

Abstract
Background and Purpose
International asthma guidelines recommend that inhaled glucocorticoids be used as a monotherapy in all patients with mild to moderate disease because of their ability to suppress airways inflammation. Current evidence suggests that the therapeutic benefit of glucocorticoids is due to the transactivation and transrepression of anti-inflammatory and pro-inflammatory genes respectively. However, the extent to which clinically relevant glucocorticoids are equivalent in their ability to modulate gene expression is unclear.
Experimental Approach
A pharmacodynamics investigation of glucocorticoid receptor (GR)-mediated gene transactivation in BEAS-2B human airway epithelial cells was performed using a glucocorticoid response element luciferase reporter coupled with an analysis of glucocorticoid-inducible genes encoding proteins with anti-inflammatory and adverse-effect potential.
Key Results
Using transactivation as a functionally relevant output, a given glucocorticoid displayed a unique, gene expression ‘fingerprint’ where intrinsic efficacy and GR density were essential determinants. We showed that depending on the gene selected for analysis, a given glucocorticoid can behave as an antagonist, partial agonist, full agonist or even ‘super agonist’. In the likely event that different, tissue-dependent gene expression profiles are reproduced in vivo, then the anti-inflammatory and adverse-effect potential of many glucocorticoids currently available as asthma therapeutics may not be equivalent.
Conclusions and Implications
The generation of gene expression ‘fingerprints’ in target and off-target human tissues could assist the rational design of GR agonists with improved therapeutic ratios. This approach could identify compounds that are useful in the management of severe asthma and other inflammatory disorders where systemic exposure is desirable.
Tables of Links
| TARGETS | |
|---|---|
| Nuclear hormone receptorsa | Enzymesb |
| Glucocorticoid receptor (GR) | 11β-hydroxysteroid dehydrogenase-1 (11β-HSD-1) |
| Pyruvate dehydrogenase kinase 4 (PDK4) |
| LIGANDS | |
|---|---|
| Carbenoxolone | Fluticasone furoate (FF) |
| Desisobutyrylciclesonide (DC) | Hydrocortisone (HC; cortisol) |
| Dexamethasone (Dex) | Mifepristone |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Introduction
Synthetic glucocorticoids, commonly referred to as corticosteroids, are structural analogues of the natural hormone hydrocortisone (HC; aka cortisol) and are remarkably effective in the treatment of a plethora of inflammatory, allergic and immunological disorders in adults and children. The primary indication for inhaled corticosteroids (ICS) is the management of asthma where they remain a mainstay in subjects with mild to moderate disease (see Newton et al., 2010 and references therein). Indeed, eosinophilic airways inflammation is a prominent pathogenic feature in many individuals with asthma and is particularly sensitive to the remedial actions of ICS (Brown, 1958; Green et al., 2002). It is generally believed that glucocorticoids suppress inflammation by inhibiting the expression of pro-inflammatory genes (Newton et al., 2010). Two general mechanisms have been described. Transrepression is, arguably, the most widely recognized of these in which the agonist-bound glucocorticoid receptor (GR) hinders the ability of certain transcription factors, such as NF-κB and AP-1, to induce pro-inflammatory gene transcription. Transrepression, via a direct interaction of the agonist-bound GR to negative glucocorticoid response elements (GREs), was also reported recently (Surjit et al., 2011). However, in simple model systems, glucocorticoids are often only partial inhibitors of gene transcription, implying that processes in addition to transrepression must also operate to explain their anti-inflammatory effects in bona fide models of inflammation (Clark, 2007; Newton et al., 2010). Indeed, compelling evidence has accumulated since the year 2000 that the induction (transactivation) of genes, many encoding proteins with anti-inflammatory potential, also constitutes a major mechanism of glucocorticoid action (Newton, 2000; Clark, 2007; Newton and Holden, 2007; Newton et al., 2010; Clark and Belvisi, 2011; King et al., 2013).
Recently, we reviewed the concept that the induction of a glucocorticoid-inducible gene represents a distinct functional response and is defined by a unique concentration–effect (E/[A] ) relationship (Newton et al., 2010). Accordingly, a given tissue should respond to a glucocorticoid with a particular gene expression ‘fingerprint’ that is governed by several tissue- and agonist-dependent factors. These include the structure of gene promoters, GR number, the complement and abundance of various obligatory co-factors, epigenetic modifications and the affinity (KA) and intrinsic efficacy of the GR agonist of interest (Zhang et al., 2007; Simons, 2008; 2010,; Newton et al., 2010). It follows that a change in the concentration of one or more tissue-dependent factors may alter the E/[A] relationship that describes the induction of a particular gene by a particular glucocorticoid. This may be reflected by a change in glucocorticoid potency and/or the degree of agonism that can be produced in a given tissue (Zhang et al., 2007; Simons, 2008; 2010,; Newton et al., 2010).
Despite these predictions, which are based on classical receptor theory wherein the binding of an agonist to its cognate receptor is assumed to follow the law of mass action, the pharmacodynamics of drugs that bind to nuclear hormone receptors, including GR, is little explored (cf. GPCRs). Indeed, the extent to which the gene transactivation potential of glucocorticoids used in clinical practice and those in development are equivalent in a given target tissue is unknown. Traditionally, high throughput screening methods are used to identify glucocorticoids for lead optimization. However, these assays are often conducted in cell-based over-expression systems that do not give accurate information on potential glucocorticoid activity in target tissues relevant to the disease of interest or those responsible for side effects.
Herein, we have compared the pharmacodynamics of a panel of seven glucocorticoids to promote gene expression in the BEAS-2B human airway epithelial cell line (see Figure 1). These include the clinically relevant compounds fluticasone furoate (FF), dexamethasone (Dex) and desisobutyrylciclesonide (DC), which are assumed to be full GR agonists, and GW 870086X (GW), a novel ligand that displays partial agonism on a number of functional outputs (Uings et al., 2013). For completeness, the naturally occurring agonist, HC, was also included in the analysis and two purported GR receptor antagonists, mifepristone (Mif; Gagne et al., 1985) and Org 34517 (Org; Peeters et al., 2004). To achieve this objective, a simple GRE luciferase reporter construct was used as a model system coupled with an analysis of a panel of glucocorticoid-inducible genes encoding proteins with both anti-inflammatory and adverse-effect potential. While glucocorticoids affect many inflammatory and immune cells to produce therapeutic benefits, airway epithelial cells were used in this study because they are believed to play a profound pathogenic role in asthma and are a primary target for ICS (reviewed in Proud and Leigh, 2011).
Figure 1.
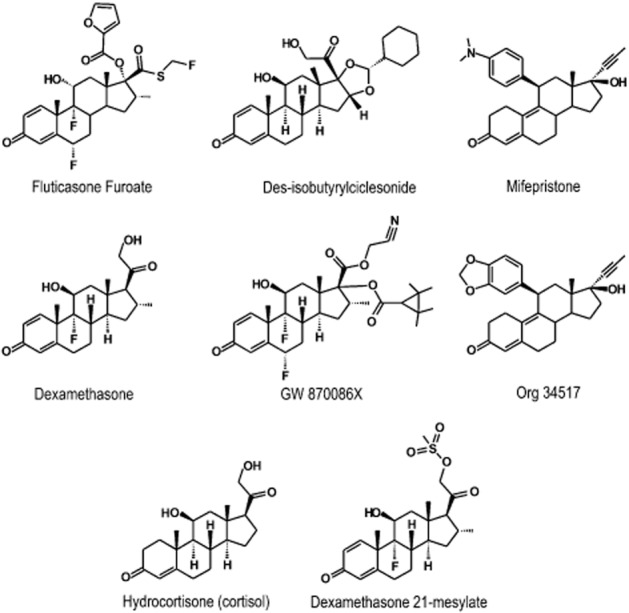
Chemical structures of the GR ligands used in this study.
Methods
Generation of a 2 × GRE reporter
Stable transfection was used to generate a simple GRE reporter cell line as described previously (Chivers et al., 2004). The construct, pGL3.neo.TATA.2GRE, contains two copies of a consensus simple GRE site (sense strand, 5′-TGT ACA GGA TGT TCT-3′) positioned upstream of a minimal β-globin promoter driving a luciferase gene and a separate neomycin gene to confer resistance to geneticin. BEAS-2B cells at ∼70% confluence in T162 flasks were transfected with 8 μg of plasmid DNA and 20 μL using Tfx-50 (Promega, Madison, WI, USA). After 24 h, geneticin (100 μg·mL−1) was added until foci of stable transfectants appeared, which were harvested to create heterogeneous populations of cells in which the site of integration was randomized.
Culture of 2 × GRE BEAS-2B reporter cells
Cells were cultured for 2 days under a 5% CO2/air atmosphere at 37°C in 24-well tissue culture plates containing DMEM/F12 (Invitrogen, Burlington, ON, Canada) supplemented with 10% FBS (Invitrogen), L-glutamine (2.5 mM) and sodium bicarbonate (0.15% v v−1). The cells were then growth-arrested for 24 h in serum-free medium (SFM). At this time, cultures were confluent and were processed for luciferase measurements or gene expression as described later.
Treatment of 2 × GRE BEAS-2B cells
Confluent, 2 × GRE BEAS-2B reporter cells (5 × 104 per well) in SFM were treated with FF, DC, Dex, GW, HC, Mif or Org (Figure 1) as indicated and incubated at 37°C under a 5% CO2 atmosphere. In some experiments, 2 × GRE BEAS-2B reporter cells were incubated for 6 h with Dex, GW or DEX and GW in combination as indicated. Alternatively, cells were pretreated with Org (60 min; Peeters et al., 2004) or the alkylating agent, Dex 21-mesylate (30 min; Dex-Mes; Simons and Thompson, 1981; Figure 1) at the concentration(s) indicated in the text. When Dex-Mes was used, cells were washed with SFM after the pre-incubation period, allowed to recover for 60 min before being exposed to glucocorticoid for 6 h. Total RNA was extracted and the expression of a panel of glucocorticoid-inducible genes was assessed (Table 2013a). Alternatively, 2 × GRE BEAS-2B cells were lysed in 100 μL 1 × firefly luciferase lysis buffer (Biotium, Hayward, CA, USA) and luciferase activity was measured using a 20/20n Luminometer (Turner Biosystems, Maddison, WI, USA) according to the manufacturer's instructions. Data are expressed as fold induction of luciferase activity relative to unstimulated cells.
Table 1.
Primer pairs for real-time PCR
| Gene | Oligonucleotide | Accession Number(s) |
|---|---|---|
| PDK4 | NM_002612.3 | |
| Forward | 5′-GCT GTC CAT GAA GCA GCT ACT G-3′ | |
| Reverse | 5′-CGC AAA AAT GCA AAA GAA GTT CT-3′ | |
| p57kip2 (CDKN1C) | NM_000076.2, NM_001122630.1, NM_001122631.1 | |
| Forward | 5′-CTG TCC GGG CCT CTG ATC T-3′ | |
| Reverse | 5′-CAT CGC CCG ACG ACT TCT-3′ | |
| GILZ (TSC22D3) | NM_198057.2, NM_004089.3, NM_001015881.1 | |
| Forward | 5′- TGG CCA TAG ACA ACA AGA TCG A-3′ | |
| Reverse | 5′- CAC AGC ATA CAT CAG ATG ATT CTT CA-3′ | |
| CRISPLD2 | NM_031476.3 | |
| Forward | 5′-CAA ACC TTC CAG CTC ATT CAT G-3′ | |
| Reverse | 5′-GGT CGT GTA GCA GTC CAA ATC C-3′ | |
| GAPDH | NM_002046.4, NM_001256799.1 | |
| Forward | 5′-ATG GAA ATC CCA TCA CCA TCT T-3′ | |
| Reverse | 5′-CAG CAT CGC CCC ACT TG-3′ | |
Forward and reverse primers for each gene are listed. Common genes symbols are shown, and where appropriate, official HUGO gene symbols are given in parentheses. Generic primers were used for genes encoding multiple isoforms.
RNA isolation, reverse transcription and real-time PCR
Total RNA was extracted from 2 × GRE BEAS-2B reporter cells using RNeasy Mini Kits (Qiagen, Inc., Mississauga, ON, Canada) and was reverse transcribed using a qscript cDNA synthesis kit according to the manufacturer's instructions (Quanta Biosciences, Gaithersburg, MD, USA). Real-time PCR analysis of cDNA was performed using the primer sequences shown in Table 2013a (designed using Primer Express® software, Applied Biosystems Inc., Foster City, CA, USA) that amplify glucocorticoid-inducible leucine zipper (GILZ; HUGO gene name: TGF–β-stimulated clone 22, domain family member 3 [TSC22D3] ), kinase inhibitor protein 2 of 57 kDa (p57kip2; HUGO gene name: cyclin-dependent kinase inhibitor 1C [CDKN1C] ), cysteine-rich secretory protein LCCL (Limulus clotting factor C, Cochlin, Lgl1) domain-containing 2 (CRISPLD2) and pyruvate dehydrogenase kinase 4 (PDK4). These reactions were performed using an ABI StepOnePlus® instrument (Applied Biosystems Inc.) on 2.5 μL of cDNA in 10 μL reactions using Fast SYBR® Green chemistry (Invitrogen) according to the manufacturer's guidelines. Relative gene expression levels were determined from a cDNA standard curve that was analysed simultaneously with the test samples and are presented as a ratio to GAPDH, whose expression was not affected by any of the glucocorticoids used in this study. Amplification conditions were: 95°C, 20 s; followed by 40 cycles of: 95°C, 3 s; 60°C, 30 s. Dissociation (melt) curves (95°C, 15 s; 60°C, 1 min; 95°C, 15 s) were constructed to confirm primer specificity.
Curve fitting
Monophasic agonist E/[A] curves were fitted by least squares, non-linear iterative regression to the following form of the Hill equation (Prism 4®, GraphPad Software, Inc., San Diego, CA, USA; Motulsky and Christopoulos, 2003):
| 1 |
where E is the effect, Emin and Emax are the lower and upper asymptote (i.e. the basal response and maximum agonist-induced response, respectively), p[A] is the negative log molar concentration of agonist, p[A]50 is a location parameter equal to the negative log molar concentration of agonist producing (Emax − Emin)/2 and n is the gradient of the E/[A] curve at the p[A]50 level.
Determination of partial agonist and antagonist equilibrium dissociation constants
Antagonist affinity values (KB) were determined by least squares, non-linear regression using a modification of the Hill and Gaddum/Schild equations (Waud et al., 1978). Concentration–effect curves were constructed to glucocorticoids in cells pretreated (60 min) with vehicle or the GR antagonist, Org (Peeters et al., 2004) at concentrations of 10, 30 and 100 nM. Each ‘family’ of E/[A] curves were then fitted simultaneously to Eq. 2013b. Thus,
 |
2 |
where [A] and [B] are the molar concentration of full agonist and partial agonist/antagonist, respectively, S is the Schild slope factor, which indicates the nature of antagonism, and pA2 is the affinity of the partial agonist and antagonist when S = 1, which is equivalent to the pKA or pKB respectively. To determine whether S deviated significantly from unity, the entire family of E/[A] curves that made up an individual experiment was fitted globally to Eq. 2013b under two conditions: one where S was constrained to a constant equal to 1 and the other where it was a shared value for all datasets. The F-test was applied to determine the equation that gave the best fit, which was used for the analysis.
In experiments designed to estimate the affinity (KA) of a partial agonist, E/[A] curves were constructed to a reference ‘full’ agonist (Dex in the present study) in the absence and presence of fixed concentrations of the partial agonist of interest, which was added concurrently. Global Schild analysis was then performed (Eq. 2013b ) under conditions where the lower asymptotes of the Dex E/[A] curves in the presence of partial agonist were unconstrained because of the inherent ability of the partial agonist to produce response. It has been shown that when the relationship between stimulus and response is rectangular hyperbolic, the experimentally derived affinity is under estimated by a factor of KA/(1 − αp), where αp is the intrinsic activity of the partial agonist. Thus, when appropriate, affinity estimates were corrected for this fact.
Determination of agonist equilibrium dissociation constants by controlled, fractional GR inactivation
The KAs of FF and Dex in 2 × GRE BEAS-2B reporter cells were estimated by ‘irreversibly’ inactivating a fraction of the total functional GR population with the alkylating agent, Dex-Mes (Simons and Thompson, 1981) according to Furchgott (1966). Agonist E/[A] curves were generated in cells treated (for 30 min) with vehicle or Dex-Mes at the concentrations indicated. Each set of E/[A] curves was then fitted simultaneously to the operational model of agonism (Eq. 1992), which describes a theoretical relationship between pharmacological effect (E) and agonist concentration (Black and Leff, 1983). Algebraically,
| 3 |
where Em is the theoretical maximum response of the tissue, [A] is the agonist concentration, n is the slope of the relationship between the concentration of agonist-receptor ( [AR] ) complexes and response (to account for E/[A] curves with gradients that are not equal to a value of 1) and τ is the operational efficacy of the agonist; this is the ratio of the total functional receptor concentration [Rt] to [AR] required to produce half-maximal effect (Leff et al., 1990). In these analyses a common value of Em, KA and n is assumed (Black and Leff, 1983; Leff et al., 1990). Only τ, which at submaximal responses decreases proportionally with the remaining fraction of non-inactivated receptors, was allowed to vary between individual E/[A] curves (Black and Leff, 1983; Leff et al., 1990). Thus, for each experiment a single estimate of Em, n and KA was calculated as well as the operational efficacy of agonist before (τ) and after receptor (τ′) inactivation. The percentage of functionally active receptors (q) remaining after treatment of cells with Dex-Mes is given by (τ′/τ) × 100.
Determination of partial agonist equilibrium dissociation constants by the comparative method
The KA of partial GR agonists was also enumerated by operational model fitting (Black and Leff, 1983) using the comparative method (Barlow et al., 1967) in which E/[A] curves of the partial agonist and full agonist are compared. Each pair of E/[A] curves (i.e. the full agonist and partial agonist E/[A] curves) were fitted simultaneously to Eqs. (1) and (3), respectively (Motulsky and Christopoulos, 2003), which yielded an estimate of KA and τ for the partial agonist, the Em and n of the tissue and a p[A]50 for each full agonist curve (Leff et al., 1990).
Determination of receptor reserve
Receptor occupancy–response curves were constructed to Dex, GW and FF using their KAs determined by Schild analysis, or receptor inactivation as indicated. At each concentration of agonist and, therefore, at each level of response, fractional GR occupancy [i.e. the ratio of agonist-occupied GR (RA) to Rt] in control cells was determined (Furchgott, 1966) assuming the binding of ligand to GR was a non-cooperative (n = 1) process. Thus,
| 4 |
Drugs and reagents
Des-isobutyrylciclesonide (2′(R)-cyclohexyl-11β,21-dihydroxy-16β-H-dioxolo [5′,4′:16, 17]pregna-1,4-diene-3,20-dione) and Org 34517 (11β-(1,3-benzodioxolo)-17β-hydroxy-17-(1-propynyl)-oestra-4,9-dien-3-one) were from Nycomed (Konstanz, Germany) and Organon Laboratories (Oss, The Netherlands) respectively. GW 870086X (6α,9α-difluoro-11β-hydroxy-16α-methyl-3-oxo-17α-(2,2,3,3-tetramethylcyclopropylcarbonyl)-oxoandrosta-1,4-diene-17β-carboxylic acid cyano-methylester) and FF (GW 685698X; 6α,9α-difluoro-17α-[ (2-furanyl carbonyl)oxy]-11β-hydroxy-16α-methyl-3-oxoandrosta-1,4-diene-17β-carbothioic acid S-fluoro methyl ester) were from GlaxoSmithKline (Stevenage, Hertfordshire, UK). Mif (RU 38486; 11β-[4-(dimethylamino)phenyl]-17β-hydroxy-17-(1-propynyl)-oestra-4,9-dien-3-one), Dex and Dex 21-mesylate were purchased from Steraloids (Newport, RI, USA). Carbenoxolone and HC were from Tocris (Bristol, UK). Glucocorticoids were dissolved in DMSO and diluted to the desired working concentrations in culture medium. The highest concentration of DMSO used in these experiments never exceeded 0.2% (v v−1) and did not affect any output measured.
Statistics
Data points, bars and values in the text and figure legends represent the mean ± SEM mean of n independent determinations. Data were analysed by Student's two-tailed t-test or repeated measures one-way anova followed, when appropriate, by Tukey's multiple comparison test. In experiments where glucocorticoid-induced gene expression was examined, all statistical analyses were performed on untransformed data. The null hypothesis was rejected when P < 0.05.
Results
Kinetics of GRE-dependent transcription
GR agonists were examined for their ability to increase luciferase activity in 2 × GRE BEAS-2B reporter cells. At a maximally effective concentration determined from preliminary experiments, FF (100 nM), Dex (1 μM), DC (100 nM) and GW (1 μM) promoted GRE-dependent transcription in a time-dependent manner (Supporting Information Fig. S1a). In each case, the time to achieve half maximum response (t1/2) was ∼3 h; luciferase activity peaked at 6 h and then waned for all agonists except GW. Irrespective of the concentration tested, maximum luciferase activity was reached 6–8 h after treatment (see Supporting Information Fig. S1b, c for FF and DC data, respectively) although the absolute, maximum fold inductions were not equal (FF > Dex > DC > GW). Based on these results, luciferase activity in all further experiments was measured in cells 6 h after exposure to GR agonist.
Comparative effects of GR ligands on GRE-dependent transcription
Concentration–effect curves were constructed to FF, Dex, DC and GW to compare their relative abilities to drive GRE-dependent transcription; two other ligands, Mif and Org, that are reported to be GR antagonists (Gagne et al., 1985; Peeters et al., 2004) were also examined (Table 2013b). Five of these ligands induced luciferase activity with a rank order of potency of FF > DC > Mif > Dex ≥ GW (Figure 2A; Table 2013b). However, the maximum response between glucocorticoids differed significantly. Relative to Dex, which was selected as a reference agonist and assigned an intrinsic activity (α) value of 1, DC, GW and Mif displayed increasing degrees of partial agonism in this system (Figure 2A; Table 2013b). In contrast, the intrinsic activity of FF was 1.21 indicating that Dex was, in fact, a partial agonist (α = 0.83 relative to FF). Org was inactive at all concentrations tested (Table 2013b).
Figure 2.
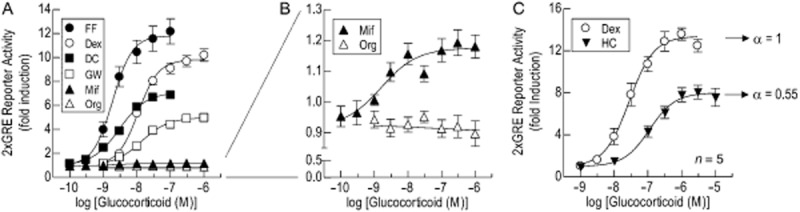
Comparative effects of a panel of GR ligands on GRE-dependent transcription. Panel A: 2 × GRE BEAS-2B reporter cells were exposed to FF (100 pM to 100 nM), Dex (1 nM to 1 μM), DC (100 pM to 100 nM), GW (1 nM to 1 μM), Mif (100 pM to 1 μM) or Org (1 nM to 1 μM). In panel B, the y axis has been expanded to illustrate the very low intrinsic activity of Mif. Panel C: E/[A] curves were constructed to Dex (1 nM to 1 μM) and HC (1 nM to 10 μM). At 6 h cells were harvested, luciferase activity was determined and E/[A] curves were constructed. Data points represent the mean ± SEM of n independent determinations (see Table 2013b).
Table 2.
Potency, Emax and intrinsic activity values of a panel of GR ligands for promoting GRE-dependent transcription in 2 × GRE BEAS-2B reporter cells
| Glucocorticoid | N | p[A]50 (M)a | Emaxa (fold induction) | Intrinsic activitya,b (α) |
|---|---|---|---|---|
| Dexc | 31 | 7.95 ± 0.03 | 9.92 ± 0.5 | 1.00 |
| FF | 13 | 8.72 ± 0.10 | 11.8 ± 1.0 | 1.21 |
| Des-ciclesonide | 25 | 8.61 ± 0.09 | 6.99 ± 0.3 | 0.55 |
| HC | 5 | 6.95 ± 0.08 | d | 0.55 |
| GW 870086X | 25 | 7.96 ± 0.07 | 4.94 ± 0.3 | 0.36 |
| Mif | 7 | 8.04 ± 0.22 | 1.26 ± 0.1 | 0.02 |
| Org 34517 | 7 | inactive | 0.89 ± 0.1 | 0 |
Parameters calculated from the graphs shown in Figure 2.
Intrinsic activity (α) = Emax(GR ligand − 1)/Emax(Dex − 1)
Dex selected as a reference agonist (α = 1).
In view of these differences in intrinsic activity, a separate experiment was conducted to compare the ability of HC, the main endogenous GR agonist, to promote GRE-dependent transcription relative to the reference glucocorticoid, Dex. As shown in Figure 2B, HC increased luciferase activity in a concentration-dependent manner, but was a partial agonist (α = 0.55) relative to Dex.
Effect of carbenoxolone on GRE-dependent transcription
Pretreatment of 2 × GRE BEAS-2B reporter cells with carbenoxolone (1 μM for 30 min), which inhibits the glucocorticoid metabolizing enzyme 11β-hydroxysteroid dehydrogenase-2 (11β-HSD-2; Monder et al., 1989), failed to affect the E/[A] curves that described FF-, Dex-, HC-, DC- and GW-induced GRE-dependent transcription (Supporting Information Fig. S2). These results are consistent with a previous report (Feinstein and Schleimer, 1999) and our own unpublished data where mRNA transcripts for 11β-HSD-2 were not detected in BEAS-2B cells, Thus, the GR agonists used in this study were metabolically stable.
Antagonism of GRE-dependent transcription by Org 34517
Pretreatment (60 min) of 2 × GRE BEAS-2B reporter cells with Org (10 nM, 30 nM and 100 nM) produced graded, dextral displacements of the E/[A] curves that described FF-, Dex, DC- and GW-induced GRE-dependent transcription (Figure 3). For each GR agonist, determination of the Schild slope factor, S, by fitting, simultaneously, to Eq. 2013b each E/[A] curve in the absence and presence of Org, indicated that this parameter did not deviate significantly from unity. Thus, Org apparently behaved in a manner consistent with surmountable, competitive antagonism (Neubig et al., 2003). Accordingly, S was constrained to a value of 1 from which mean pKB values of 8.34, 8.38, 8.12 and 8.46 for Org were derived with FF, Dex, DC and GW respectively (Figure 3). It should be noted that there was a tendency for the upper asymptote of agonist E/[A] curves to be suppressed in the presence of Org. However, because this effect of Org was not concentration-related and seemingly dependent on the glucocorticoid, its significance is unclear (Figure 3).
Figure 3.
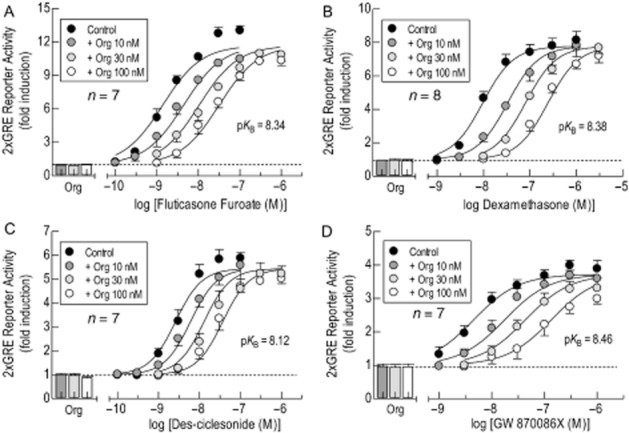
Schild analysis of the antagonism of GRE-dependent transcription by Org. In 2 × GRE BEAS-2B reporter cells, E/[A] curves were constructed to FF (panel A), Dex (panel B), DC (panel C) and GW (panel D) alone and after pretreatment (60 min) with Org (10, 30 and 100 nM) as described in the legend to Figure 2. Each family of E/[A] curves was then fitted simultaneously to Eq. 2013b from which the affinity (pKB) of Org was derived. The bars show the effect on luciferase activity of each concentration of Org alone; the horizontal dashed line in each panel defines baseline luciferase activity. Data points and bars represent the mean ± SEM of n independent determinations.
Antagonism of Dex-induced, GRE-dependent transcription by GW 870086X
On 2 × GRE BEAS-2B reporter cells, the partial agonist, GW, increased luciferase activity by 4.5- and 5.6-fold at 100 nM and 1 μM respectively (Figure 4A). At the same concentrations, GW produced graded, dextral displacements of the E/[A] curves that described Dex-induced, GRE-dependent transcription (11.5- and 62-fold at 100 nM and 1 μM respectively). Subjecting the resulting family of Dex E/[A] curves in the absence and presence of GW to a modification of classical Schild analysis showed that GW behaved as a surmountable, competitive antagonist (S = 1) with a mean pKB value of 8.08 (Figure 4A).
Figure 4.
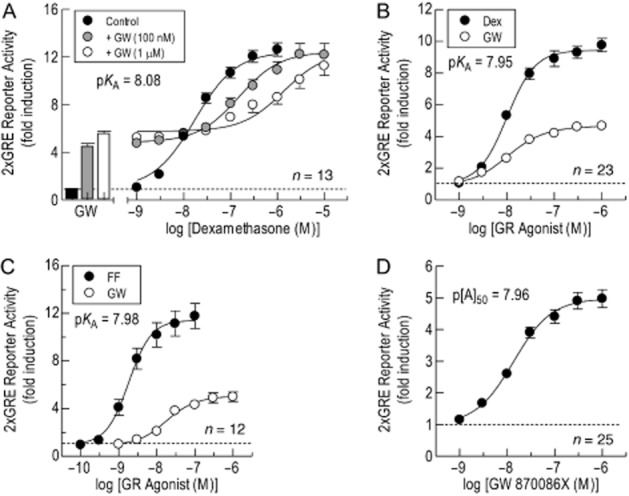
Determination of the affinity of GW by Schild analysis and the comparative method. Panel A: In 2 × GRE BEAS-2B reporter cells, E/[A] curves were constructed to Dex alone and in the presence of GW (100 nM and 1 μM) as described in the legend to Figure 2. The entire family of E/[A] curves was then fitted simultaneously to Eq. 2013b from which a pKA of 8.08 was derived. The bars in panel A show the effect of GW alone on luciferase activity. Panels B and C: E/[A] curves were constructed to GW and Dex, and GW and FF, respectively, and the resulting pairs of curves fitted simultaneously to Eqs. (1) and (3) from which estimates of KA, τ, n, Em and p[A]50 (of the reference agonist curve) were derived (see Table 1992). Panel D shows the E/[A] relationship for GW-induced, GRE-dependent transcription. The horizontal dashed line in each panel defines baseline luciferase activity. Data points represent the mean ± SEM of N independent determinations.
The affinity of GW was also determined by operational model fitting using the comparative method with FF and Dex as reference agonists (Figure 4B & C). This experimental approach yielded pKA values of ∼8.0 that were very similar to the affinity of GW derived by Schild analysis (Figure 4; Table 1992). Furthermore, inspection of the data in Figure 4D shows that the pKA of GW was equivalent to its p[A]50 for driving GRE-dependent transcription as would be expected for a low-efficacy partial agonist where response approximates to a linear function of receptor occupancy. Similarly, the affinity of the partial agonist, DC, calculated using the comparative method yielded pKA values of 8.52 and 8.59 when FF and Dex were used as reference agonists respectively (Figure 5; Table 1992). Again, these values were very similar to the potency of DC (p[A]50 = 8.61) for driving GRE-dependent transcription (Figure 5A & B; Table 1992) and are consistent with a lack of GR reserve for this glucocorticoid in BEAS-2B reporter cells.
Figure 5.
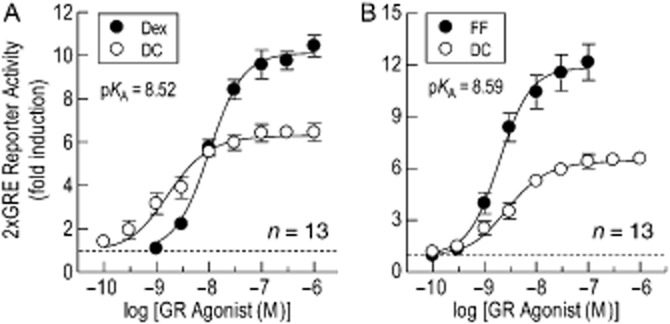
Determination of operational parameter estimates for DC-induced, GRE-dependent transcription by the comparative method. In 2 × GRE BEAS-2B reporter cells, E/[A] curves were constructed to DC (100 pM to 1 μM) and compared against those constructed to Dex (1 nM to 1 μM; panel A) and FF (100 pM to 100 nM; panel B) as described in the legend to Figure 2. The resulting pairs of curves were fitted simultaneously to Eqs. (1) and (3), respectively, from which estimates of KA, τ, n, Em and p[A]50 (of the reference agonist curve) were derived (see Table 1992). The horizontal dashed line in each panel indicates baseline luciferase activity. Data points represent the mean ± SEM of n independent determinations.
Table 3.
Operational parameter estimates that define the ability of a panel of GR agonists for promoting GRE-dependent transcription in 2 × GRE BEAS-2B reporter cells
| Parameter estimatesa | |||||
|---|---|---|---|---|---|
| Agonist | Method | pKA (M) | Em (fold) | n | τ |
| Dex | GR inactivation | 7.93 ± 0.27 | 21.7 ± 3.6 | 1.90 ± 0.58 | 1.07 ± 0.34 |
| FF | GR inactivation | 8.69 ± 0.17 | 32.2 ± 4.3 | 1.56 ± 0.38 | 0.74 ± 0.12 |
| Des-ciclesonide | ComparativeDex | 8.52 ± 0.21 | 10.1 ± 0.5b | 1.53 ± 0.08 | 1.83 ± 0.33b |
| Des-ciclesonide | ComparativeFF | 8.59 ± 0.17 | 11.8 ± 1.0b | 1.99 ± 0.13 | 1.18 ± 0.10b |
| GW 870086X | ComparativeDex | 7.95 ± 0.10 | 9.5 ± 0.4b | 1.53 ± 0.07 | 1.09 ± 0.17b |
| GW 870086X | ComparativeFF | 7.98 ± 0.07 | 11.4 ± 1.0b | 1.98 ± 0.13 | 0.88 ± 0.09b |
Dex and FF E/[A] curves were constructed in the absence and presence of Dex-Mes (3, 10 and 30 nM; 30 min pre-incubation followed by washout), and the resultant sets of curves were analysed simultaneously by operational model fitting from which estimates of KA, τ, n and Em were derived. These parameters were also derived for DC and GW using the comparative method with FF and Dex as reference agonists.
Em values calculated by the comparative method are equivalent to the Emax values determined directly from the agonist E/[A] curve (see Table 2013b) by logistic curve fitting (Eq. 2013a ). Accordingly, values of τ assume that Dex and FF are full agonists and generate a response that is equivalent to the Em. However, GR inactivation indicates that this assumption is incorrect. Thus, in the comparative method, Em and τ values are significantly underestimated and overestimated, respectively, and are italicized for that reason.
Relationship between GR occupancy and GRE-dependent transcription
The relationship between GR occupancy and response was determined by constructing agonist E/[A] curves before and after controlled inactivation of a fraction of the total GR population with an irreversible alkylating agent, Dex-Mes (Simons and Thompson, 1981), according to the method of Furchgott (1966). As shown in Supporting Information Fig. S3, Dex-Mes, did not promote luciferase activity in 2 × GRE BEAS-2B reporter cells at concentrations up to 10 μM under conditions where Dex gave a robust response (p[A]50 = 8.12 ± 0.12; max fold induction = 18.6 ± 1.0). At the outset of the study, it was assumed that Dex and FF were high-efficacy GR agonists in driving GRE-dependent transcription (i.e. where [A]50 < KA). However, in BEAS-2B reporter cells treated with Dex-Mes (3, 10 and 30 nM for 30 min), the maximal asymptote of the E/[A] curves that described FF- and Dex-induced, GRE-dependent transcription was reduced in a graded fashion in the absence of any significant change in agonist potency (Figure 6A & B; Table 1967). Applying the operational model yielded pKA values of 7.93 and 8.69 for Dex and FF, respectively, which were very close to their p[A]50 values in the absence of Dex-Mes (i.e. KA/[A]50 ∼ 1; Table 1967). Accordingly, the relationships between GR occupancy and response were essentially linear and did not deviate appreciably from the line of identity (Figure 6C & D). The linearity between the concentration of agonist-occupied receptors ( [AR] ) and response is unusual in pharmacology because it excludes a receptor reserve and, as shown by Black et al. (1985), represents the limiting case of a hyperbola where KE (i.e. the value of [AR] that produces half maximum response) > > > [AR]. Thus, in BEAS-2B reporter cells Dex and FF were partial agonists in promoting GRE-dependent transcription (vide infra).
Figure 6.
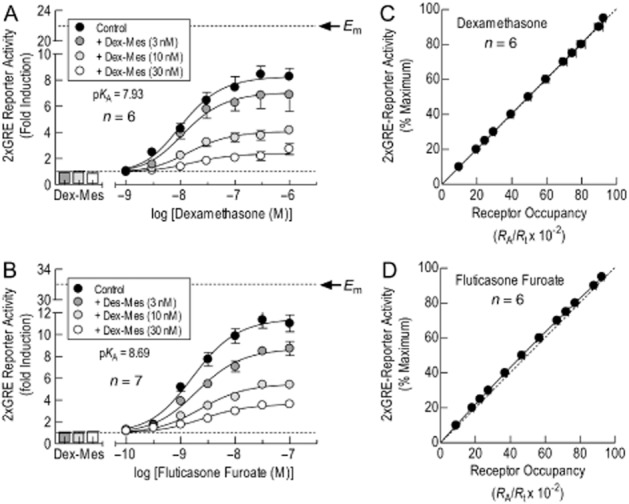
Relationship between GR occupancy and GRE-dependent transcription. 2 × GRE BEAS-2B reporter cells were treated with Dex-Mes (3, 10 or 30 nM for 30 min) or vehicle. The cells were washed in Dex-Mes-free medium and E/[A] curves constructed to Dex (panel A) or FF (panel B) as described in the legend to Figure 2. The resulting sets of curves were analysed by operational model fitting from which estimates of KA, τ, n, Em (indicated by the arrow) and p[A]50 (of the control curve) were derived (Table 1967). Data points represent the mean ± SEM of n independent determinations. In panels C and D, the KAs of Dex and FF derived by GR inactivation were used to calculate the relationship between fractional GR occupancy (RA/Rt) and GRE-dependent transcription according to Eq. 1967. The horizontal dashed line in panels A and B indicates baseline luciferase activity. The dashed line in panels C and D is the line of identity where reporter activation is a linear function of GR occupancy.
Table 4.
Relationship between the potency of FF and GW in promoting GRE-dependent transcription in 2 × GRE BEAS-2B reporter cells and their affinity for GR determined by fractional, irreversible GR inactivation with Dex-Mes
The operational model also provides estimates of τ (the efficacy of an agonist in a given tissue), Em (the maximum response that a given tissue can produce) and n (the slope of the curve that describes the relationship between the formation of AR complexes and response). Analyses of the data shown in Figure 6A and B, indicate that FF and Dex had very low efficacies in 2 × GRE BEAS-2B reporter cells (mean τ values = 0.74 and 1.07 respectively; Table 1992). This finding was consistent with estimates of Em (dashed lines in Figure 6A & B) predicted by the operational model, which were significantly greater (approximately threefold) than the maximum measured responses (Emax) produced by FF and Dex in the absence of Dex-Mes determined by logistic curve fitting (filled circles in Figure 6A & B). As the Em is, by definition, produced by a full agonist, these data suggest that FF and Dex were partial agonists in promoting GRE-dependent transcription.
Comparative effects of GR agonists on gene expression
The ability of FF, Dex, DC, GW and HC to induce a panel of genes in 2 × GRE BEAS-2B cells was assessed at concentrations that maximally increased luciferase activity (Table 2008). Mif (1 μM) and Org (1 μM) were also included in the analysis to evaluate their potential as transcriptional activators. The genes studied were GILZ, p57kip2 and CRISPLD2, which have anti-inflammatory potential (Samuelsson et al., 1999; Eddleston et al., 2007; Wang et al., 2009; Vásárhelyi et al., 2014), and an adverse-effect, metabolic gene, PDK, that regulates blood glucose (Sugden and Holness, 2002). As shown in Figure 7, the panel of glucocorticoids studied displayed distinct degrees of agonism that varied in a gene-dependent manner. On p57kip2 and PDK4, the profile of gene expression was similar to that found with the 2 × GRE reporter with DC, GW and HC, behaving as partial agonists (Figure 7A & C; Table 2008). Nevertheless, there were also noticeable differences. In particular, FF was significantly more effective at inducing PDK4 (1.9-fold) than was Dex, whereas on p57kip2 there was no such discrepancy (cf. Figure 7A & B; Table 2008). Conversely, GW, HC and DC were weak agonists at inducing PDK4 (Figure 7B), but equi-effective with FF and Dex on CRISPLD2 (Figure 7D; Table 2008). A similar pattern was found on GILZ (Figure 7C). Mif and Org, which were essentially inactive on the 2 × GRE reporter (Figure 2B), significantly, albeit weakly, induced p57kip2 and GILZ relative to the other GR agonists studied (Figure 7A & C; Table 2008). Org 34517 also significantly increased the expression of PDK4 and CRISPLD2, but again, these effects were extremely modest (Figure 7B & D).
Figure 7.
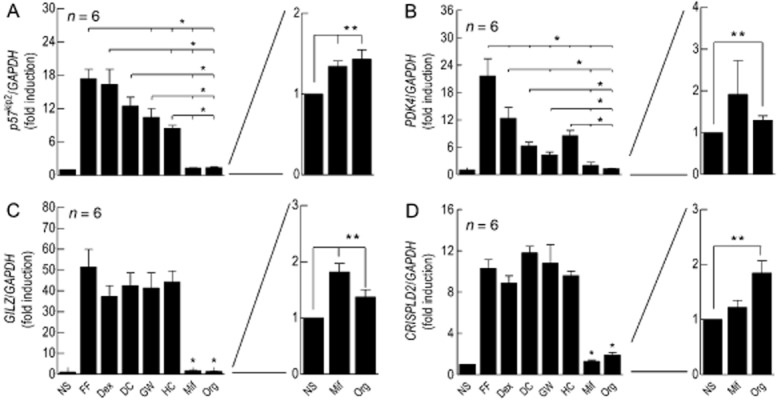
Comparative effects of a panel of GR ligands on gene expression. 2 × GRE BEAS-2B reporter cells were treated with FF (100 nM), Dex (1 μM), DC (100 nM), GW (1 μM), HC (3 μM), Mif (1 μM) or Org (1 μM). At 6 h, total RNA was extracted, reverse transcribed and the resulting cDNA subjected to real-time PCR using primer pairs specific for p57kip2 (panel A), PDK4 (panel B), GILZ (panel C) and CRISPLD2 (panel D). Data are the mean ± SEM of n independent determinations and are expressed as a ratio to GAPDH. *P < 0.05, significant difference in gene expression relative to FF. **P < 0.05, significant induction relative to untreated cells. Data were analysed by repeated measures, one-way anova/Tukey's multiple comparisons test using untransformed data.
Table 5.
Relative activities of glucocorticoids for the induction of p57kip2, GILZ, PDK4 and CRISPLD2 in 2 × GRE BEAS-2B reporter cells
| Glucocorticoida | Relative gene expression (Dex = 1) | |||
|---|---|---|---|---|
| p57kip2 | GILZ | PDK4 | CRISPLD2 | |
| Dex (1 μM)b | 1 | 1 | 1 | 1 |
| FF (100 nM) | 1.07 | 1.39 | 1.88 | 1.18 |
| Des-ciclesonide (100 nM) | 0.75 | 1.14 | 0.48 | 1.37 |
| GW 870086X (1 μM) | 0.62 | 1.11 | 0.30 | 1.25 |
| HC (3 μM) | 0.48 | 1.19 | 0.69 | 1.09 |
| Mif (1 μM) | 0.02 | 0.02 | 0.09 | 0.04 |
| Org 34517 (1 μM) | 0.03 | 0.01 | 0.03 | 0.11 |
Glucocorticoids were used at a concentration that maximally activated the 2 × GRE reporter.
For each gene, data are expressed relative to the fold induction produced by Dex, which was assigned a value of 1.
Effect of GR inactivation with Dex-Mes on GW 870086X- and FF-induced gene expression
Controlled, fractional GR inactivation was achieved by exposing 2 × GRE BEAS-2B reporter cells for 30 min to Dex-Mes at a concentration (10 nM) that produced no, or only a very modest (albeit statistically insignificant), effect on gene expression (Supporting Information Fig. S4). The fraction (q) of functional GR remaining after Dex-Mes was ∼33% (where q = τ′/τ × 100; see Methods). Gene expression induced by FF and GW was determined before and after GR inactivation at concentrations 100 times greater than their respective KA values (300 nM and 1 μM, respectively), which according to the law of mass action, will achieve >99% GR occupancy. Under these conditions, the expression of GILZ, CRISPLD2, p57kip2 and PDK4 induced by FF was inhibited by 13, 38, 41 and 56% respectively (Figure 8A–D). Dex-Mes produced a similar profile of inhibition when GW was the agonist (GILZ: 19%; CRISPLD2: 41%; p57kip2: 35%; PDK4: 58%; Figure 8E–H) indicating that the relationship between a given level of GR occupancy and magnitude of response is gene dependent.
Figure 8.

Effect of controlled, GR inactivation on FF- and GW-induced gene expression. 2 × GRE BEAS-2B reporter cells were treated with Dex-Mes (10 nM for 30 min) or vehicle. The cells were washed in Dex-Mes-free medium, left to recover for 60 min and exposed to FF (300 nM) or GW (1 μM) at concentrations that produce >99% GR occupancy. At 6 h, total RNA was extracted, reverse transcribed and the resulting cDNA subjected to real-time PCR using primer pairs specific for GILZ (panels A and E), and CRISPLD2 (panels B and F), p57kip2 (panels C and G) and PDK4 (panels D and H). Data are the mean ± SEM of n independent determinations and are expressed as a ratio to GAPDH. The dashed line in each panel defines basal gene expression. The percentage change (Δ) in gene expression effected by Dex-Mes is given above each panel. *P < 0.05, significant attenuation of gene expression; repeated measures, one-way anova/Tukey's multiple comparisons test. NSD, not significantly different.
Relationship between GR occupancy and gene expression
FF and GW increased the expression of GILZ, PDK4, CRISPLD2 and p57kip2 in a concentration-dependent manner (Figure 9A–D). Consistent with the 2 × GRE reporter data, FF was more potent (4.9- to 6.6-fold) than GW although the rank order of sensitivity of these genes to the two ligands was identical (GILZ > CRISPLD2 > p57kip2 > PDK4; Table 2009). However, while both glucocorticoids were equi-effective at inducing CRISPLD2 and GILZ, GW was a partial agonist on p57kip2 and PDK4 with α values of 0.71 and 0.26, respectively, relative to FF (Figure 9A–D; Table 2009). Assuming that the affinity of a ligand is invariant at a given receptor, these data indicate that the number of active GRs required to promote a fixed level of gene expression was dependent on both the agonist and the gene of interest. These relationships were quantified empirically using the KA values of GW (8.3 nM) and FF (2.04 nM) determined by Schild analysis (Figure 4A) and controlled GR inactivation (Figure 6B) respectively. For both glucocorticoids, occupancy–response relationships were hyperbolic confirming the presence of a GR ‘reserve’ for gene induction (Figure 9E–H; Table 1983). However, significant differences were apparent. Thus, consistent with variations in the calculated KA/A50 ratios (Table 2009), the proportion of ‘spare’ receptors required to produce a given level of response varied in gene-dependent manner (GILZ > CRISPLD2 > p57kip2 > PDK4; Table 1983). Comparative studies showed that there was a greater GR ‘reserve’ for the induction of all four genes by FF than by GW (Figure 9E–H). Notably, on PDK4, the occupancy–response relationship was very shallow and approached linearity, which is the limiting (and rare) situation of a hyperbola where [A]50 → KA (Figure 9H). Indeed, 100% GR occupancy by GW-induced PDK4 expression by only 26% of the maximum FF-induced response, which itself required only 18% GR occupancy (Figure 9H). Similar data were found for p57kip2, CRISPLD2 and GILZ (Figure 9E–G).
Figure 9.
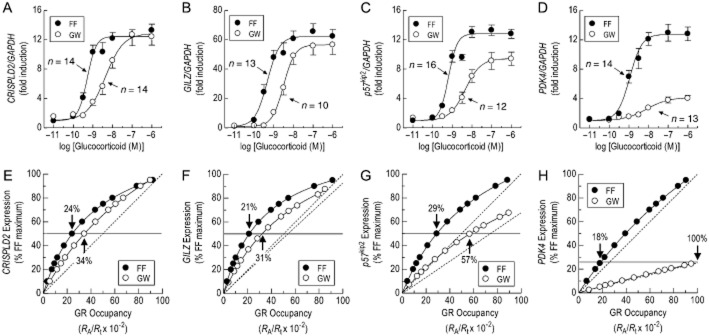
Comparative effects of FF and GW on gene expression. 2 × GRE BEAS-2B reporter cells were treated with FF and GW (both 100 pM to 1 μM). At 6 h, total RNA was extracted, reverse transcribed and the resulting cDNA subjected to real-time PCR using primer pairs specific for CRISPLD2, GILZ, p57kip2 and PDK4. Panels A to D show, as a ratio to GAPDH, the E/[A] relationship for the induction of these genes by FF and GW where each data point represents the mean ± SEM of n independent determinations. In panels E–H, mean gene expression is shown as a linear function of fractional GR occupancy according to Eq. 1967 using KA values for FF (2.04 nM) and GW (8.30 nM) derived from the experiments shown in Figures 4A and 6B. The dashed line(s) in each panel represents is the line of identity where gene expression is a linear function of GR occupancy. The solid black line in each panel defines a given level of gene expression (50% for CRISPLD2, GILZ, p57kip2; 27% for PDK4). The point at which these lines bisect the occupancy–response curves indicated the percentage of GRs required to produce this level of response. *P < 0.05, FF and GW maximum fold inductions significantly different. Student's two-tailed, unpaired, t-test.
Table 6.
Pharmacodynamic parameters of FF and GW for inducing GILZ, CRISPLD2, p57kip2 and PDK4 in 2 × GRE BEAS-2B reporter cells
| Gene | FF (p[A]50) | FF (max fold induction) | GW 870086X (p[A]50) | GW 870086X (max fold induction) | A50GW/A50FF | KAFF/A50FW | KAGW/A50GW |
|---|---|---|---|---|---|---|---|
| GILZ | 9.26 ± 0.08 (13) | 61.2 ± 3.8 | 8.57 ± 0.07 (10) | 56.5 ± 6.2 | 4.9 | 3.7 | 3.1 |
| CRISPLD2 | 9.21 ± 0.07 (14) | 12.6 ± 0.7 | 8.45 ± 0.06 (14) | 12.6 ± 1.3 | 5.8 | 3.3 | 2.3 |
| p57kip2 | 9.09 ± 0.05 (16) | 13.2 ± 0.6a | 8.27 ± 0.07 (12) | 9.4 ± 0.9 | 6.6 | 2.5 | 1.5 |
| PDK4 | 8.88 ± 0.08 (14) | 13.1 ± 0.9b | 8.08 ± 0.09 (13) | 4.3 ± 0.5 | 6.3 | 1.5 | 1.0 |
Table 7.
Relationship between GR occupancy and gene expression in 2 × GRE BEAS-2B reporter cells
| Gene | Agonist | Percentage GR occupancy required to produce: | |||
|---|---|---|---|---|---|
| 25% response | 50% response | 75% response | 95% response | ||
| GILZ | FF | 8.2 | 21.4 | 46.5 | 91.5 |
| GW 870086X | 11.7 | 28.5 | 54.7 | 85.1 | |
| CRISPLD2 | FF | 9.5 | 24.3 | 50.3 | 92.0 |
| GW 870086X | 14.9 | 34.3 | 60.7 | 89.7 | |
| p57kip2 | FF | 12.0 | 29.0 | 53.0 | 88.5 |
| GW 870086X | 16.1 | 36.3 | 62.4 | 89.5 | |
| PDK4 | FF | 18.0 | 39.4 | 65.3 | 90.5 |
| GW 870086X | 22.5 | 46.7 | 72.7 | 95.0 | |
The KA of GW and FF were derived by Schild analysis and controlled GR inactivation, respectively (Figures 5A and 7B), and used to calculate the relationship between fractional GR occupancy and gene expression using the mean E/[A] curves shown in Figure 10A–D. Data show the fraction (%) of GR occupied by FF and GW that produce 25, 50, 75 and 95% of maximum gene expression.
Antagonism of Dex-induced gene expression by GW 870086X
Treatment (6 h) of 2 × GRE BEAS-2B cells with GW (1 μM) or Dex (1 μM) increased the expression of PDK4 by 4.5- and 17.5-fold respectively (Figure 10A). When both of these ligands were added concurrently, the magnitude of Dex-induced PDK4 induction was significantly antagonized (to 5.1-fold). A similar, but less dramatic, effect was obtained for the induction of p57kip2 (Figure 10B). In contrast, Dex and GW induced CRISPLD2 and GILZ to similar degrees, which were not significantly affected when the two glucocorticoids were used in combination (Figure 10C & D).
Figure 10.

Antagonism of Dex-induced gene expression by GW. 2 × GRE BEAS-2B reporter cells were treated with Dex (1 μM), GW (1 μM) or Dex and GW in combination. At 6 h, total RNA was extracted, reverse transcribed and the resulting cDNA subjected to real-time PCR using primers pairs specific for PDK4 (panel A), p57kip2 (panel B), CRISPLD2 (panel C) and GILZ (panel D). Individual data sets and the mean ± SEM of n independent determinations for each gene are shown and are expressed as a ratio to GAPDH. The dashed line in each panel defines baseline gene expression. *P < 0.05, significant inhibition of Dex-induced gene induction. Data were analysed by repeated measures one-way anova/Tukey's multiple comparisons test using untransformed data.
Discussion
There is compelling evidence that the anti-inflammatory effects of glucocorticoids involve the induction (transactivation) of anti-inflammatory genes that occurs in parallel with the process of transrepression (King et al., 2013). Herein, we report that a panel of glucocorticoids did not behave uniformly when gene transactivation was used as a functionally relevant output. Thus, in BEAS-2B human airway epithelial cells, which were used as a model system, the intrinsic activity values of these glucocorticoids varied markedly in a gene-dependent manner. Indeed, our data identified glucocorticoids that behaved as super agonists, full agonists, partial agonists and antagonists depending on the gene of interest. As discussed later, these findings may apply to all cell types that express GR and may have significant implications for asthma and other inflammatory disorders for which glucocorticoids are a mainstay therapy.
Effect of glucocorticoids on GRE-dependent transcription
On 2 × GRE BEAS-2B reporter cells, the seven glucocorticoids examined displayed a wide spectrum of intrinsic activities. Thus, Dex, DC, HC, GW and Mif demonstrated increasing degrees of partial agonism when compared with FF, whereas Org was inactive. This profile of agonism was unaffected by carbenoxolone, an inhibitor of 11β-HSD, indicating that it was not the result of variable metabolic glucocorticoid inactivation. Moreover, although many glucocorticoids, including some of those studied here, have activity at other nuclear hormone receptors (Salter et al., 2007), GRE-dependent transcription induced by FF, Dex, DC, and GW was antagonized by Org with an affinity (KB ∼ 5 nM) consistent with GR activation (Peeters et al., 2004).
Classical receptor theory has evolved predominantly from the study of GPCRs and predicts that a partial agonist will antagonize the effect of a full agonist at a given receptor (Kenakin, 1987). Our data demonstrate that this principle also describes the behaviour of a partial agonist at a nuclear hormone receptor. Two ligands used here, DC and GW, acted as partial agonists in driving the GRE reporter with intermediate intrinsic activity values. Consistent with theory and a recent study (Uings et al., 2013), the more partial of these ligands, GW, produced a competitive and surmountable antagonism of Dex-induced, GRE-dependent transcription with a pKB of 8.08. This value is very similar to the affinity of GW determined by operational model fitting using the comparative method and its potency for driving GRE-dependent transcription (Figure 4). Thus, on 2 × GRE BEAS-2B reporter cells, these data are consistent with the behaviour of a weak, partial agonist where response approaches a linear function of receptor occupancy (i.e. [A]50 → KA). It is noteworthy, that GR exists as multiple isoforms that are derived from mRNA splicing and alternative translation initiation (reviewed in Oakley and Cidlowski, 2013). In the analyses performed here, agonist affinity was assumed to be invariant across these different receptor subtypes, which is supported by available data (Lu and Cidlowski, 2005; Lu et al., 2007).
To interrogate the relationship between receptor number and the ability of FF and Dex to induce luciferase activity, E/[A] curves were constructed before and after controlled, fractional, irreversible GR inactivation with Dex-Mes (Simons and Thompson, 1981). Operational model fitting revealed several unanticipated, but related, findings. Similar to the behaviour of GW, the potency of Dex and FF in the absence of Dex-Mes was comparable with their respective affinities for GR where KA/[A]50 ∼ 1 (Figure 5). For ‘full’ agonists, this behaviour is rare in pharmacology and indicates that response approximates to a linear function of GR occupancy rather than the typical hyperbolic relationship, which signifies a receptor reserve (Kenakin, 1998). However, operational model fitting also indicated that the predicted system maximum, Em, was greater than the upper asymptote of the control glucocorticoid E/[A] curves and that efficacy estimates (τ) were less than a value of 1 [τ < 1 indicates that 100% GR occupancy is required to produce half maximal response (Motulsky and Christopoulos, 2003) ]. Thus, although FF and Dex had higher intrinsic activity values than GW, these data indicate that they were, nevertheless, partial agonists.
Effect of glucocorticoids on gene expression
The 2 × GRE BEAS-2B reporter is an artificial system and is unlikely to faithfully model the process of gene transactivation. This limitation prompted us to measure the induction of several genes that have anti-inflammatory (p57kip2, CRISPLD2, GILZ) or adverse-effect (PDK4) potential (see Giembycz and Newton, 2014, Moodley et al., 2013, Sugden and Holness, 2002 for a description of these genes). At a concentration of each glucocorticoid that maximally activated the reporter, distinct patterns of agonism were revealed that were both ligand- and gene-dependent. p57kip2 and PDK4 were induced with a rank order of agonism that mirrored, qualitatively, the induction of the 2 × GRE BEAS-2B reporter. Thus, on both genes, GW and DC were partial agonists. Moreover, consistent with theory, GW antagonized gene expression induced by the stronger agonist, Dex. However, despite these similarities, marked differences were also apparent. Strikingly, on PDK4, Dex and the endogenous ligand, HC, were partial agonists (α = 0.53 and 0.37, respectively, compared with FF) and DC and GW displayed only modest activity (α < 0.25). Conversely, FF and Dex were equi-effective at inducing p57kip2 whereas DC and GW displayed significant agonism. Studies on GILZ and CRISPLD2 revealed yet another profile of gene expression where FF, Dex, HC, DC and GW displayed comparable degrees of agonism. Collectively, therefore, an analysis of just four genes illustrates that a given glucocorticoid expresses a unique gene expression ‘fingerprint’. These findings may be relevant therapeutically when one considers that 0.1–1.0% (but, potentially, up to 10%) of the human genome in any given cell type is regulated in a positive manner by glucocorticoids (Galon et al., 2002; Wan and Nordeen, 2002; Planey et al., 2003; Rogatsky et al., 2003; Leclerc et al., 2004; Donn et al., 2007; James et al., 2007). Moreover, GR density varies considerably between different human tissues (Pujols et al., 2002; Su et al., 2004). This variation has important pharmacodynamic implications because efficacy (i.e. the ability to produce response), in its simplest form, is the product of intrinsic efficacy (a sole property of the agonist) and receptor number (a tissue-dependent parameter). Thus, GR density in a target tissue will dictate whether a given glucocorticoid will behave as a full agonist, partial agonist or antagonist on the expression of a particular gene.
The gene-dependent differences in glucocorticoid intrinsic activity suggested that the relationship between GR occupancy and transcription was not uniform. To test this hypothesis empirically, gene expression was measured in 2 × GRE BEAS-2B reporter cells subjected to controlled, fractional GR inactivation with Dex-Mes. As shown in Figure 8, gene expression induced by FF and GW exhibited different sensitivities to Dex-Mes (PDK4 > p57kip2 > CRISPLD2 > GILZ). These findings provide strong evidence that the ability of agonist-bound GR to promote transcription is, indeed, gene-dependent. Experiments published by Uings et al. (2013) revealed that the potency of Dex to induce a panel of genes in A549 cells varied up to 20-fold, which is entirely consistent with this conclusion. The construction of occupancy–response plots corroborated those data and showed, in addition, that FF had a higher intrinsic efficacy (i.e. there was a greater GR ‘reserve’) than GW for the induction of GILZ, CRISPLD2, PDK4 and p57kip2. Furthermore, the intrinsic efficacy of each glucocorticoid varied in a gene-dependent manner indicating that a given cell interprets equivalent degrees of GR occupancy differently. In its simplest form, this may relate to variations in the ability of agonist-bound GR to interact with DNA in the promoter(s) of target genes. Indeed, it is believed that the 3D-conformation adopted by activated nuclear hormone receptors including GR is agonist-dependent (Allan et al., 1992; Biggadike et al., 2008; Biggadike et al., 2009; Wagner et al., 1996). Clearly, this could influence its interaction with obligatory co-activators and/or co-repressors and, therefore, its ability to bind DNA and promote gene transcription. Similarly, it is well established that glucocorticoid-inducible promoter regions through which ligand-bound GR increases gene transcription are variable (Newton et al., 2010). Thus, notwithstanding epigenetic and additional undefined regulatory mechanisms, it seems probable that the 3D-conformation of activated GR and the promoter context in the gene of interest will dictate the ‘transcriptional competency’ of a given glucocorticoid.
Clinical implications
Gene-dependent differences in glucocorticoid transactivation potential, as shown here, raise an important clinical question: how much GR agonism is required for benefit in asthma to be realized with an acceptable therapeutic ratio? At least two possibilities can be considered. First, high-efficacy GR agonists such as FF may be desirable (or even necessary) because they will induce most, if not all, glucocorticoid-inducible, anti-inflammatory genes. Clearly, a potential disadvantage of such an ICS could be the unwanted expression of metabolic genes such as PDK4. This could be particularly problematic if there is significant systemic exposure although this could be mitigated through pulmonary retention, significant first pass hepatic metabolism and high plasma protein binding. Alternatively, perhaps GR agonists with moderate intrinsic efficacy will be sufficient (e.g. GW). One would predict that transactivation of many therapeutically relevant genes (e.g. CRISPLD2, GILZ, p57kip2) would still occur, albeit perhaps less robustly, but the number of, and degree to which, adverse-effect genes are expressed could be minimized providing a ‘safer’ glucocorticoid.
Perhaps the most interesting finding of this study was that FF was significantly more effective (∼2.7-fold) at inducing PDK4 than the endogenous ligand, HC. This observation is consistent with the behaviour of a ‘super agonist’, which, by definition, produces a greater effect at a given receptor than the natural, endogenous ligand (Smith et al., 2011; Langmead and Christopoulos, 2013). FF was identified as a ‘super agonist’ because GR density in 2 × GRE BEAS-2B reporter cells for promoting the transcription of PDK4 was limiting rendering HC a partial agonist. Taken together, these data demonstrate that GR agonists can be synthesized with higher intrinsic efficacies than an endogenous agonist. However, whether they would be superior anti-inflammatory drugs or more prone to cause adverse events, despite optimized pharmacokinetics, is unclear.
A final point to mention is that it could be unwise to combine different ICS in asthma therapy. Although this is not recommended clinical practice, the data herein warn against considering this approach to treatment. Indeed, the ability of GW to antagonize Dex-induced p57kip2 and PDK4 expression suggests that co-administration of different glucocorticoids may not achieve greater asthma control and could even be counter-productive.
Conclusions
In the present study, we applied a pharmacodynamics approach to study GR-mediated gene expression in human airway epithelial cells using a diverse panel of glucocorticoids. Our primary finding was that a given glucocorticoid displays a unique, gene expression profile that is governed by its intrinsic efficacy and GR density. Although only four genes were studied, the recent report that GW exhibits intrinsic activity values relative to Dex that vary from approximately 0.1 to 0.9 for eight genes in a different (A549) cell type (Uings et al., 2013) lends additional support to the overall conclusion presented here. Thus, depending on the gene of interest and variations in GR density in target as well as off-target tissues, a glucocorticoid may behave as an antagonist, partial agonist, full agonist or even ‘super agonist’. We submit that these results provide a tractable means to rationally design new GR agonists through the generation of gene expression fingerprints. However, for this concept to constitute a viable drug discovery platform, compounds would have to be screened in target and off-target human tissues, and the physiological role(s) of genes in the fingerprint defined. With this knowledge, novel GR agonists could then be identified, and theoretically, optimized to preferentially induce genes with anti-inflammatory activity at the expense of those that mediate adverse effects. While the separation of ‘beneficial’ from ‘detrimental’ genes is unlikely to be absolute, such approaches could identify drug candidates with improved therapeutic ratios that could be useful in the management of severe asthma and other inflammatory disorders where systemic exposure is warranted.
Acknowledgments
The authors thank Mr. Michael Yu for generating the data shown in Supporting Information Fig. S3.
Glossary
Abbreviations
- CRISPLD2
cysteine-rich secretory protein LCCL (Limulus clotting factor C, Cochlin, Lgl1) domain-containing 2
- DC
desisobutyrylciclesonide
- Dex
dexamethasone
- FF
fluticasone furoate
- GILZ
glucocorticoid-inducible leucine zipper
- GR
glucocorticoid receptor
- GRE
glucocorticoid response element
- GW
GW 870086X (6α,9α-difluoro-11β-hydroxy-16α-methyl-3-oxo-17α-(2,2,3,3-tetramethylcyclopropylcarbonyl)-oxo-androsta-1,4-diene-17β-carboxylic acid cyanomethylester)
- HC
hydrocortisone
- HSD
hydroxysteroid dehydrogenase
- Mif
mifepristone
- Org
Org 34517 (11β-(1,3-benzodioxolo)-17β-hydroxy-17-(1-propynyl)-oestra-4,9-dien-3-one)
- p57kip2
kinase inhibitor protein 2 of 57 kDa
- PDK
pyruvate dehydrogenase kinase
- SFM
serum-free medium
Author contributions
M. G., T. J. and R. N. participated in research design. T. J. conducted experiments. M. J. provided novel reagents. M. G. and T. J. performed data analysis. All authors wrote or contributed to the writing of the paper.
Conflict of interest
T. J., M. J., R. N. and M. G. have nothing to declare. T. J. is a recipient of an Alberta Lung Association Graduate Studentship. R. N. is an Alberta Innovates – Health Solutions Senior Scholar. M. G. holds a Tier 1 Canada Research Chair in Pulmonary Pharmacology. Work in the laboratories of R. N. and M. G. is supported by operating grants awarded by the CIHR (MOP 68828 and MOP 93742 respectively).
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Kinetics of GRE-dependent transcription. Panel A: 2 × GRE BEAS-2B reporter cells were treated with fluticasone furoate (FF; 100 nM), dexamethasone (Dex; 1 μM), des-ciclesonide (DC; 100 nM), GW 870086X (GW; 1 μM) or vehicle (NS). Panels B and C: cells were treated with FF, DC (at 1 nM, 3 nM or 100 nM) or vehicle (NS). At the times indicated, cells were harvested for the determination of luciferase activity. Data points represent the mean ± SEM of n independent determinations. The dashed line in each panel defines baseline luciferase expression.
Figure S2 Effect of carbenoxolone on GRE-dependent transcription. 2 × GRE BEAS-2B reporter cells were pretreated with carbenoxolone (CBX; 1 μM for 30 min) or its vehicle and E/[A] curves were constructed to fluticasone furoate (A), dexamethasone (B), hydrocortisone (C), des-ciclesonide (D) and GW-870086X (E). After 6 h, cells were harvested for the determination of luciferase activity. Data points represent the mean ± SEM of n independent determinations. The dashed line in each panel defines baseline luciferase expression.
Figure S3 Effect of dexamethasone (Dex) and dexamethasone mesylate (Dex-Mes) on GRE-dependent transcription. 2 × GRE BEAS-2B reporter cells were treated with Dex or Dex-Mes at the concentrations indicated. At 6 h cells were harvested for the determination of luciferase activity. Data points represent the mean ± SEM of n independent determinations.
Figure S4 Effect of dexamethasone 21-mesylate (Dex-Mes) on gene expression. 2 × GRE BEAS-2B reporter cells were treated with Dex-Mes at the concentrations indicated. At 6 h, total RNA was extracted, reverse transcribed and the resulting cDNA subjected to real-time PCR using primer pairs specific for GILZ (panel A), CRISPLD2 (panel B), p57kip2 (panel C) and PDK4 (panel D). Data are expressed as the mean ± SEM of n independent determinations and are expressed as a ratio to GAPDH. The dashed line in each panel defines baseline gene expression. *P < 0.05, significant induction relative to untreated cells; one-way anova/Tukey's multiple comparison test on untransformed data.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Nuclear hormone receptors. Br J Pharmacol. 2013a;170:1632–1675. doi: 10.1111/bph.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan GF, Leng X, Tsai SY, Weigel NL, Edwards DP, Tsai MJ, et al. Hormone and antihormone induce distinct conformational changes which are central to steroid receptor activation. J Biol Chem. 1992;267:19513–19520. [PubMed] [Google Scholar]
- Barlow RB, Scott NC, Stephenson RP. The affinity and efficacy of onium salts on the frog rectus abdominis. Br J Pharmacol Chemother. 1967;31:188–196. doi: 10.1111/j.1476-5381.1967.tb01989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggadike K, Bledsoe RK, Hassell AM, Kirk BE, McLay IM, Shewchuk LM, et al. X-Ray crystal structure of the novel enhanced-affinity glucocorticoid agonist fluticasone furoate in the glucocorticoid receptor-ligand binding domain. J Med Chem. 2008;51:3349–3352. doi: 10.1021/jm800279t. [DOI] [PubMed] [Google Scholar]
- Biggadike K, Bledsoe RK, Coe DM, Cooper TWJ, House D, Iannone MA, et al. Design and x-ray crystal structures of high-potency nonsteroidal glucocorticoid agonists exploiting a novel binding site on the receptor. Proc Natl Acad Sci U S A. 2009;106:18114–18119. doi: 10.1073/pnas.0909125106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JW, Leff P. Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P, Shankley NP, Wood J. An operational model of pharmacological agonism: the effect of E/[A] curve shape on agonist dissociation constant estimation. Br J Pharmacol. 1985;84:561–571. doi: 10.1111/j.1476-5381.1985.tb12941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown HM. Treatment of chronic asthma with prednisolone; significance of eosinophils in the sputum. Lancet. 1958;2:1245–1247. doi: 10.1016/s0140-6736(58)91385-0. [DOI] [PubMed] [Google Scholar]
- Chivers JE, Cambridge LM, Catley MC, Mak JC, Donnelly LE, Barnes PJ, et al. Differential effects of RU486 reveal distinct mechanisms for glucocorticoid repression of prostaglandin E release. Eur J Biochem. 2004;271:4042–4052. doi: 10.1111/j.1432-1033.2004.04342.x. [DOI] [PubMed] [Google Scholar]
- Clark AR. Anti-inflammatory functions of glucocorticoid-induced genes. Mol Cell Endocrinol. 2007;275:79–97. doi: 10.1016/j.mce.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Clark AR, Belvisi MG. Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol Ther. 2011;134:54–67. doi: 10.1016/j.pharmthera.2011.12.004. [DOI] [PubMed] [Google Scholar]
- Donn R, Berry A, Stevens A, Farrow S, Betts J, Stevens R, et al. Use of gene expression profiling to identify a novel glucocorticoid sensitivity determining gene, BMPRII. FASEB J. 2007;21:402–414. doi: 10.1096/fj.06-7236com. [DOI] [PubMed] [Google Scholar]
- Eddleston J, Herschbach J, Wagelie-Steffen AL, Christiansen SC, Zuraw BL. The anti-inflammatory effect of glucocorticoids is mediated by glucocorticoid-induced leucine zipper in epithelial cells. J Allergy Clin Immunol. 2007;119:115–122. doi: 10.1016/j.jaci.2006.08.027. [DOI] [PubMed] [Google Scholar]
- Feinstein MB, Schleimer RP. Regulation of the action of hydrocortisone in airway epithelial cells by 11β-hydroxysteroid dehydrogenase. Am J Respir Cell Mol Biol. 1999;21:403–408. doi: 10.1165/ajrcmb.21.3.3560. [DOI] [PubMed] [Google Scholar]
- Furchgott RF. The use of β-haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor–agonist complexes. Adv Drug Res. 1966;3:21–55. [Google Scholar]
- Gagne D, Pons M, Philibert D. RU 38486: a potent antiglucocorticoid in vitro and in vivo. J Steroid Biochem. 1985;23:247–251. doi: 10.1016/0022-4731(85)90401-7. [DOI] [PubMed] [Google Scholar]
- Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 2002;16:61–71. doi: 10.1096/fj.01-0245com. [DOI] [PubMed] [Google Scholar]
- Giembycz MA, Newton R. How phosphodiesterase 4 inhibitors work in patients with chronic obstructive pulmonary disease of the severe, bronchitic, frequent exacerbator phenotype. Clin Chest Med. 2014;35:203–217. doi: 10.1016/j.ccm.2013.09.007. [DOI] [PubMed] [Google Scholar]
- Green RH, Brightling CE, Woltmann G, Parker D, Wardlaw AJ, Pavord ID. Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax. 2002;57:875–879. doi: 10.1136/thorax.57.10.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James CG, Ulici V, Tuckermann J, Underhill TM, Beier F. Expression profiling of dexamethasone-treated primary chondrocytes identifies targets of glucocorticoid signalling in endochondral bone development. BMC Genomics. 2007;8:205. doi: 10.1186/1471-2164-8-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, editor. Current Protocols in Pharmacology. Hoboken, NJ: Wiley, Inc; 1998. Receptor Theory; pp. 1.2.1–1.2.27. (ed.) (. In: [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Pharmacologic Analysis of Drug–Receptor Interaction. New York: Raven Press; 1987. [Google Scholar]
- King EM, Chivers JE, Rider CF, Minnich A, Giembycz MA, Newton R. Glucocorticoid repression of inflammatory gene expression shows differential responsiveness by transactivation- and transrepression-dependent mechanisms. PLoS ONE. 2013;8:e53936. doi: 10.1371/journal.pone.0053936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead CJ, Christopoulos A. Supra-physiological efficacy at GPCRs: superstition or super agonists? Br J Pharmacol. 2013;169:353–356. doi: 10.1111/bph.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc N, Luppen CA, Ho VV, Nagpal S, Hacia JG, Smith E, et al. Gene expression profiling of glucocorticoid-inhibited osteoblasts. J Mol Endocrinol. 2004;33:175–193. doi: 10.1677/jme.0.0330175. [DOI] [PubMed] [Google Scholar]
- Leff P, Prentice DJ, Giles H, Martin GR, Wood J. Estimation of agonist affinity and efficacy by direct, operational model-fitting. J Pharmacol Methods. 1990;23:225–237. doi: 10.1016/0160-5402(90)90066-t. [DOI] [PubMed] [Google Scholar]
- Lu NK, Cidlowski JC. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18:331–342. doi: 10.1016/j.molcel.2005.03.025. [DOI] [PubMed] [Google Scholar]
- Lu NK, Collins JB, Grissom SF, Cidlowski JA. Selective regulation of bone cell apoptosis by translational isoforms of the glucocorticoid receptor. Mol Cell Biol. 2007;27:7143–7160. doi: 10.1128/MCB.00253-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monder C, Stewart PM, Lakshmi V, Valentino R, Burt D, Edwards CRW. Licorice inhibits corticosteroid 11β-dehydrogenase of rat kidney and liver: in vivo and in vitro studies. Endocrinology. 1989;125:1046–1053. doi: 10.1210/endo-125-2-1046. [DOI] [PubMed] [Google Scholar]
- Moodley T, Wilson SM, Joshi T, Rider CF, Sharma P, Dong Y, et al. Phosphodiesterase 4 inhibitors augment the ability of formoterol to enhance glucocorticoid-dependent gene transcription in human airway epithelial cells: a novel mechanism for the clinical efficacy of roflumilast in severe COPD. Mol Pharmacol. 2013;83:894–906. doi: 10.1124/mol.112.083493. [DOI] [PubMed] [Google Scholar]
- Motulsky HJ, Christopoulos A. Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting. San Diego, CA: GraphPad Software, Inc; 2003. [Google Scholar]
- Neubig RR, Spedding M, Kenakin T, Christopoulos A. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol Rev. 2003;55:597–606. doi: 10.1124/pr.55.4.4. [DOI] [PubMed] [Google Scholar]
- Newton R. Molecular mechanisms of glucocorticoid action: what is important? Thorax. 2000;55:603–613. doi: 10.1136/thorax.55.7.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton R, Holden NS. Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol Pharmacol. 2007;72:799–809. doi: 10.1124/mol.107.038794. [DOI] [PubMed] [Google Scholar]
- Newton R, Leigh R, Giembycz MA. Pharmacological strategies for improving the efficacy and therapeutic ratio of glucocorticoids in inflammatory lung diseases. Pharmacol Ther. 2010;125:286–327. doi: 10.1016/j.pharmthera.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signalling mechanism in health and disease. J Allergy Clin Immunol. 2013;132:1033–1044. doi: 10.1016/j.jaci.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters BW, Tonnaer JA, Groen MB, Broekkamp CL, van der Voort HA, Schoonen WG, et al. Glucocorticoid receptor antagonists: new tools to investigate disorders characterized by cortisol hypersecretion. Stress. 2004;7:233–241. doi: 10.1080/10253890400019672. [DOI] [PubMed] [Google Scholar]
- Planey SL, Abrams MT, Robertson NM, Litwack G. Role of apical caspases and glucocorticoid-regulated genes in glucocorticoid-induced apoptosis of pre-B leukemic cells. Cancer Res. 2003;63:172–178. [PubMed] [Google Scholar]
- Proud D, Leigh R. Epithelial cells and airway diseases. Immunol Rev. 2011;242:186–204. doi: 10.1111/j.1600-065X.2011.01033.x. [DOI] [PubMed] [Google Scholar]
- Pujols L, Mullol J, Roca-Ferrer J, Torrego A, Xaubet A, Cidlowski JA, et al. Expression of glucocorticoid receptor α- and β-isoforms in human cells and tissues. Am J Physiol Cell Physiol. 2002;283:C1324–C1331. doi: 10.1152/ajpcell.00363.2001. [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq CM, et al. Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc Natl Acad Sci USA. 2003;100:13845–13850. doi: 10.1073/pnas.2336092100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter M, Biggadike K, Matthews JL, West MR, Haase MV, Farrow SN, et al. Pharmacological properties of the enhanced-affinity glucocorticoid fluticasone furoate in vitro and in an in vivo model of respiratory inflammatory disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L660–L667. doi: 10.1152/ajplung.00108.2007. [DOI] [PubMed] [Google Scholar]
- Samuelsson MK, Pazirandeh A, Davani B, Okret S. p57kip2, a glucocorticoid-induced inhibitor of cell cycle progression in HeLa cells. Mol Endocrinol. 1999;13:1811–1822. doi: 10.1210/mend.13.11.0379. [DOI] [PubMed] [Google Scholar]
- Simons SS. What goes on behind closed doors: physiological versus pharmacological steroid hormone actions. Bioessays. 2008;30:744–756. doi: 10.1002/bies.20792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons SS. Glucocorticoid receptor cofactors as therapeutic targets. Curr Opin Pharmacol. 2010;10:613–619. doi: 10.1016/j.coph.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons SS, Thompson EB. Dexamethasone 21-mesylate: an affinity label of glucocorticoid receptors from rat hepatoma tissue culture cells. Proc Natl Acad Sci USA. 1981;78:3541–3545. doi: 10.1073/pnas.78.6.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NJ, Bennett KA, Milligan G. When simple agonism is not enough: emerging modalities of GPCR ligands. Mol Cell Endocrinol. 2011;331:241–247. doi: 10.1016/j.mce.2010.07.009. [DOI] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden MC, Holness MJ. Therapeutic potential of the mammalian pyruvate dehydrogenase kinases in the prevention of hyperglycaemia. Curr Drug Targets Immune Endocr Metabol Disord. 2002;2:151–165. [PubMed] [Google Scholar]
- Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011;145:224–241. doi: 10.1016/j.cell.2011.03.027. [DOI] [PubMed] [Google Scholar]
- Uings I, Needham D, Matthews J, Haase M, Austin R, Angell D, et al. Discovery of GW870086: a potent anti-inflammatory steroid with a unique pharmacological profile. Br J Pharmacol. 2013;169:1389–1403. doi: 10.1111/bph.12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vásárhelyi V, Trexler M, Patthy L. Both LCCL-domains of human CRISPLD2 have high affinity for lipid A. Biochimie. 2014;97:66–71. doi: 10.1016/j.biochi.2013.09.021. [DOI] [PubMed] [Google Scholar]
- Wagner BL, Pollio G, Leonhardt S, Wani MC, Lee DY, Imhof MO, et al. 16α-Substituted analogs of the antiprogestin RU486 induce a unique conformation in the human progesterone receptor resulting in mixed agonist activity. Proc Natl Acad Sci USA. 1996;93:8739–8744. doi: 10.1073/pnas.93.16.8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Nordeen SK. Overlapping but distinct gene regulation profiles by glucocorticoids and progestins in human breast cancer cells. Mol Endocrinol. 2002;16:1204–1214. doi: 10.1210/mend.16.6.0848. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Xing WM, Fan HH, Wang KS, Zhang HK, Wang QW, et al. The novel lipopolysaccharide-binding protein CRISPLD2 is a critical serum protein to regulate endotoxin function. J Immunol. 2009;183:6646–6656. doi: 10.4049/jimmunol.0802348. [DOI] [PubMed] [Google Scholar]
- Waud DR, Son SL, Waud BE. Kinetic and empirical analysis of dose–response curves illustrated with a cardiac example. Life Sci. 1978;22:1275–1285. doi: 10.1016/0024-3205(78)90096-6. [DOI] [PubMed] [Google Scholar]
- Zhang S, Jonklaas J, Danielsen M. The glucocorticoid agonist activities of mifepristone (RU486) and progesterone are dependent on glucocorticoid receptor levels but not on EC50 values. Steroids. 2007;72:600–608. doi: 10.1016/j.steroids.2007.03.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Kinetics of GRE-dependent transcription. Panel A: 2 × GRE BEAS-2B reporter cells were treated with fluticasone furoate (FF; 100 nM), dexamethasone (Dex; 1 μM), des-ciclesonide (DC; 100 nM), GW 870086X (GW; 1 μM) or vehicle (NS). Panels B and C: cells were treated with FF, DC (at 1 nM, 3 nM or 100 nM) or vehicle (NS). At the times indicated, cells were harvested for the determination of luciferase activity. Data points represent the mean ± SEM of n independent determinations. The dashed line in each panel defines baseline luciferase expression.
Figure S2 Effect of carbenoxolone on GRE-dependent transcription. 2 × GRE BEAS-2B reporter cells were pretreated with carbenoxolone (CBX; 1 μM for 30 min) or its vehicle and E/[A] curves were constructed to fluticasone furoate (A), dexamethasone (B), hydrocortisone (C), des-ciclesonide (D) and GW-870086X (E). After 6 h, cells were harvested for the determination of luciferase activity. Data points represent the mean ± SEM of n independent determinations. The dashed line in each panel defines baseline luciferase expression.
Figure S3 Effect of dexamethasone (Dex) and dexamethasone mesylate (Dex-Mes) on GRE-dependent transcription. 2 × GRE BEAS-2B reporter cells were treated with Dex or Dex-Mes at the concentrations indicated. At 6 h cells were harvested for the determination of luciferase activity. Data points represent the mean ± SEM of n independent determinations.
Figure S4 Effect of dexamethasone 21-mesylate (Dex-Mes) on gene expression. 2 × GRE BEAS-2B reporter cells were treated with Dex-Mes at the concentrations indicated. At 6 h, total RNA was extracted, reverse transcribed and the resulting cDNA subjected to real-time PCR using primer pairs specific for GILZ (panel A), CRISPLD2 (panel B), p57kip2 (panel C) and PDK4 (panel D). Data are expressed as the mean ± SEM of n independent determinations and are expressed as a ratio to GAPDH. The dashed line in each panel defines baseline gene expression. *P < 0.05, significant induction relative to untreated cells; one-way anova/Tukey's multiple comparison test on untransformed data.