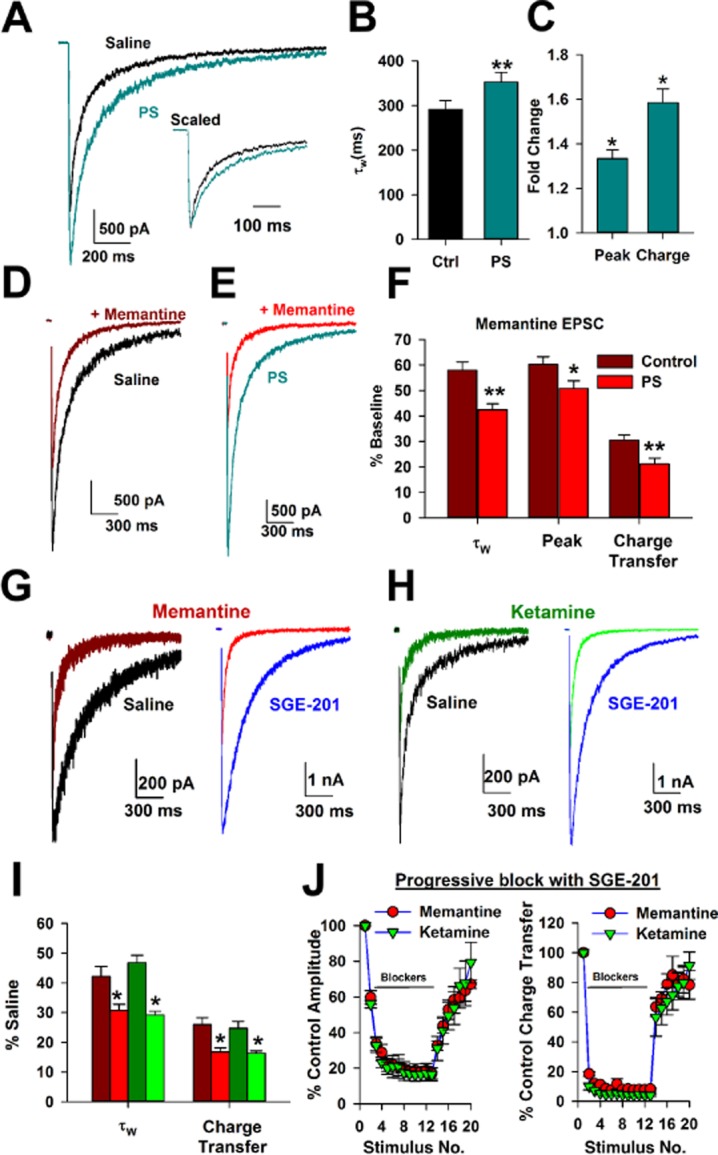
Positive allosteric modulation does not differentiate between actions of memantine and ketamine during synaptic activation. Representative NMDA receptor EPSCs in saline and with 50 μM PS. Inset: scaled traces to highlight the prolonged EPSC decay. B. Summary of PS effect on the weighted time constant of EPSC decay (τw; **P < 0.01 paired t-test, n = 9). C. PS potentiated the NMDA receptor EPSC peak amplitude and total charge (*P < 0.05, unpaired t-test relative to baseline, n = 9). D–E: Representative traces depicting effect of memantine (10 μM) on NMDA receptor-EPSCs in control conditions (D) or with PS (E). F. PS increased the effect of memantine on NMDA receptor EPSC decay kinetics (i.e., accelerated the decay), peak and charge transfer, measured relative to the effect of memantine in the absence of PS (n = 10–11, **P < 0.01, *P < 0.05). G–H. EPSCs evoked in saline or in SGE-201 (1 μM) with or without memantine or ketamine. Saline and SGE-201 traces are from separate cells, and SGE-201 was continuously present throughout recording. I. SGE-201 augmented the effect of memantine and ketamine on τW and charge transfer of the NMDA EPSC compared with baseline effects. There was no difference in the effects of memantine and ketamine (n = 15–19, *P < 0.05 unpaired t-test, within blocker comparison; P > 0.05 unpaired t-test, between blocker comparison). J. Cumulative blocking plots in the continuous presence of memantine (10 μM) or ketamine (10 μM) during 0.03 Hz stimulation of EPSCs. Memantine and ketamine reached maximal block, measured by peak amplitude (left) or charge transfer (right), with indistinguishable time courses in the presence of 1 μM SGE-201 and also recovered similarly, implying a similar degree of trapping. P > 0.05, unpaired t-test, n = 6.
SGE-201 has an apparently different mechanism of action compared with PS (Paul et al., 2013). We previously showed that SGE-201 does not have a prominent effect on EPSC decay time course (Paul et al., 2013). Consistent with this, in the present dataset, SGE-201 marginally prolonged EPSC decays (weighted τ = 347 ± 17 ms, n = 33, vs. 408 ± 25 ms, n = 34; P = 0.049). Conversely, SGE-201 enhanced peak amplitude roughly twofold (control cells vs. SGE-201 cells: −2.7 vs. −4.6 nA, P = 0.01, n = 33–34). Thus, unlike PS, SGE-201 affected EPSC decay only weakly, with primary effects on EPSC peak amplitude. Nevertheless, like PS, SGE-201 enhanced memantine's effects on EPSC decay (Figure 3G and I).
Positive allosteric modulation by SGE-201 fails to distinguish memantine from ketamine
The interaction between PAMs and memantine on EPSCs shows that blocker function is altered by increasing channel Popen. Does this alteration extend to other blockers, particularly ketamine? We previously showed that biophysical differences between memantine and ketamine were not detectable when receptors were activated briefly and transiently by synaptic release in the absence of a PAM, and blocker differences were masked by low-channel Popen (Emnett et al., 2013). At a fixed membrane potential of −70 mV, we found that SGE-201 also augmented the actions of ketamine (Figure 3H). However, effects of ketamine and memantine were still indistinguishable (Figure 3I). The inability to distinguish memantine from ketamine persisted in a paradigm of progressive block (stimulation frequency 0.03 Hz in the presence of a blocker) and recovery from block (Figure 3J). The similar recovery time course suggests that any differences in partial trapping between memantine and ketamine (Kotermanski et al., 2009b) are not readily evident with synaptic activation of NMDA receptors.
PAM shifts voltage-dependent blocker re-equilibration to a physiologically relevant time domain
In the absence of NMDA receptor modulation, a difference between ketamine and memantine emerges in the rate constants of re-equilibration during strong depolarization. Furthermore, increasing channel Popen by chelating a negative modulator (Zn2+) or raising glycine concentration accelerated blocker re-equilibration (Emnett et al., 2013). Accordingly, we found that SGE-201 accelerated re-equilibration kinetics of both memantine and ketamine during a voltage pulse ( Figure 4). In the absence of modulator, kinetics are sufficiently slow that both ketamine and memantine remain trapped during physiological activity, leading to indistinguishable blocker effects during baseline activity. However, SGE-201 reduced re-equilibration time constants to values that could be influenced by excitatory post-synaptic potentials (EPSPs; Figure 4C).
Figure 4.

Positive modulation accelerates blocker re-equilibration during voltage jumps to time domains relevant to synaptic transmission. A–B. Representative traces depicting baseline NMDA responses at −70 and +50 mV (300 μM) and NMDA plus SGE-201 (1 μM). Currents in the absence of NMDA have been digitally subtracted. The right panels show responses in the presence of memantine (A, 10 μM) and ketamine (B, 10 μM). Right traces represent fractional responses calculated by dividing currents in the presence of blocker by currents in the absence of blocker, shown as a percentage of each baseline. Dotted lines indicate 0 and 100% of original NMDA current as indicated. Solid black lines in right traces indicate least-squares bi-exponential fits to re-equilibration of the blocker at +50 mV. C. The time course of re-equilibration after the voltage jump from −70 to +50 mV was estimated from fits like those in right panels of A and B for memantine and ketamine in the presence and absence of SGE-201. SGE-201 accelerated the rate constant of re-equilibration to a time domain potentially relevant to EPSPs. **P < 0.01, paired t-test, n = 19 each.
In summary, we found that PAMs influence non–steady-state inhibition, but not steady-state NMDA receptor inhibition, by clinically interesting NMDA receptor channel blockers. PAM activity alone at a fixed membrane potential near rest was insufficient to reveal a difference between memantine and ketamine. However, oxysterol PAM activity accelerated voltage-dependent re-equilibration. In this case, oxysterols shift the kinetics of blocker actions into a time domain possibly relevant for synaptic potentials and activity. Thus, we examined the differences between the drugs in measures of network activity and pathological depolarization to discern if oxysterol presence could expose latent blocker differences (Emnett et al., 2013).
Oxysterols expose physiologically relevant differences between memantine and ketamine in neuronal networks
We have previously shown that memantine and ketamine suppress physiological network activity indistinguishably, measured by ASDR in MEA recordings. We were able to expose differences between memantine and ketamine only during strong pathophysiological depolarization with oxygen–glucose deprivation (Emnett et al., 2013). In the present work, we evaluated whether the acceleration of voltage-dependent blocker re-equilibration in the presence of a PAM could also expose the differences between the blockers.
First, we tested whether SGE-201 increased network activity in a solely NMDA receptor-dependent manner (Figure 5A–B). SGE-201 (1 μM) approximately doubled ASDR, and the effect was entirely reversed by the high-affinity, non-competitive NMDA receptor antagonist MK-801 (15 μM) (Figure 5B). This suggests that SGE-201's neuromodulatory effects were specific for NMDA receptor function. This is unlike PAMs such as PS, Zn2+ chelators, arachidonic acid and other modulators, virtually all of which have important off-target effects (Deutsch et al., 1992; Fraser et al., 1993). Thus, SGE-201 offers a selective tool to evaluate the role of NMDA receptor efficacy in network function. In contrast to previous results in which memantine and ketamine had indistinguishable effects on ASDR (Emnett et al., 2013) (replicated independently in Figure 5), in the presence of SGE-201, ketamine was clearly more effective than memantine on ASDR, which was consistent with the faster voltage-dependent re-equilibration kinetics of memantine (Figure 5D).
Figure 5.
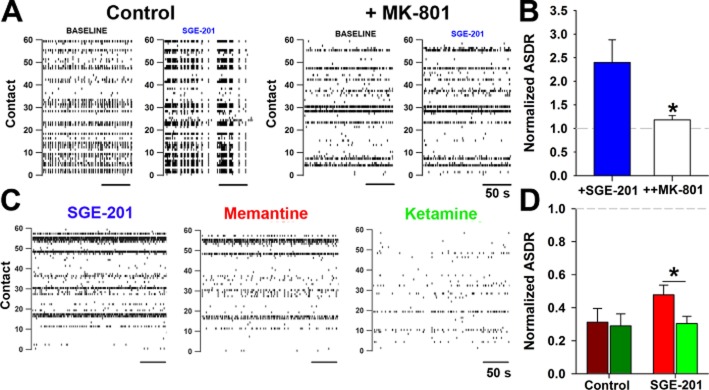
SGE-201 exposes memantine versus ketamine differences in suppression of network activity. A. Representative raster plots from MEAs depicting baseline and SGE-201 (1 μM) activity in normal medium and with medium containing 15 μM MK-801. B. SGE-201 potentiated MEA ASDR (baseline: 19.3 ± 8.2 s−1 without SGE-201 vs. 53.1 ± 33 s−1 with SGE-201). This effect was blocked by MK-801. *P < 0.05 compared with SGE-201 alone, n = 6. C. Representative raster plots depicting baseline, memantine (10 μM), and ketamine (10 μM) conditions in the presence of SGE-201 (1 μM). Blocker recordings (30 min) were interleaved between baseline and wash periods (both 30 min). SGE-201 was included in the baseline, drug, and wash conditions. D. Normalized ASDR with memantine (10 μM) and ketamine (10 μM) compared with baseline response (average of the baseline and wash conditions). Dark red and green bars are normalized values in SGE-201-free conditions (baseline: 48 ± 9 s−1, n = 10, P > 0.05, unpaired t-test, n = 5 paired experiments) and represent an independent replication of earlier results (Emnett et al., 2013). Bright red and green bars are in the continual presence of SGE-201 (baseline: 56 ± 6 s−1, n = 22). *P < 0.05, unpaired t-test, n = 11 paired experiments.
This difference could be disclosed as a direct result of the enhanced NMDA receptor channel Popen caused by SGE-201, or it could result from the more intense activity (depolarization), a secondary consequence of SGE-201 treatment. To test whether enhanced activity alone is sufficient, we increased network firing without SGE-201 by applying CYZ, a PAM for AMPA receptors and a pre-synaptic potentiator (10 μM, Figure 6A) (Partin et al., 1993; Yamada and Tang, 1993; Ishikawa and Takahashi, 2001). Average ASDR increased in the presence of CYZ similar to the effect of SGE-201 (Figure 6B). As with SGE-201 potentiation, the increase in activity exposed a significant difference between memantine and ketamine (Figure 6C). This suggests that sufficiently strong activity per se will make obvious the biophysical difference between the NMDA receptor blockers.
Figure 6.
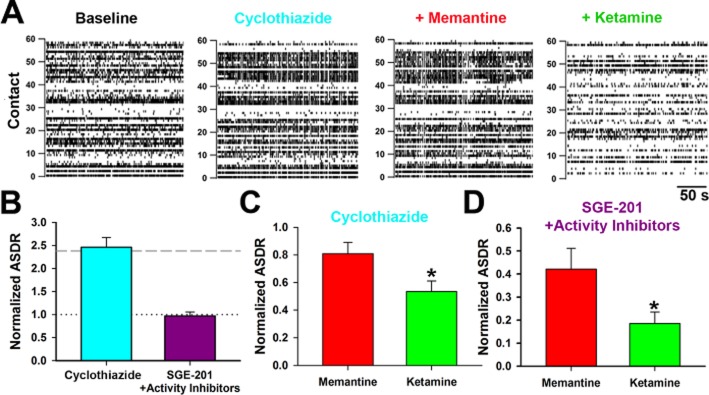
Excessive activity alone or basal activity plus SGE-201 differentiates the effects of NMDA receptor blockers on network function. A. Representative rasters depicting baseline conditions and 10 μM CYZ, CYZ + memantine and CYZ + ketamine conditions. B. ASDR normalized to baseline following addition of CYZ or SGE-201 (1 μM) + activity blockers (10 nM TTX or 100 nM NBQX – see Methods). The dashed grey line indicates average potentiation achieved with SGE-201 alone. The dotted grey line represents no potentiation. Average raw ASDR values with CYZ were 86 ± 15 s−1 compared with 42 ± 11 s−1 before treatment. Raw ASDR values with SGE-201 and activity blockers were 38 ± 7 s−1 compared with 42 ± 7 s−1 before treatment. C. In the presence of CYZ, memantine and ketamine, each at 10 μM, strongly diverge in their ability to suppress ASDR. *P < 0.05, n = 7. D. The presence of SGE-201 induced a difference between memantine and ketamine, even in the presence of activity suppressors. *P < 0.05, n = 11.
We then examined the effect of increasing channel gating alone with SGE-201, while counteracting the secondary increase in network activity with subsaturating concentrations of either 10 nM TTX or 100 nM NBQX. Both inhibitors yielded similar trends so results were pooled. In cultures in which the increase in ASDR by SGE-201 was inhibited (Figure 6B), the difference between memantine and ketamine in the presence of SGE-201 was preserved (Figure 6D). We conclude that either increased activity or positive NMDA receptor modulation in the context of basal activity is sufficient to reveal differences between NMDA receptor blocker in terms of network functioning.
We also examined the implications of PAM activity for NMDA receptor-dependent excitotoxicity elicited by hypoxia. We titrated the duration of hypoxia to yield ∼50% cell death in the absence of SGE-201. Under these conditions, there was only a trend-level difference between the neuroprotective effects of memantine and ketamine (Figure 7A and B). This contrasts with a more severe insult, which yielded a more pronounced difference between blockers consistent with the difference in voltage-dependent re-equilibration (Emnett et al., 2013). In the presence of SGE-201 (0.2–1.0 μM), which increased hypoxic damage, ketamine neuroprotection was significantly greater than that of memantine (Figure 7C). As with changes in network activity, we explored whether stronger depolarization or increased channel Popen was sufficient for the effects observed. To address this, we repeated the experiment using exogenous NMDA as the excitotoxin in the presence of 0.5 μM TTX. This allowed us to control more precisely the level of depolarization experienced by cells in the presence of SGE-201 and to limit excitotoxic contributions of endogenous glutamate. Pilot experiments demonstrated that 8 μM NMDA for 2.5 h alone produced cell death, similar to that observed with hypoxia. Further experiments showed that 8 μM NMDA yielded depolarizing currents equivalent to those generated by 4–5 μM NMDA in the presence of 0.2 μM SGE-201 (Figure 8A). Treating cells with 8 μM NMDA or with 4–5 μM NMDA plus SGE-201 resulted in similar levels of cell death, verifying that we achieved the desired matching. Nevertheless, the effects of ketamine and memantine were markedly different in the two conditions. While both drugs alone protected equivalently against NMDA toxicity, there was a clear difference when SGE-201 was present (Figure 8B). This suggests that when depolarization is matched, augmented channel function is sufficient to expose a neuroprotective difference between memantine and ketamine.
Figure 7.

SGE-201 presence reveals differences between memantine and ketamine neuroprotection during hypoxia. A. Hippocampal cultures were exposed to hypoxia (95% N2, 5% CO2, see Methods), for 2.5 h. Cell death was assessed 24 h post insult using PI (3 μM). Images depict representative fields (10×) of control, hypoxia, and memantine and ketamine dishes; red dots indicate dead (i.e. PI positive) neurons. For analysis, Hoescht stain (5 μM) was also included to identify the total neuronal population (not shown). B. Without SGE-201, memantine and ketamine protected similarly against the hypoxic insult. P = 0.035, paired t-test, n = 12. C. In the presence of 0.2–1.0 μM SGE-201, a significantly stronger difference emerged between memantine and ketamine neuroprotection. *P < 0.05, within experiment paired t-test; P < 0.001 for a significant interaction between SGE-201 and blocker neuroprotection: two-way anova, n = 8.
Figure 8.

SGE-201 presence alone distinguishes neuroprotective effects with matched depolarization. A. Neuronal response to the indicated concentrations of NMDA and SGE-201. 4–5 μM NMDA in the presence of 0.2 μM SGE-201 was equivalent to 8 μM NMDA alone. B. Normalized survival, relative to untreated cultures, is indicated for the indicated concentrations of NMDA, SGE-201 and memantine or ketamine. Although the amount of cell death achieved under baseline conditions and with SGE-201 alone was indistinguishable, suggesting similar depolarization, ketamine protected significantly more than memantine in the presence of SGE-201. *P < 0.001, two-way anova, n = 5. TTX (500 nM) was present throughout the entire toxicity experiment to prevent action potentials.
Our data from both network arrays and from excitotoxic insults suggest that either positive allosteric modulation in the context of basal depolarization or strong activity/depolarization alone will expose differences between memantine and ketamine. Importantly, these modulators are capable of distinguishing between memantine and ketamine in models of network and pathological activity in vitro.
Discussion
Memantine and ketamine are of interest because of their clinical differences, but similar underlying pharmacodynamic effects. We previously demonstrated that the low Popen of the NMDA receptor channel masks pharmacodynamic differences between the two drugs under basal conditions, and we found differences only when cells were subjected to extreme depolarization during oxygen–glucose deprivation (Emnett et al., 2013). However, it was unclear whether positive modulation of NMDA receptors could expose differences detectable during physiological network activity. Here, we show that increasing NMDA receptor channel Popen with PAMs separates drug actions in measures of network activity and neuroprotection. These PAM-induced differences emerge only under conditions of dynamic depolarization, not during steady-state agonist presentation or at constant voltages near rest. Our results suggest that regions of the brain that exhibit strong endogenous modulation of NMDA receptors and/or particularly strong activity patterns may be a focus for understanding differences between the drugs in vivo.
Previous work is equivocal on the point of whether blocker IC50 is affected by channel Popen, as manipulated with agonist concentration (Chen and Lipton, 1997; Gilling et al., 2007). As an uncompetitive antagonist, memantine requires channel opening for binding. While increasing agonist concentration certainly alters blocker access to the channel and accelerates kinetics (Chen and Lipton, 1997), the effect on steady-state block is less clear. A simple model of trapping block (Blanpied et al., 1997; Emnett et al., 2013) leads to a predicted shift in blocker IC50 from 0.27 to 0.11 μM with a change in agonist concentration from EC50 to EC90 (Supporting Information Fig. S1), in agreement with early work (Chen and Lipton, 1997). However, the model may not include complexities important for blocker action. Later work failed to find any effect of agonist concentration on blocker IC50 (Gilling et al., 2007). Using a different approach by manipulating channel behaviour with PAMs, we also found that IC50 concentrations are unaffected even up to a threefold increase in NMDA current (Figure 1). In addition, oxysterols accelerated kinetics of channel block, in agreement with previous work using agonist concentration to manipulate channel Popen (Chen and Lipton, 1997; Gilling et al., 2007; 2009). Because our approach employed a baseline of relatively high agonist concentration, our results reduce the chances of underestimating steady-state block at low agonist concentrations resulting from extremely slow blocker onset kinetics (Gilling et al., 2007; 2009).
The present work demonstrates different mechanisms for two lipophilic PAMs. We previously demonstrated that SGE-201 and PS exhibited additive potentiation at saturating modulator concentrations, but SGE-201 and the endogenous oxysterol 24S-hydroxycholesterol occluded each other (Paul et al., 2013). This implies different NMDA receptor sites for PS and oxysterols. In addition, with prolonged agonist presentation, PS exhibited mixed potentiation and inhibition. It is also a potent antagonist at GABAA receptors, and it has pre-synaptic effects at some synapses (Park-Chung et al., 1999; Shen et al., 2000; Eisenman et al., 2003; Horak et al., 2004; Zamudio-Bulcock and Valenzuela, 2011). Despite their biophysical differences at NMDA receptors, the two PAMs had a similar effect on NMDA receptor trapping blockers.
Because of its specificity for NMDA receptors and its purer PAM activity, SGE-201 is a better choice than PS to experimentally address the interaction between PAMs and NMDA receptor blockers during spontaneous network activity and hypoxic damage. Our experiments in these two paradigms revealed that PAM activity alone, while holding depolarization constant, exposed differences between memantine and ketamine that were normally only evident with biophysical manipulations, such as prolonged voltage pulses (Emnett et al., 2013). In the case of MEAs, this implied that normal synaptic depolarization was sufficient to expose such differences, but low basal channel Popen prevented the difference from emerging. Although channel Popen is increased by temperature, the effect alone is apparently not sufficiently strong to reveal the differences between memantine and ketamine in MEA recordings and toxicity assays, both of which were performed at 37°. As blocker access to its binding site in the channel pore increases further, voltage-dependent differences are revealed. In both of our paradigms, ketamine consistently had greater effect than memantine, including in excitotoxic neuroprotection. This is surprising given memantine's role as a clinical neuroprotectant, but consistent with our previous findings (Emnett et al., 2013). Memantine's primary clinical use is in Alzheimer's disease, where NMDA hyperfunction is likely to have only a limited role in damage (Newcomer et al., 2000). Because toxicity in Alzheimer's disease probably involves multiple mechanisms (Swomley et al., 2014), memantine's neuroprotective effect in Alzheimer's disease could be unrelated to NMDA receptor function. In preliminary experiments, we tried to examine the effect of memantine and ketamine on cell death induced by Aβ-oligomers, but we were unable to demonstrate toxicity under our culture conditions, using incubation for 48 h in 0.5–1.0 μM Aβ-oligomer (data not shown).
We previously demonstrated that a difference between memantine and ketamine was detectable during prolonged pathological depolarization during oxygen–glucose deprivation (Emnett et al., 2013). Here we show, in MEAs, that neuronal activity alone can also reach levels that expose drug differences, even in the absence of added PAM. We found that a twofold increase in spiking from baseline in the presence of CYZ differentiated the blockers. Interestingly, CYZ was also associated with weaker suppression of network activity by both blockers, in comparison with blocker effects in the presence of SGE-201 (Figures 7). CYZ, which prolongs AMPA receptor EPSCs (Partin et al., 1993), could result in a stronger envelope of subthreshold depolarization or intense bursting not evident in ASDR measures. Either explanation could explain the reduced effectiveness of the voltage-dependent blockers. Consistent with the former explanation, we previously showed that EPSP prolongation (achieved with voltage-clamp waveforms) is predicted to expose biophysical differences between memantine and ketamine (Emnett et al., 2013).
A recent study showed ketamine blocks spontaneous NMDA receptor miniature EPSCS more than memantine, but only in the presence of physiological magnesium (Gideons et al., 2014). This recent study further extends the idea that blockade of spontaneous transmission underlies ketamine's antidepressant behavioural effect (Autry et al., 2011; Kavalali and Monteggia, 2012). However, the basis for selective Mg2+-dependent reduction in memantine's effect is unclear as Mg2+ shifts the effects of both channel blockers similarly (Kotermanski and Johnson, 2009a). In the present work, MEA and toxicity studies were performed in Mg2+-containing solutions, and yet we were unable to discern a difference between the blockers during basal, non-potentiated, activity (Figures 6 and 8). This suggests that any differential effects of the drugs on spontaneous, quantal, synaptic transmission do not strongly influence network activity under our conditions, even when physiological levels of Mg2+ are present. On the other hand, differences among drugs on spontaneous transmission evident in the Gideons et al. study could have effects that were undetected in the time frame of our experiments. As it stands, our results offer an alternative explanation for the different effects of memantine and ketamine. Of course, we cannot exclude the possibility that both explanations converge in vivo.
There are a variety of factors that can influence NMDA receptor channel Popen and thereby NMDA receptor blocker actions in vivo. Different receptor subunits (GluN2A-D) are known to have different open probabilities and could influence NMDA receptor blocker effects given different regional expression patterns of the subunits (Monyer et al., 1994). Additionally, co-agonist (glycine, D-serine) concentrations are potentially subject to modulation (Papouin et al., 2012). Locally or regionally varied levels of co-agonist could modulate effects of channel blockers such as memantine and ketamine (Emnett et al., 2013). Zn2+, pH, and arachidonic acid are other allosteric modulators that could vary and influence the cellular effects of NMDA receptor channel blockers. PS has been investigated for many years as a possibly physiologically relevant PAM (Majewska and Schwartz, 1987; Schumacher et al., 2008). However, its concentrations in situ are unlikely to reach those that affect NMDA receptor function (Liere et al., 2004).
In the present work, we focused on a new class of PAM. The experimental tractability and selectivity of SGE-201 allowed us to provide proof of principle that allosteric NMDA receptor modulation can directly affect NMDA receptor blocker actions during dynamic network function. This proof was not previously forthcoming because other PAMS, such as PS, have notable off-target effects. 24S-hydroxycholesterol is quite potent and, endogenously, may achieve local concentrations in mature tissue that modulate NMDA receptor function (McDonald et al., 2007). Tests to date suggest that this class of modulator may be much more selective for NMDA receptors than the modulators cited earlier (Paul et al., 2013).
24S-hydroxycholesterol is produced endogenously by cholesterol 24-hydroxylase (CYP46A1). Studies of transcript and protein distribution reveal that the enzyme is neuron specific. Despite the high cholesterol content of myelin, it is not expressed in oligodendrocytes or other support cells, and its expression is mainly restricted to somatodendritic (post-synaptic) regions of principal cells and only some interneuron classes. Labelling varies regionally (Lund et al., 1999; Lein et al., 2007; Ramirez et al., 2008). For instance, within the hippocampal formation, CA1 pyramidal neurons show stronger protein expression than dentate granule cells (Ramirez et al., 2008). Therefore, control of NMDA receptor function by endogenous oxysterol may vary by region and by cell type within a region. Together with the other classes of modulators already discussed, there could be a complicated system of controls over NMDA receptor gating that varies by brain area. Future work could focus on these regional differences to better understand differences between memantine and ketamine in vivo.
In conclusion, if the pharmacodynamics of NMDA receptor channel blockers contributes to their clinical differences, we propose that endogenous PAMs could contribute to such differences. Modulators could include, but are not limited to the novel modulator 24S-hydroxycholesterol. In the presence of PAMs, blocker differences are more obvious in dynamic networks, but not under steady-state conditions. Either PAM actions combined with basal depolarization/activity or strong depolarization/activity alone are sufficient to expose differences between blockers.
Acknowledgments
This study was supported by the National Institutes of Health grants R01MH078823, RO1MH101874, R01MH077791, R01AA017413 and T32GM008151-27 and a gift from the Bantly Foundation. GluN1 and GluN2A subunits were gifts from Jon Johnson, University of Pittsburgh. We are grateful to Ann Benz for culture support and to lab members for discussion.
Glossary
Abbreviations
- ASDR
array-wide spike detection rate
- D-APV
D-2-amino-5-phosphonovalerate
- EPSC
excitatory post-synaptic current
- EPSP
excitatory post-synaptic potential
- MEA
multi-electrode array
- NBQX
2,3-dihydroxy-6-nitro-7-sulphonyl-benzo[f]quinoxaline
- PAM
positive allosteric modulator
- PI
propidium iodide
- Popen
open probability
- PS
pregnenolone sulphate
- SGE-201
analogue of 24S-hydroxycholesterol
- TTX
tetrodotoxin
Author contributions
C. E., L. N. E. and S. M. designed studies. C. E., J. M. and A. T. performed studies. C. E., L. N. E., A. T. and S. M. analysed the data. C. E. and S. M. drafted the paper. J. J. D. and S. M. P. provided SGE-201. C. E., L. N. E., J. M., A. T., J. D., S. M. P., C. F. Z. and S. M. edited the paper.
Conflict of interest
S. M. P. and J. J. D. are employees of Sage Therapeutics, which has intellectual property interest in SGE-201. C. F. Z. is a consultant to Sage Therapeutics.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Predicted effects of agonist concentration on blocker IC50 from a simple trapping block model. The model is taken from previous work (Blanpied et al., 1997. J Neurophysiol 77: 309–323). A represents agonist, and B represents blocker. EC50 and EC90 concentrations of agonist were chosen to simulate the approximately twofold increase in Popen typically observed with SGE-201 in the present work. Simulations were performed with NEURON (Carnevale and Hines 2006. The Neuron Book. Cambridge University Press). Control was taken as the probability of A2R* during application of a simulated agonist (30 μM and 300 μM) in the absence of blocker (kon = 0). Rate constants were the following, from Emnett et al. 2013. Mol Pharmacol 84:935: ka+ = 2 μM−1 s−1, ka− = 40 s−1, β = 5.2 s−1, α = 130 s−1, kon = 2 μM−1 s−1, koff = 7.6 s−1, β′ = 0.3 s−1, α′ = 35.4 s−1, ka+′ = 0.2 μM−1 s−1, ka-′ = 0.02 s−1.
References
- Aan Het Rot M, Zarate CA, Jr, Charney DS, Mathew SJ. Ketamine for depression: where do we go from here? Biol Psychiatry. 2012;72:537–547. doi: 10.1016/j.biopsych.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ligand-Gated Ion Channels. Br J Pharmacol. 2013;170:1582–1606. doi: 10.1111/bph.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu CS, Ramanathan M. Pre-ischemic treatment with memantine reversed the neurochemical and behavioural parameters but not energy metabolites in middle cerebral artery occluded rats. Pharmacol Biochem Behav. 2009;92:424–432. doi: 10.1016/j.pbb.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Richerson GB, Stevens CF. Origin of variability in quantal size in cultured hippocampal neurons and hippocampal slices. Proc Natl Acad Sci USA. 1990;87:5359–5362. doi: 10.1073/pnas.87.14.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpied TA, Boeckman FA, Aizenman E, Johnson JW. Trapping channel block of NMDA-activated responses by amantadine and memantine. J Neurophysiol. 1997;77:309–323. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- Chen H, Pellegrini J, Aggarwal S, Lei S, Warach S, Jensen F, et al. Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci. 1992;12:4427–4436. doi: 10.1523/JNEUROSCI.12-11-04427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Lipton SA. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J Physiol. 1997;499(Pt 1):27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HSV, Wang YF, Rayudu PV, Edgecomb P, Neill JC, Segal MM, et al. Neuroprotective concentrations of the N-methyl-aspartate open-channel blocker memantine are effective without cytoplasmic vacuolation following post-ischemic administration and do not block maze learning or long-term potentiation. Neuroscience. 1998;86:1121–1132. doi: 10.1016/s0306-4522(98)00163-8. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Junker V, Kremers W, Thal S, Plesnila N, Krieglstein J. Combination therapy in ischemic stroke: synergistic neuroprotective effects of memantine and clenbuterol. Stroke. 2004;35:1197–1202. doi: 10.1161/01.STR.0000125855.17686.6d. [DOI] [PubMed] [Google Scholar]
- Deutsch SI, Mastropaolo J, Hitri A. GABA-active steroids: endogenous modulators of GABA-gated chloride ion conductance. Clin Neuropharmacol. 1992;15:352–364. [PubMed] [Google Scholar]
- Eisenman LN, He Y, Fields C, Zorumski CF, Mennerick S. Activation-dependent properties of pregnenolone sulfate inhibition of GABAA receptor-mediated current. J Physiol. 2003;550(Pt 3):679–691. doi: 10.1113/jphysiol.2003.043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emnett CM, Eisenman L, Taylor AM, Izumi Y, Zorumski CF, Mennerick SJ. Indistinguishable synaptic pharmacodynamics of the NMDAR channel blockers memantine and ketamine. Mol Pharmacol. 2013;84:935–947. doi: 10.1124/mol.113.089334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser DD, Hoehn K, Weiss S, MacVicar BA. Arachidonic acid inhibits sodium currents and synaptic transmission in cultured striatal neurons. Neuron. 1993;11:633–644. doi: 10.1016/0896-6273(93)90075-3. [DOI] [PubMed] [Google Scholar]
- Gideons ES, Kavalali ET, Monteggia LM. Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc Natl Acad Sci U S A. 2014;111:8649–8654. doi: 10.1073/pnas.1323920111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilling KE, Jatzke C, Parsons CG. Agonist concentration dependency of blocking kinetics but not equilibrium block of N-methyl-D-aspartate receptors by memantine. Neuropharmacology. 2007;53:415–420. doi: 10.1016/j.neuropharm.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Gilling KE, Jatzke C, Hechenberger M, Parsons CG. Potency, voltage-dependency, agonist concentration-dependency, blocking kinetics and partial untrapping of the uncompetitive N-methyl-D-aspartate (NMDA) channel blocker memantine at human NMDA (GluN1/GluN2A) receptors. Neuropharmacology. 2009;56:866–875. doi: 10.1016/j.neuropharm.2009.01.012. [DOI] [PubMed] [Google Scholar]
- Horak M, Vlcek K, Petrovic M, Chodounska H, Vyklicky L., Jr Molecular mechanism of pregnenolone sulfate action at NR1/NR2B receptors. J Neurosci. 2004;24:10318–10325. doi: 10.1523/JNEUROSCI.2099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE, Baughman RW. Primary culture of identified neurons from the visual cortex of postnatal rats. J Neurosci. 1986;6:3044–3060. doi: 10.1523/JNEUROSCI.06-10-03044.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Takahashi T. Mechanisms underlying presynaptic facilitatory effect of cyclothiazide at the calyx of Held of juvenile rats. J Physiol. 2001;533(Pt 2):423–431. doi: 10.1111/j.1469-7793.2001.0423a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karu K, Hornshaw M, Woffendin G, Bodin K, Hamberg M, Alvelius G, et al. Liquid chromatography–mass spectrometry utilizing multi-stage fragmentation for the identification of oxysterols. J Lipid Res. 2007;48:976–987. doi: 10.1194/jlr.M600497-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Monteggia LM. Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am J Psychiatry. 2012;169:1150–1156. doi: 10.1176/appi.ajp.2012.12040531. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotermanski SE, Johnson JW. Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer's drug memantine. J Neurosci. 2009a;29:2774–2779. doi: 10.1523/JNEUROSCI.3703-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotermanski SE, Wood JT, Johnson JW. Memantine binding to a superficial site on NMDA receptors contributes to partial trapping. J Physiol. 2009b;587:4589–4604. doi: 10.1113/jphysiol.2009.176297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotermanski SE, Johnson JW, Thiels E. Comparison of behavioral effects of the NMDA receptor channel blockers memantine and ketamine in rats. Pharmacol Biochem Behav. 2013;109:67–76. doi: 10.1016/j.pbb.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Liere P, Pianos A, Eychenne B, Cambourg A, Liu S, Griffiths W, et al. Novel lipoidal derivatives of pregnenolone and dehydroepiandrosterone and absence of their sulfated counterparts in rodent brain. J Lipid Res. 2004;45:2287–2302. doi: 10.1194/jlr.M400244-JLR200. [DOI] [PubMed] [Google Scholar]
- Linsenbardt AJ, Taylor A, Emnett CM, Doherty JJ, Krishnan K, Covey DF, et al. Different oxysterols have opposing actions at N-methyl-D-aspartate receptors. Neuropharmacology. 2014;85:232–242. doi: 10.1016/j.neuropharm.2014.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund EG, Guileyardo JM, Russell DW. cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci U S A. 1999;96:7238–7243. doi: 10.1073/pnas.96.13.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM, Russell DW. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem. 2003;278:22980–22988. doi: 10.1074/jbc.M303415200. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Schwartz RD. Pregnenolone-sulfate: an endogenous antagonist of the gamma-aminobutyric acid receptor complex in brain? Brain Res. 1987;404:355–360. doi: 10.1016/0006-8993(87)91394-1. [DOI] [PubMed] [Google Scholar]
- Marland S, Ellerton J, Andolfatto G, Strapazzon G, Thomassen O, Brandner B, et al. Ketamine: use in anesthesia. CNS Neurosci Ther. 2013;19:381–389. doi: 10.1111/cns.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JG, Thompson BM, McCrum EC, Russell DW. Extraction and analysis of sterols in biological matrices by high performance liquid chromatography electrospray ionization mass spectrometry. Methods Enzymol. 2007;432:145–170. doi: 10.1016/S0076-6879(07)32006-5. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Que J, Benz A, Zorumski CF. Passive and synaptic properties of hippocampal neurons grown in microcultures and in mass cultures. J Neurophysiol. 1995;73:320–332. doi: 10.1152/jn.1995.73.1.320. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Chisari M, Shu HJ, Taylor A, Vasek M, Eisenman LN, et al. Diverse voltage-sensitive dyes modulate GABAA receptor function. J Neurosci. 2010;30:2871–2879. doi: 10.1523/JNEUROSCI.5607-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170:1134–1142. doi: 10.1176/appi.ajp.2013.13030392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomer JW, Farber NB, Olney JW. NMDA receptor function, memory, and brain aging. Dialogues Clin Neurosci. 2000;2:219–232. doi: 10.31887/DCNS.2000.2.3/jnewcomer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papouin T, Ladepeche L, Ruel J, Sacchi S, Labasque M, Hanini M, et al. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150:633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- Park-Chung M, Malayev A, Purdy RH, Gibbs TT, Farb DH. Sulfated and unsulfated steroids modulate gamma-aminobutyric acidA receptor function through distinct sites. Brain Res. 1999;830:72–87. doi: 10.1016/s0006-8993(99)01381-5. [DOI] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A. Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin A. Neuron. 1993;11:1069–1082. doi: 10.1016/0896-6273(93)90220-l. [DOI] [PubMed] [Google Scholar]
- Paul SM, Doherty JJ, Robichaud AJ, Belfort GM, Chow BY, Hammond RS, et al. The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J Neurosci. 2013;33:17290–17300. doi: 10.1523/JNEUROSCI.2619-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Andersson S, Russell DW. Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J Comp Neurol. 2008;507:1676–1693. doi: 10.1002/cne.21605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Feltz A, Westbrook G. Synaptic NMDA receptor channels have a low open probability. J Neurosci. 1995;15:2788–2795. doi: 10.1523/JNEUROSCI.15-04-02788.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher M, Liere P, Akwa Y, Rajkowski K, Griffiths W, Bodin K, et al. Pregnenolone sulfate in the brain: a controversial neurosteroid. Neurochem Int. 2008;52:522–540. doi: 10.1016/j.neuint.2007.08.022. [DOI] [PubMed] [Google Scholar]
- Shen W, Mennerick S, Covey DF, Zorumski CF. Pregnenolone sulfate modulates inhibitory synaptic transmission by enhancing GABA(A) receptor desensitization. J Neurosci. 2000;20:3571–3579. doi: 10.1523/JNEUROSCI.20-10-03571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swomley AM, Forster S, Keeney JT, Triplett J, Zhang Z, Sultana R, et al. Abeta, oxidative stress in Alzheimer disease: evidence based on proteomics studies. Biochim Biophys Acta. 2014;1842:1248–1257. doi: 10.1016/j.bbadis.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291:317–324. doi: 10.1001/jama.291.3.317. [DOI] [PubMed] [Google Scholar]
- Tong G, Jahr CE. Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron. 1994;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- Wagenaar DA, Pine J, Potter SM. An extremely rich repertoire of bursting patterns during the development of cortical cultures. BMC Neurosci. 2006;7:11. doi: 10.1186/1471-2202-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroge CM, Hogins J, Eisenman L, Mennerick S. Synaptic NMDA receptors mediate hypoxic excitotoxic death. J Neurosci. 2012;32:6732–6742. doi: 10.1523/JNEUROSCI.6371-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu FS, Gibbs TT, Farb DH. Pregnenolone sulfate: a positive allosteric modulator at the N-methyl-D-aspartate receptor. Mol Pharmacol. 1991;40:333–336. [PubMed] [Google Scholar]
- Xia P, Chen H-S, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KA, Tang CM. Benzothiadiazides inhibit rapid glutamate receptor desensitization and enhance glutamatergic synaptic currents. J Neurosci. 1993;13:3904–3915. doi: 10.1523/JNEUROSCI.13-09-03904.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamudio-Bulcock PA, Valenzuela CF. Pregnenolone sulfate increases glutamate release at neonatal climbing fiber-to-Purkinje cell synapses. Neuroscience. 2011;175:24–36. doi: 10.1016/j.neuroscience.2010.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zar JH. Biostatistical Analysis. 4th edn. Upper Saddle River, NJ: Prentice-Hall, Inc; 1999. [Google Scholar]
- Zorumski CF, Yang J, Fischbach GD. Calcium-dependent, slow desensitization distinguishes different types of glutamate receptors. Cell Mol Neurobiol. 1989;9:95–104. doi: 10.1007/BF00711446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Predicted effects of agonist concentration on blocker IC50 from a simple trapping block model. The model is taken from previous work (Blanpied et al., 1997. J Neurophysiol 77: 309–323). A represents agonist, and B represents blocker. EC50 and EC90 concentrations of agonist were chosen to simulate the approximately twofold increase in Popen typically observed with SGE-201 in the present work. Simulations were performed with NEURON (Carnevale and Hines 2006. The Neuron Book. Cambridge University Press). Control was taken as the probability of A2R* during application of a simulated agonist (30 μM and 300 μM) in the absence of blocker (kon = 0). Rate constants were the following, from Emnett et al. 2013. Mol Pharmacol 84:935: ka+ = 2 μM−1 s−1, ka− = 40 s−1, β = 5.2 s−1, α = 130 s−1, kon = 2 μM−1 s−1, koff = 7.6 s−1, β′ = 0.3 s−1, α′ = 35.4 s−1, ka+′ = 0.2 μM−1 s−1, ka-′ = 0.02 s−1.