

Abstract
Background and Purpose
Treatment with selective oestrogen receptor modulators (SERMs) reduces low-density lipoprotein (LDL) cholesterol levels. We assessed the effect of tamoxifen, raloxifene and toremifene and their combinations with lovastatin on LDL receptor activity in lymphocytes from normolipidaemic and familial hypercholesterolaemic (FH) subjects, and human HepG2 hepatocytes and MOLT-4 lymphoblasts.
Experimental Approach
Lymphocytes were isolated from peripheral blood, treated with different compounds, and 1,1′-dioctadecyl-3,3,3,3′-tetramethylindocarbocyanine perchlorate (DiI)-labelled LDL uptake was analysed by flow cytometry.
Key Results
Tamoxifen, toremifene and raloxifene, in this order, stimulated DiI-LDL uptake by lymphocytes by inhibiting LDL-derived cholesterol trafficking and subsequent down-regulation of LDL receptor expression. Differently to what occurred in HepG2 and MOLT-4 cells, only tamoxifen consistently displayed a potentiating effect with lovastatin in primary lymphocytes. The SERM-mediated increase in LDL receptor activity was not altered by the anti-oestrogen ICI 182 780 nor was it reproduced by 17β-oestradiol. However, the tamoxifen-active metabolite endoxifen was equally effective as tamoxifen. The SERMs produced similar effects on LDL receptor activity in heterozygous FH lymphocytes as in normal lymphocytes, although none of them had a potentiating effect with lovastatin in heterozygous FH lymphocytes. The SERMs had no effect in homozygous FH lymphocytes.
Conclusions and Implications
Clinically used SERMs up-regulate LDL receptors in primary human lymphocytes. There is a mild enhancement between SERMs and lovastatin of lymphocyte LDLR activity, the potentiation being greater in HepG2 and MOLT-4 cells. The effect of SERMs is independent of oestrogen receptors but is preserved in the tamoxifen-active metabolite endoxifen. This mechanism may contribute to the cholesterol-lowering action of SERMs.
Tables of Links
| LIGANDS | |
|---|---|
| 17β-oestradiol (E2) | Lovastatin |
| Acetic acid | Oleic acid |
| Cholesterol | Raloxifene |
| Glutamine | Tamoxifen |
| ICI 182780 | Toremifene |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,b,c,).
Introduction
The low-density lipoprotein (LDL) receptor (LDLR) is a 160 kDa cell surface glycoprotein that binds and mediates internalization of LDL and very low-density lipoprotein remnants. LDLR activity is a critical determinant of plasma cholesterol levels. Thus, mutations in the LDLR gene cause familial hypercholesterolemia (FH), the most common and severe form of monogenic hypercholesterolaemia, with an autosomal codominant pattern of inheritance, and which highly increases the risk for cardiovascular disease (Goldstein et al., 2001). LDLR-bound lipoproteins are transported to acidic endocytic compartments where cholesteryl esters, the major component of LDL, are hydrolysed to free cholesterol by lysosomal acid lipase. The subsequent egress of cholesterol from the endosomal compartment allows cholesterol to be available to other intracellular organelles. The cholesterol that reaches the endoplasmic reticulum can be re-esterified by acyl-coenzyme A:cholesterol acyltransferase (ACAT) for storage in lipid droplets (Ikonen, 2008).
Cells maintain their cholesterol content within a narrow range, LDLR-mediated cholesterol uptake and endogenous cholesterol biosynthesis being subject to negative feedback regulation by intracellular cholesterol. The cholesterol-sensing machinery, a complex formed by the sterol regulatory element-binding protein (SREBP), the SREBP cleavage-activating protein (Scap) and the insulin-induced gene product (Insig), resides in the endoplasmic reticulum. When cells are depleted of cholesterol, Scap escorts SREBP from the endoplasmic reticulum to the Golgi apparatus where the active form of SREBP is produced. This is transported to the nucleus to activate transcription of the LDLR and cholesterol biosynthetic genes. When cholesterol levels rise, Scap binds to Insig, which retains the SREBP–Scap complex in the endoplasmic reticulum, thus preventing SREBP activation and gene induction (Brown and Goldstein, 2009). There are three forms of SREBPs, designed SREBP-1a, -1c and -2. The two former, which are encoded by a single gene, preferentially activate genes for fatty acid and acylglycerol biosynthesis, whereas SREBP-2 principally activates genes for cholesterol biosynthesis and the LDLR gene (Horton et al., 2002). This regulatory mechanism is essential for the action of statins, which are competitive inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme in cholesterol biosynthesis. By inhibiting this pathway, statins induce a compensatory increase in the expression of LDLR, effectively lowering LDL cholesterol levels (Baigent et al., 2005). However, there are large variations in interindividual plasma cholesterol responses to statins (Maggo et al., 2011).
Selective oestrogen receptor modulators (SERMs) are non-steroidal molecules that bind to oestrogen receptors (ERs) and are widely prescribed for the treatment and prevention of breast cancer, osteoporosis and ovulatory dysfunction (Jordan, 2007; Pickar et al., 2010). SERMs display an oestrogen–agonist or oestrogen–antagonist effect depending on the tissue targeted. This property derives from the unique conformational change in the ER induced by each SERM as well as cell and gene context-dependent factors (Riggs and Hartmann, 2003; Ali et al., 2011). Additionally, ER-independent effects have been reported (Gundimeda et al., 1996; Poirot et al., 2012; Silvente-Poirot and Poirot, 2014). Because of this complexity, different SERMs can exhibit a distinct effect on a given gene in a given cell type.
Tamoxifen and toremifene, a first- and a second-generation SERM, respectively, derived from triphenylethylene, are used to treat breast cancer. Raloxifene is a benzothiophene derivative indicated for the treatment and prevention of osteoporosis in postmenopausal women (Jordan, 2007; Pickar et al., 2010). Treatment with SERMs affects a variety of parameters related to the cardiovascular system and, actually, the cardioprotective effect of these drugs is subject to debate (Grainger and Schofield, 2005; Regitz-Zagrosek et al., 2007). Among these actions, SERMs consistently decrease plasma and LDL cholesterol levels (Riggs and Hartmann, 2003), which suggests an important effect of these drugs on the liver. The mechanism for this effect has not been fully established, but both the inhibition of cholesterol biosynthesis (Gylling et al., 1995; Holleran et al., 1998; Kedjouar et al., 2004; Suarez et al., 2004) and increase in LDLR activity (Brüning et al., 2003; Suarez et al., 2004) may be involved. SERMs can induce the accumulation of several cholesterol precursors, such as zymosterol, zymostenol and desmosterol (Gylling et al., 1995; Holleran et al., 1998; Poirot et al., 2012), which accumulate in cells as multilamellar bodies (de Medina et al., 2009). In particular, it has been reported that SERMs are able to bind to and inhibit the anti-oestrogen binding site (AEBS), a microsomal complex composed of two enzymes of cholesterol biosynthesis: 3β-hydroxysterol-Δ8-Δ7-isomerase and 3β-hydroxysterol-Δ7-reductase (Kedjouar et al., 2004). Moreover, tamoxifen inhibits ACAT-mediated cholesterol esterification in macrophages (de Medina et al., 2004), a mechanism that may contribute to free sterol accumulation. As regards LDLR, we have shown that tamoxifen increases LDLR expression and activity in the MOLT-4 cell line (Suarez et al., 2004). In these cells, tamoxifen interfered with the egress of LDL-derived cholesterol from the late endosomal/lysosomal compartment, thus preventing the LDL-induced down-regulation of LDLR expression. Moreover, when tamoxifen and lovastatin were combined the stimulation of LDLR activity was synergistic (Suarez et al., 2004).
Cell lines are proliferating cells that require a high provision of cholesterol for membrane biosynthesis (Lasuncion et al., 2012). In the present work, we studied whether the stimulating effect of tamoxifen and its combination with lovastatin on LDLR activity can be reproduced in non-proliferating primary human cells. For this, we used human peripheral blood lymphocytes, which constitute a suitable model system to examine LDLR function and regulation (Cuthbert et al., 1986; 1989,), and compared them with HepG2 hepatocytes and MOLT-4 lymphoblasts. Moreover, we aimed to ascertain whether raloxifene and toremifene are comparable with tamoxifen in its effect on LDLR activity. Finally, the effectiveness of the SERMs in LDLR-defective cells was determined, for which the lymphocytes from FH subjects were studied. The results indicate that the three SERMs up-regulate LDLR in lymphocytes from both normolipidaemic (NL) and heterozygous familial hypercholesterolaemia (HeFH) subjects, but a milder potentiating effect with lovastatin was observed relative to HepG2 and MOLT-4 cells. Moreover, we provide some insights into the mechanisms underlying the effects of such SERMs.
Methods
Subjects
Healthy NL volunteers (mean age: 35 years; 20–58 years) and hypercholesterolaemic patients with clinical diagnosis of FH (mean age: 42 years; 14–64 years) were studied. FH patients were recruited in lipid clinics at the Instituto de Investigación Sanitaria-Fundación Jiménez Díaz and Hospital Universitario Ramón y Cajal, Madrid, Spain. The FH diagnosis was based on the Make Early Diagnosis to Prevent Early Deaths criteria (Mata et al., 2002). All these patients were receiving cholesterol-lowering therapy when they entered the study. The subjects on hormone replacement therapy were excluded. Moreover, three previously diagnosed homozygous FH patients (10, 31 and 46 years old) were included in the study. The investigation conformed to the Declaration of Helsinki. Informed consent was obtained from each patient and the protocol was approved by the Ethics Committee of the Hospital Universitario Ramón y Cajal. The genetic diagnosis was made as previously described (Alonso et al., 2008). Briefly, samples were analysed using a DNA microarray (Lipochip, version 4, Progenika, Derio, Spain), and capillary sequencing was conducted using multiplex PCR. Negative samples for the DNA array or sequencing were also analysed for copy number variations using an adapted quantitative fluorescent multiplex PCR methodology (Alonso et al., 2008).
Primary human lymphocyte, MOLT-4 and HepG2 cell cultures
Lymphocytes were isolated from blood from fasting subjects by density gradient centrifugation as detailed in the Supporting Information. To up-regulate LDLR, lymphocytes were incubated in RPMI 1640 containing 2 mM glutamine, antibiotics (see the Supporting Information) and 10% heat-inactivated lipoprotein-deficient serum (LPDS) for 72 h at 37°C in a humidified atmosphere of 5% CO2. For the experiments assessing the effects of ICI 182 780, 17β-oestradiol (E2) and/or endoxifen, RPMI 1640 without phenol red and charcoal/dextran-treated LPDS were used. Cell viability was 87–92% as judged by trypan blue exclusion test and was not significantly affected by the different treatments.
MOLT-4 cells were maintained in RPMI 1640 containing 10% heat-inactivated FBS and the abovementioned additives. HepG2 were maintained in DMEM supplemented with 10% FBS, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate and the abovementioned additives. For experiments, MOLT-4 and HepG2 cells were pretreated with medium containing 10% LPDS for 24 h.
LDL receptor activity assays
Human LDL was isolated from a single donor and labelled with the fluorescence probe DiI as reported previously (Calvo et al., 1998). Lymphocytes (0.5 × 106 cells·mL−1), MOLT-4 cells (0.5 × 106 cells·mL−1) or HepG2 cells (80% confluent) were cultured in the corresponding fresh media (see above) supplemented with 60 μg·mL−1 DiI-labelled LDL (DiI-LDL) plus the different SERMs, lovastatin, E2, endoxifen and/or ICI 182 780 dissolved in DMSO (final concentration 0.044%) or vehicle, and incubated at 37°C for different times as indicated. When the expression of active LDLR was measured, lymphocytes were treated or not with 60 μg·mL−1 LDL in the presence or the absence of different SERMs and vitamin E (in ethanol, final concentration 0.4%) at 37°C; subsequently, cells were washed and resuspended in fresh medium containing 30 μg·mL−1 DiI-LDL and incubated at 37°C for 2 h. Specific uptake was calculated by subtracting non-specific values determined in the presence of 30-fold excess of unlabelled LDL. All determinations were performed in duplicate. At the end of the incubations, cells were washed, resuspended in PBS and analysed by flow cytometry (FACScalibur, Becton Dickinson, Franklin Lakes, NJ, USA). Forward scatter and side scatter gates were established to exclude dead cells and cell debris. The acquisition number of cells was set at 104. We estimated the specific median intensity of fluorescence after subtracting cell autofluorescence.
LDL binding and uptake in lymphocytes
Bmax values were calculated from saturation binding and uptake curves and were expressed relative to the respective values of a single inter-assay control (Supporting Information Figure S1). See the Supporting Information for detailed information.
Analysis of sterol biosynthesis and content
To determine the content of sterols, these were extracted from 1.5 × 107 cells and analysed by GC-MS as described in detail by Canfran-Duque et al. (2013). To study sterol biosynthesis, 107 cells were incubated in medium containing 10% LPDS and supplemented with 40 μCi of [2-14C]-acetic acid (53.3 mCi·mmol−1, Perkin Elmer, Waltham, MA, USA) for 10 h. Subsequently, cells were processed to determine 14C incorporation into sterols by HPLC as described previously (Canfran-Duque et al., 2013).
Measurement of [3H]-oleic acid incorporation into cholesteryl esters
The cells (1.5 × 107) were incubated in the presence of the SERMs or vehicle for 24 h. Two h after staring these treatments, an emulsion containing 12 μCi [9,10-3H]-oleic acid (50 Ci·mmol−1, Hartmann Analytic, Braunschweig, Germany) – 0.15% human serum albumin was added to the media. Then the cells were processed for TLC as described in the Supporting Information.
Western blot analysis
Protein expression levels were determined by Western blotting using SDS-PAGE as detailed in the Supporting Information.
Filipin staining and fluorescence microscopy
Cells were cultured on glass coverslips previously treated with poly-D-lysine, stained with filipin for free cholesterol as described previously (Canfran-Duque et al., 2013) and examined on an Olympus BX51 microscope (Olympus Optical España, Barcelona, Spain).
Quantitative real-time reverse transcription-PCR
Lymphocytes were treated or not with 60 μg·mL−1 LDL in the presence or the absence of SERMs for 8 h. Total RNA was extracted with the TriPure isolation reagent (Roche, Basel, Switzerland) and reverse transcribed with the PrimeScript RT reagent kit (Takara, Otsu, Japan). Real-time PCR amplification was performed on a LightCycler 480 using the SYBR Green I Master kit (Roche) according to the procedure previously described (Canfran-Duque et al., 2013) and using RPLP0 (coding for ribosomal protein, large, P0) as the invariant control. Primer sequences for different genes are shown in Supporting Information Table S1.
Statistical analyses
The effects of SERMs were analysed by one-way repeated measures (RM) anova. For data assessed at various doses or time points, main and interactive effects were analysed by two-way RM anova. Similarly, to assess the interaction of SERMs with other drugs or with subject groups, two-way RM anova was used. Different subject groups were compared by one-way anova. Post hoc multiple comparisons were performed by Student–Newman–Keuls test. The analyses were performed using the SigmaStat 2.0 software (Jandel Corporation, San Rafael, CA, USA).
Materials
HepG2 human hepatocytes (ATCC CRL-11997) and MOLT-4 human lymphoblastoid cells (ATCC CLR 1582) were obtained from the American Type Culture Collection. Tamoxifen, endoxifen and 7α-[9(4,4,5,5,pentafluoropentyl-sulphinyl)nonyl]oestra-1,3,5,(10)-triene-3,17β-diol (ICI 182 780) were purchased from Tocris Bioscience (Bristol, UK). Raloxifene, toremifene and lovastatin were kindly provided by Lilly (Indianapolis, IN, USA), Orion Pharma (Espoo, Finland) and Merck, Sharp and Dohme (MSD, Madrid, Spain), respectively. Lymphoprep was purchased from Nycomed Pharma AS (Zürich, Switzerland). FBS, charcoal/dextran-treated FBS, DMEM, RPMI 1640 with or without phenol red, antibiotics, non-essential amino acids, sodium pyruvate and L-glutamine were from Gibco (Life Technologies, Carlsbad, CA, USA). LPDS was prepared from FBS or charcoal/dextran-treated FBS by ultracentrifugation at a density of 1.21 kg·L−1. 1,1′-Dioctadecyl-3,3,3,3′-tetramethylindocarbocyanine perchlorate (DiI) was from Molecular Probes (Life Technologies). E2 vitamin E, filipin and poly-D-lysine were obtained from Sigma-Aldrich (St. Louis, MO, USA). All other chemicals were of analytical grade.
Results
Dose and time effects of SERMs on LDL receptor activity
To characterize the effect of tamoxifen, raloxifene and toremifene on LDLR activity, lymphocytes from NL male donors were used. First, we analysed the effect of different doses of SERMs on DiI-LDL uptake over 24 h of treatment. As shown in Figure 1A, the three SERMs dose-dependently increased DiI-LDL uptake, tamoxifen being the most active followed by toremifene, whereas raloxifene had no appreciable effect below a 5 μM dose. To ascertain the effect of the incubation time, SERMs were used at 5 μM. The three SERMs increased DiI-LDL uptake with the time of treatment (Figure 1B). Tamoxifen was again the most effective, whereas the effect of raloxifene was negligible, its trend not being significantly different from that of the control condition (Figure 1B). We also tested the effect of SERMs on DiI-LDL uptake by lymphocytes from NL female donors (Supporting Information Figure S2) and found equivalent results to those with NL male donors.
Figure 1.
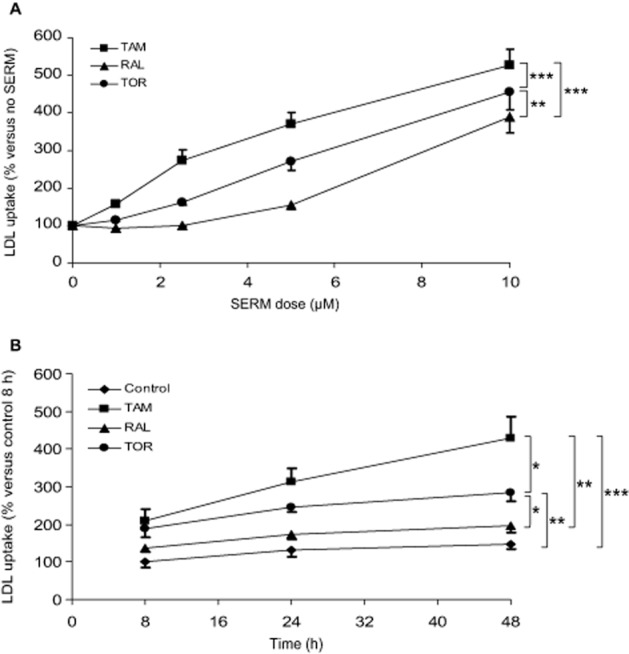
Dose and time-dependent effects of tamoxifen, raloxifene and toremifene on DiI-LDL uptake by lymphocytes from normolipidaemic men. (A) Lymphocytes from five male donors were treated with DiI-LDL and vehicle (control), tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR) for 24 h as indicated. Data (mean ± SEM) are expressed as % of the control of the same cell preparation. (B) Lymphocytes from five male donors were treated with DiI-LDL and vehicle (control) or 5 μM SERMs for the indicated times. Data (mean ± SEM) are expressed as % of the control at 8 h of the same cell preparation. The trends were compared by two-way RM anova and post hoc by Student–Newman–Keuls test. *P < 0.05, **P < 0.01, ***P < 0.001.
Effect of the SERMs on LDL receptor expression
Two different approaches were undertaken. First, lymphocytes from NL men were treated with LDL, LDL plus SERMs at a 5 μM concentration or unsupplemented LPDS as a reference for different times, then all these media were replaced by medium containing DiI-LDL and an additional 2 h incubation was performed to measure the specific DiI-LDL uptake. By this procedure, the resulting DiI-LDL uptake reflects the amount of active LDLR at the end of the treatment (Suarez et al., 2004). As shown in Figure 2A, the addition of LDL alone caused a progressive decline in DiI-LDL uptake as compared with the incubation with LPDS, consistently with the known cholesterol-induced down-regulation of LDLR expression. Treatment with any of the SERMs opposed the LDL-mediated repression throughout all the time studied, the corresponding curves being parallel to that of drug-untreated cells. Tamoxifen was the most effective in opposing the LDL effect followed by toremifene (Figure 2A).
Figure 2.

Effects of tamoxifen, raloxifene and toremifene on LDL receptor expression. (A) Lymphocytes from four male donors were treated with 10% LPDS alone or supplemented with 60 μg·mL−1 LDL plus vehicle (control) or 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR) for 6, 22 or 46 h, then the media were replaced with fresh medium containing 30 μg·mL−1 DiI-LDL and DiI-LDL uptake was measured after an additional 2 h incubation. Data (mean ± SEM) are expressed as % of the LPDS condition at the corresponding time of treatment of the same cell preparation. DiI-LDL uptake (mean ± SEM) with LPDS was 17.7 ± 4.7, 21.3 ± 4.7 and 24.1 ± 5.4 arbitrary units of fluorescence at 8, 24 and 48 h respectively. The trends were compared by two-way RM anova and post hoc by the Student–Newman–Keuls test. *P < 0.05, **P < 0.01, ***P < 0.001. (B) Representative Western blot of lymphocytes from a male donor (left). Lymphocytes were treated with LPDS or LDL plus vehicle or 5 μM SERMs for the indicated times. Then, cells were lysed and 80 μg of protein were subjected to SDS-PAGE. The right panel represents the ratio (mean ± SEM) between the densities of LDLR and GAPDH bands after 24 h of treatment for lymphocytes from four male donors. Data are expressed as % of the LPDS condition of the same cell preparation. Statistical analysis was performed by one-way RM anova and post hoc by Student–Newman–Keuls test. *P < 0.05, **P < 0.01, ***P < 0.001 versus control; #P < 0.05 versus RAL.
As a second approach, we analysed the effect of SERMs on LDLR protein levels by Western blot (Figure 2B). The results were in complete agreement with those obtained by measuring the 2 h DiI-LDL uptake as LDL markedly diminished LDLR protein levels from the earliest time point relative to LPDS (Figure 2B, left panel), and the SERMs opposed this effect, tamoxifen and raloxifene being the most and least effective respectively.
Tamoxifen has been shown to increase LDLR gene expression through a mechanism involving the production of reactive oxygen species (ROS) and inhibition of cholesterol-5,6-epoxide hydrolase, an effect that is blocked by vitamin E (Segala et al., 2013). We examined the effect of up to 500 μM vitamin E on the expression of active LDLR in the presence or the absence of the SERMs. Increasing concentrations of vitamin E showed a tendency to reduce the effect of the SERMs, but this trend was not statistically significant (Figure 3). When the effect of individual vitamin E concentrations was examined, it was found that only the dose of 250 μM significantly interacted with the SERMs (P = 0.036); this dose of vitamin E decreases the effect of raloxifene (P = 0.003) but not that of tamoxifen or toremifene.
Figure 3.
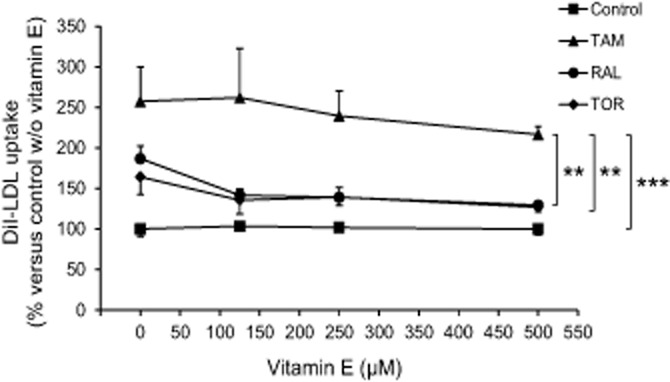
Effect of vitamin E on the SERM-mediated stimulation of the expression of active LDL receptor. Lymphocytes from four male donors were treated with 60 μg·mL−1 LDL plus vehicle (control) or 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR) and combined or not with different concentrations of vitamin E for 22 h as indicated, then the media were replaced with fresh medium containing 30 μg·mL−1 DiI-LDL and DiI-LDL uptake was measured after an additional 2 h incubation. Data (mean ± SEM) are expressed as % of the condition with LDL alone of the same cell preparation. The trends were compared by two-way RM anova and post hoc by the Student–Newman–Keuls test. **P < 0.01, ***P < 0.001.
Effect of the SERMs on cellular cholesterol distribution and content
Primary lymphocytes were treated with the SERMs for 24 h, stained with filipin and analysed by fluorescence microscopy. As shown in Figure 4, treatment with any SERM at 5 μM caused the appearance of bright peripheral granules indicative of cytoplasmic accumulation of free sterols. These granules co-localized with DiI fluorescence (Figure 4), which indicates that the accumulated cholesterol derives from internalized LDL and is retained in late endosomes/lysosomes.
Figure 4.
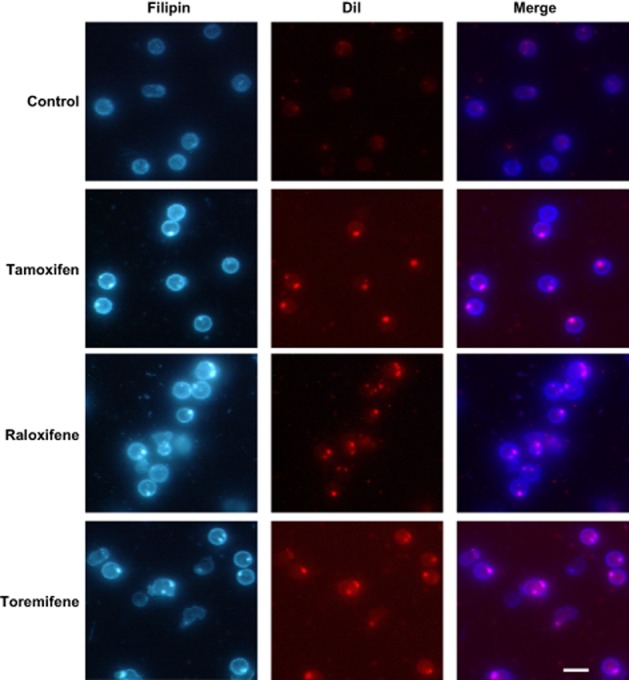
Effects of tamoxifen, raloxifene and toremifene on cholesterol distribution in lymphocytes. Lymphocytes from male donors were treated with DiI-LDL and vehicle (control) or 5 μM SERMs for 24 h. Then, cells were fixed, stained with filipin and examined for filipin and DiI fluorescence. Photographs correspond to lymphocytes from one subject representative of three. Bar, 10 μm.
The sterol content of lymphocytes was analysed. LDL addition increased cellular cholesterol levels relative to LPDS (Supporting Information Figure S3). The treatment with any of the three SERMs further increased cellular cholesterol content, although moderately, the differences not reaching statistical significance as compared with LDL alone (Supporting Information Figure S3). This is consistent with the rather few free sterol granules accumulating in SERM-treated lymphocytes (Figure 4) as compared with the great amount appearing in different cell lines (Suarez et al., 2004; de Medina et al., 2009). On the other hand, cholesterol precursors were undetectable under any condition, which is consistent with negligible cholesterol biosynthesis rates in primary lymphocytes. Actually, cholesterol biosynthesis from [14C]-acetate (acetic acid) when these cells were incubated with LPDS, which maximally stimulates such a pathway, was about 5% of that in MOLT-4 cells cultured in the same conditions. Treatment of lymphocytes with 5 μM tamoxifen barely changed [14C]-acetate incorporation into cholesterol (data not shown).
Effect of the SERMs on the SREBP pathway and ACAT activity
The increased expression of LDLR in association with an accumulation of LDL-derived cholesterol in late endosomes/lysosomes prompted us to measure the expression of SREBP target genes, which is controlled by the amount of cholesterol reaching the endoplasmic reticulum. For this, we determined the mRNA levels of LDLR and other SREBP-2 and SREBP-1 target genes, including HMGCR, FASN, SREBF2 and SREBF1 (coding for HMG-CoA reductase, fatty acid synthase, SREBP-2 and SREBP-1, respectively) in lymphocytes treated with LDL, LDL plus the SERMs or LPDS for 8 h. As shown in Figure 5, LDL addition repressed the expression of LDLR, HMGCR, FASN and SREBF2 to different extents relative to LPDS. The SERMs opposed this repression, raloxifene being the most effective. By contrast, SREBF1 expression was not reduced by LDL nor was altered by the SERMs (Figure 5). For comparison, we measured the expression of ABCA1 (coding for ATP-binding cassette, subfamily A, member 1), a liver X receptor (LXR) target (Kalaany and Mangelsdorf, 2006). LDL stimulated ABCA1 expression 20-fold and the three SERMs greatly attenuated this effect (Figure 5).
Figure 5.

Effects of tamoxifen, raloxifene and toremifene on gene expression. Lymphocytes from four male donors were treated with 10% LPDS alone or supplemented with 60 μg·mL−1 LDL plus vehicle (control) or 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR) for 8 h. The mRNA levels of the indicated genes are expressed as the relative amount of mRNA compared with that under the LPDS condition of the same cell preparation, which was set to 1. Statistical analysis was performed by one-way RM anova and post hoc by Student–Newman–Keuls test. *P < 0.05, **P < 0.01, ***P < 0.001 versus control; ##P < 0.01, ###P < 0.001 versus TAM; §§P < 0.01, §§§P < 0.001 versus TOR.
To ascertain whether the changes in SREBP target gene expression were associated with changes in the amount of active SREBP, the levels of nuclear SREBP-2 were analysed in lymphocyte nuclear extracts. The three SERMs opposed the LDL-mediated reduction of nuclear SREBP-2 (Supporting Information Figure S4).
Like SREBPs, ACAT resides in the endoplasmic reticulum, where this enzyme catalyses the esterification of cholesterol in response to an expanded cholesterol pool. The effect of the SERMs on the incorporation of [3H]-oleic acid into cholesteryl esters by lymphocytes was determined after 24 h of treatment. As shown in Table 2013a, the three SERMs inhibited ACAT activity.
Table 1.
Effect of SERMs on the incorporation of [3H]-oleic acid into cholesteryl esters by lymphocytes
| Subject 1 | Subject 2 | Subject 3 | P versus control | |
|---|---|---|---|---|
| Control | 100.0 | 100.0 | 100.0 | – |
| TAM | 32.3 | 48.8 | 60.8 | 0.001 |
| RAL | 42.0 | 70.1 | 61.7 | 0.002 |
| TOR | 52.1 | 70.1 | 80.8 | 0.003 |
Lymphocytes from three male donors were treated with 60 μg·mL−1 LDL plus vehicle (control) or 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR) for 24 h. [3H] oleic acid was added 2 h after adding the treatments. 3H-dpm·mg−1 cell protein was calculated, and the results are expressed as % of the control of the same cell preparation. Statistical analysis was performed by one-way RM anova and post hoc by Student–Newman–Keuls test.
Analysis of the effect of the combined treatment with SERMs and lovastatin on LDL receptor activity: comparison with MOLT-4 and HepG2 cells
Tamoxifen exerts a synergistic effect with lovastatin on the stimulation of LDLR activity in MOLT-4 cells (Suarez et al., 2004), but whether this property is shared with other SERMs is unknown. To test this possibility, MOLT-4 cells were treated or not with the SERMs (5 μM), lovastatin (1 μM) or both kinds of drugs combined, and the uptake of DiI-LDL over a 24 h treatment period was determined. As shown in Figure 6A, each SERM significantly increased DiI-LDL uptake, tamoxifen and toremifene surpassing the effect of raloxifene. Lovastatin addition resulted in a differential effect depending on the presence of SERMs as evidenced by the statistically significant interaction between both variables (P < 0.001). Lovastatin alone did not change DiI-LDL uptake as compared with that in the absence of any drug, which is attributable to the counter-regulatory effect of LDL (Suarez et al., 2004). However, when lovastatin was combined with the different SERMs a significantly greater uptake was observed in each case relative to the corresponding SERM alone (Figure 6A).
Figure 6.
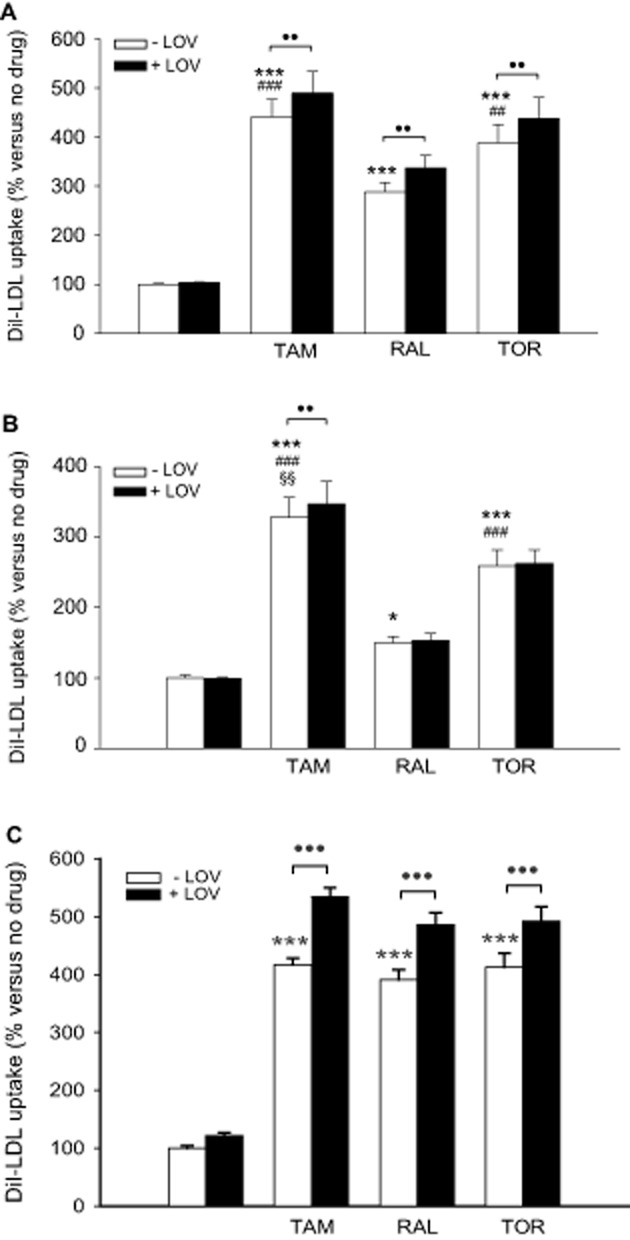
Effects of tamoxifen, raloxifene, toremifene and lovastatin on DiI-LDL uptake by MOLT-4 cells, lymphocytes and HepG2 cells. (A) MOLT-4 cells were treated with DiI-LDL and vehicle, 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR), 1 μM lovastatin (LOV) or the combinations of these drugs for 24 h as indicated. Data (mean ± SEM) correspond to five independent experiments and are expressed as % of the corresponding condition without any drug added. (B) Lymphocytes from 10 male donors were treated as indicated for MOLT-4 cells. Data (mean ± SEM) are expressed as % of the condition without any drug added of the same cell preparation. (C) HepG2 cells were treated as indicated for MOLT-4 cells and lymphocytes. Data (mean ± SEM) correspond to five independent experiments and are expressed as % of the corresponding condition without any drug added. Statistical analyses were performed by two-way RM anova and post hoc by the Student–Newman–Keuls test. ●●P < 0.01, ●●●P < 0.001 between presence and absence of LOV; *P < 0.05, ***P < 0.001 versus control; ##P < 0.01, ###P < 0.001 versus RAL; §§P < 0.01 versus TOR. For simplicity, the statistical differences between the conditions within the presence of LOV were omitted.
We next questioned whether this enhancement between SERMs and lovastatin is reproduced in primary lymphocytes treated equally as MOLT-4 cells. Also in primary lymphocytes, the SERMs significantly interacted with lovastatin (P = 0.029), and lovastatin alone did not alter DiI-LDL uptake. However, the enhancement was milder, and only the combination of lovastatin with tamoxifen resulted in a significant increase relative to the SERM alone (Figure 6B).
Given that the liver is crucial for the control of plasma cholesterol and that hepatocyte and lymphocyte cholesterol homeostasis are not identical, we studied the responses of a human hepatocyte cell line to the SERMs and lovastatin, for which HepG2 cells were used. The three SERMs were equally effective at stimulating DiI-LDL uptake (Figure 6C). Again, lovastatin was ineffective in the absence of any SERM but it potentiated the effect of each SERM (P for interaction = 0.007) to a higher extent than in lymphocytes and MOLT-4 cells (Figure 6).
Effect of ICI 182 780, 17β-oestradiol and endoxifen on LDL receptor activity
To discern the involvement of ERs in the effect of SERMs on LDLR activity, lymphocytes were treated with different SERMs or E2 in the absence or the presence of ICI 182 780, a selective ER down-regulator (Robertson, 2001). ICI 182 780 has been reported to reduce ERα protein levels (Pink and Jordan, 1996) as we observed in lymphocytes with each of the different treatments, although very slightly in the presence of E2 (Supporting Information Figure S5). ICI 182 780 did not alter the stimulating effect of any SERM on DiI-LDL uptake by lymphocytes (Figure 7A). Consistently, E2, at a wide range of concentrations, was unable to appreciably influence DiI-LDL uptake as compared with untreated cells, and ICI 182 780 addition also produced no effect (Figure 7A). Moreover, when E2 was combined with SERMs, it did not alter the effect of any of these drugs (Figure 7A).
Figure 7.
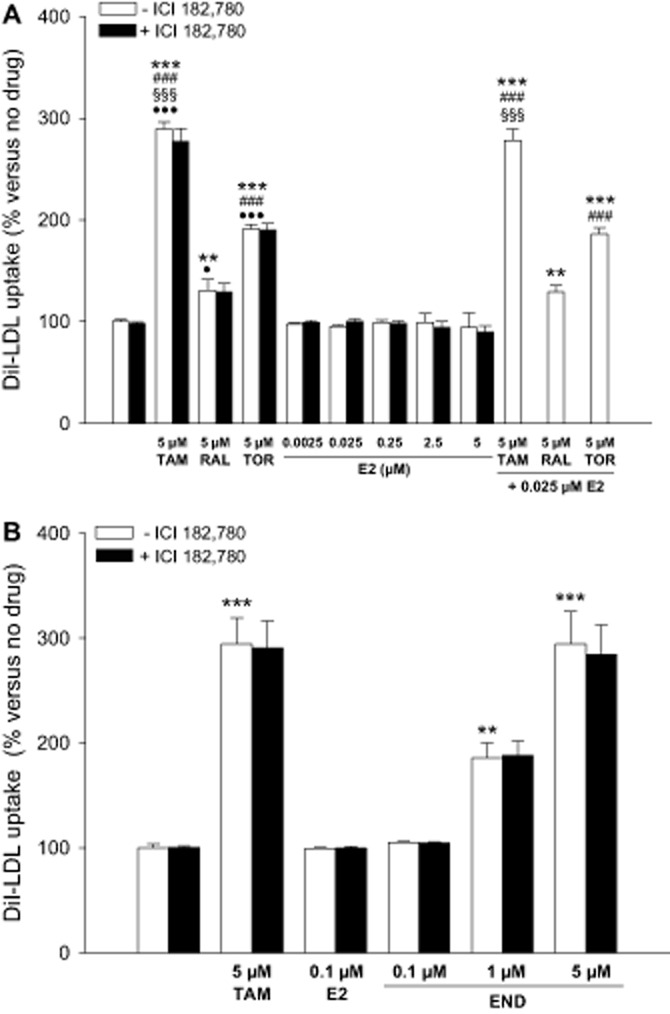
Effects of 17β-oestradiol, ICI 182 780 and endoxifen on DiI-LDL uptake by lymphocytes. (A) Lymphocytes from four male donors were treated with DiI-LDL and vehicle, tamoxifen (TAM), raloxifene (RAL), toremifene (TOR), 17β-oestradiol (E2), 1 μM ICI 182 780 or the combinations of these drugs for 24 h as indicated. Data (mean ± SEM) are expressed as % of the condition without any drug added of the same cell preparation. (B) Lymphocytes from five male donors were treated with DiI-LDL and vehicle, TAM, E2, endoxifen (END), 1 μM ICI 182 780 or the combinations of these drugs for 24 h as indicated. Data (mean ± SEM) are expressed as % of the condition without any drug added of the same cell preparation. Statistical analyses were performed by two-way RM anova and post hoc by Student–Newman–Keuls test. **P < 0.01, ***P < 0.001 versus control; ###P < 0.001 versus RAL; §§§P < 0.001 versus TOR; ●P < 0.05, ●●●P < 0.001 versus 5 μM E2. For simplicity, the statistical differences between the conditions within the presence of ICI 182 780 were omitted.
The anti-tumoural benefit of tamoxifen is thought to arise from its conversion to 4-hydroxytamoxifen and 4-hydroxy-N-desmethyltamoxifen, or endoxifen, their affinities for binding to ERs being equivalent to that of E2 and much higher than that of tamoxifen (Stearns and Rae, 2008). Because endoxifen is present at several fold higher concentrations than 4-hydroxytamoxifen in patients taking tamoxifen, the former is considered the most important active metabolite of tamoxifen (Stearns and Rae, 2008). We explored whether endoxifen stimulates, like tamoxifen, or not, like E2, lymphocyte LDLR activity. As shown in Figure 7B, endoxifen dose dependently increased DiI-LDL uptake so that at a 5 μM concentration its effect equalled that of tamoxifen at the same dose. Moreover, the effect of endoxifen was not inhibited by ICI 182 780 (Figure 7B).
Effect of the SERMs on LDL receptor activity in lymphocytes from FH patients
The effects of SERMs on LDLR activity in lymphocytes from FH patients were assessed. These were compared with the effects on lymphocytes from a group of NL subjects analysed simultaneously and who were independent of the NL subjects previously studied. Both men and women were included in this substudy (see below). As expected, lymphocytes from heterozygous carriers of LDLR mutations (HeFH; Supporting Information Table S2) had, on average, lower Bmax values for DiI-LDL binding and uptake than those from NL subjects (Supporting Information Figure S6). Additionally, three homozygous FH subjects (HoFH; Supporting Information Table S2) with residual Di-LDL uptake (Supporting Information Figure S6) were studied. One of these carried the class 4 mutation Asn825Lys, which preserves lipoprotein binding (Martinez-Botas et al., 1999) (Supporting Information Figures S1 and S6 and Table S2).
The effect of SERMs on DiI-LDL uptake by the lymphocytes from these NL, HeFH and HoFH subjects was analysed by treating the cells as above. Freshly isolated lymphocytes from the same single NL donor were included in each set of analyses (inter-assay control) to normalize the results across assays. Because both men and women were studied, we first assessed the effect of sex on DiI-LDL uptake by NL, untreated lymphocytes, and found a non-significant difference (P = 0.345) between male (110.0 ± 5.8, mean ± SEM; n = 7) and female cells (99.1 ± 7.9, n = 12). Moreover, sex had no influence on the effect of SERMs (P for interaction = 0.899), thus confirming previous results. Therefore, individuals of both sexes were analysed together in this substudy.
Consistent with the above findings, tamoxifen and, to a lesser extent, toremifene increased DiI-LDL uptake by lymphocytes from NL subjects as compared with their untreated cells, whereas raloxifene had a non-significant effect (Figure 8A). In HeFH lymphocytes (Figure 8B), tamoxifen and toremifene augmented DiI-LDL uptake, both drugs displaying a similar potency; raloxifene again caused a milder increase, although this effect reached statistical significance. On the other hand, when the effect of each SERM relative to untreated lymphocytes was compared between NL and HeFH subjects, it was found that the degree of stimulation of DiI-LDL uptake by tamoxifen was similar in both groups of subjects (250.7 ± 18.4 vs. 250.4 ± 19.2%, respectively; mean ± SEM; P = 0.988), and the same occurred with the stimulation by toremifene (189.1 ± 14.3 vs. 216.7 ± 22.5%, P = 0.285) and raloxifene (122.3 ± 7.4 vs. 138.8 ± 12.2%, P = 0.246). Actually, there was no interaction between SERMs and the subject group (P = 0.262). As regard HoFH lymphocytes, none of the SERMs exhibited an appreciable effect on DiI-LDL uptake (Figure 8C).
Figure 8.
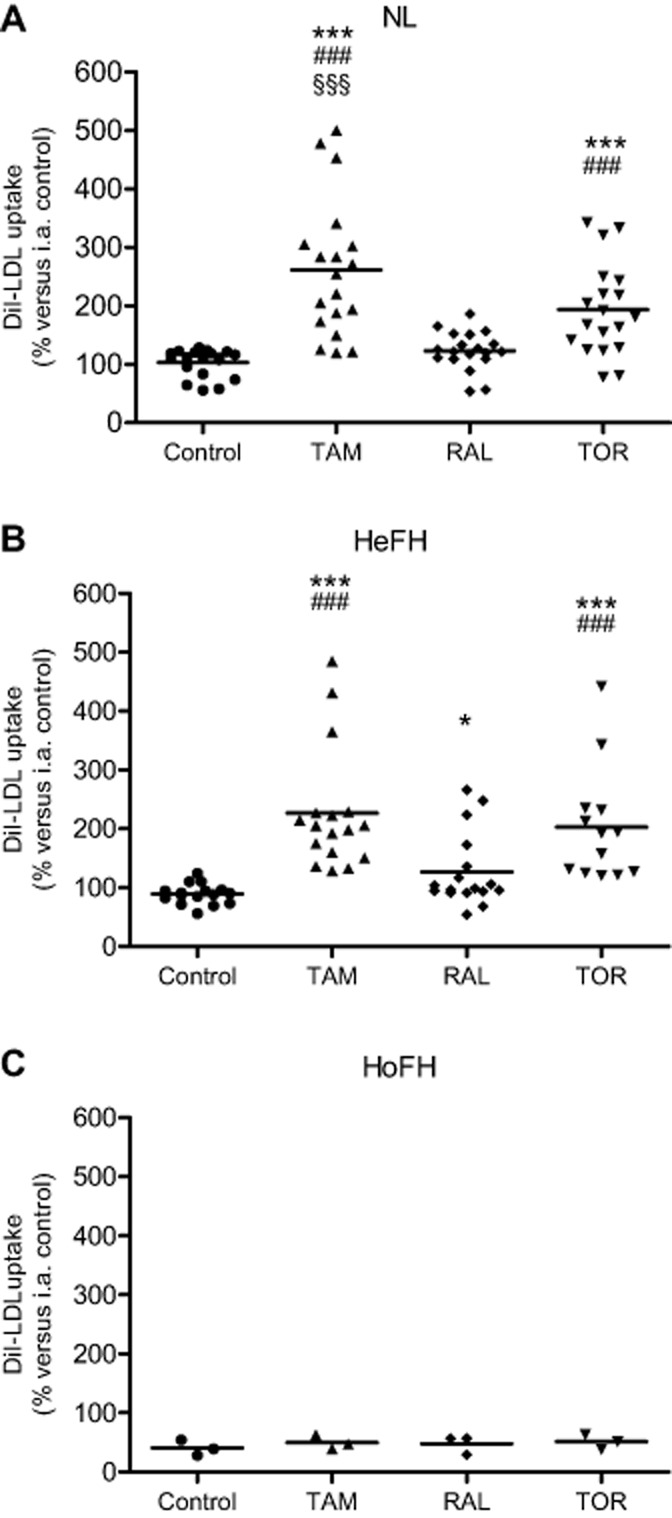
Effects of tamoxifen, raloxifene and toremifene on DiI-LDL uptake by lymphocytes from normolipidaemic and FH subjects. Lymphocytes were treated with DiI-LDL and vehicle (control) or 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR) for 24 h. (A) NL subjects. (B) HeFH patients. (C) HoFH patients. Data are expressed as % of the inter-assay (i.a.) control. Horizontal lines depict the mean value for each treatment. Statistical analysis was performed by one-way RM anova and post hoc by Student–Newman–Keuls test. *P < 0.05, ***P < 0.001 versus control; ###P < 0.001 versus RAL; §§§P < 0.001 versus TOR.
Finally, we assessed the effect of the combinations of SERMs with lovastatin on DiI-LDL uptake by NL and HeFH lymphocytes. The results obtained with NL lymphocytes essentially confirmed the previous ones. Thus, the SERMs significantly interacted with lovastatin (P = 0.026). Lovastatin alone had no effect as compared with the control condition, but the combination with tamoxifen and, in this occasion, raloxifene resulted in a slight but significant effect relative to the SERMs alone (Supporting Information Figure S7A). This analysis could be performed with the lymphocytes from 13 HeFH patients. In these cells, lovastatin was also unable to increase DiI-LDL uptake (Supporting Information Figure S7B), but there was no interaction with the SERMs (P = 0.212).
Discussion
In the present work we assessed the effect of tamoxifen, raloxifene and toremifene on LDLR activity in lymphocytes from NL and FH subjects. The results show that the three SERMs stimulate LDLR activity and that this effect is exerted by inhibiting the LDL-induced repression of LDLR expression. On a molar basis, tamoxifen was the most effective, whereas raloxifene was the least potent. The increasing effect of SERMs on LDLR expression and activity is associated with the accumulation of LDL-derived cholesterol in late endosomes/lysosomes, the stimulation of the SREBP pathway and the inhibition of both ACAT activity and the expression of an LXR target. These effects can be explained by inhibition of the transport of LDL-derived cholesterol from late endosomes/lysosomes to other organelles, especially the endoplasmic reticulum, which is the crucial regulatory compartment in cholesterol homeostasis. In essence, these findings confirm in human primary cells the previous ones obtained with tamoxifen in MOLT-4 cells (Suarez et al., 2004) and extend them to raloxifene and toremifene.
Although we studied primary lymphocytes as markers related to the lowering of plasma cholesterol levels by SERMs, these cells, in contrast to hepatocytes, are not responsible for regulating plasma cholesterol. The fact that, as found herein, the three SERMs increase LDLR activity in HepG2 hepatocytes, in which tamoxifen also causes vesicular accumulation of LDL-derived cholesterol (Suarez et al., 2004), adds to the relevance of the observations.
In lymphocytes from FH subjects, we found that, except in those from HoFH patients, SERMs also increase LDLR activity, tamoxifen and toremifene having a greater effect than raloxifene. The direct comparison of the magnitude of their effects on lymphocytes from HeFH and NL subjects supports the concept that the degree of stimulation of LDLR activity by each drug is not influenced by the LDLR deficiency of HeFH lymphocytes. The lack of effect of any SERM on HoFH cells indicates that the presence of functional LDLR is required for SERMs to be effective, thus confirming that these drugs stimulate LDL uptake through the LDLR pathway specifically. Therefore, in LDLR-defective lymphocytes that have part of their receptors functional, as occurs in HeFH subjects, SERMs are similarly effective as in lymphocytes from NL subjects.
Despite the SERM-mediated increase in LDL uptake, LDLR expression and activity remained higher than in the control condition throughout the treatment period (Figures 1B and 2A), which indicates an impairment of the LDL-induced down-regulation of LDLR. Thus, by blocking the egress of LDL-derived cholesterol from late endosomes/lysosomes, SERMs prevent its arrival to the endoplasmic reticulum where SREBPs reside. As our results indicate, in the presence of these drugs exogenous cholesterol is unable to inhibit the processing of SREBPs and, hence, the expression of SREBP-2-targeted genes, including LDLR and HMGCR, and SREBP-1-targeted genes, including FASN (Horton et al., 2002; Brown and Goldstein, 2009). These features resemble those seen in cells deficient in Niemann–Pick type C (NPC)1 or NPC2, two proteins required for cholesterol export from the lysosome and whose dysfunction causes NPC disease (Liscum and Faust, 1987). However, it is worth noting that LDLR expression in SERM-treated lymphocytes declined with time of exposure to LDL, which suggests that some cholesterol exits the lysosome and reaches the regulatory compartment. The inhibition of cholesterol esterification by the SERMs is also consistent with a failure in the transport of cholesterol to the endoplasmic reticulum, where ACAT is also located. However, a direct inhibition of this enzyme may also contribute to the lower cholesterol esterification rates, as has been demonstrated for tamoxifen in rat liver microsomal extracts, although raloxifene displayed a much weaker effect than tamoxifen in this system (de Medina et al., 2004) and no data are available for toremifene.
The prevention of LDL-induced ABCA1 expression by SERMs suggests an impairment of signalling through the LXR pathway. LXR is a nuclear receptor activated by cholesterol-derived oxysterols that stimulates the transcription of ABCA1, a gene encoding a cell surface transporter that exports cholesterol (Kalaany and Mangelsdorf, 2006). The impaired signalling through LXR in SERM-treated lymphocytes resembles the deficient generation of LDL cholesterol-derived oxysterols and subsequent activation of LXR found in NPC fibroblasts (Frolov et al., 2003). However, a role for SERM-induced transcription of SREBF2 in reducing ABCA1 mRNA levels cannot be ruled out. SREBF2, whose transcription is regulated by SREBP-2, encodes microRNA-33a, which targets the mRNA of ABCA1 (Najafi-Shoushtari et al., 2010; Rayner et al., 2010). Together, these alterations induced by SERM treatment highlight the importance of intracellular cholesterol trafficking for cholesterol homeostasis, even in the face of limited changes in cellular cholesterol content. On the other hand, a cholesterol trafficking-independent effect may also be involved in the SERM-mediated repression of ABCA1 expression. Tamoxifen has been shown to influence this expression by increasing the levels of cholesterol-5α,6α-epoxide, a cell and gene context-dependent modulator of LXR (Berrodin et al., 2010; Segala et al., 2013).
ERs appear not to be involved in the SERM-mediated up-regulation of lymphocyte LDLR. This is in agreement with present and previous (Suarez et al., 2004) results showing that SERMs increase LDLR activity in MOLT-4 cells, which do not express significant amounts of ERs (Danel et al., 1985). However, Brüning et al. (2003) reported that tamoxifen and E2 are able to enhance LDLR transcription through the binding of an ERα/Sp1 complex to a Sp1-cis-element present in the promoter. This was found using luciferase reporter gene assays in ERα-cotransfected HepG2 cells, while in non-cotransfected or ERβ-cotransfected cells both tamoxifen and E2 failed to induce transcription of the reporter gene (Brüning et al., 2003). This suggests that a high expression of ERα is required in order to observe an ER-mediated effect of these agents on LDLR transcription. The fact that, as observed herein, the SERMs increase LDLR activity in intact HepG2 cells reinforces the concept that these drugs are able to up-regulate LDLR via a mechanism that bypasses ERs.
It has been demonstrated that, although less potently than lovastatin, SERMs are able to inhibit cholesterol biosynthesis (Gylling et al., 1995; Holleran et al., 1998; Kedjouar et al., 2004; Suarez et al., 2004), which may contribute to the increase in LDLR expression. Particularly, Kedjouar et al. (2004), working with MCF-7 human breast cancer cells, found that tamoxifen and raloxifene bind to and inhibit the AEBS, a microsomal complex containing two enzymes of cholesterol biosynthesis: 3β-hydroxysterol-Δ8-Δ7-isomerase and 3β-hydroxysterol-Δ7-reductase. However, this effect is unlikely to have a significant role in primary lymphocytes as these cells have negligible cholesterol biosynthesis and no cholesterol precursors could be detected with any treatment. These findings are consistent with the very low cholesterol biosynthetic rates usually found in non-proliferating cells (Lasuncion et al., 2012).
Tamoxifen has been shown to influence LXR-dependent gene expression through a mechanism involving the production of ROS and the inhibition of cholesterol-5,6-epoxide hydrolase (Segala et al., 2013), an enzymatic activity carried out by the AEBS (de Medina et al., 2010). This leads to the accumulation of, among other cholesterol epoxides, cholesterol-5α,6α-epoxide and, indirectly, to increased LDLR gene expression in breast cancer cells (Segala et al., 2013). Because vitamin E attenuates these effects, we addressed the effect of vitamin E on the SERM-mediated increase in active LDLR expression in primary lymphocytes. Our findings suggest some involvement of ROS in the effect of the SERMs, especially that of raloxifene, but this does not appear to be the major mechanism common to these drugs in primary lymphocytes, differently to what occurs in breast cancer cells (Segala et al., 2013). It is likely that the effects of the SERMs on lymphocyte intracellular cholesterol trafficking and LDLR expression lie in the chemical properties conferred by their cationic amphiphilic structure as it has been reported that other cationic amphiphilic amines, such as U18666A, AY-9944 (Issandou et al., 2004) and antipsychotic drugs (Kristiana et al., 2010; Canfran-Duque et al., 2013), also induce lysosomal retention of lipoprotein cholesterol and up-regulation of SREBP-targeted genes. On the other hand, the higher effects of tamoxifen and toremifene relative to raloxifene on lymphocyte LDLR activity may relate to their chemical structures: tamoxifen and toremifene are triphenylethylene derivatives, only differing in the presence of a chlorine atom at position 4 in toremifene, whereas raloxifene is a benzothiophene derivative (Riggs and Hartmann, 2003).
The conversion of tamoxifen to its active metabolites 4-hydroxytamoxifen and endoxifen requires cytochrome P-450 2D6 (CYP2D6) (Stearns and Rae, 2008). Given that lymphocytes do not express detectable CYP2D6 activity (McConnachie et al., 2003), the formation of such tamoxifen derivatives is not required to up-regulate LDLR. Nevertheless, we questioned whether endoxifen, considered to be the most important active metabolite of tamoxifen (Stearns and Rae, 2008), is similar to either its precursor or E2 in their effects on LDLR activity. In contrast with E2, endoxifen elicited equal responses to tamoxifen. This suggests that the conversion of tamoxifen to endoxifen in CYP2D6-containing cells preserves the ability to increase LDLR activity through an ER-independent mechanism. It is likely that 4-hydroxytamoxifen and N-desmethyltamoxifen, the two alternative intermediates in that conversion (Stearns and Rae, 2008), also have the ability to enhance LDLR activity. All these derivative compounds share with tamoxifen the characteristic of being cationic amphiphiles with tertiary or secondary amines.
The SERM-mediated increase in LDLR activity may contribute to the cholesterol-lowering effect of these drugs in vivo. Consistently, another SERM, acolbifene, has been shown to increase LDLR expression in rat liver in association with a robust hypocholesterolaemic action (Lemieux et al., 2005). It should be noted that raloxifene, although less potent than tamoxifen and toremifene on lymphocyte LDLR activity, is effective in reducing LDL cholesterol levels (Riggs and Hartmann, 2003), suggesting that raloxifene may be more active at enhancing LDLR activity in other cell types, such as the hepatocytes. This hypothesis is in agreement with the similar magnitude of the effects of the three SERMs in HepG2 cells. The concentrations of tamoxifen we used herein are within the range of those achieved in the plasma of treated patients (Etienne et al., 1989). It is also worth mentioning that raloxifene, as well as toremifene, is usually administered to patients at higher doses than tamoxifen (Vogel et al., 2006), which may offset possible differences between their molar potencies in vivo (see Figure 1A). Other mechanisms may also contribute to the hypocholesterolaemic action of SERMs. As mentioned above, besides inhibiting cholesterol biosynthesis (Gylling et al., 1995; Holleran et al., 1998; Kedjouar et al., 2004; Suarez et al., 2004), SERMs are able to suppress ACAT activity (present results and de Medina et al., 2004) and to modulate LXR-targeted gene expression (Segala et al., 2013).
Similar to what was previously demonstrated for tamoxifen (Suarez et al., 2004), raloxifene and toremifene are able to stimulate LDLR activity in MOLT-4 cells, this effect being potentiated by lovastatin. Similar responses were observed in HepG2 cells. Despite this, lovastatin alone failed to increase DiI-LDL uptake in such cell lines, which is attributable to the predominance of the LDL-mediated repression of LDLR (Suarez et al., 2004). Interestingly, in the presence of any of the SERMs, lovastatin addition significantly increased DiI-LDL uptake above the levels attained with the corresponding SERM alone, supporting the notion that SERMs limit the availability of lipoprotein cholesterol, thus allowing lovastatin to up-regulate the receptor through the effective inhibition of cholesterol biosynthesis. As regards primary lymphocytes, lovastatin alone also produced no effect on LDLR activity. However, in these cells the expression of the enhancement between the SERMs and lovastatin was milder, and only observed with tamoxifen and, less consistently, raloxifene. The reason for this differential response to the combined treatments is unknown, but it cannot be simply explained as a function of the magnitude of the effect of SERMs on LDLR activity, lower for raloxifene than for toremifene. In contrast with NL lymphocytes, the enhancement of LDLR activity was not found in HeFH lymphocytes. It is unknown whether or not the lack of response of HeFH lymphocytes to the combined treatments relates to the LDLR deficiency of these cells.
The slight enhancement between SERMs and lovastatin of lymphocyte LDLR activity may relate to the low cholesterol biosynthesis rates in these cells. Hence, a lower susceptibility to inhibition by lovastatin is expected in circulating lymphocytes compared with MOLT-4 and HepG2 cells. Nevertheless, primary lymphocytes are a mix of cells consisting of a majority of quiescent cells, a small proportion of activated cells and a variety of lymphocyte subsets, which are variously dependent on cholesterol availability for cell expansion or renewal. It is then likely that different lymphocytes have different magnitudes of response to the combinations of these drugs. In the same line of reasoning, the possibility exists that the magnitude of the effect of the combined treatments in vivo is higher in cells and tissues with an active cholesterol metabolism, like the liver. Although the response of HepG2 cells to these treatments was stronger than in the other cells studied herein, the lack of a significant effect of lovastatin alone is not in keeping with what occurs in hepatocytes in vivo. It has been documented that statin administration to humans inhibits cholesterol biosynthesis (Naoumova et al., 1996) and increases hepatic LDLR expression (Tobert, 2003). This indicates that hepatic cholesterol biosynthesis remains active in the face of physiological concentrations of LDL, thus making this pathway susceptible to statin inhibition and, in turn, increasing LDLR expression. In this situation, the simultaneous administration of SERM may further up-regulate LDLR. Supporting this hypothesis, one study assessing the efficacy of raloxifene and low-dose simvastatin coadministration to postmenopausal women found that this treatment was more effective in reducing LDL cholesterol and apolipoprotein B levels than either monotherapy (Insull et al., 2005).
In conclusion, tamoxifen, toremifene and raloxifene, in this order, stimulate LDLR activity and expression in primary human lymphocytes from both NL and HeFH subjects by interfering with the cholesterol homeostatic mechanisms, although only a mild enhancement was found with lovastatin. The potentiation with this drug was greater in HepG2 and MOLT-4 cells. The effect of these SERMs is ER-independent but is preserved in the tamoxifen-active metabolite endoxifen. This mechanism may contribute to the LDL cholesterol lowering action these SERMs have on patients.
Acknowledgments
We thank Angela Murúa, Lorena Crespo and Gema de la Peña for excellent technical support, and Lilly, Orion Pharma and MSD for providing raloxifene, toremifene and lovastatin respectively. This work was supported by the Instituto de Salud Carlos III (PI 11/2077 to D. G.-C.) and the Ministerio de Economía y Competitividad (SAF2011-29951 to M. A. L.). The CIBERobn is an initiative of the Instituto de Salud Carlos III.
Glossary
Abbreviations
- ACAT
acyl-coenzyme A:cholesterol acyltransferase
- AEBS
anti-oestrogen binding site
- CYP2D6
cytochrome P-450 2D6
- DiI
1,1′-dioctadecyl-3,3,3,3′-tetramethylindocarbocyanineperchlorate
- E2
17β-oestradiol
- ER
oestrogen receptor
- FH
familial hypercholesterolaemia
- HeFH
heterozygous familial hypercholesterolaemia
- HMG-CoA
3-hydroxy-3-methylglutaryl-coenzyme A
- ICI 182 780
7α-[9(4,4,5,5,pentafluoropentyl-sulphinyl)nonyl]oestra-1,3,5,(10)-triene-3,17β-diol
- Insig
insulin-induced gene product
- LDL
low-density lipoprotein
- LDLR
low-density lipoprotein receptor
- LPDS
lipoprotein-deficient serum
- LXR
liver X receptor
- NL
normolipidaemic
- NPC
Niemann–Pick type C
- RAL
raloxifene
- RM
repeated measure
- ROS
reactive oxygen species
- Scap
SREBP cleavage-activating protein
- SERM
selective oestrogen receptor modulator
- SREBP
sterol regulatory element-binding protein
Author contributions
F. C. performed the experiments and analysed the data. M. E. F.-S. performed the experiments. O. P. measured the sterol content of lymphocytes. R. A., M. A., C. V. and P. M. diagnosed and recruited the patients. M. A. L. and D. G.-C. conceived the study and wrote the manuscript.
Conflict of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Exemplars of saturation binding and uptake curves used to determine the corresponding Bmax values. The saturation binding (A) and uptake (B) curves of lymphocytes from the inter-assay (i.a.) control and the FH patient homozygous for the class 4 mutation Asn825Lys (Asn825Lys Ho) are shown. Binding and uptake assays were performed as indicated in Supporting Methods. MIF, median intensity of fluorescence; AUF, arbitrary units of fluorescence.
Figure S2 Effects of tamoxifen, raloxifene and toremifene on DiI-LDL uptake by lymphocytes from normolipidaemic women. Lymphocytes from five female donors were treated with DiI-LDL and vehicle (control) or 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR) for 24 h. Data (mean ± SEM) are expressed as percentage of the corresponding control condition. Statistical analysis was performed by one-way RM anova and post hoc by the Student–Newman–Keuls test. *P < 0.05, ***P < 0.001 versus control; ##P < 0.01, ###P < 0.001 versus RAL; §P < 0.05 versus TOR.
Figure S3 Effects of tamoxifen, raloxifene and toremifene on cellular cholesterol levels in lymphocytes. Lymphocytes from five male donors were treated with 10% LPDS alone or supplemented with 60 μg·mL−1 LDL plus vehicle (control) or 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR) for 24 h. Data (mean ± SEM) are expressed in μg cholesterol·mg−1 cell protein. Statistical analysis was performed by one-way RM anova and post hoc by the Student–Newman–Keuls test. *P < 0.05, **P < 0.01 versus LPDS condition.
Figure S4 Effect of tamoxifen, raloxifene and toremifene on nuclear SREBP-2 levels in lymphocytes. Representative Western blot of lymphocytes from a male donor out of two. Lymphocytes were treated with LPDS or LDL plus vehicle or 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR) for 8 h. Then, the nuclear extracts were isolated and 40 μg of protein were subjected to SDS-PAGE. nSREBP-2, nuclear SREBP-2.
Figure S5 Effect of ICI 182 780 on the protein levels of ERα in the presence or the absence of tamoxifen, raloxifene, toremifene or 17β-oestradiol in lymphocytes. Representative Western blot of lymphocytes from a male donor out of two. Lymphocytes were treated with 60 μg·mL−1 LDL plus vehicle, or 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR), or 25 nM 17β-oestradiol (E2) and in the absence or the presence of 1 μM ICI 182 780 for 24 h. Then, cells were lysed and 100 μg of protein were subjected to SDS-PAGE.
Figure S6 Bmax values of DiI-LDL binding and uptake by lymphocytes from normolipidaemic and FH subjects. Lymphocytes were incubated with different concentrations of DiI-LDL for 2 h at 4 or 37°C to determine the Bmax values for DiI-LDL binding (A) and uptake (B), respectively, as described in Methods. Data are expressed as percentage of the inter-assay (i.a.) control. Horizontal lines depict the mean value of each group. NL, normolipidaemic; HeFH, heterozygous FH; HoFH, homozygous FH. Statistical analyses were performed by one-way anova and post hoc by the Student–Newman–Keuls test. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure S7 Effects of the combinations of tamoxifen, raloxifene and toremifene with lovastatin on DiI-LDL uptake by lymphocytes from normolipidaemic and heterozygous FH subjects. Lymphocytes from normolipidaemic (NL) (A) and heterozygous FH (HeFH) subjects (B) were treated with DiI-LDL and vehicle (control), 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR), 1 μM lovastatin (LOV) or the combination of these drugs for 24 h as indicated. Data are expressed as percentage of the inter-assay (i.a.) control. Horizontal lines depict the mean value for each treatment. Statistical analysis was performed by two-way RM anova and post hoc by the Student–Newman–Keuls test. *P < 0.05. For simplicity, the statistical differences between the conditions within the absence and within the presence of lovastatin were omitted.
Table S1 Primers for quantitative real-time RT-PCR.
Table S2 LDLR gene mutations and Bmax values of LDL binding and uptake in heterozygous and homozygous FH subjects.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Nuclear hormone receptors. Br J Pharmacol. 2013a;170:1652–1675. doi: 10.1111/bph.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013b;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali S, Buluwela L, Coombes RC. Antiestrogens and their therapeutic applications in breast cancer and other diseases. Annu Rev Med. 2011;62:217–232. doi: 10.1146/annurev-med-052209-100305. [DOI] [PubMed] [Google Scholar]
- Alonso R, Mata N, Castillo S, Fuentes F, Saenz P, Muniz O, et al. Cardiovascular disease in familial hypercholesterolaemia: influence of low-density lipoprotein receptor mutation type and classic risk factors. Atherosclerosis. 2008;200:315–321. doi: 10.1016/j.atherosclerosis.2007.12.024. [DOI] [PubMed] [Google Scholar]
- Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- Berrodin TJ, Shen Q, Quinet EM, Yudt MR, Freedman LP, Nagpal S. Identification of 5alpha, 6alpha-epoxycholesterol as a novel modulator of liver X receptor activity. Mol Pharmacol. 2010;78:1046–1058. doi: 10.1124/mol.110.065193. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Cholesterol feedback: from Schoenheimer's bottle to Scap's MELADL. J Lipid Res. 2009;50:S15–S27. doi: 10.1194/jlr.R800054-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüning JC, Lingohr P, Gillette J, Hanstein B, Avci H, Krone W, et al. Estrogen receptor-alpha and Sp1 interact in the induction of the low density lipoprotein-receptor. J Steroid Biochem Mol Biol. 2003;86:113–121. doi: 10.1016/s0960-0760(03)00263-2. [DOI] [PubMed] [Google Scholar]
- Calvo D, Gomez-Coronado D, Suarez Y, Lasuncion MA, Vega MA. Human CD36 is a high affinity receptor for the native lipoproteins HDL, LDL, and VLDL. J Lipid Res. 1998;39:777–788. [PubMed] [Google Scholar]
- Canfran-Duque A, Casado ME, Pastor O, Sanchez-Wandelmer J, de la Pena G, Lerma M, et al. Atypical antipsychotics alter cholesterol and fatty acid metabolism in vitro. J Lipid Res. 2013;54:310–324. doi: 10.1194/jlr.M026948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert JA, East CA, Bilheimer DW, Lipsky PE. Detection of familial hypercholesterolemia by assaying functional low-density-lipoprotein receptors on lymphocytes. N Engl J Med. 1986;314:879–883. doi: 10.1056/NEJM198604033141404. [DOI] [PubMed] [Google Scholar]
- Cuthbert JA, Russell DW, Lipsky PE. Regulation of low density lipoprotein receptor gene expression in human lymphocytes. J Biol Chem. 1989;264:1298–1304. [PubMed] [Google Scholar]
- Danel L, Menouni M, Cohen JH, Magaud JP, Lenoir G, Revillard JP, et al. Distribution of androgen and estrogen receptors among lymphoid and haemopoietic cell lines. Leuk Res. 1985;9:1373–1378. doi: 10.1016/0145-2126(85)90125-0. [DOI] [PubMed] [Google Scholar]
- Etienne MC, Milano G, Fischel JL, Frenay M, François E, Formento JL, et al. Tamoxifen metabolism: pharmacokinetic and in vitro study. Br J Cancer. 1989;60:30–35. doi: 10.1038/bjc.1989.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov A, Zielinski SE, Crowley JR, Dudley-Rucker N, Schaffer JE, Ory DS. NPC1 and NPC2 regulate cellular cholesterol homeostasis through generation of low density lipoprotein cholesterol-derived oxysterols. J Biol Chem. 2003;278:25517–25525. doi: 10.1074/jbc.M302588200. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. New York: McGraw-Hill; 2001. pp. 2863–2913. . In: (eds). [Google Scholar]
- Grainger DJ, Schofield PM. Tamoxifen for the prevention of myocardial infarction in humans: preclinical and early clinical evidence. Circulation. 2005;112:3018–3024. doi: 10.1161/CIRCULATIONAHA.104.531178. [DOI] [PubMed] [Google Scholar]
- Gundimeda U, Chen ZH, Gopalakrishna R. Tamoxifen modulates protein kinase C via oxidative stress in estrogen receptor-negative breast cancer cells. J Biol Chem. 1996;271:13504–13514. doi: 10.1074/jbc.271.23.13504. [DOI] [PubMed] [Google Scholar]
- Gylling H, Pyrhönen S, Mäntylä E, Mäenpää H, Kangas L, Miettinen TA. Tamoxifen and toremifene lower serum cholesterol by inhibition of delta 8-cholesterol conversion to lathosterol in women with breast cancer. J Clin Oncol. 1995;13:2900–2905. doi: 10.1200/JCO.1995.13.12.2900. [DOI] [PubMed] [Google Scholar]
- Holleran AL, Lindenthal B, Aldaghlas TA, Kelleher JK. Effect of tamoxifen on cholesterol synthesis in HepG2 cells and cultured rat hepatocytes. Metabolism. 1998;47:1504–1513. doi: 10.1016/s0026-0495(98)90078-6. [DOI] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol. 2008;9:125–138. doi: 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- Insull W, Jr, Davidson MH, Kulkarni PM, Siddhanti S, Ciaccia AV, Keech CA. Effects of raloxifene and low-dose simvastatin coadministration on plasma lipids in postmenopausal women with primary hypercholesterolemia. Metabolism. 2005;54:939–946. doi: 10.1016/j.metabol.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Issandou M, Guillard R, Boullay AB, Linhart V, Lopez-Perez E. Up-regulation of low-density lipoprotein receptor in human hepatocytes is induced by sequestration of free cholesterol in the endosomal/lysosomal compartment. Biochem Pharmacol. 2004;67:2281–2289. doi: 10.1016/j.bcp.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Jordan VC. SERMs: meeting the promise of multifunctional medicines. J Natl Cancer Inst. 2007;99:350–356. doi: 10.1093/jnci/djk062. [DOI] [PubMed] [Google Scholar]
- Kalaany NY, Mangelsdorf DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol. 2006;68:159–191. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- Kedjouar B, de Medina P, Oulad-Abdelghani M, Payre B, Silvente-Poirot S, Favre G, et al. Molecular characterization of the microsomal tamoxifen binding site. J Biol Chem. 2004;279:34048–34061. doi: 10.1074/jbc.M405230200. [DOI] [PubMed] [Google Scholar]
- Kristiana I, Sharpe LJ, Catts VS, Lutze-Mann LH, Brown AJ. Antipsychotic drugs upregulate lipogenic gene expression by disrupting intracellular trafficking of lipoprotein-derived cholesterol. Pharmacogenomics J. 2010;10:396–407. doi: 10.1038/tpj.2009.62. [DOI] [PubMed] [Google Scholar]
- Lasuncion MA, Martin-Sanchez C, Canfran-Duque A, Busto R. Post-lanosterol biosynthesis of cholesterol and cancer. Curr Opin Pharmacol. 2012;12:717–723. doi: 10.1016/j.coph.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Lemieux C, Gelinas Y, Lalonde J, Labrie F, Cianflone K, Deshaies Y. Hypolipidemic action of the SERM acolbifene is associated with decreased liver MTP and increased SR-BI and LDL receptors. J Lipid Res. 2005;46:1285–1294. doi: 10.1194/jlr.M400448-JLR200. [DOI] [PubMed] [Google Scholar]
- Liscum L, Faust JR. Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts. J Biol Chem. 1987;262:17002–17008. [PubMed] [Google Scholar]
- Maggo SD, Kennedy MA, Clark DW. Clinical implications of pharmacogenetic variation on the effects of statins. Drug Saf. 2011;34:1–19. doi: 10.2165/11584380-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Martinez-Botas J, Suarez Y, Reshef A, Carrero P, Ortega H, Gomez-Coronado D, et al. Impact of different low-density lipoprotein (LDL) receptor mutations on the ability of LDL to support lymphocyte proliferation. Metabolism. 1999;48:834–839. doi: 10.1016/s0026-0495(99)90214-7. [DOI] [PubMed] [Google Scholar]
- Mata P, Alonso R, Castillo S, Pocovi M. MEDPED and the Spanish Familial Hypercholesterolemia Foundation. Atheroscler Suppl. 2002;2:9–11. doi: 10.1016/s1567-5688(01)00014-9. [DOI] [PubMed] [Google Scholar]
- McConnachie LA, Phillips B, Bajpai M, Shen DD, Ho RJ. Only truncated, not complete cytochrome p450 2D6 RNA transcript and no detectable enzyme activity are expressed in human lymphocytes. Drug Metab Dispos. 2003;31:1103–1107. doi: 10.1124/dmd.31.9.1103. [DOI] [PubMed] [Google Scholar]
- de Medina P, Payre BL, Bernad J, Bosser I, Pipy B, Silvente-Poirot S, et al. Tamoxifen is a potent inhibitor of cholesterol esterification and prevents the formation of foam cells. J Pharmacol Exp Ther. 2004;308:1165–1173. doi: 10.1124/jpet.103.060426. [DOI] [PubMed] [Google Scholar]
- de Medina P, Payre B, Boubekeur N, Bertrand-Michel J, Terce F, Silvente-Poirot S, et al. Ligands of the antiestrogen-binding site induce active cell death and autophagy in human breast cancer cells through the modulation of cholesterol metabolism. Cell Death Differ. 2009;16:1372–1384. doi: 10.1038/cdd.2009.62. [DOI] [PubMed] [Google Scholar]
- de Medina P, Paillasse MR, Segala G, Poirot M, Silvente-Poirot S. Identification and pharmacological characterization of cholesterol-5,6-epoxide hydrolase as a target for tamoxifen and AEBS ligands. Proc Natl Acad Sci U S A. 2010;107:13520–13525. doi: 10.1073/pnas.1002922107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, et al. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naoumova RP, Marais AD, Mountney J, Firth JC, Rendell NB, Taylor GW, et al. Plasma mevalonic acid, an index of cholesterol synthesis in vivo, and responsiveness to HMG-CoA reductase inhibitors in familial hypercholesterolaemia. Atherosclerosis. 1996;119:203–213. doi: 10.1016/0021-9150(95)05649-1. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickar JH, MacNeil T, Ohleth K. SERMs: progress and future perspectives. Maturitas. 2010;67:129–138. doi: 10.1016/j.maturitas.2010.05.009. [DOI] [PubMed] [Google Scholar]
- Pink JJ, Jordan VC. Models of estrogen receptor regulation by estrogens and antiestrogens in breast cancer cell lines. Cancer Res. 1996;56:2321–2330. [PubMed] [Google Scholar]
- Poirot M, Silvente-Poirot S, Weichselbaum RR. Cholesterol metabolism and resistance to tamoxifen. Curr Opin Pharmacol. 2012;12:683–689. doi: 10.1016/j.coph.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regitz-Zagrosek V, Wintermantel TM, Schubert C. Estrogens and SERMs in coronary heart disease. Curr Opin Pharmacol. 2007;7:130–139. doi: 10.1016/j.coph.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Riggs BL, Hartmann LC. Selective estrogen-receptor modulators – mechanisms of action and application to clinical practice. N Engl J Med. 2003;348:618–629. doi: 10.1056/NEJMra022219. [DOI] [PubMed] [Google Scholar]
- Robertson JF. ICI 182,780 (Fulvestrant) – the first oestrogen receptor down-regulator – current clinical data. Br J Cancer. 2001;85(Suppl. 2):11–14. doi: 10.1054/bjoc.2001.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segala G, de Medina P, Iuliano L, Zerbinati C, Paillasse MR, Noguer E, et al. 5,6-Epoxy-cholesterols contribute to the anticancer pharmacology of tamoxifen in breast cancer cells. Biochem Pharmacol. 2013;86:175–189. doi: 10.1016/j.bcp.2013.02.031. [DOI] [PubMed] [Google Scholar]
- Silvente-Poirot S, Poirot M. Cholesterol and cancer, in the balance. Science. 2014;343:1445–1446. doi: 10.1126/science.1252787. [DOI] [PubMed] [Google Scholar]
- Stearns V, Rae JM. Pharmacogenetics and breast cancer endocrine therapy: CYP2D6 as a predictive factor for tamoxifen metabolism and drug response? Expert Rev Mol Med. 2008;10:e34. doi: 10.1017/S1462399408000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez Y, Fernandez C, Gomez-Coronado D, Ferruelo AJ, Davalos A, Martinez-Botas J, et al. Synergistic upregulation of low-density lipoprotein receptor activity by tamoxifen and lovastatin. Cardiovasc Res. 2004;64:346–355. doi: 10.1016/j.cardiores.2004.06.024. [DOI] [PubMed] [Google Scholar]
- Tobert JA. Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nat Rev Drug Discov. 2003;2:517–526. doi: 10.1038/nrd1112. [DOI] [PubMed] [Google Scholar]
- Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA. 2006;295:2727–2741. doi: 10.1001/jama.295.23.joc60074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Exemplars of saturation binding and uptake curves used to determine the corresponding Bmax values. The saturation binding (A) and uptake (B) curves of lymphocytes from the inter-assay (i.a.) control and the FH patient homozygous for the class 4 mutation Asn825Lys (Asn825Lys Ho) are shown. Binding and uptake assays were performed as indicated in Supporting Methods. MIF, median intensity of fluorescence; AUF, arbitrary units of fluorescence.
Figure S2 Effects of tamoxifen, raloxifene and toremifene on DiI-LDL uptake by lymphocytes from normolipidaemic women. Lymphocytes from five female donors were treated with DiI-LDL and vehicle (control) or 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR) for 24 h. Data (mean ± SEM) are expressed as percentage of the corresponding control condition. Statistical analysis was performed by one-way RM anova and post hoc by the Student–Newman–Keuls test. *P < 0.05, ***P < 0.001 versus control; ##P < 0.01, ###P < 0.001 versus RAL; §P < 0.05 versus TOR.
Figure S3 Effects of tamoxifen, raloxifene and toremifene on cellular cholesterol levels in lymphocytes. Lymphocytes from five male donors were treated with 10% LPDS alone or supplemented with 60 μg·mL−1 LDL plus vehicle (control) or 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR) for 24 h. Data (mean ± SEM) are expressed in μg cholesterol·mg−1 cell protein. Statistical analysis was performed by one-way RM anova and post hoc by the Student–Newman–Keuls test. *P < 0.05, **P < 0.01 versus LPDS condition.
Figure S4 Effect of tamoxifen, raloxifene and toremifene on nuclear SREBP-2 levels in lymphocytes. Representative Western blot of lymphocytes from a male donor out of two. Lymphocytes were treated with LPDS or LDL plus vehicle or 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR) for 8 h. Then, the nuclear extracts were isolated and 40 μg of protein were subjected to SDS-PAGE. nSREBP-2, nuclear SREBP-2.
Figure S5 Effect of ICI 182 780 on the protein levels of ERα in the presence or the absence of tamoxifen, raloxifene, toremifene or 17β-oestradiol in lymphocytes. Representative Western blot of lymphocytes from a male donor out of two. Lymphocytes were treated with 60 μg·mL−1 LDL plus vehicle, or 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR), or 25 nM 17β-oestradiol (E2) and in the absence or the presence of 1 μM ICI 182 780 for 24 h. Then, cells were lysed and 100 μg of protein were subjected to SDS-PAGE.
Figure S6 Bmax values of DiI-LDL binding and uptake by lymphocytes from normolipidaemic and FH subjects. Lymphocytes were incubated with different concentrations of DiI-LDL for 2 h at 4 or 37°C to determine the Bmax values for DiI-LDL binding (A) and uptake (B), respectively, as described in Methods. Data are expressed as percentage of the inter-assay (i.a.) control. Horizontal lines depict the mean value of each group. NL, normolipidaemic; HeFH, heterozygous FH; HoFH, homozygous FH. Statistical analyses were performed by one-way anova and post hoc by the Student–Newman–Keuls test. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure S7 Effects of the combinations of tamoxifen, raloxifene and toremifene with lovastatin on DiI-LDL uptake by lymphocytes from normolipidaemic and heterozygous FH subjects. Lymphocytes from normolipidaemic (NL) (A) and heterozygous FH (HeFH) subjects (B) were treated with DiI-LDL and vehicle (control), 5 μM tamoxifen (TAM), raloxifene (RAL) or toremifene (TOR), 1 μM lovastatin (LOV) or the combination of these drugs for 24 h as indicated. Data are expressed as percentage of the inter-assay (i.a.) control. Horizontal lines depict the mean value for each treatment. Statistical analysis was performed by two-way RM anova and post hoc by the Student–Newman–Keuls test. *P < 0.05. For simplicity, the statistical differences between the conditions within the absence and within the presence of lovastatin were omitted.
Table S1 Primers for quantitative real-time RT-PCR.
Table S2 LDLR gene mutations and Bmax values of LDL binding and uptake in heterozygous and homozygous FH subjects.