

Abstract
Chronic thromboembolic pulmonary hypertension (CTEPH) is one of the primary causes of severe pulmonary hypertension. In order to identify long noncoding RNAs (lncRNAs) that may be involved in the development of CTEPH, comprehensive lncRNA and messenger RNA (mRNA) profiling of endothelial tissues from the pulmonary arteries of CTEPH patients was conducted with microarray analysis. Differential expression of 185 lncRNAs was observed in the CTEPH tissues compared with healthy control tissues. Further analysis identified 464 regulated enhancer-like lncRNAs and overlapping, antisense or nearby mRNA pairs. Coexpression networks were subsequently constructed and investigated. The expression levels of the lncRNAs, NR_036693, NR_027783, NR_033766 and NR_001284, were significantly altered. Gene ontology and pathway analysis demonstrated the potential role of lncRNAs in the regulation of central process, including inflammatory response, response to endogenous stimulus and antigen processing and presentation. The use of bioinformatics may help to uncover and analyze large quantities of data identified by microarray analyses, through rigorous experimental planning, statistical analysis and the collection of more comprehensive data regarding CTEPH. The results of the present study provided evidence which may be helpful in future studies on the diagnosis and management of CTEPH.
Keywords: chronic thromboembolic pulmonary hypertension, mRNA, long noncoding RNA, microarray, gene ontology
Introduction
Pulmonary endarterectomy (PEA) can prevent mortality due to right ventricle failure (1), which occurs in chronic thromboembolic pulmonary hypertension (CTEPH), a type-4 pulmonary hypertension (2). A number of gene expression studies conducted in postmortem lung tissue samples from patients with CTEPH have indicated that aberrant processing of the messenger RNA (mRNA) transcriptome in CTEPH may provide a mechanistic convergence between the diverse genetic heritability of this disease and the disruption of fundamental signaling pathways, resulting in the common CTEPH phenotype. Studies conducted in pulmonary endothelial cells have identified differentially expressed genes, which may be involved in the pathogenesis of CTEPH (3,4). However, gene expression regulation is a complex process, which involves an interplay between DNA sequence variation, chromatin and epigenetic modifications, protein transcription factors and regulatory noncoding RNAs.
A previous study by our group examining the role of the transcriptome in pulmonary artery endomembrane samples demonstrated that the abnormal expression of mRNA transcripts may represent a point of convergence in the otherwise heterogeneous genomics, underlying the development of CTEPH (5). However, the regulatory RNAs inducing the aberrant mRNA expression levels observed in CTEPH have not been concurrently assessed. To the best of our knowledge, only a single study conducted in the tissues of CTEPH patients has demonstrated that miRNA-759 may influence the susceptibility to the development of CTEPH (6). Long noncoding RNAs (lncRNAs), a novel class of regulatory RNAs, have been shown to be involved in certain fundamental events in gene regulation. However, the role of these molecules in the pathogenesis of CTEPH remains unclear (7).
lncRNAs are noncoding RNA molecules that are longer than 200 nucleotides. lncRNAs were originally considered to be ‘transcriptional noise’; however, their involvement in important mechanisms controlling the gene expression regulation has been demonstrated. These mechanisms include targeting transcription factors, initiating chromatin remodeling, directing methylation complexes and blocking proximate transcription (8). Aberrant regulation of lncRNAs has been shown to be associated with a number of diseases, including certain forms of cancer (9,10). Numerous lncRNAs have been identified through large-scale analyses of full-length cDNA sequences in humans, mice and flies. lncRNA molecules have been shown to play an important role in the control of imprinting, cell differentiation, immune response, pathogenesis of various human diseases, tumorigenesis and other biological processes (11–15). However, the expression and biological function of lncRNAs in CTEPH remains to be elucidated.
The aim of the present study was to determine whether the dysregulation of the lncRNA expression is involved in the molecular pathogenesis of CTEPH. The lncRNAs expression profiles of five CTEPH patients were compared with healthy control individuals (normal tissues). In addition, an assessment of the transcriptional differences in all known protein-coding mRNAs was conducted.
Materials and methods
Patient samples
In total, five patients diagnosed with CTEPH (male, 2; female, 3; mean age, 38.2 years; age range, 17–52 years) who had been referred to Beijing Chaoyang Hospital (Capital Medical University, Beijing, China) were recruited to the study. The study was approved by the relevant ethics committee of Beijing Chaoyang University. All the patients provided informed written consent prior to participation in the study. Pulmonary angiography and right heart catheterization were used in the diagnosis of CTEPH and determination of cardiopulmonary hemodynamics (16). Mean pulmonary artery pressure >25 mmHg at rest or >30 mmHg during exercise was considered to indicate the presence of pulmonary hypertension. Pulmonary vascular resistance (PVR) and the six-minute walk test (6-MWT) were hemodynamic variables applied to assess the cardiopulmonary function and prognosis of the CTEPH patients. At inclusion, all the patients received oral anticoagulants for a minimum of 6 months and underwent PEA in accordance with the guidelines of the Beijing Chao-Yang Hospital (Beijing, China). In addition, healthy control samples were obtained from five lung transplant donors. The control subjects and patients were matched according to age and gender. Written informed consent was obtained from the healthy controls or their families.
RNA extraction
To prepare the samples for microarray profiling, total RNA was isolated from the CTEPH patient and normal tissue samples using TRIzol™ reagent (Invitrogen Life Technologies, Inc., Burlington, ON, Canada) and purified using an RNeasy Mini kit (Qiagen, Hilden, German), including a DNase digestion treatment. RNA concentrations were determined by measuring the sample absorbance at 260 nm with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA, USA). A260/A280 ratio values of 1.8–2.1 were set as the quality control standard.
Microarray profiling
cDNA was generated via reverse transcription of RNA obtained from pulmonary artery endothelium samples using a reverse transcription kit (miScript II RT kit; Qiagen). cDNA obtained from pulmonary artery endothelium samples of the CTEPH patients or normal controls was hybridized to GeneChip® Human Gene 2.0 ST arrays (Affymetrix, Inc., Santa Clara, CA, USA), according to the manufacturer’s instructions. Affymetrix Expression Console™ software (version 1.2.1; Affymetrix, Inc.) was used for microarray analysis. The raw data (CEL files) were normalized at the transcript level using the robust multiarray average method (17), followed by median summarization of the transcript expression levels. Subsequently, gene-level data were filtered to include only the probe sets derived from the ‘core’ metaprobe list, representing the reference sequence (RefSeq) genes.
Significant differential gene analysis
The random variance model (RVM) t-test was used to filter differentially expressed genes in the control and CTEPH groups. This test was selected since it can effectively raise the degrees of freedom when investigating small samples. Following significance analysis of microarrays and false discovery rate (FDR) analysis, the differentially expressed genes were identified according to the predetermined P-value threshold using BRB-ArrayTools (version 4.3.0 Beta 1; National Cancer Institute, Bethesda, MD, USA). P<0.05 was considered to indicate a statistically significant difference (18–20). The differentially expressed genes were subjected to unsupervised hierarchical clustering (Cluster 3.0; Stanford University, Stanford, CA, USA) and TreeView version 3.0 analysis (Stanford University).
Coexpression network
Gene coexpression networks were constructed based on the normalized signal intensity of the specific expression of genes, in order to identify the interactions among genes (21). For each pair of genes, the Pearson correlation coefficient was calculated in order to identify pairs with a significant correlation, thus enabling construction of the network (22).
When conducting a network analysis, the simplest and most important measure of gene centrality within a network is degree centrality. Degree centrality is defined as the number of links a particular node has to other nodes in the network (23). Furthermore, k-cores were introduced using graph theory in order to simplify the graph topology analysis and investigate various network properties. A k-core of a network consists of a subnetwork where all the nodes are connected to at least k other genes. A k-core of a protein-protein interaction network usually contains cohesive groups of proteins with similar functions (23,24).
Network structure analysis aims to locate core regulatory factors (genes). Within a network, core regulatory factors connect the majority of nearby genes and have the highest degree centralities. When evaluating different networks, core regulatory factors are determined by the degree differences between the CTEPH and normal tissue samples (25), since they show the highest degree differences. The network was constructed using Cytoscape software version 2.8.3 (Cytoscape Consortium, San Diego, CA, USA).
Gene ontology (GO) analysis
Based on the gene ontology database (http://www.geneontology.org/; accessed on January 15, 2014), the significance level of GO terms for the CTEPH-associated differentially expressed genes was analyzed by two-side Fisher’s exact test and χ2 test using the Database for Annotation, Visualization and Integrated Discovery (DAVID) software version 6.7 (http://david.abcc.ncifcrf.gov/home.jsp; accessed on January 11, 2014; National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA) (26). Differentially expressed genes were analyzed independently according to whether they were upregulated or downregulated. P-values were calculated for the differentially expressed genes in all GO categories. P<0.01 and FDR<0.01 were considered to indicate statistically significant results.
Pathway analysis
Based on the Kyoto Encyclopedia of Genes and Genomes database (http://www.genome.jp/kegg/; accessed on January 12, 2014; Kanehisa Laboratories, Kyoto, Japan), the significance levels of CTEPH-associated differentially expressed gene pathways were analyzed using Pathway-Express version 1.0 (Intelligent Systems and Bioinformatics Laboratory, Detroit, MI, USA) (27,28). The occurrence of significant differences from the expected values was assessed using a two-sided binomial distribution. The number of differentially expressed genes corresponding to each pathway category was counted and compared with the number of genes expected for each pathway category. All the signaling pathways were analyzed using γ P<0.05, provided by the impact analysis, as the threshold indicating a statistically significant difference.
Results
Overview of lncRNA profiles
Based on the lncRNAs expression profiles, a number of differentially expressed lncRNAs were identified between the CTEPH and healthy control samples. The expression profiles of lncRNAs in the paired samples were determined by calculating the log fold change of CTEPH/control samples. Due to the limited sample size, FDR and P-values were calculated from normalized expression levels. Hundreds of differentially expressed human lncRNAs were identified using the RefSeq (www.ncbi.nlm.nih.gov/refseq), Ensembl (www.ensembl.org), lncRNAdb (www.lncrnadb.org), Broad Institute, Human Body Map lincRNAs (www.broadinstitute.org/genome_bio/human_lincrnas/) and transcripts of uncertain coding potential catalog (http://www.broadinstitute.org/genome_bio/human_lincrnas/?q=TUCP_transcripts_catalog) databases in the CTEPH patients and healthy controls.
Using the microarray data, the expression levels of lncRNAs in the five CTEPH tissue samples were compared with the matched normal tissue samples. In total, 185 lncRNAs were identified in which the expression levels were found to be significantly different between the two groups. The characteristics of the five CTEPH patients and five normal controls are shown in Table I. The procedure used to obtain a sequence may vary depending on the database used for lncRNA classification. While tens of thousands lncRNAs were investigated in normal and diseased tissues, only a few hundreds lncRNAs were found to be significantly upregulated or downregulated. Thus, the upregulation or downregulation of lncRNAs was used to distinguish the CTEPH patient tissues from healthy tissues (Fig. 1). Compared with normal tissues, NR_033766 was the most evidently downregulated lncRNA, whereas TCONS_l2_00010131-XLOC_l2_005462 was upregulated to the greatest degree (Table II). Therefore, downregulated lncRNAs were shown to be more prevalent compared with upregulated lncRNAs in the CTEPH group.
Table I.
Clinical characteristics of study participants.
| Group | n | Age, years (mean ± SD) | Gender, n (M/F) | Median mPAP (range), mmHg | PVR, dyn × sec × cm−5 | 6WMT, m |
|---|---|---|---|---|---|---|
| Healthy control | 5 | 35.3±10.6 | 2/3 | - | - | - |
| CTEPH patients | 5 | 38.2±14.7 | 2/3 | 55 (33–78) | 1,075.4 | 454.6667 |
SD, standard deviation; M, male; F, female; mPAP, mean pulmonary artery pressure; PVR, pulmonary vascular resistance; 6MWT, six-minute walk test; CTEPH, chronic thromboembolic pulmonary hypertension.
Figure 1.
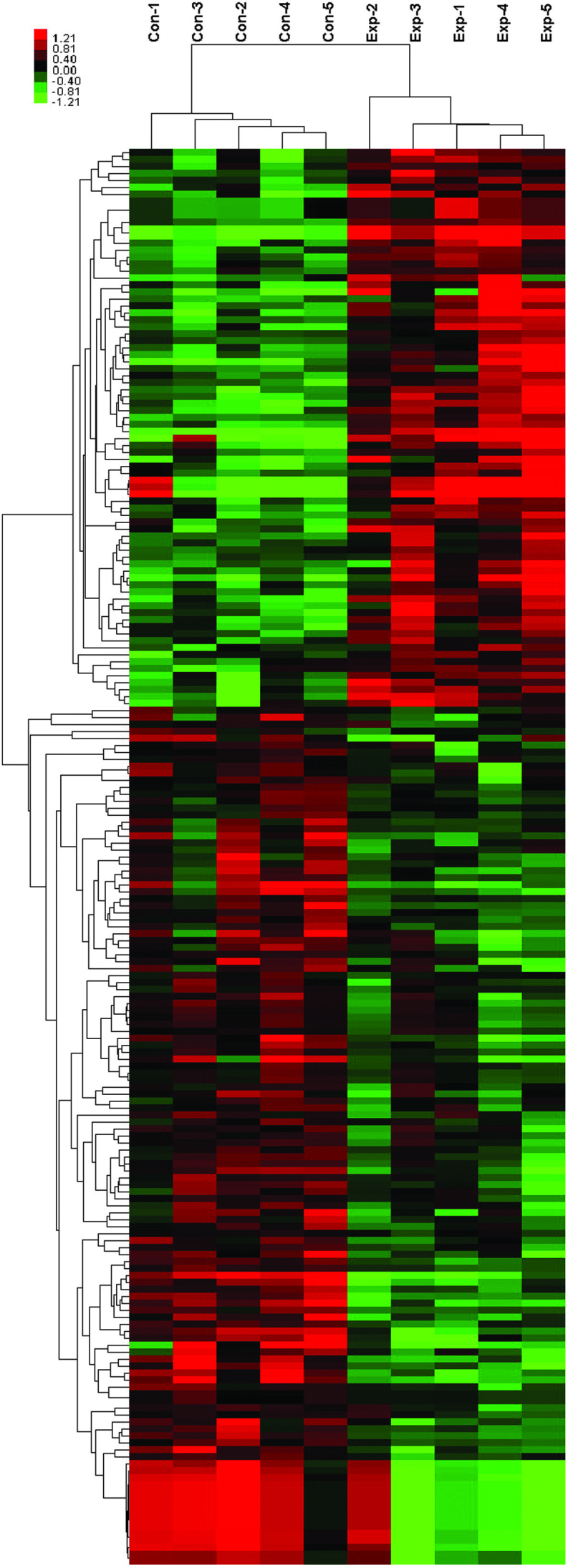
Unsupervised classification of samples from chronic thromboembolic pulmonary hypertension patients and healthy controls, based on long noncoding RNA expression profiling. The data are depicted as a data matrix with rows representing the probes and columns representing the samples. The expression levels are presented according to the color scale shown at the top. Red and green indicate the expression levels above and below the median, respectively. The magnitude of deviation from the median is represented by the color saturation. Con, control group; Exp, experimental group.
Table II.
Collection of the top ten deregulated lncRNAs detected using microarray analysis in ten CTEPH and control samples.
| Downregulated in CTEPH tissues | Upregulated in CTEPH tissues | ||||
|---|---|---|---|---|---|
|
|
|
||||
| lncRNA | P-value | Fold changea | lncRNA | P-value | Fold changea |
| NR_003679 | 5.70×10−6 | 0.45 | TCONS_l2_00010131-XLOC_l2_005462 | 3.00×10−5 | 5.89 |
| NR_033766 | 2.14×10−5 | 0.16 | TCONS_l2_00011539-XLOC_l2_005705 | 3.00×10−5 | 5.89 |
| TCONS_l2_00004769-XLOC_l2_002469 | 3.52×10−5 | 0.41 | NR_026544 | 1.16×10−4 | 2.61 |
| TCONS_00023959-XLOC_011280 | 4.21×10−5 | 0.49 | TCONS_00009277-XLOC_004803 | 1.18×10−4 | 1.82 |
| TCONS_00023957-XLOC_011280 | 4.96×10−5 | 0.52 | TCONS_l2_00016084-XLOC_l2_008434 | 1.24×10−4 | 2.02 |
| NR_026799 | 6.78×10−5 | 0.39 | NR_028406 | 2.36×10−4 | 2.17 |
| TCONS_l2_00020176-XLOC_l2_010319 | 9.89×10−5 | 0.32 | TCONS_00000337-XLOC_000468 | 4.50×10−4 | 2.42 |
| NR_026985 | 1.29×10−4 | 0.46 | NR_002433 | 5.00×10−4 | 2.46 |
| TCONS_00000192-XLOC_000173 | 1.42×10−4 | 0.28 | TCONS_00028854-XLOC_013966 | 5.18×10−4 | 1.89 |
| NR_026597 | 1.56×10−4 | 0.45 | NR_033652 | 5.66×10−4 | 1.91 |
Fold change vs. healthy control tissues.
lncRNA, long noncoding RNA; CTEPH, chronic thromboembolic pulmonary hypertension.
Overview of mRNA profiles
In total, ≤30,654 coding transcripts were detected in the ten samples that were examined. Using the RVM t-test method, 880 genes were found to be upregulated and 734 genes were found to be downregulated in the CTEPH samples, compared with the healthy controls. The results supported the hypothesis that CTEPH is a metabolic disease as the mRNA expression level of oxidized low-density lipoprotein receptor 1 showed the greatest upregulation, while the mRNA expression level of chordin-like 1 showed the greatest downregulation. Therefore, upregulation of mRNA expression levels was more prevalent compared with downregulation in the CTEPH group.
Analysis of nearby lncRNAs and mRNAs
Previous studies have used chromatin-state maps to identify 3,019 lncRNAs with a clear evolutionary conservation, which are associated with distinct and diverse biological processes (such as cell proliferation), RNA binding complexes, immune surveillance, embryonic stem cell pluripotency, neuronal processes, morphogenesis, gametogenesis and muscle development (29,30). Among the 185 differentially expressed lncRNAs identified in the present study, 74 lncRNAs were shown to have differentially expressed mRNAs that were overlapping, antisense or nearby. Further analysis resulted in the identification of nine pairs of differentially expressed lncRNAs overlapping with mRNAs, nine pairs of lncRNAs and antisense mRNAs, 340 lncRNAs located upstream of mRNAs (distance, <300 kb) and 106 lncRNAs located downstream of mRNAs (distance, <300 kb) in each comparison between CTEPH and normal control tissues. Among the 464 lncRNA-mRNA pairs, the expression regulation of 442 lncRNAs and nearby coding genes was in the same direction (up or down), whereas the expression of 22 pairs was regulated in opposite directions.
Construction of the coding-noncoding gene coexpression network
The correlation analysis among differentially expressed lncRNAs and mRNAs was used to construct a coding-noncoding (CNC) gene coexpression network. LncRNAs and mRNAs having Pearson correlation coefficients ≥0.97 were selected and the network was constructed using the Cytoscape software. Within this coexpression network, the CTEPH-CNC network node consisted of 129 lncRNAs and 275 mRNAs, whereas the normal control-CNC network node consisted of 134 lncRNAs and 294 mRNAs (Fig. 2). A total of 832 network nodes in the two networks formed 3,239 coexpression pairs of lncRNAs and mRNAs, with positively correlated expression in the majority of pairs. Investigation of the CNC network indicated that one mRNA may correlate with numerous lncRNAs and vice versa.
Figure 2.

Coding-noncoding gene coexpression network of (A) chronic thromboembolic pulmonary hypertension and (B) normal control groups. Blue represents downregulation and red represents upregulation. Circle nodes represent messenger RNA (mRNA), while rim nodes represent long noncoding RNA (lncRNA). Solid lines represent a positive regulatory association and dashed lines represent a negative regulatory association between lncRNA and mRNA.
In order to identify the most significant RNA molecules in CTEPH, the degree of certain RNAs (k-core) in each network was normalized and the difference in connectivity (diffK), representing the differences between the two networks, was calculated. NR_036693 and NR_027783 were found to be the most differentially expressed lncRNAs, whilst the expression of the arginine vasopressin receptor 1A gene showed the greatest significant difference in the genes examined. The CNC network presented here, may implicate the inter-regulation of lncRNAs and mRNAs in the development of CTEPH.
Coexpressed coding gene function analysis
An important part of research into lncRNAs involves inferring the possible functions of nearby protein-coding genes (29,31). In the current study, the differentially expressed mRNAs of the two CNC networks were combined. Subsequently, the DAVID functional annotation chart (32,33) and pathway analysis results were used to perform functional enrichment analysis of the differentially regulated protein-coding gene and lncRNA pairs. The annotation terms showing the greatest significant differences (with the lowest P-values) were found to be immune response, inflammatory response, defense response and response to wounding (Table III). In addition, the most important signaling pathways relevant to CTEPH were found to be antigen processing and presentation, cytokine-cytokine receptor interaction and leukocyte transendothelial migration (Fig. 3). Therefore, lncRNAs are hypothesized to modulate the host response through their effect on nearby protein-coding genes.
Table III.
GO analysis.
| GO ID | Term | Regulation | P-value | FDR |
|---|---|---|---|---|
| GO:0006955 | Immune response | Up | 4.21×10−59 | 7.32×10−56 |
| GO:0006952 | Defense response | Up | 1.24×10−49 | 2.16×10−46 |
| GO:0006954 | Inflammatory response | Up | 5.61×10−44 | 9.77×10−41 |
| GO:0009611 | Response to wounding | Up | 2.88×10−43 | 5.01×10−40 |
| GO:0002684 | Positive regulation of immune system process | Up | 7.37×10−39 | 1.28×10−35 |
| GO:0001775 | Cell activation | Up | 3.95×10−29 | 6.88×10−26 |
| GO:0002696 | Positive regulation of leukocyte activation | Up | 1.01×10−27 | 1.76×10−24 |
| GO:0050867 | Positive regulation of cell activation | Up | 4.41×10−27 | 7.68×10−24 |
| GO:0045321 | Leukocyte activation | Up | 1.56×10−26 | 2.72×10−23 |
| GO:0050865 | Regulation of cell activation | Up | 9.23×10−26 | 1.61×10−22 |
| GO:0042110 | T cell activation | Up | 2.35×10−25 | 4.10×10−22 |
| GO:0002694 | Regulation of leukocyte activation | Up | 2.58×10−25 | 4.49×10−22 |
| GO:0046649 | Lymphocyte activation | Up | 6.86×10−24 | 1.19×10−20 |
| GO:0051249 | Regulation of lymphocyte activation | Up | 5.15×10−22 | 8.98×10−19 |
| GO:0051251 | Positive regulation of lymphocyte activation | Up | 5.97×10−22 | 1.04×10−18 |
| GO:0048584 | Positive regulation of response to stimulus | Up | 1.77×10−21 | 3.09×10−18 |
| GO:0042330 | Taxis | Up | 4.76×10−21 | 8.30×10−18 |
| GO:0006935 | Chemotaxis | Up | 4.76×10−21 | 8.30×10−18 |
| GO:0050870 | Positive regulation of T cell activation | Up | 4.71×10−19 | 8.21×10−16 |
| GO:0050863 | Regulation of T cell activation | Up | 1.18×10−18 | 2.06×10−15 |
| GO:0042981 | Regulation of apoptosis | Up | 5.68×10−18 | 9.89×10−15 |
| GO:0043067 | Regulation of programmed cell death | Up | 8.67×10−18 | 1.51×10−14 |
| GO:0010941 | Regulation of cell death | Up | 1.01×10−17 | 1.77×10−14 |
| GO:0001817 | Regulation of cytokine production | Up | 2.11×10−17 | 3.67×10−14 |
| GO:0050778 | Positive regulation of immune response | Up | 1.73×10−16 | 3.89×10−13 |
| GO:0007626 | Locomotory behavior | Up | 1.26×10−15 | 2.13×10−12 |
| GO:0006928 | Cell motion | Up | 2.59×10−15 | 4.45×10−12 |
| GO:0051240 | Positive regulation of multicellular organismal process | Up | 4.47×10−15 | 7.74×10−12 |
| GO:0030098 | Lymphocyte differentiation | Up | 4.87×10−15 | 8.50×10−12 |
| GO:0030217 | T cell differentiation | Up | 5.86×10−15 | 1.02×10−11 |
| GO:0007166 | Cell surface receptor linked signal transduction | Up | 7.85×10−15 | 1.37×10−11 |
| GO:0051094 | Positive regulation of developmental process | Up | 1.49×10−14 | 2.59×10−11 |
| GO:0010033 | Response to organic substance | Up | 3.12×10−14 | 5.43×10−11 |
| GO:0002521 | Leukocyte differentiation | Up | 4.00×10−14 | 6.96×10−11 |
| GO:0002252 | Immune effector process | Up | 6.25×10−14 | 1.09×10−10 |
| GO:0045619 | Regulation of lymphocyte differentiation | Up | 7.29×10−14 | 1.27×10−10 |
| GO:0045058 | T cell selection | Up | 1.03×10−13 | 1.79×10−10 |
| GO:0042127 | Regulation of cell proliferation | Up | 1.56×10−13 | 2.71×10−10 |
| GO:0007610 | Behavior | Up | 3.16×10−13 | 5.51×10−10 |
| GO:0009617 | Response to bacterium | Up | 9.56×10−13 | 1.66×10−9 |
| GO:0001819 | Positive regulation of cytokine production | Up | 1.36×10−12 | 2.36×10−9 |
| GO:0034097 | Response to cytokine stimulus | Up | 2.55×10−12 | 4.44×10−9 |
| GO:0002253 | Activation of immune response | Up | 2.73×10−12 | 4.76×10−9 |
| GO:0050670 | Regulation of lymphocyte proliferation | Up | 5.42×10−12 | 9.44×10−9 |
| GO:0045061 | Thymic T cell selection | Up | 5.98×10−12 | 1.04×10−8 |
| GO:0050900 | Leukocyte migration | Up | 6.39×10−12 | 1.11×10−8 |
| GO:0070663 | Regulation of leukocyte proliferation | Up | 6.50×10−12 | 1.13×10−8 |
| GO:0032944 | Regulation of mononuclear cell proliferation | Up | 6.50×10−12 | 1.13×10−8 |
| GO:0030097 | Hemopoiesis | Up | 7.70×10−12 | 1.34×10−8 |
| GO:0002443 | Leukocyte mediated immunity | Up | 9.29×10−12 | 1.62×10−8 |
| GO:0045087 | Innate immune response | Up | 1.23×10−11 | 2.15×10−8 |
| GO:0007243 | Protein kinase cascade | Up | 1.71×10−11 | 2.99×10−8 |
| GO:0042102 | Positive regulation of T cell proliferation | Up | 2.24×10−11 | 3.90×10−8 |
| GO:0002460 | Adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains | Up | 2.59×10−11 | 4.51×10−8 |
| GO:0002250 | Adaptive immune response | Up | 2.59×10−11 | 4.51×10−8 |
| GO:0045597 | Positive regulation of cell differentiation | Up | 2.99×10−11 | 5.20×10−8 |
| GO:0016477 | Cell migration | Up | 3.00×10−11 | 5.22×10−8 |
| GO:0002757 | Immune response-activating signal transduction | Up | 3.64×10−11 | 6.34×10−8 |
| GO:0010647 | Positive regulation of cell communication | Up | 3.66×10−11 | 6.38×10−8 |
| GO:0008284 | Positive regulation of cell proliferation | Up | 4.73×10−11 | 8.23×10−8 |
| GO:0048534 | Hemopoietic or lymphoid organ development | Up | 5.60×10−11 | 9.76×10−8 |
| GO:0050671 | Positive regulation of lymphocyte proliferation | Up | 7.41×10−11 | 1.29×10−7 |
| GO:0070665 | Positive regulation of leukocyte proliferation | Up | 9.30×10−11 | 1.62×10−7 |
| GO:0032946 | Positive regulation of mononuclear cell proliferation | Up | 9.30×10−11 | 1.62×10−7 |
| GO:0002764 | Immune response-regulating signal transduction | Up | 9.30×10−11 | 1.62×10−7 |
| GO:0002449 | Lymphocyte mediated immunity | Up | 1.02×10−10 | 1.78×10−7 |
| GO:0007242 | Intracellular signaling cascade | Up | 1.11×10−10 | 1.94×10−7 |
| GO:0002237 | Response to molecule of bacterial origin | Up | 1.23×10−10 | 2.14×10−7 |
| GO:0045089 | Positive regulation of innate immune response | Up | 1.61×10−10 | 2.80×10−7 |
| GO:0045621 | Positive regulation of lymphocyte differentiation | Up | 1.62×10−10 | 2.82×10−7 |
| GO:0031349 | Positive regulation of defense response | Up | 1.78×10−10 | 3.10×10−7 |
| GO:0002520 | Immune system development | Up | 1.86×10−10 | 3.25×10−7 |
| GO:0048870 | Cell motility | Up | 2.74×10−10 | 4.76×10−7 |
| GO:0051674 | Localization of cell | Up | 2.74×10−10 | 4.76×10−7 |
| GO:0009615 | Response to virus | Up | 3.09×10−10 | 5.39×10−7 |
| GO:0042129 | Regulation of T cell proliferation | Up | 3.30×10−10 | 5.74×10−7 |
| GO:0032496 | Response to lipopolysaccharide | Up | 3.58×10−10 | 6.24×10−7 |
| GO:0007159 | Leukocyte adhesion | Up | 4.19×10−10 | 7.29×10−7 |
| GO:0045580 | Regulation of T cell differentiation | Up | 5.34×10−10 | 9.31×10−7 |
| GO:0043065 | Positive regulation of apoptosis | Up | 5.62×10−10 | 9.78×10−7 |
| GO:0043068 | Positive regulation of programmed cell death | Up | 6.60×10−10 | 1.15×10−6 |
| GO:0009967 | Positive regulation of signal transduction | Up | 6.94×10−10 | 1.21×10−6 |
| GO:0010942 | Positive regulation of cell death | Up | 7.34×10−10 | 1.28×10−6 |
| GO:0051092 | Positive regulation of NF-κB transcription factor activity | Up | 9.07×10−10 | 1.58×10−6 |
| GO:0019882 | Antigen processing and presentation | Up | 9.44×10−10 | 1.64×10−6 |
| GO:0045088 | Regulation of innate immune response | Up | 1.03×10−9 | 1.79×10−6 |
| GO:0019221 | Cytokine-mediated signaling pathway | Up | 1.44×10−9 | 2.52×10−6 |
| GO:0019724 | B cell mediated immunity | Up | 1.55×10−9 | 2.71×10−6 |
| GO:0002504 | Antigen processing and presentation of peptide or polysaccharide antigen via MHC class II | Up | 2.18×10−9 | 3.79×10−6 |
| GO:0002221 | Pattern recognition receptor signaling pathway | Up | 5.25×10−9 | 9.14×10−6 |
| GO:0032844 | Regulation of homeostatic process | Up | 5.62×10−9 | 9.78×10−6 |
| GO:0008219 | Cell death | Up | 8.22×10−9 | 1.43×10−5 |
| GO:0032101 | Regulation of response to external stimulus | Up | 8.42×10−9 | 1.47×10−5 |
| GO:0033077 | T cell differentiation in the thymus | Up | 8.44×10−9 | 1.47×10−5 |
| GO:0006874 | Cellular calcium ion homeostasis | Up | 9.61×10−9 | 1.67×10−5 |
| GO:0016265 | Death | Up | 9.82×10−9 | 1.71×10−5 |
| GO:0002683 | Negative regulation of immune system process | Up | 1.09×10−8 | 1.90×10−5 |
| GO:0060326 | Cell chemotaxis | Up | 1.10×10−8 | 1.91×10−5 |
| GO:0002758 | Innate immune response-activating signal transduction | Up | 1.32×10−8 | 2.30×10−5 |
| GO:0002218 | Activation of innate immune response | Up | 1.32×10−8 | 2.30×10−5 |
| GO:0051090 | Regulation of transcription factor activity | Up | 1.43×10−8 | 2.49×10−5 |
| GO:0055074 | Calcium ion homeostasis | Up | 1.44×10−8 | 2.51×10−5 |
| GO:0008283 | Cell proliferation | Up | 1.52×10−8 | 2.65×10−5 |
| GO:0016064 | Immunoglobulin mediated immune response | Up | 1.60×10−8 | 2.79×10−5 |
| GO:0006915 | Apoptosis | Up | 1.80×10−8 | 3.14×10−5 |
| GO:0048545 | Response to steroid hormone stimulus | Up | 1.98×10−8 | 3.44×10−5 |
| GO:0012501 | Programmed cell death | Up | 2.53×10−8 | 4.41×10−5 |
| GO:0006875 | Cellular metal ion homeostasis | Up | 2.69×10−8 | 4.68×10−5 |
| GO:0042035 | Regulation of cytokine biosynthetic process | Up | 3.36×10−8 | 5.85×10−5 |
| GO:0045582 | Positive regulation of T cell differentiation | Up | 3.73×10−8 | 6.49×10−5 |
| GO:0051091 | Positive regulation of transcription factor activity | Up | 4.64×10−8 | 8.07×10−5 |
| GO:0001816 | Cytokine production | Up | 5.16×10−8 | 8.98×10−5 |
| GO:0055065 | Metal ion homeostasis | Up | 5.22×10−8 | 9.09×10−5 |
| GO:0042325 | Regulation of phosphorylation | Up | 5.84×10−8 | 1.02×10−4 |
| GO:0045060 | Negative thymic T cell selection | Up | 6.40×10−8 | 1.11×10−4 |
| GO:0040017 | Positive regulation of locomotion | Up | 7.34×10−8 | 1.28×10−4 |
| GO:0007249 | I-κB kinase/NF-κB cascade | Up | 7.52×10−8 | 1.31×10−4 |
| GO:0042108 | Positive regulation of cytokine biosynthetic process | Up | 7.63×10−8 | 1.33×10−4 |
| GO:0006917 | Induction of apoptosis | Up | 8.00×10−8 | 1.39×10−4 |
| GO:0012502 | Induction of programmed cell death | Up | 8.50×10−8 | 1.48×10−4 |
| GO:0051101 | Regulation of DNA binding | Up | 1.01×10−7 | 1.75×10−4 |
| GO:0002697 | Regulation of immune effector process | Up | 1.03×10−7 | 1.80×10−4 |
| GO:0030595 | Leukocyte chemotaxis | Up | 1.27×10−7 | 2.22×10−4 |
| GO:0044093 | Positive regulation of molecular function | Up | 1.28×10−7 | 2.22×10−4 |
| GO:0051174 | Regulation of phosphorus metabolic process | Up | 1.29×10−7 | 2.24×10−4 |
| GO:0019220 | Regulation of phosphate metabolic process | Up | 1.29×10−7 | 2.24×10−4 |
| GO:0050864 | Regulation of B cell activation | Up | 1.33×10−7 | 2.31×10−4 |
| GO:0043383 | Negative T cell selection | Up | 1.42×10−7 | 2.47×10−4 |
| GO:0050730 | Regulation of peptidyl-tyrosine phosphorylation | Up | 1.59×10−7 | 2.77×10−4 |
| GO:0007155 | Cell adhesion | Up | 1.65×10−7 | 2.87×10−4 |
| GO:0022610 | Biological adhesion | Up | 1.70×10−7 | 2.97×10−4 |
| GO:0043388 | Positive regulation of DNA binding | Up | 2.11×10−7 | 3.68×10−4 |
| GO:0030005 | Cellular di-, tri-valent inorganic cation homeostasis | Up | 2.29×10−7 | 3.99×10−4 |
| GO:0002822 | Regulation of adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains | Up | 2.62×10−7 | 4.55×10−4 |
| GO:0007204 | Elevation of cytosolic calcium ion concentration | Up | 2.67×10−7 | 4.65×10−4 |
| GO:0002819 | Regulation of adaptive immune response | Up | 3.07×10−7 | 5.35×10−4 |
| GO:0006916 | Anti-apoptosis | Up | 3.21×10−7 | 5.58×10−4 |
| GO:0055066 | Di-, tri-valent inorganic cation homeostasis | Up | 4.78×10−7 | 8.33×10−4 |
| GO:0051480 | Cytosolic calcium ion homeostasis | Up | 5.76×10−7 | 1.00×10−3 |
| GO:0006468 | Protein amino acid phosphorylation | Up | 5.91×10−7 | 1.03×10−3 |
| GO:0051099 | Positive regulation of binding | Up | 5.97×10−7 | 1.04×10−3 |
| GO:0042592 | Homeostatic process | Up | 7.89×10−7 | 1.37×10−3 |
| GO:0050871 | Positive regulation of B cell activation | Up | 9.20×10−7 | 1.60×10−3 |
| GO:0051050 | Positive regulation of transport | Up | 9.32×10−7 | 1.62×10−3 |
| GO:0001932 | Regulation of protein amino acid phosphorylation | Up | 1.09×10−6 | 1.90×10−3 |
| GO:0030003 | Cellular cation homeostasis | Up | 1.11×10−6 | 1.94×10−3 |
| GO:0045086 | Positive regulation of interleukin-2 biosynthetic process | Up | 1.37×10−6 | 2.39×10−3 |
| GO:0051098 | Regulation of binding | Up | 1.54×10−6 | 2.68×10−3 |
| GO:0051047 | Positive regulation of secretion | Up | 1.86×10−6 | 3.23×10−3 |
| GO:0002224 | Toll-like receptor signaling pathway | Up | 2.11×10−6 | 3.68×10−3 |
| GO:0045637 | Regulation of myeloid cell differentiation | Up | 2.16×10−6 | 3.76×10−3 |
| GO:0045059 | Positive thymic T cell selection | Up | 2.46×10−6 | 4.29×10−3 |
| GO:0002429 | Immune response-activating cell surface receptor signaling pathway | Up | 3.05×10−6 | 5.30×10−3 |
| GO:0009725 | Response to hormone stimulus | Up | 3.08×10−6 | 5.37×10−3 |
| GO:0051241 | Negative regulation of multicellular organismal process | Up | 3.36×10−6 | 5.85×10−3 |
| GO:0040012 | Regulation of locomotion | Up | 3.76×10−6 | 6.55×10−3 |
| GO:0070482 | Response to oxygen levels | Up | 3.85×10−6 | 6.71×10−3 |
| GO:0051270 | Regulation of cell motion | Up | 4.00×10−6 | 6.96×10−3 |
| GO:0010740 | Positive regulation of protein kinase cascade | Up | 4.10×10−6 | 7.15×10−3 |
| GO:0006793 | Phosphorus metabolic process | Up | 4.14×10−6 | 7.20×10−3 |
| GO:0006796 | Phosphate metabolic process | Up | 4.14×10−6 | 7.20×10−3 |
| GO:0045577 | Regulation of B cell differentiation | Up | 4.48×10−6 | 7.80×10−3 |
| GO:0002495 | Antigen processing and presentation of peptide antigen via MHC class II | Up | 4.86×10−6 | 8.47×10−3 |
| GO:0019886 | Antigen processing and presentation of exogenous peptide antigen via MHC class II | Up | 4.86×10−6 | 8.47×10−3 |
| GO:0051272 | Positive regulation of cell motion | Up | 4.98×10−6 | 8.67×10−3 |
| GO:0002768 | Immune response-regulating cell surface receptor signaling pathway | Up | 5.12×10−6 | 8.92×10−3 |
| GO:0055080 | Cation homeostasis | Up | 5.58×10−6 | 9.72×10−3 |
| GO:0044057 | Regulation of system process | Down | 1.01×10−13 | 1.67×10−10 |
| GO:0009719 | Response to endogenous stimulus | Down | 1.25×10−12 | 2.07×10−9 |
| GO:0009725 | Response to hormone stimulus | Down | 2.19×10−11 | 3.62×10−8 |
| GO:0007267 | Cell-cell signaling | Down | 2.88×10−11 | 4.77×10−8 |
| GO:0010033 | Response to organic substance | Down | 1.52×10−10 | 2.52×10−7 |
| GO:0007166 | Cell surface receptor linked signal transduction | Down | 3.97×10−10 | 6.57×10−7 |
| GO:0048878 | Chemical homeostasis | Down | 6.67×10−10 | 1.10×10−6 |
| GO:0050678 | Regulation of epithelial cell proliferation | Down | 8.15×10−10 | 1.35×10−6 |
| GO:0042127 | Regulation of cell proliferation | Down | 8.51×10−10 | 1.41×10−6 |
| GO:0050679 | Positive regulation of epithelial cell proliferation | Down | 7.87×10−9 | 1.30×10−5 |
| GO:0007610 | Behavior | Down | 8.57×10−9 | 1.42×10−5 |
| GO:0042592 | Homeostatic process | Down | 1.18×10−8 | 1.95×10−5 |
| GO:0050801 | Ion homeostasis | Down | 6.17×10−8 | 1.02×10−4 |
| GO:0055065 | Metal ion homeostasis | Down | 9.62×10−8 | 1.59×10−4 |
| GO:0006873 | Cellular ion homeostasis | Down | 1.39×10−7 | 2.30×10−4 |
| GO:0055082 | Cellular chemical homeostasis | Down | 1.69×10−7 | 2.80×10−4 |
| GO:0032870 | Cellular response to hormone stimulus | Down | 2.14×10−7 | 3.54×10−4 |
| GO:0007169 | Transmembrane receptor protein tyrosine kinase signaling pathway | Down | 2.36×10−7 | 3.91×10−4 |
| GO:0019725 | Cellular homeostasis | Down | 3.35×10−7 | 5.53×10−4 |
| GO:0007167 | Enzyme linked receptor protein signaling pathway | Down | 3.42×10−7 | 5.65×10−4 |
| GO:0008284 | Positive regulation of cell proliferation | Down | 4.78×10−7 | 7.91×10−4 |
| GO:0040012 | Regulation of locomotion | Down | 5.05×10−7 | 8.35×10−4 |
| GO:0006875 | Cellular metal ion homeostasis | Down | 6.10×10−7 | 1.01×10−3 |
| GO:0008016 | Regulation of heart contraction | Down | 6.95×10−7 | 1.15×10−3 |
| GO:0048511 | Rhythmic process | Down | 1.92×10−6 | 3.17×10−3 |
| GO:0055080 | Cation homeostasis | Down | 2.63×10−6 | 4.36×10−3 |
| GO:0019932 | Second-messenger-mediated signaling | Down | 3.16×10−6 | 5.23×10−3 |
| GO:0040017 | Positive regulation of locomotion | Down | 3.58×10−6 | 5.92×10−3 |
| GO:0055074 | Calcium ion homeostasis | Down | 3.90×10−6 | 6.45×10−3 |
| GO:0010863 | Positive regulation of phospholipase C activity | Down | 4.14×10−6 | 6.84×10−3 |
| GO:0007202 | Activation of phospholipase C activity | Down | 4.14×10−6 | 6.84×10−3 |
| GO:0007242 | Intracellular signaling cascade | Down | 4.31×10−6 | 7.14×10−3 |
| GO:0051969 | Regulation of transmission of nerve impulse | Down | 5.40×10−6 | 8.93×10−3 |
| GO:0010518 | Positive regulation of phospholipase activity | Down | 5.88×10−6 | 9.73×10−3 |
GO, gene ontology; FDR, false discovery rate; NF, nuclear factor; MHC, major histocompatibility complex.
Figure 3.
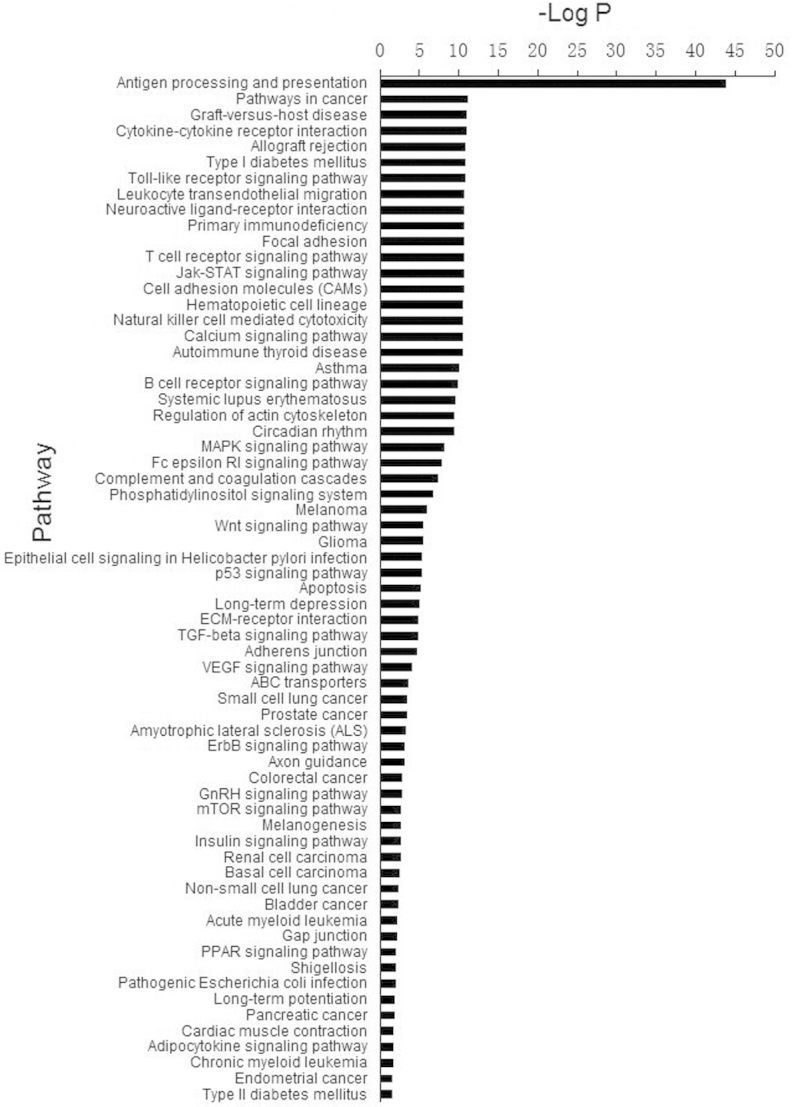
Histogram of signaling pathways that were found to be significantly different between the chronic thromboembolic pulmonary hypertension and normal control groups. −log P, negative logarithm of P-value (larger −log P values indicate smaller P-values).
Discussion
To the best of our knowledge, no previous studies have investigated the lncRNA expression profiles in CTEPH or the association of lncRNA expression with clinical characteristics and outcomes in CTEPH patients. CTEPH is a polygenic disorder, resulting from genetic alterations. The description of thousands of genomic sequences, along with technological developments enabling the identification of gene expression profiles on a large scale, have improved the understanding of the pathogenesis of a number of diseases, including cancer (34). These advances may also facilitate the development of novel therapeutic targets, as well as diagnostic and prognostic markers. Thus, a concerted effort to genetically characterize CTEPH may provide an improved understanding of the pathogenesis and development of the disease, as well as help identify novel personalized treatments. A number of studies have demonstrated that the expression levels of lncRNAs are dysregulated in numerous human diseases, including lung cancer and hepatocellular carcinoma (35–37,38). In addition, lncRNAs, such as HOTAIR, are involved in the development and progression of tumors, such as breast cancer (39).
In the current study, differentially expressed lncRNAs and nearby coding gene pairs were described. The silencing or reduced expression of particular lncRNAs has been previously demonstrated to result in a concomitant reduction in the expression of nearby protein-coding genes, including numerous proteins which are known to govern the regulation of cellular differentiation. In addition, the lncRNAs and nearby coding genes may share upstream regulation or local transcriptional effects (29,31,40–42). Certain lncRNAs have been reported to increase gene expression. For instance, Evf-2 ncRNA forms a complex with the homeodomain-containing protein Dlx2, which leads to transcriptional enhancement (43). In addition, heat-shock RNA-1 ncRNA binding to heat-shock transcription factor 1 has been found to result in the induction of heat-shock proteins (44). Furthermore, an isoform of the ncRNA steroid receptor RNA activator is known to be associated with steroid receptor responsiveness (45). Finally, Ørom et al (46) recently identified that noncoding RNA-activators 1–7 can enhance the expression of nearby genes. Thus, analyzing the genes nearby to lncRNAs may assist in understanding the involvement of lncRNAs in CTEPH. The majority of lncRNAs have a distinct spatial and temporal specificity in the process of organismal differentiation and development (47). A previous study, which investigated 1,300 lncRNAs in mice, demonstrated that in particular areas of the brain, there are different expression patterns of lncRNAs (48). These lncRNA expression signatures have been detected in prostate carcinoma and hepatic tumors (49). Thus, differential expression patterns of lncRNAs may be present in the pulmonary artery tissues of CTEPH patients, and lncRNAs that are differentially expressed may result in alterations in cellular function, which may be associated with the pathogenesis of CTEPH. In the present study, 464 pairs of differentially expressed enhancer-like lncRNAs and mRNAs, of which 95.3% (442/464) were regulated in the same direction (up or down). Therefore, the present study hypothesized that a number of these lncRNAs enhance the activation of nearby genes.
Molecular networks are useful in the investigation of biological processes and can be constructed using results obtained from co-immunoprecipitation experiments (50) or from algorithmic predictions based on gene function correlation and expression profiles (51). Network models based on algorithmic predictions from high throughput gene expression tests may be used to construct images of the networks regulating gene expression and metabolic pathways in the groups analyzed. The networks intrinsic to the CTEPH phenotype are hypothesized to be involved in the normal functioning of the pulmonary artery endothelium. Based on the information obtained regarding the expression of lncRNAs and mRNAs, Pearson correlation coefficients were calculated. Pairs found to have a significant correlation were selected and used to construct a network. These results demonstrated an association between lncRNAs and mRNAs, indicating that lncRNAs may regulate specific mRNAs, or vice versa. mRNAs are likely to be directly involved in the pathogenesis of CTEPH, while lncRNAs function through the epigenetic modification of mRNAs.
Based on previous studies and the results of the computer analysis conducted in the present study, four lncRNAs with the largest diffK were further investigated. lncRNA NR_036693 is a 5,255 bp transcript variant 6 of Homo sapiens C-type lectin domain family 2, member D. This gene encodes a member of the natural killer cell receptor C-type lectin family. The natural killer cell has been found to be involved in vascular remodeling, which may lead to pulmonary arterial hypertension (52,53). NR_027783 is a 1,199 bp transcript variant 2 from Homo sapiens spermidine/spermine N1-acetyltransferase 1 (SAT1). The protein encoded by this gene is a member of the acetyltransferase family and a rate-limiting enzyme in polyamine metabolism. Numerous studies have demonstrated that the polyamine regulatory pathway is a pharmacological target in pulmonary arterial hypertension (54,55). NR_033766 is a 6,384 bp transcript variant 7 from Homo sapiens forkhead box P2 (FOXP2). The FOXP2 gene is involved in the normal development of the areas of the brain controlling speech and language during embryogenesis. In addition, FOXP2 may be associated with a number of biological pathways and cascades, which also influence the development of language. Mutations in this gene result in speech-language disorder 1 (SPCH1), also termed as autosomal dominant speech and language disorder with orofacial dyspraxia. NR_001284 is a 2,783 bp pseudogene, Homo sapiens tenascin XA, the biological function of which remains unclear.
Identification of the putative functions of genes associated with lncRNAs may improve the understanding of the functional role of these molecules (30,32). Peng et al (56) performed a functional enrichment analysis on protein-coding genes nearby to differentially expressed lncRNAs in SARS-CoV infected mice. The authors identified that the most significant functional group consisted of annotation terms associated with gene expression, including transcription regulation, nuclear and DNA-binding transcription factor activity, as well as the regulation of RNA metabolism. In the present study, GO functional enrichment analysis of differentially expressed mRNAs with their coexpressed differentially expressed lncRNA partners, demonstrated that these genes were functionally associated with immune response, inflammatory response, response of wounding and response to endogenous stimulus. Furthermore, pathway analysis revealed that antigen processing and presentation, cytokine-cytokine receptor interaction and leukocyte transendothelial migration may be involved in the development of CTEPH. Although the function of lncRNAs in CTEPH still requires further investigation, the present study hypothesized that the formation of CTEPH may be caused by certain lncRNAs.
To the best of our knowledge, this is the first study describing the expression profiles of human lncRNAs in CTEPH by microarray. The expression levels of a number of lncRNAs were found to be aberrant in tissue samples from CTEPH patients, compared with the healthy control tissues. These deregulated lncRNAs may function as activators or suppressors of genes involved in the development and progression of the disease. Further investigation is required to determine whether these lncRNAs may serve as novel therapeutic targets and diagnostic biomarkers in CTEPH.
Acknowledgements
This study was supported by a grant from the High-Level Technic Personnel Training Plan of Beijing Health System (no. 2011-3-017).
References
- 1.Dartevelle P, Fadel E, Mussot S, et al. Chronic thromboembolic pulmonary hypertension. Eur Respir J. 2004;23:637–648. doi: 10.1183/09031936.04.00079704. [DOI] [PubMed] [Google Scholar]
- 2.Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S43–S54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 3.Wynants M, Quarck R, Ronisz A, et al. Effects of C-reactive protein on human pulmonary vascular cells in chronic thromboembolic pulmonary hypertension. Eur Respir J. 2012;40:886–894. doi: 10.1183/09031936.00197511. [DOI] [PubMed] [Google Scholar]
- 4.Quarck R, Wynants M, Ronisz A, et al. Characterization of proximal pulmonary arterial cells from chronic thromboembolic pulmonary hypertension patients. Respir Res. 2012;13:27. doi: 10.1186/1465-9921-13-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gu S, Su P, Yan J, et al. Comparison of gene expression profiles and related pathways in chronic thromboembolic pulmonary hypertension. Int J Mol Med. 2014;33:277–300. doi: 10.3892/ijmm.2013.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Z, Nakajima T, Tanabe N, et al. Susceptibility to chronic thromboembolic pulmonary hypertension may be conferred by miR-759 via its targeted interaction with polymorphic fibrinogen alpha gene. Hum Genet. 2010;128:443–452. doi: 10.1007/s00439-010-0866-8. [DOI] [PubMed] [Google Scholar]
- 7.Spizzo R, Almeida MI, Colombatti A, Calin GA. Long non-coding RNAs and cancer: a new frontier of translational research? Oncogene. 2012;31:4577–4587. doi: 10.1038/onc.2011.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 10.Ziats MN, Rennert OM. Aberrant expression of long noncoding RNAs in autistic brain. J Mol Neurosci. 2013;49:589–593. doi: 10.1007/s12031-012-9880-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ota T, Suzuki Y, Nishikawa T, et al. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat Genet. 2004;36:40–45. doi: 10.1038/ng1285. [DOI] [PubMed] [Google Scholar]
- 12.Okazaki Y, Furuno M, Kasukawa T, et al. FANTOM Consortium and RIKEN Genome Exploration Research Group Phase I & II Team: Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- 13.Tupy JL, Bailey AM, Dailey G, et al. Identification of putative noncoding polyadenylated transcripts in Drosophila melanogaster. Proc Natl Acad Sci USA. 2005;102:5495–5500. doi: 10.1073/pnas.0501422102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. Non-coding RNAs: regulators of disease. J Pathol. 2010;220:126–139. doi: 10.1002/path.2638. [DOI] [PubMed] [Google Scholar]
- 15.Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23:1494–1504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Auger WR, Fedullo PF, Moser KM, Buchbinder M, Peterson KL. Chronic major-vessel thromboembolic pulmonary artery obstruction: appearance at angiography. Radiology. 1992;182:393–398. doi: 10.1148/radiology.182.2.1732955. [DOI] [PubMed] [Google Scholar]
- 17.Kim Y, Doan BQ, Duggal P, Bailey-Wilson JE. Normalization of microarray expression data using within-pedigree pool and its effect on linkage analysis. BMC Proc. 2007;1(Suppl 1):S152. doi: 10.1186/1753-6561-1-s1-s152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19:2448–2455. doi: 10.1093/bioinformatics/btg345. [DOI] [PubMed] [Google Scholar]
- 19.Yang H, Crawford N, Lukes L, et al. Metastasis predictive signature profiles pre-exist in normal tissues. Clin Exp Metastasis. 2005;22:593–603. doi: 10.1007/s10585-005-6244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke R, Ressom HW, Wang A, et al. The properties of high-dimensional data spaces: implications for exploring gene and protein expression data. Nat Rev Cancer. 2008;8:37–49. doi: 10.1038/nrc2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pujana MA, Han JD, Starita LM, et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet. 2007;39:1338–1349. doi: 10.1038/ng.2007.2. [DOI] [PubMed] [Google Scholar]
- 22.Prieto C, Risueño A, Fontanillo C, De las Rivas J. Human gene coexpression landscape: confident network derived from tissue transcriptomic profiles. PLoS One. 2008;3:e3911. doi: 10.1371/journal.pone.0003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barabási AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 24.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabási AL. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- 25.Carlson MR, Zhang B, Fang Z, et al. Gene connectivity, function, and sequence conservation: predictions from modular yeast co-expression networks. BMC Genomics. 2006;7:40. doi: 10.1186/1471-2164-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dennis G, Jr, Sherman BT, Hosack DA, et al. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- 27.Khatri P, Draghici S, Ostermeier GC, Krawetz SA. Profiling gene expression using onto-express. Genomics. 2002;79:266–270. doi: 10.1006/geno.2002.6698. [DOI] [PubMed] [Google Scholar]
- 28.Draghici S, Khatri P, Martins RP, Ostermeier GC, Krawetz SA. Global functional profiling of gene expression. Genomics. 2003;81:98–104. doi: 10.1016/s0888-7543(02)00021-6. [DOI] [PubMed] [Google Scholar]
- 29.Guttman M, Amit I, Garber M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khalil AM, Guttman M, Huarte M, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ponjavic J, Oliver PL, Lunter G, Ponting CP. Genomic and transcriptional co-localization of protein-coding and long non-coding RNA pairs in the developing brain. PLos Genet. 2009;5:e1000617. doi: 10.1371/journal.pgen.1000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 33.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dall’Oglio MF, Coelho RF, Leite KR, et al. Gene expression profile of renal cell carcinoma clear cell type. Int Braz J Urol. 2010;36:410–419. doi: 10.1590/S1677-55382010000400004. [DOI] [PubMed] [Google Scholar]
- 35.Tang Y, Wang Y, Teng X. Sequence-dependent effect of gemcitabine and cisplatin on A549 non-small-cell lung cancer cells. Mol Med Rep. 2013;8:221–226. doi: 10.3892/mmr.2013.1495. [DOI] [PubMed] [Google Scholar]
- 36.Huang JF, Guo YJ, Zhao CX, et al. Hepatitis B virus X protein (HBx)-related long noncoding RNA (lncRNA) down-regulated expression by HBx (Dreh) inhibits hepatocellular carcinoma metastasis by targeting the intermediate filament protein vimentin. Hepatology. 2013;57:1882–1892. doi: 10.1002/hep.26195. [DOI] [PubMed] [Google Scholar]
- 37.Yang Y, Li H, Hou S, et al. The noncoding RNA expression profile and the effect of lncRNA AK126698 on cisplatin resistance in non-small-cell lung cancer Cell. PLos One. 2013;8:e65309. doi: 10.1371/journal.pone.0065309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu G, Yao W, Wang J, et al. LncRNAs expression signatures of renal clear cell carcinoma revealed by microarray. PLoS One. 2012;7:e42377. doi: 10.1371/journal.pone.0042377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta RA, Shah N, Wang KC, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huarte M, Guttman M, Feldser D, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ebisuya M, Yamamoto T, Nakajima M, Nishida E. Ripples from neighbouring transcription. Nat Cell Biol. 2008;10:1106–1113. doi: 10.1038/ncb1771. [DOI] [PubMed] [Google Scholar]
- 42.Sproul D, Gilbert N, Bickmore WA. The role of chromatin structure in regulating the expression of clustered genes. Nat Rev Genet. 2005;6:775–781. doi: 10.1038/nrg1688. [DOI] [PubMed] [Google Scholar]
- 43.Feng J, Bi C, Clark BS, et al. The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 2006;20:1470–1484. doi: 10.1101/gad.1416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shamovsky I, Ivannikov M, Kandel ES, Gershon D, Nudler E. RNA-mediated response to heat shock in mammalian cells. Nature. 2006;440:556–560. doi: 10.1038/nature04518. [DOI] [PubMed] [Google Scholar]
- 45.Lanz RB, McKenna NJ, Onate SA, et al. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell. 1999;97:17–27. doi: 10.1016/S0092-8674(00)80711-4. [DOI] [PubMed] [Google Scholar]
- 46.Ørom UA, Derrien T, Beringer M, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yin Z, Guan D, Fan Q, et al. lncRNA expression signatures in response to enterovirus 71 infection. Biochem Biophys Res Commun. 2013;430:629–633. doi: 10.1016/j.bbrc.2012.11.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS. Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci USA. 2008;105:716–721. doi: 10.1073/pnas.0706729105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ren S, Peng Z, Mao JH, et al. RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell Res. 2012;22:806–821. doi: 10.1038/cr.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smidtas S, Yartseva A, Schächter V, Képès F. Model of interactions in biology and application to heterogeneous network in yeast. C R Biol. 2006;329:945–952. doi: 10.1016/j.crvi.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 51.Nikiforova VJ, Willmitzer L. Network visualization and network analysis. EXS. 2007;97:245–275. doi: 10.1007/978-3-7643-7439-6_11. [DOI] [PubMed] [Google Scholar]
- 52.Perros F, Cohen-Kaminsky S, Gambaryan N, et al. Cytotoxic cells and granulysin in pulmonary arterial hypertension and pulmonary veno-occlusive disease. Am J Respir Crit Care Med. 2013;187:189–196. doi: 10.1164/rccm.201208-1364OC. [DOI] [PubMed] [Google Scholar]
- 53.Ormiston ML, Chang C, Long LL, et al. Impaired natural killer cell phenotype and function in idiopathic and heritable pulmonary arterial hypertension. Circulation. 2012;126:1099–1109. doi: 10.1161/CIRCULATIONAHA.112.110619. [DOI] [PubMed] [Google Scholar]
- 54.Gillespie MN, Olson JW. Polyamine regulatory pathways as pharmacologic targets in pulmonary arterial hypertension. Adv Exp Med Biol. 2010;661:375–389. doi: 10.1007/978-1-60761-500-2_24. [DOI] [PubMed] [Google Scholar]
- 55.Babal P, Ruchko M, Ault-Ziel K, et al. Regulation of ornithine decarboxylase and polyamine import by hypoxia in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;282:L840–L846. doi: 10.1152/ajplung.00347.2001. [DOI] [PubMed] [Google Scholar]
- 56.Peng X, Gralinski L, Armour CD, et al. Unique signatures of long noncoding RNA expression in response to virus infection and altered innate immune signaling. MBio. 2010;1:e00206–e00210. doi: 10.1128/mBio.00206-10. [DOI] [PMC free article] [PubMed] [Google Scholar]