

Abstract
Podophyllotoxin (PPT), as well as its congeners and derivatives, exhibits pronounced biological activities, especially antineoplastic effects. Its strong inhibitory effect on tumor cell growth led to the development of three of the most highly prescribed anticancer drugs in the world, etoposide, teniposide, and the water-soluble prodrug etoposide phosphate. Their clinical success as well as intriguing mechanism of action stimulated great interest in further modification of PPT for better antitumor activity. The C-4 position has been a major target for structural derivatization aimed at either producing more potent compounds or overcoming drug resistance. Accordingly, numerous PPT derivatives have been prepared via hemisynthesis and important structure–activity relationship (SAR) correlations have been identified. Several resulting compounds, including GL-331, TOP-53, and NK611, reached clinical trials. Some excellent reviews on the distribution, sources, applications, synthesis, and SAR of PPT have been published. This review focuses on a second generation of new etoposide-related drugs and provides detailed coverage of the current status and recent development of C-4-modified PPT analogs as anticancer clinical trial candidates.
Keywords: podophyllotoxin, cytotoxic agents, C-4 position, reviews
1. INTRODUCTION
Podophyllotoxin (PPT, 1, Fig. 1), a naturally occurring aryltetralin lignan, holds a unique position among natural products having been known for approximately 1000 years from its first application in folk medicines to its most recent developments in PPT-derived antitumor agents.1–8 Interest in PPT was initiated by Kaplan,9 who demonstrated its curative effect against tumor growth (Condylomata acuminata), and subsequently by King and Sullivan,10,11 who found its antiproliferative effect to be similar to that of colchicine at the cellular level. However, the initially high expectation for clinical use of PPT declined rapidly due to its unacceptable side effects, including nausea, vomiting, and damage to normal tissues. Nevertheless, due to its remarkable biological activity and extensive use in traditional medicine, PPT has remained an important starting point in the development of new therapeutic agents.
Figure 1.

Structures of podophyllotoxin (1, PPT), epipodophyllotoxin (2, EPPT), 4′-demethylepipodophyllotoxin (3, DEPPT), etoposide (4, ETO), teniposide (5), etopophos (6), NK-611(7), GL-331(8), NPF (9), TOP-53 (10), and tafluposide (11).
During early chemical modification studies, stereotransformation of α to β (epipodophyllotoxin, EPPT, 2) at the C-4 position, together with 4′-O-demethylation, gave 4′-demethylepipodophyllotoxin (DEPPT, 3). Investigation of semisynthetic glucoconjugates based on DEPPT led to two anticancer drugs, etoposide (ETO, 4) and teniposide (5), as well as etopophos (6), a water-soluble prodrug of ETO (Fig. 1).12–22 These compounds are currently used as drugs, alone or in combination with other agents, in clinical cancer chemotherapy against small cell lung cancer, acute leukemia, lymphoma, testicular carcinoma, and Kaposi’s sarcoma. Notably, the two structural modifications leading to DEPPT-type compounds also led to a different mechanism of action (MOA). While PPT acts as antimicrotubule agent, ETO and 5 function as topoisomerase II (topo II) inhibitors.23–25 Their clinical success and intriguing MOA stimulated great interest in further exploration of DEPPT derivatives with better antitumor activity. However, during the almost 50 years since clinical trials on ETO began in 1967, intense research efforts have resulted in both enthusiasm and setbacks. Studies showed that some nonsugar substituted analogs, particularly N-linked congeners, such as NK-611 (7), GL-331 (8), and NPF (9), as well as C-linked congeners, such as TOP-53 (10), exhibited superior pharmacological properties compared to ETO.26–32 These compounds were brought into clinical evaluations; however, they did not proceed further. Recently, tafluposide (F11782) (10), a novel catalytic inhibitor of topo I and II, has also been obtained (Fig. 1).33 Overall, these variants displayed improved water solubility and cytotoxic activity, as well as drug resistance and antitumor profiles, indicating that rational C-4 modifications can further optimize the activity of DEPPT. In in silico studies, both a composite pharmacophore model and comparative molecular field analysis34 further demonstrated that the C-4 molecular area could accommodate considerable structural diversity. These results suggested valuable directions for structural modification and hence, many researchers focused their studies on the design and synthesis of C-4 modified analogs.
Excellent reviews35–42 on PPT derivatives from a historical point of view and recent reviews43–49 on the distribution, sources, applications, total synthesis, and structure–activity relationship (SAR) correlations of PPT by us and other groups are available. However, an updated review focused on the important C-4 position is needed to facilitate the progress of future research for developing PPT-based new drugs. Herein, we describe PPT-related analogs containing carbon-, sulfur-, selenium-, oxygen-, and nitrogen-linked substituents at the C-4 position, as well as analogs containing spin labels in the C-4 substituent and at other positions. Orientations at the C-4 position include both α (as in PPT) and β (as in EPPT, DEPPT, and ETO), and substitutions at C-4′ include both methoxy (as in PPT and EPPT) and hydroxyl (as in DEPPT and ETO).
2. BIOLOGICAL ACTIVITIES AND MEDICAL APPLICATIONS
PPT-containing extracts have been widely used as folk remedies in traditional oriental medicine. They were commonly used in China, Japan, and the Eastern world as purgatives and to treat snake bites, periodontitis, skin disorders, coughs, various intestinal worm diseases, venereal warts (C. acuminata), lymphadenopathy, and certain tumors.1,8,50 Today, PPT is still an effective and comparatively safe drug choice in the treatment of venereal warts. Besides antitumor effects, PPT analogs exhibit diverse activities, including reverse transcriptase (RT) inhibition and anti-HIV activity; immunomodulatory activity; effects on the cardiovascular system; antileishmaniasis properties; 5-lipoxygenase inhibition; insecticidal activity; phytogrowth inhibitory activity; ichthyotoxic activity; antimelanocortin-4 receptor (MC4R) activity; and antirheumatic, antipsoriatic, antimalarial, and antiasthmatic properties.40,42,51
Among the plethora of physiological activities and agricultural applications, the anti-neoplastic and antiviral properties of PPT congeners are arguably the most eminent from a pharmacological perspective. An alcohol extract of podophyllin was first cited in 1942 as a topical treatment for venereal warts, an ailment caused by a papilloma virus.9 This study was one of the first to report the antiviral activity of podophyllin. In the early 1980s, both Bedows and Hatfield52 and Markkanen et al.53 found that PPT and related lignans showed antiviral activity against measles and herpes simplex type I. Other researchers54–58 surveyed the effects of PPT analogs against multiple viruses, including Sindbis virus (RNA virus), murine cytomegalovirus (herpes DNA virus), vesicular stomatitis virus, and HIV. The antiviral effects of PPT analogs appear due to their ability to bind tubulin, disrupt the cellular cytoskeleton, and interfere with viral replication. In addition to tubulin binding, synthetic PPT analogs also inhibit viral RT, which may be exploited to selectively combat RNA viruses, such as HIV.58 PPT is also effective in the treatment of anogenital warts in children and against Molluscum contagiosum, generally a self-limiting benign skin disease that affects mostly children, young adults, and HIV patients.59
Antitumor activity is probably the most well-known effect of PPT analogs. PPT-derived anticancer drugs are widely used in the treatment of Wilms’ tumor, various genital tumors, non-Hodgkin’s and other lymphomas, as well as lung cancers. Their powerful anticancer properties result from either inhibition of microtubule assembly or inhibition of DNA-topo II enzymatic activity. Combination therapies of ETO with other chemotherapeutic agents or techniques are currently being implemented.41,45,60
Studies on penetration of PPT into human bioengineered skin have demonstrated that PPT analogs induce acantholysis and cytolysis in this skin-equivalent model.61 We reported that PPT derivatives exhibit insecticidal activity against some economically important insects, more promising and pronounced activity as compared with PPT.62–64 Additionally, antifeedant and phytogrowth inhibitory activities of PPT have been described, which have application to pesticides.65,66 Other biological activities of PPT analogs are receiving increased current interest, for example, antioxidative properties, prevention of carcinogen production from estrogens, and inhibition of aromatase enzymatic activity, which could contribute to the prevention of dependent cancers.67 A benzylidated PPT glycoside (CPH82) induced clinical improvement in patients with rheumatoid arthritis and interfered with the cell cycle in rapidly proliferating cells with accumulation of bone marrow cells in mitosis.1,68 In addition, PPT analogs possess immunosuppressive activity and are seen as candidates for use in organ transplantation.69–71
3. MECHANISM OF ACTION
Cytotoxic PPT derivatives can be divided into two main types, tubulin polymerization inhibitors for “PPT-like” compounds and topo II inhibitors for “ETO-like” compounds. In 1976, Loike and Horwitz72 reported that “PPT-like” compounds exert cytotoxic activity by inhibiting tubulin polymerization that prevents microtubule formation and destabilizes microtubules, as well as arresting cell division in metaphase. More recent studies have further explored the MOA of PPT-derived cyclolignans. Gordaliza et al.73 proposed that PPT cyclolignans might work as alkylating agents at the C-2 methylene, rather than as acylating agents. Schönbrunn et al.74 cocrystallized PPT with a tubulin fragment in 1999 and described the effects of microtubule damaging agents, such as PPT and colchicine,75 on DNA and the cell cycle.43,76 López-Pérez et al.77 further described the role of dipole moment in the activity of PPT-related cyclolignans.
In contrast to PPT, 4′-demethylation and introduction of a β-glycosidic moiety at the C-4 position (“ETO-like” compounds) converts the resulting DEPPT derivatives into potent irreversible inhibitors of DNA topo II. Their action is based on formation of a nucleic acid–drug–enzyme ternary complex that blocks DNA strand relegation, generates DNA breaks, and blocks cell cycle progression in late S and G2 phases.78–81 The cytotoxic effects of DEPPT analogs have also been strongly linked to metabolic activation of the dimethoxyphenol ring (E-ring), producing metabolites that form chemical adducts with and inactivate DNA.82–84 These investigations pointed out that an ETO ortho-quinone is relevant to the MOA. 3′,4′-Catechol derivatives of ETO formed in the presence of cytochrome P-450 were further oxidized to 3′,4′-ortho-quinones in the presence of oxygen or under the influence of horseradish peroxidase or prostaglandin E synthase. Both catechol and ortho-quinone bound strongly to purified calf thymus DNA through formation of free radicals or even through direct binding of the quinone to the DNA, which may contribute to ETO’s cytotoxic activity.82–84
During the period of 2001–2010, many investigators reported various effects of PPT and DEPPT derivatives on cellular proteins and their signaling pathways. Lin et al.85 showed that GL-331 decreased extracellular signal-regulated kinase (ERK) phosphorylation and subsequently inhibited cyclin D1 transcription. In studies by Tseng et al.,86 PPT induced c-jun N-terminal kinase (JNK) phosphorylation at the molecular level, while ETO activated ERK, JNK, and p38 in selected tumor cell lines, as shown by Boldt et al.87 Qi et al. reported that GP-7, a new spin-labeled derivative of PPT, activated the caspase signaling pathway by releasing cytochrome c.88,89 In tumor necrosis factor α (TNF-α) induced human aortic smooth muscle cells, deoxy-PPT strongly inhibited matrix metalloproteinases (MMP-9) expression and migration, as well as mRNA transcription of MMP-9 gene expression and phosphorylation of ERK 1, ERK 2, p38, and JNK, as reported by Suh et al.90 Furthermore, Yong et al. described the involvement of deoxy-PPT in inhibition of tubulin polymerization and dysregulation of cyclin A and cyclin B1 expression, resulting in mitotic cell cycle arrest and activation of caspase-3 and caspase-7 to promote apoptotic cell death.91 The same studies also demonstrated that deoxy-PPT caused cell cycle arrest of HeLa cells at the G2/M phase, followed by induction of apoptosis. Shin et al.92 found that deoxy-PPT-induced apoptosis could involve the activation of p53 and ataxia-telangiectasia mutated and checkpoint kinase 2, possibly through a mitochondria-mediated pathway. Lastly, PPT can induce cAMP response element-binding protein (CREB) activation and CRE-driven gene expression via protein kinase An activation by a cAMP-independent mechanism, as shown by Chen and Xie.93
4. HEMISYNTHESIS AND STRUCTURE–ACTIVITY RELATIONSHIPS
A. C-4-C Derivatives
In an attempt to obtain topo II inhibitors with higher potency and greater distribution in lung tissue, scientists at Taiho Pharmaceutical Co. Ltd. synthesized modified 4-deoxy-DEPPT derivatives with various C-4β alkyl (12–18), amidomethyl (19–25), and aminoethyl (26–38) groups (Fig. 2, Table I).31,94 The new compounds with carbon rather than oxygen at the C-4 position were screened for cytotoxic activity against P388 mouse leukemia in vitro. Although the 4β-alkyl derivatives (12–18) did not inhibit topo II, their cytotoxicity was equal to that of ETO. Compounds 19–25 with various 4β-amidomethyl groups showed decreased inhibition of topo II, but did exhibit cytotoxic effects. Remarkably, 4β-aminoethyl derivatives TOP53, 34, 35, and 38 exhibited significant cytotoxic activity against P-388 cells with IC50 values ranging from 0.001 to 0.0043 μM. In addition, these compounds also displayed potent inhibitory effects on topo II with either improved or similar potency compared with ETO as illustrated in Table I. Among this series, TOP-53 was selected for further evaluation.95 Compared with ETO, TOP-53 displayed twofold greater topo II inhibitory activity and superior in vivo antitumor activity against several cancer types, especially metastatic lung tumors. In view of its high potency and good properties, TOP-53 progressed to phase II clinical trials, but did not reach clinical use.
Figure 2.
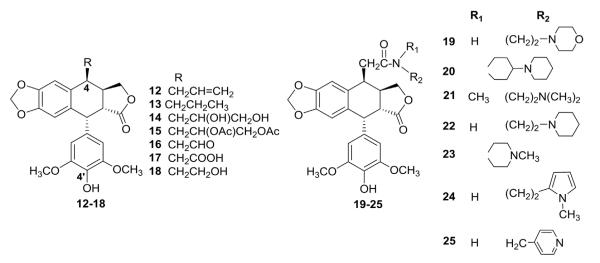
Structures of alkyl and amidomethyl analogs 12–25.
Table I.
Biological Data for Aminoethyl Analogs TOP53 and 26–38
| ||
|---|---|---|
| Compound | Cytotoxicity P388 (IC50, μM) | Topo II (IC50, μM) |
| ETO | 0.01 | 59.2 (1.0)a |
| 26 | 0.07 | 17.2 (0.29) |
| 27 | 0.66 | 25.1 (0.42) |
| 28 | 0.019 | 61.4 (1.03) |
| 29 | 0.15 | 97.3 (1.64) |
| 30 | 0.02 | 58.3 (0.98) |
| 31 | 1.0 | NTb |
| TOP53 | 0.001 | 32.5 (0.54) |
| 32 | 0.055 | 26.9 (0.45) |
| 33 | 0.037 | 30.0 (0.50) |
| 34 | 0.0033 | 29.8 (0.53) |
| 35 | 0.0030 | 33.6 (0.56) |
| 36 | 0.26 | 115.7 (1.95) |
| 37 | 0.10 | 31.3 (0.52) |
| 38 | 0.0043 | 32.3 (0.54) |
The value in parentheses is the ratio of IC50 of compound/IC50 of ETO.
NT, not tested.
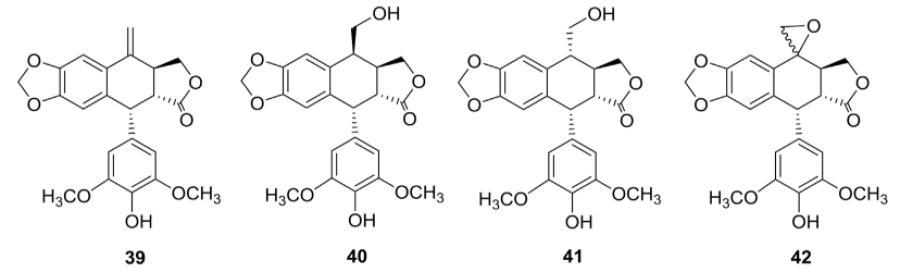
Roulland et al.96 used Takai olefination to introduce a methylene moiety at the C-4 position. The resulting compound 39 was hydrolyzed and further oxidized to give compounds 40–42 (Table II). Among these four compounds, the greatest cytotoxicity was shown by 39 (IC50 35 nM). The cell cycle perturbation induced by these compounds was studied in the same L1210 cell line. Compounds 40 and 41 induced partial accumulation of cells in the G2/M phase of the cell cycle (42–69% vs. 24% for untreated control cells) at relatively moderate concentrations.
Table II.
Biological Data for New Compounds 39–42
| |||||
|---|---|---|---|---|---|
| Cell cyclea |
Inhibition of microtubule assemblyb (IC50, μM) |
||||
| Compound | L1210 (IC50, μM) |
Percent of G2/M |
Percent of 8N |
Concentration (μM) |
|
| 39 | 0.035 | 60 | 27 | 0.1 | 2.3 |
| 40 | 0.24 | 42 | 44 | 0.25 | NTc |
| 41 | 0.10 | 69 | 15 | 0.5 | 7.3 |
| 42 | 1.30 | 69 | 20 | 5 | 8.3 |
Untreated control cells: 24% G2/M, 1% 8N.
4-Deoxypodophyllotoxin IC50 = 1.1 μM.
NT, not tested.
4β-Cyano-4-deoxy-DEPPT was synthesized by reaction of DEPPT with trimethylsilyl cyanide in the presence of boron trifluoride etherate.97 Hydrolysis of this intermediate in acetic acid gave 4β-carboxy-4-deoxy-DEPPT (43), from which a series of 4β-alkoxylcarbonyl (43–68), thionylcarbonyl (69–82), and carbamoyl (83–96) derivatives (Fig. 3) were synthesized and evaluated for inhibitory activity against L-1210 and KB cell lines in vitro.98,99 However, most of the compounds exhibited IC50 values ranging from 0.05 to 1 μM and thus were less active than ETO. Compound 53 exhibited the highest potency.
Figure 3.

Structures of compounds 43–96.
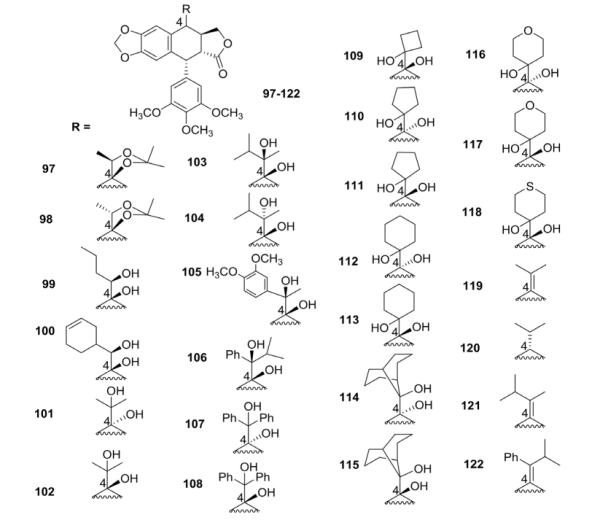
Recently, pinacol PPT analogs 97–122 bearing different side chains and functional groups at C-4 were synthesized through reductive cross-coupling of PPT with different aldehydes and ketones (Table III).100 In general, these compounds were fairly cytotoxic, and some of them (e.g., 115, 117) displayed better activity than the reference EPPT. Among all compounds, 4β-OH (epipodo) derivatives showed greater cytotoxicity than their corresponding 4α-OH (podo) analogs. Similarly, to PPT, the former compound series arrested the cellular cycle of A-549 cells at the G2/M phase, with differences only in potency. Significantly, 4-isopropyl-4-deoxypodophyllotoxin (120) was identified as a promising lead compound for a novel type of tubulin polymerization inhibitor.
Table III.
Cytotoxicity Data for Pinacol, Alkylidene, and Alkyl Analogs 97–122
| |||
|---|---|---|---|
| GI50 ± SD (nM)a |
|||
| Compound | A-549 | HT-29 | SK-BR3 |
| EPPT | 60 | 60 | NT |
| 97 | 6220 ± 562 | 5230 ± 443 | 5870 ± 126 |
| 98 | 6020 ± 526 | 6560 ± 456 | 6140 ± 786 |
| 99 | 279 ± 12 | 404 ± 75 | 288 ± 8 |
| 100 | 2030 ± 15 | 2510 ± 413 | 2430 ± 9 |
| 101 | 1480 ± 564 | 659 ± 43 | 511 ± 65 |
| 102 | 577 ± 61 | 598 ± 83 | 457 ± 24 |
| 103 | 254 ± 6 | 204 ± 2 | 181 ± 7 |
| 104 | 1150 ± 27 | 1730 ± 87 | 1030 ± 70 |
| 105 | 2620 ± 65 | 4750 ± 126 | 4990 ± 345 |
| 106 | 139 ± 5 | 159 ± 16 | 148 ± 15 |
| 107 | 874 ± 68 | 528 ± 20 | 524 ± 36 |
| 108 | 376 ± 25 | 281 ± 32 | 313 ± 57 |
| 109 | 191 ± 18 | 198 ± 18 | 219 ± 40 |
| 110 | 1690 ± 356 | 2720 ± 315 | 2420 ± 154 |
| 111 | 444 ± 12 | 451 ± 8 | 439 ± 29 |
| 112 | 547 ± 71 | 859 ± 13 | 610 ± 24 |
| 113 | 90.8 ± 1.3 | 97.5 ± 4.6 | 64.9 ± 2.8 |
| 114 | 5680 ± 6 | 6420 ± 85 | 6720 ± 41 |
| 115 | 6.39 ± 0.15 | 5.14 ± 0.50 | 7.39 ± 0.6 |
| 116 | 8920 ± 317 | 15440 ± 491 | 8010 ± 140 |
| 117 | 29.0 ± 1.1 | 26.8 ± 0.3 | 25.9 ± 0.4 |
| 118 | 3320 ± 213 | 2620 ± 53 | 1780 ± 187 |
| 119 | 6440 ± 477 | 8280 ± 386 | 3780 ± 82 |
| 120 | 79.5 ± 9.0 | 83.1 ± 1.4 | 84.1 ± 3.0 |
| 121 | 7700 ± 137 | 10200 ± 221 | 4530 ± 141 |
| 122 | 6210 ± 326 | 11400 ± 582 | 14300 ± 589 |
Cytotoxicity results are expressed as GI50 values, the compound concentration producing a 50% cell growth inhibition and represent the mean ± SD of three independent experiments. Values under 100 nM are highlighted for easier comparison.
B. C-4-S Derivatives
Showalterk et al.101 reported compounds 123–126, which are thioglucose-derived analogs of ETO (Fig. 4). They expected that substitution of sulfur in various oxidation states for the glycosidic oxygen would not only alter lipophilicity, but also vary the geometric disposition of the sugar moiety on the aglycone. Although this change should affect the biological activity remarkably, no biological data were reported.
Figure 4.
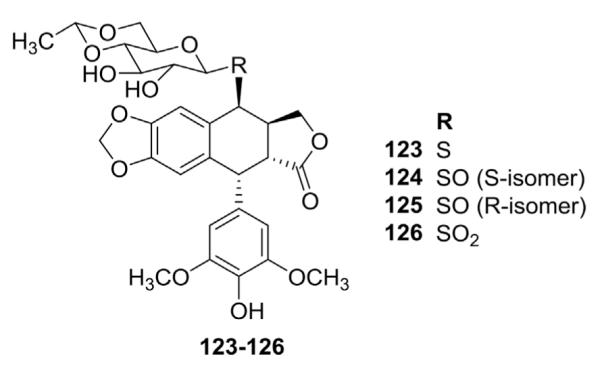
Structures of thioglucose ETO analogs 123–126.
Moreover, Wang et al.102–104 developed two efficient methods employing the BF3·Et2O/H2S and CH3COSH/NH3 reagent system to synthesize 4-sulfanyl-DEPPT. This compound then served as a valuable building block for the preparation of versatile 4S-substituted-DEPPT analogs, including various alkylthio (127–135) and 4-(2-aminoethylthio) (136–154) derivatives (Fig. 5). Most of the compounds showed comparable or better activity than ETO against L1210 and KB cells in vitro. A large activity range for 127–154 indicated that the substituents on 4S-derivatives can markedly affect the activity profiles of this compound class.
Figure 5.

Structures of thioether analogs 127–154.
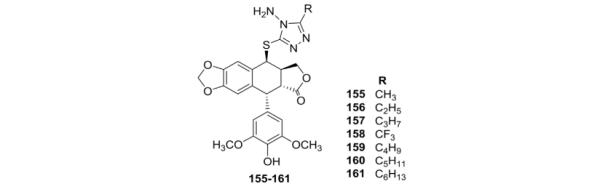
Many triazoles exhibit a wide range of biological activities, such as antifungal, antiviral, antiphlogistic, antitumor, and other effects. Therefore, Lu et al.105 prepared seven DEPPT analogs linked through the sulfur of various 4-amino-5-alkyl-4H-1,2,4-triazole-3-thiol compounds. When screened in vitro against HL-60 and K562 cells, compounds 155–161 were more potent against the latter cell line (Table IV).
Table IV.
Biological Data for S-Linked Analogs 155–161
| |||
|---|---|---|---|
| Inhibition rate (%)a |
|||
| Compound | Concentration (mol/L) | HL-60 | K562 |
| 155 | 10−6 | 37.9 | 68.76 |
| 10−5 | 48.2 | 77.24 | |
| 10−4 | 58.6 | 80.24 | |
| 10−6 | 48.2 | 76.15 | |
| 156 | 10−5 | 55.1 | 83.10 |
| 10−4 | 58.6 | 86.83 | |
| 10−6 | 24.1 | 68.32 | |
| 157 | 10−5 | 55.1 | 73.95 |
| 10−4 | 55.1 | 86.32 | |
| 10−6 | 10.3 | 52.30 | |
| 158 | 10−5 | 55.1 | 62.61 |
| 10−4 | 62.0 | 80.90 | |
| 10−6 | 22.3 | 60.71 | |
| 159 | 10−5 | 49.6 | 72.86 |
| 10−4 | 58.6 | 79.07 | |
| 10−6 | 40.0 | NT | |
| 160 | 10−5 | 65.0 | NT |
| 10−4 | 67.5 | NT | |
| 10−6 | 20.6 | NT | |
| 161 | 10−5 | 58.6 | NT |
| 10−4 | 62.0 | 66.86 | |
Results obtained after 72 hr.
C. C-4-Se Derivatives
Se-derived PPT analogs have received very little attention, even though such molecules could be potential new pharmaceutical agents, as Se compounds are promising molecules in cancer prevention and have potential in cancer treatment.106 Accordingly, Wang et al.107 synthesized six 4-alkylselenyl-DEPPTs (162–167, Fig. 6) from 4-bromo-DEPPT and selenourea in the presence of Et3N. All six compounds showed potent cytotoxic activity against L-1210 and KB cells. Miao et al.108 found that 4-phenylselenyl-DEPPT (168, Fig. 6) suppressed the proliferation of human hepatoma SMMC-7721 cells in a dose- and time-dependent manner and induced SMMC-7721 cell apoptosis by translocation of Bax, activating the mitochondrial pathway of apoptosis through release of proapoptotic factors, such as cytochrome c. Four related 4-selenyl-DEPPT derivatives (169–171, Fig. 6) synthesized by Zhang and Cao109 showed cytotoxicity against HO-8910 cells.
Figure 6.
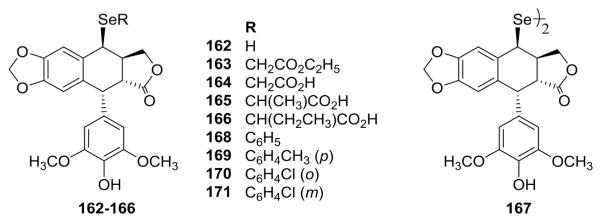
Structures of Se-linked analogs 162–171.
D. C-4-O Derivatives
While ETO and 5 are O-glycosides of DEPPT, other nonglycosidic ether substituents have been incorporated into DEPPT in order to lower toxicity, while maintaining or enhancing activity. Terada et al.110 synthesized various DEPPT ethers (172–186, Fig. 7) by reaction of protected 4′-O-benzyloxycarbonyl-DEPPT with alcohols in the presence of boron trifluoride etherate,followed by deprotection. The introduction of an aminoalkoxy group at the C-4 position increased both DNA topo II inhibitory and cytotoxic activities.110
Figure 7.
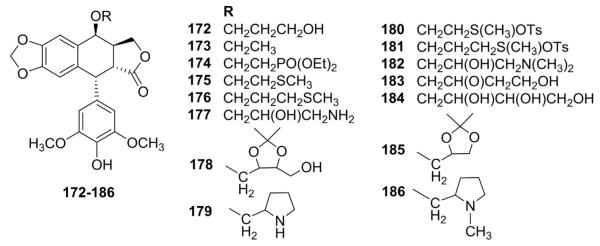
Structures of ether analogs 172–186.
Ma and co-workers111,112 also reported a series of 4β-ether-DEPPT (187–199) and five 4,4′-O-bis(2-hydroxy-3-substituted-amino)-propyl-DEPPT (200–204) analogs that were screened for cytotoxic activity against L-1210 and KB cells (Fig. 8). Most compounds showed equivalent or greater potency compared to ETO, in particular, certain compounds with side chains containing a hydroxyl or an amino group.111,112 Recently, Bathini et al.113 synthesized 4β-O-propenylethers (205–209, Fig. 8) from 4-chloro-4-deoxy-PPT and allylic alcohols in the presence of barium carbonate. The cytotoxicity of these compounds against KB cells was comparable to that of ETO.
Figure 8.

Structures of ether analogs 187–209.
Because an ester group can be important to the cytotoxicity and antileukemic activity of compounds in certain classes, Lee and co-workers 114 prepared a series of PPT esters (210–221, Fig. 9) and examined their in vivo antileukemic activity against P-388 lymphocytic leukemia cell growth in mice. The biological data indicated that the introduction of an ester moiety into PPT did not enhance antileukemic activity, but instead generally caused a loss of activity.
Figure 9.
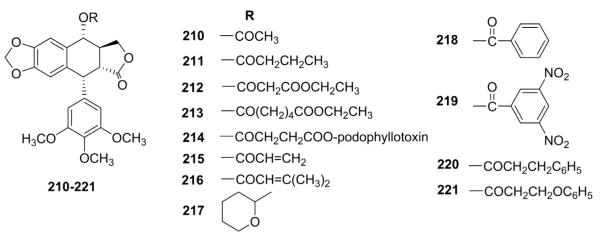
Structures of ester analogs 210–221.
In order to determine whether the presence of a glucosidic moiety on the C-4 position is essential, Gupta and Chenchaiah115 synthesized different C-4 ester DEPPT analogs (222–235, Fig. 10) through the same general procedure described by Lee and co-workers114 In biological studies, these analogs possessed similar biological activity to PPT, and none acted in the same manner as ETO. Treatment of CHO cells with these esters caused a large increase in the mitotic index of cells, and the SAR on these esters provided information regarding the role of substituents at the C-4 position on PPT-like activity.
Figure 10.
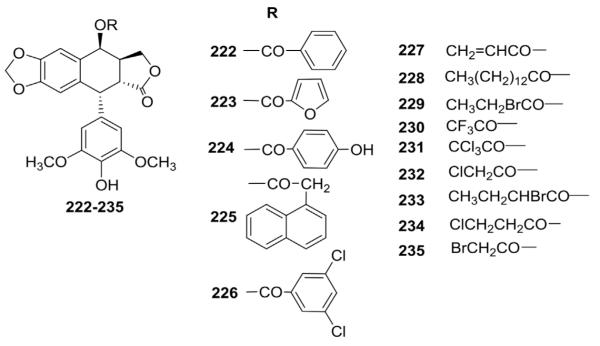
Structures of ester analogs 222–235.
More recently, López-Pérez et al.116 synthesized various hydrophobic esters of PPT, including norbornene-carboxylate esters prepared through Diels-Alder cycloaddition by treating dienophilic acrylates of cyclolignans with cyclopentadiene. Compounds 236–241 (Fig. 11) were tested for in vitro cytotoxicity against four neoplastic cell lines (P-388, A-549, HT-29, MEL-28). The results showed that the introduction of a linear or aromatic C-4 acyl moiety in PPT analogs induced either loss or no effect on the cytotoxicity of the parent hydroxy derivatives, while C-4α-PPT norbornene-carboxylates 241a,b showed improved potency compared with PPT. The introduction of one additional methylene unit between the bicyclic system and the carboxyl group (240) generated more hydrophobic esters, but led to diminished activity. These results indicated that the cytotoxic activity is not primarily due to a lipophilic factor and the precise spatial arrangement of a bulky moiety may contribute to additional nonpolar interactions that can enhance binding to the target site.
Figure 11.
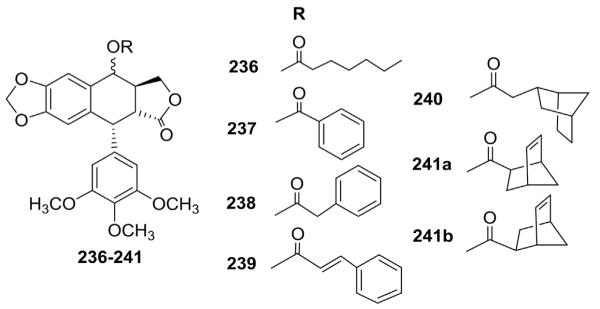
Structures of ester analogs 236–241.
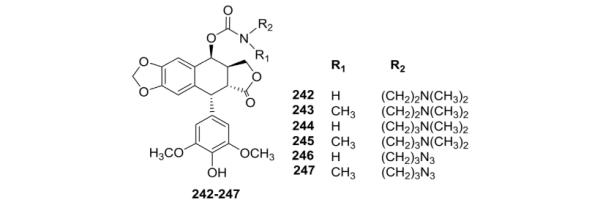
Because TOP-53 showed promising potential in the treatment of several types of cancer compared with ETO, Duca et al.117 introduced a carbamate substituted with a N-alkylamino or N-alkylazido group in the C-4β-position of DEPPT to give DEPPT-4-amino/azidoalkylcarbamates (242–247, Table V). In particular, compound 247 with an N-methyl-N-azidopropylcarbamate group displayed potent cytotoxicity against the L-1210 cellline (IC50, 0.038 μM compared with 0.83 μM for ETO) and proved to be a more potent topo II poison than ETO.
Table V.
Biological Data for Carbamoyl Analogs 242–247
| |||
|---|---|---|---|
| Compound | Topo IIα inhibition (% linear DNA)a |
Cytotoxicity IC50 (μM)b |
Cell cycle effectc |
| ETO | 50 | 0.83 | 76% (2.5 μM) |
| 242 | 56 | 0.038 | 67% (0.1 μM) |
| 243 | 27 | 1.7 | 77% (2.5 μM) |
| 244 | 58 | 0.34 | 72% (1 μM) |
| 245 | 17 | 0.96 | NTd |
| 246 | 38 | 1.3 | NT |
| 247 | 8 | 0.24 | NT |
Average value of at least three independent experiments at 20 μM of drug.
IC50, concentration of drug required to cause 50% reduction in L1210 cell growth.
Percentage of L1210 cells in the G2/M phase at the specified drug concentration.
NT, not tested.
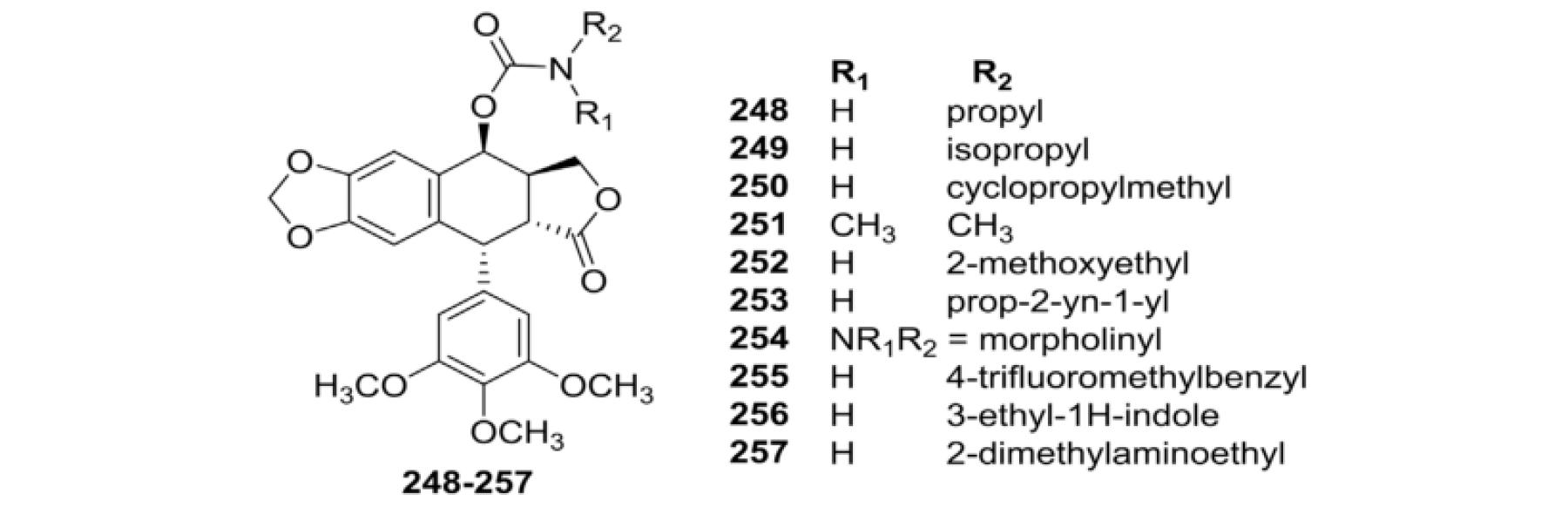
Recently, a series of new 4β-carbamoyl-PPT analogs were prepared by Kamal et al.118 (248–257, Table VI) and evaluated for cytotoxic activity against 11 cancer cell lines. As shown, most of the compounds exhibited better growth-inhibition activity than ETO against the tested cell lines. Compounds 254 and 256 were also evaluated for DNA topo II inhibitory activity and showed significant inhibition comparable to that of ETO.
Table VI.
Cytotoxic Data for Carbamoyl Analogs 248–257
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| GI50 (μM) |
|||||||||||
| Compound | Zr-75–1 | MCF7 | KB | Gurav | DWD | Colo 205 | A549 | Hop62 | PC3 | SiHa | A2780 |
| ETO | 0.2 | 2.1 | 0.3 | 0.5 | 0.6 | 0.13 | 3.08 | 0.80 | 2.6 | 3.1 | 1.3 |
| 248 | 0.19 | 2.0 | 2.1 | 0.17 | 2.0 | 2.7 | <0.1 | 0.16 | 01.7 | 1.2 | 0.19 |
| 249 | 0.15 | 0.19 | 0.18 | 0.16 | <0.1 | 2.7 | <0.1 | 2.1 | 0.19 | 0.19 | 0.14 |
| 250 | 0.19 | 2.3 | 2.4 | 0.18 | 2.0 | 2.5 | <0.1 | 2.5 | 2.3 | 2.3 | 2.0 |
| 251 | 0.12 | 0.16 | 0.18 | 0.17 | <0.1 | 2.1 | <0.1 | 0.13 | 0.15 | 2.1 | 0.18 |
| 252 | <0.1 | 0.15 | 0.15 | 0.14 | <0.1 | 2.2 | <0.1 | 0.12 | 0.16 | 2.4 | 0.15 |
| 253 | 0.14 | 0.19 | 0.17 | 0.14 | <0.1 | 2.7 | <0.1 | 0.14 | 0.16 | 1.9 | 0.16 |
| 254 | 0.14 | 0.16 | 0.12 | 0.14 | <0.1 | 2.7 | <0.1 | 0.13 | 0.16 | 1.4 | 0.13 |
| 255 | 01.6 | 0.18 | 0.18 | 0.19 | 2.0 | 0.15 | <0.1 | 2.3 | 2.1 | 2.0 | 0.18 |
| 256 | 0.13 | 0.16 | 0.18 | 0.12 | <0.1 | 0.12 | <0.1 | 2.1 | 0.16 | 1.8 | 0.11 |
| 257 | <0.1 | 0.11 | 0.16 | 0.15 | <0.1 | 2.1 | <0.1 | 0.12 | 0.15 | 2.1 | 0.12 |
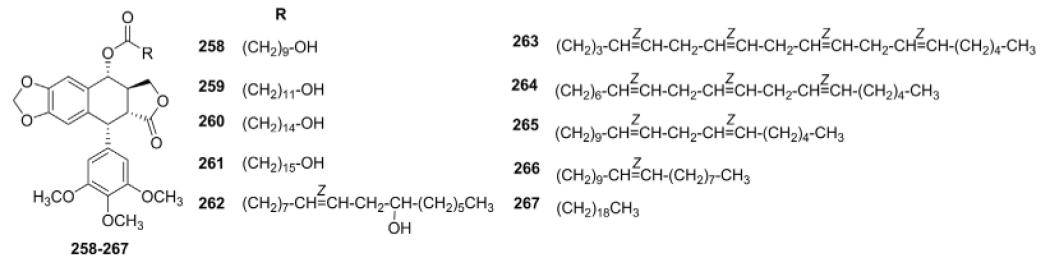
In view of the significance of long-chain fatty acid (FA) in the treatment of cancer, Mustafa et al.119 prepared a series of FA analogs of PPT (258–267, Table VII) by coupling unusual C-10 to C-20 FAs with the 4α-hydroxyl of PPT. These compounds were investigated for in vitro cytotoxic activity against a panel of human cancer cell lines, including SK-MEL, KB, BT-549, SK-OV-3, and HL-60 cells. All of the compounds showed significant cytotoxicity against all tumor cells tested. With IC50 values ranging from 0.07 μM for HL-60 cells to 0.4 μM for KB cells, compounds 258 and 263 were the most active analogs. Interestingly, they did not affect the growth of noncancerous mammalian cells (VERO cells) up to the highest concentration (15 μM) used in the assay, demonstrating promising selectivity toward tumor cells.
Table VII.
Cytotoxicity Data for Ester Analogs 258–267
| ||||||
|---|---|---|---|---|---|---|
| IC50 (μM) |
Kidney fibroblast VERO cells |
|||||
| Compound | SK-MEL | KB | BT-549 | SK-OV-3 | HL-60 | |
| PPT | 0.22 | 0.24 | 0.36 | 0.19 | 0.01 | 0.55 |
| 258 | 0.21 | 0.31 | 0.22 | 0.26 | 0.07 | NA |
| 259 | 0.41 | 1.19 | 0.29 | 0.44 | 0.19 | NA |
| 260 | 0.89 | NA | 1.19 | 0.76 | 0.24 | NA |
| 261 | 0.90 | 2.39 | 0.85 | 0.81 | 0.18 | NA |
| 262 | 4.0 | 4.30 | 2.59 | 3.75 | 0.47 | NA |
| 263 | 0.34 | 0.40 | 0.27 | 0.27 | 0.07 | NA |
| 264 | 1.17 | 1.35 | 1.08 | 1.08 | 0.24 | NA |
| 265 | 1.42 | 1.42 | 0.89 | 0.79 | 0.85 | NA |
| 266 | NA | NA | NA | NA | NA | NA |
| 267 | NA | NA | NA | NA | NA | NA |
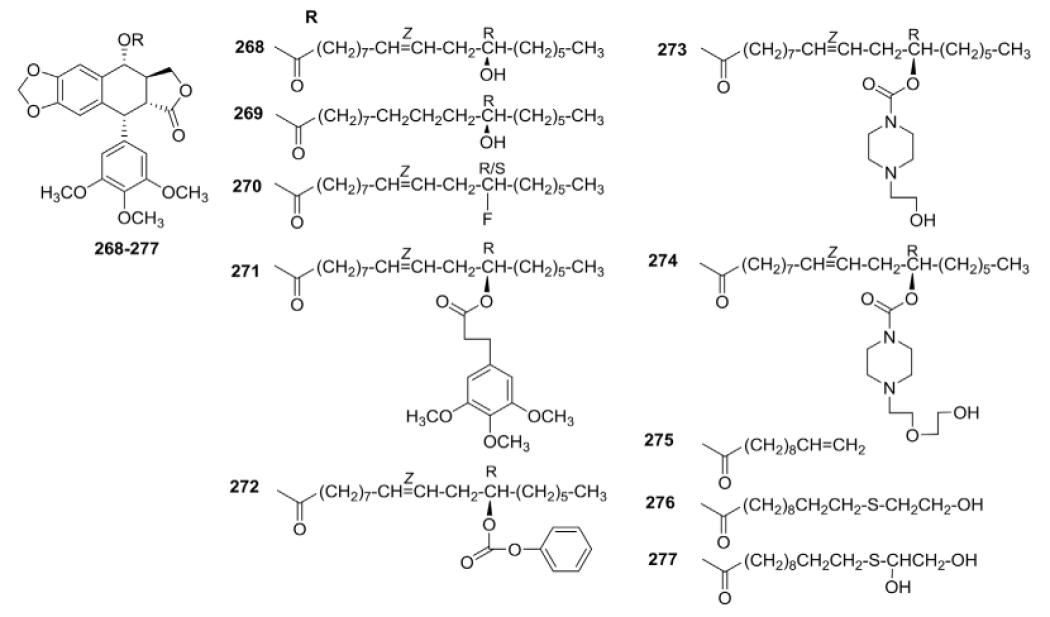
Based on these preliminary results, the same group further synthesized additional PPT-FA adducts (268–277, Table VIII).120 These compounds were assayed in vitro against four human solid tumors (SK-MEL, KB, BT-549, SK-OV-3, and HL-60) and noncancerous VERO cell lines. The SAR indicated that a “12-hydroxy-Z-ene” system was probably important for activity against the human neoplastic cell line panel. The FA thioether analogs 276 and 277, derived from 275, showed promising activity against all five cancer cell lines, with one exemption; 277 was not active against ovary carcinoma (SK-OV-3) cells. Unlike PPT, none of these compounds were toxic toward normal mammalian cells, demonstrating selectivity for cancer cells over normal cells.
Table VIII.
Cytotoxicity Data for Ester Analogs 268–277
| ||||||
|---|---|---|---|---|---|---|
| IC50 (μM) |
||||||
| Compound | SK-MEL | KB | BT-549 | SK-OV-3 | HL-60 | VERO |
| PPT | 0.22 | 0.24 | 0.36 | 0.19 | 0.01 | 0.55 |
| 268 | 0.45 | 1.26 | 4.76 | 0.17 | 0.66 | NA |
| 269 | 8.12 | NA | 7.50 | 10.64 | 0.22 | NA |
| 270 | NA | NA | NA | NA | 0.73 | NA |
| 271 | NA | NA | NA | NA | NA | NA |
| 272 | NA | NA | NA | NA | NA | NA |
| 273 | 0.72 | 2.4 | 1.2 | 0.84 | 0.58 | NA |
| 274 | 1.87 | 2.2 | 2.53 | 0.77 | 0.55 | NA |
| 275 | NA | 0.93 | 1.33 | NA | 0.08 | NA |
| 276 | 0.30 | 0.45 | 0.30 | 0.30 | 0.10 | NA |
| 277 | 2.9 | 0.35 | 0.81 | NA | 0.07 | NA |
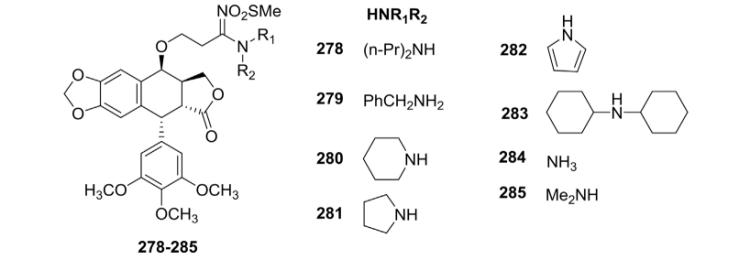
Recently, we reported121 the synthesis of novel sulfonylamidine PPT derivatives (278–285, Table IX) via a Cu-catalyzed reaction along with their in vitro evaluation against a panel of human cancer cell lines, including K562, SGC, Hela, and HepG.121 MOA studies were also investigated. Compound 282 displayed significant antiproliferative activity against all four cell lines and strong tubulin polymerization inhibitory effects. The results indicated that these compounds effectively interfered with tubulin dynamics and prevented mitosis in cancer cells, thus leading to cell cycle arrest and, eventually, dose-dependent apoptosis. In addition, docking analysis and molecular dynamics showed that the binding of 282 to tubulin was mainly stabilized by hydrophobic interactions, together with hydrogen-bonding interactions with β-tubulin’s Cys241 residue.
Table IX.
Cytotoxicity Data for Analogs 278–285
| ||||
|---|---|---|---|---|
| IC50 (μM)a |
||||
| Compound | K562 | SGC-7901 | HeLa | HepG2 |
| Colchicine | 0.55 ± 0.02 | 0.05 ± 0.02 | 0.10 ± 0.02 | 0.02 ± 0.03 |
| PPT | 4.51 ± 0.03 | 0.34 ± 0.02 | 0.14 ± 0.04 | 0.51 ± 0.02 |
| 278 | 2.96 ± 0.03 | 2.87 ± 0.03 | 2.63 ± 0.03 | 2.14 ± 0.04 |
| 279 | 12.07 ± 0.04 | 19.16 ± 0.02 | 11.23 ± 0.02 | 17.88 ± 0.02 |
| 280 | 9.01 ± 0.03 | 18.20 ± 0.03 | 10.37 ± 0.02 | 57.14 ± 0.01 |
| 281 | 8.34 ± 0.02 | 11.21 ± 0.04 | 5.99 ± 0.03 | 20.79 ± 0.02 |
| 282 | 1.01 ± 0.02 | 1.36 ± 0.01 | 0.75 ± 0.03 | 0.79 ± 0.01 |
| 283 | 2.61 ± 0.02 | 3.21 ± 0.02 | 4.33 ± 0.01 | 1.95 ± 0.02 |
| 284 | 3.48 ± 0.03 | 4.31 ± 0.01 | 3.75 ± 0.02 | 2.87 ± 0.03 |
| 285 | 5.18 ± 0.02 | 3.71 ± 0.03 | 2.58 ± 0.02 | 4.17 ± 0.02 |
Cytotoxicity results are expressed as IC50 values, the compound concentration producing a 50% cell growth inhibition and represent the mean ± SD of three independent experiments.
To overcome multidrug resistance (MDR) and lower the toxicity of PPT derivatives, Yu et al.122 synthesized 4-O- and 4-N-indol-3-yl-glyoxyl-substituted derivatives of PPT and tested them against a panel of four human cancer cell lines, including HeLa (cervix), SKOV3 (ovary), K562 (leukemia), and K562ADR (adriamycin-resistant leukemia) in vitro. Generally, the O-linked derivatives (esters) of PPT showed greater potency than the corresponding N-linked congeners (amides). Compound 286 (L1EPO, Fig. 12) down-regulated the mdr-1 gene, reduced the expression of P-gp, and displayed dose-dependent cytotoxicity. Moreover, it was less cytotoxic against normal human cell lines (fibroblast, VEC, GI50 > 10 μM) than cancer cell lines. L1EPO has the potential to overcome P-glycoprotein-mediated MDR in the K562/A02 cell line.123
Figure 12.
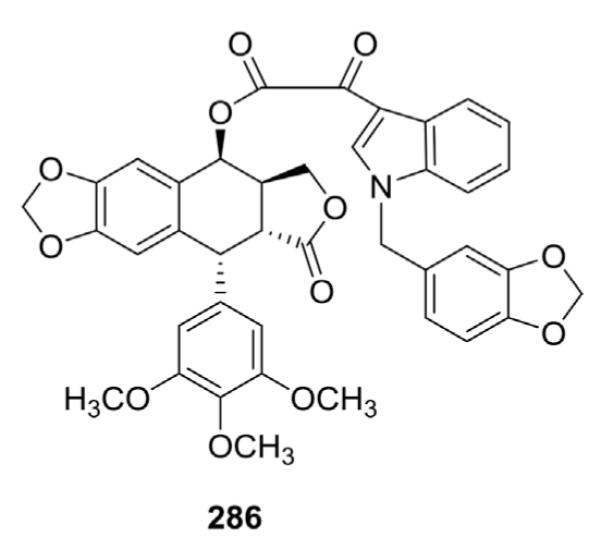
Structure of L1EPO (286).
As shown in Figure 13, Gupta et al.124 prepared three DEPPT–lexitropsin conjugates (287–289) with the aim of conferring higher affinity for DNA and improving cellular uptake and metabolic stability. Investigation of the biological effects of these bifunctional hybrids demonstrated that conjugation with minor groove-binding moieties could alter or increase the number of topo II–induced cleavable sites.
Figure 13.
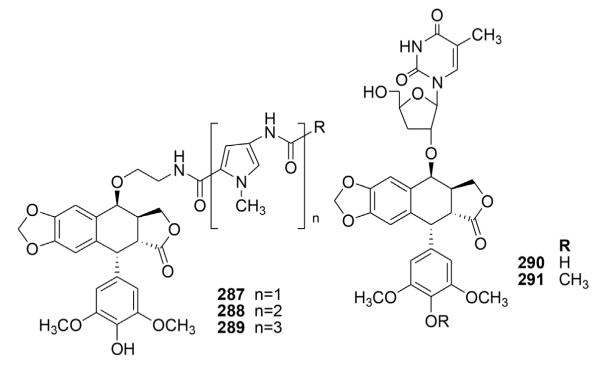
Structures of compounds 287–291.
Since nucleosides are biologically active moieties, Derry et al.125 synthesized two novel derivatives of DEPPT and EPPT (290 and 291, respectively, Fig. 13), in which the nucleoside thymidine was conjugated with the parent compounds at the C-4 position using boron trifluoride etherate. The observed cross-resistance patterns of the thymidine derivatives suggested that these compounds display PPT-like activity without ETO-like activity. The two thymidine derivatives exhibited much lower activity in comparison with PPT and DEPPT, suggesting that the thymidine moiety interferes with the compounds’ interaction with the receptor site on the tubulin molecule.
In order to improve the therapeutic efficacy of PPT, a novel PPT conjugate, 3,6-endo-methylene-1,2,3,6-tetrahydrophthalimido-acetamidoglycinylglycine PPT ester (ETPA-gly-gly-PPT, 292, Fig. 14), as well as its homo- and copolymer with acrylic acid were prepared by photopolymerization using 2,2-dimethoxy-2-phenylacetophenone as a photoinitiator.126 Compared with PPT (IC50 1.4–4.0 ng/mL), compound 292 showed decreased cytotoxicity (IC50 13–100 ng/mL) against A375, KM12, PC3P, and CEM cancer cell lines.
Figure 14.
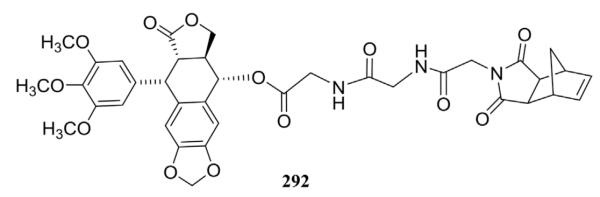
Structure of compound 292.
Pharmacomodulation of biologically active compounds via conjugation approaches has become an appealing research area in different fields of medicinal chemistry. Also, the concept of a bivalent molecule is now accepted as an effective strategy for designing ligands, inhibitors, and drugs that influence biological systems. Thus, Passarella’s group127–130 recently reported a series of novel PPT-based hybrids. Some of these compounds exerted significant antiproliferative activity, having a marked ability to inhibit tubulin polymerization in vitro and to disrupt the microtubule network in vivo. Among the synthesized disulfide dimers of PPT, increased spacer length resulted in progressive reduction of antiproliferative activity against the NCI-H460 human nonsmall cell lung carcinoma cell line. For example, the IC50 values of compounds 293 and 294 were 0.03 and 0.3 μg/mL, respectively, compared with 0.005 μg/mL for PPT.130 Simultaneously, our group131 also described disulfide dimers of PPT (293–297; Fig. 15), in which two PPT moieties were linked with different dithiodicarboxylic acid spacers.
Figure 15.
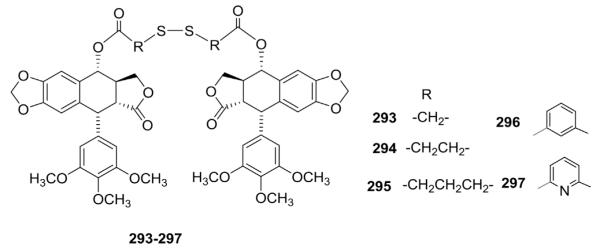
Structures of novel bis-ester-linked analogs 293–297.
E. C-4-N Derivatives
Allevi et al.132 condensed 4-bromo-4-deoxy-DEPPT with 4,6-O-ethylidene-2,3-di-O-trimethylsilyl-β-d-glucopyranosylamine in the presence of Hg(CN)2 to produce a 4-aminoglucose analog (298, Fig. 16) of ETO. However, no biological data were reported.
Figure 16.
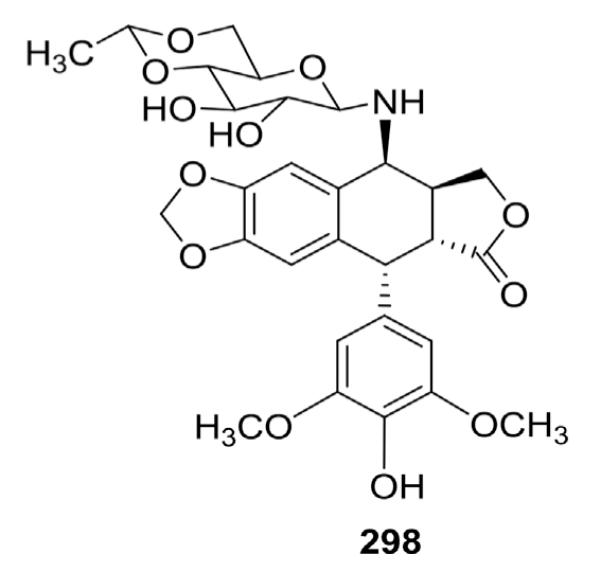
Structure of 4-aminoglucose ETO analog 298.
In recent years, C-4-N-substituted analogs have occupied a significant position in the development of DEPPT-derived antitumor agents. Lee and co-workers performed pioneering work in this field and have studied C4-N-substituted congeners of DEPPT for many years.133–141 The various projects below illustrate several aspects of the development process. First, C-4α and C-4β–aliphatic amino derivatives (299–306, Fig. 17) were synthesized by direct nucleophilic substitution of 4-bromo-4-desoxy-DEPPT with appropriate alkylamines. Replacement of the glycosidic moiety of ETO with a 2″-hydroxyethylamino or 2″-methoxyethylamino moiety at the C-4β position resulted in potent inhibition of human DNA topo II, as well as strong ability to cause cellular protein-linked DNA strand breakage (compounds 299, 303, and 305).133 The C-4β isomers were more potent than the C-4α isomers, which indicated that the C-4 stereochemistry is quite important in determining the inhibitory potency.
Figure 17.
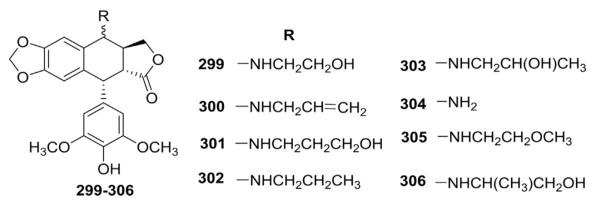
Structures of alkylamino analogs 299–306.
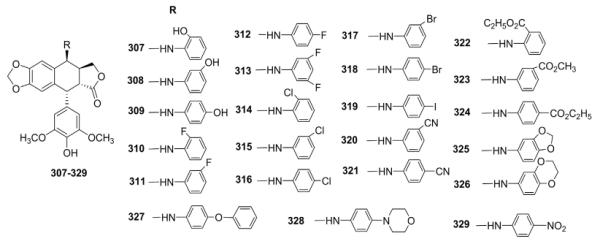
In subsequent studies,134 numerous substituted-4β-anilino side chains were introduced into the DEPPT structure by nucleophilic substitution reaction of 4β-bromo-4′-demethyl-4-desoxyPPT with various substituted arylamines (Table X). The modification resulted in substantially increased activity. Most of the 4β-arylamino-DEPPTs were as or more potent than ETO in topo II inhibition and/or cellular protein–DNA complex formation assays. In most cases, para substitution in the phenyl ring resulted in the greatest activity.29,30,134 Compared with ETO, compounds 308, 312, and 324 were tenfold more active in inhibiting DNA topo II and caused two to three times more protein–DNA complex formation. As a highlight, GL331 (329) was selected as the optimal drug candidate. GL-331 functions as a highly potent topo II inhibitor, causing DNA double-strand breakage and G2 phase arrest. It could also induce cell death by stimulating protein tyrosine phosphatase activity and apoptotic DNA formation. GL-331 was also shown to be active in many MDR cancer cell lines. Because of its good stability and biocompatability, as well as favorable pharmacokinetic profiles, GL331 successfully reached clinical trials against several forms of cancers, especially ETO-resistant malignancies, but has not reached clinical status.
Table X.
Biological Data for Arylamino Analogs 307–329
| |||
|---|---|---|---|
| Compound | Cytotoxicitya ID50 KB (μM) |
Inhibition of DNA topo II activity ID50,b μM |
Cellular protein–DNA complex formation % (10 μM) |
| ETO | 0.2 | 50 | 100 |
| 307 | 4.54 | 25 | 151 |
| 308 | 0.45 | 5 | 290 |
| 309 | 2.26 | 25 | 211 |
| 310 | 0.25 | – | 121 |
| 311 | 0.23 | 50 | 158 |
| 312 | 0.24 | 10 | 213 |
| 313 | 1.08 | 50 | 115 |
| 314 | 2.34 | – | 32 |
| 315 | 2.29 | – | 51 |
| 316 | 0.22 | 50 | 99 |
| 317 | 2.36 | – | 62 |
| 318 | 0.22 | 100 | 179 |
| 319 | 0.34 | – | 64 |
| 320 | 0.69 | 25 | 137 |
| 321 | 0.64 | 10 | 211 |
| 322 | 1.0 | >100 | 4 |
| 323 | 2.7 | 50 | 249 |
| 324 | 0.84 | 5 | 207 |
| 325 | <1.0 | 50 | 164 |
| 326 | 0.68 | 10 | 279 |
| 327 | 1.0 | 25 | 97 |
| 328 | 0.66 | 10 | 140 |
| 329 | 0.49 | 10 | 323 |
ID50, concentration of drug that afforded 50% reduction in cell number after a 3-day incubation.
Each compound was examined at 25, 50, and 100 μM. The ID50 value was established on the basis of the degree of inhibition at these three concentrations.
The synthesis and biological evaluation of a series of 4β-benzylamino (330–343) and 4β-benzoylamino (344–355) derivatives were then reported by the same group (Fig. 18).135,136 These compounds were less potent than the 4β-arylamino derivatives, but most were as active or more active than ETO. In topo II inhibition and protein–DNA assays, the activity was observed in the order of 4β-arylamino > 4β-benzylamino > 4β-benzoylamino.
Figure 18.

Structures of benzyl- and benzoyl-amino analogs 330–355.
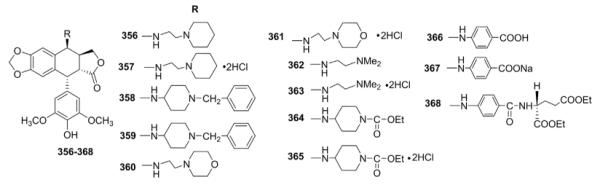
Subsequently, Lee and co-workers137 synthesized another series of 4β-amino derivatives of DEPPT (Table XI) that could form water-soluble salts. Most compounds showed excellent activity in KB cell cytotoxicity and protein–DNA complex formation assays. In general, the salts showed comparable or greater activity than their parent-free bases. Compared with ETO, compounds 357 and 359–365 showed comparable or greater inhibition of human DNA topo II. In a dose–response study of protein–DNA complex formation, compound 359 was 20 times more active than ETO. Furthermore, both compound 359 and its free base 358 were highly active against ETO-resistant KB cell lines.
Table XI.
Biological Data for Amino Analogs 356–368
| |||
|---|---|---|---|
| Compound | Cytotoxicitya ID50 KB, μM |
Inhibition of DNA topo II activity ID50,b μM |
Cellular protein–DNA complex formation % (20 μM) |
| ETO | 0.20 | 50 | 100 |
| 356 | 1.4 | 25 | 190 |
| 357 | 1.6 | 50 | 183 |
| 358 | 0.027 | 100 | 83 |
| 359 | 0.021 | 50 | 172 |
| 360 | 2.0 | >100 | 77 |
| 361 | 4.0 | 50 | 140 |
| 362 | 0.4 | 25 | 203 |
| 363 | – | 25 | 183 |
| 364 | 0.74 | 25 | 17 |
| 365 | 1.13 | 25 | 138 |
| 366 | >4.0 | >100 | 1.9 |
| 367 | >4.0 | >100 | 6.9 |
| 368 | <0.4 | 100 | 83 |
ID50, concentration of drug that afforded 50% reduction in cell number after a 3-day incubation.
Each compound was examined at 25, 50, and 100 μM. The ID50 was established on the basis of the degree of inhibition at these three concentrations.
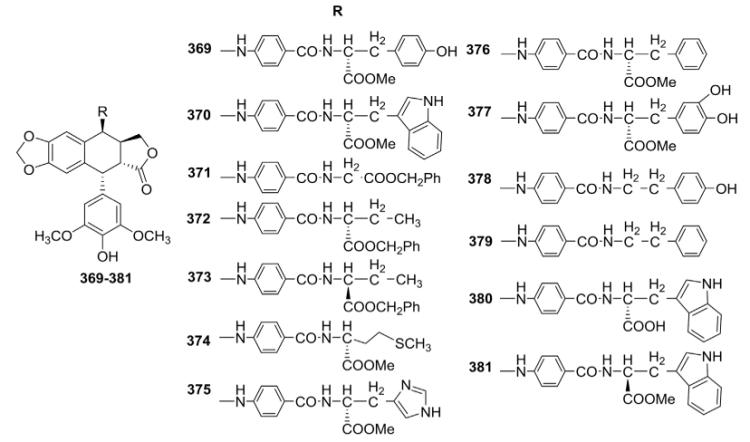
To further address poor water solubility, a problem associated with ETO, Lee and coworkers138 introduced protected α-amino acids into a 4β-[(p-benzamido)-amino] side chain of new DEPPT derivatives (Table XII). Compounds 369 and 370 exhibited better preclinical activity profiles, including cell growth inhibition, cell killing, and in vitro topo II inhibition, as compared to the prototype molecule ETO, while retaining the superior drug-resistance profile of GL-331. This observation indicated that introduction of bulky substituents at the para position of an anilino moiety would enhance topo II inhibition and still maintain superior cell growth inhibition and drug-resistance profiles. Other members in this series of 4β-[(p-benzamido)-amino]-DEPPT derivatives were also reported in detail by Lee and co-workers,139 revealing important SAR information. The large activity range of compounds 369–381 (Table XII) indicated that the substituents on the α-carbon of the amino acids markedly affected the activity profiles of this compound class. The impressive ability of compounds 372, 374, and 376 to induce intracellular protein-linked DNA breaks suggested that a hydrophobic interaction might exist between the enzyme/DNA complex and this molecular area of the compounds. Inclusion of moieties containing nitrogen or oxygen atoms in the amino acid side chains (e.g., compounds 369, 370, 375, 377, and 380) decreased protein-linked DNA breakage. Adding hydroxy groups on the phenyl ring sharply decreased the activity (e.g., compounds 369, 376, and 377). Many factors could possibly contribute to this decrease. First, hydrogen bonding between the nitrogen or oxygen atom and the enzyme/DNA complex might cause the activity loss, presumably by preventing the molecules from assuming an optimal conformation. Second, it is also possible that relatively polar (e.g., hydroxyl in compounds 369 and 377, imidazole in compound 375) and sterically bulky (e.g., indolyl in compounds 370 and 380) moieties might impede an important hydrophobic interaction between the molecules and enzyme/DNA complex. The orientation of the amino acid also showed a stereo preference (compare 372 vs. 373; 381 vs. 370). Hydrolysis of the methyl ester in 370 to give the free acid in 380 resulted in a dramatic reduction in cellular protein–DNA complex formation and cell growth inhibition. Interestingly, changing the amino acid to an alkylamine (i.e., deletion of the COOMe) significantly increased the inhibitory potency against KB cell growth; however, it also led to unfavorable drug-resistance profiles (compounds 378 vs. 369; 379 vs. 376). The unique activity profiles of compounds 378 and 379 implied that their action mode or cellular uptake mechanisms might be different from those of their amino acid congeners.139
Table XII.
Biological Data for 4″-Benzamido-Amino Analogs 369–381
| ||||
|---|---|---|---|---|
| Compound | Percent PLDB formationa |
KB ED50b (μg/mL) |
KB-7d ED50b (μg/mL) |
Relative resistancec (fold) |
| ETO | 100 (100) | 0.5 | >10 | >20 |
| 369 | 76 | 1.9 | 5 | 2.6 |
| 370 | 58 | 0.5 | 2.5 | 5 |
| 371 | 178 | 0.5 | 0.3 | 0.6 |
| 372 | 210 | 0.5 | 0.25 | 0.5 |
| 373 | 133 | 0.1 | 0.25 | 2.5 |
| 374 | 295 | 0.8 | 2.4 | 3 |
| 375 | 122 | 0.23 | 2.2 | 9.6 |
| 376 | 226 | 0.55 | 1.0 | 1.8 |
| 377 | 33 | 1 | 5.5 | 5.5 |
| 378 | 368 (275) | 0.025 | 0.8 | 32 |
| 379 | (227) | 0.035 | 0.5 | 14 |
| 380 | 7 | 4 | 8 | 2 |
| 381 | 121 | 0.065 | 0.4 | 6.2 |
Percent protein-linked DNA breaks (PLDB) formation was determined by the SDS/potassium precipitation method. Percentage values are levels of protein-linked DNA breaks induced by drug treatment relative to the ETO control set arbitrarily at 100%. Values in parentheses reflect effects at a concentration of 5 μg/mL. Other values reflect effects at a concentration of 10 μg/mL.
ED50 is the concentration of drug that afforded 50% reduction in cell number after a 3-day incubation.
Relative resistance (fold) values are the ED50 values against KB-7d over those against KB cells.
Lee and co-workers also employed a conjugation strategy to overcome drug resistance or enhance cytotoxic activity.140,141 They linked 4β-amino-DEPPT derivatives with camptothecin (a topo I inhibitor) or taxol derivatives (microtubule assembly promoters) through aromatic bridges (Fig. 19). Surprisingly, the DEPPT-camptothecin hybrids 382 and 383 did not show cross-resistance in ETO-resistant cells. The two compounds inhibited topo I in a concentration-dependent manner, but only 383 was active against topo II. In a nude mouse model with human prostate DU-145 tumor cells, compound 383 showed better antitumor activity than ETO, camptothecin, and 382. The cytotoxic potencies of the taxoid conjugates 384 and 385 were between those of ETO and the parent taxoid. Taxoid conjugate 386, which has an additional DEPPT moiety at the taxoid 7-hydroxyl, was least active, suggesting that the 7-hydroxyl was important to the cytotoxic activity. Against paclitaxel-resistant cells, conjugates 384 and 385 showed enhanced activity. In contrast to the cytotoxic results, compound 386 was a better topo II inhibitor than 384 and 385. The authors postulated a role for the 7-hydroxy group in drug transport.
Figure 19.
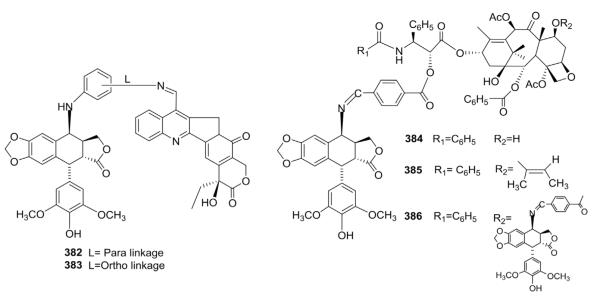
Structures of amino conjugates (382–386) with camptothecin or taxoids.
The increased activity incurred by replacing the glycosidic moiety of ETO with 4-amino groups also led to the synthesis and testing of numerous 4-anilino-, 4-amido-, 4-arylamino-, 4-sulfonylamino-, and 4-alkylamino-DEPPT derivatives by other groups.142–148 For example, Kamal et al.149 synthesized 4β-amido or 4β-sulfonamido derivatives of DEPPT. All five 4β-amido-2-substituted benzophenone analogs (387–391) showed moderate cytotoxicity against the tested cell lines. Among the 4β-benzenesulfonamido derivatives, 4′-methylated analog 396 was highly cytotoxic, but the corresponding 4′-demethylated analog (398) was about 100-fold less potent. A methyl substituent on the phenyl ring at the para-position (399) further decreased activity by tenfold. Two (392, 394) of the four 4β-nicotinylamido substituted analogs (392–395) showed high cytotoxicity. Overall, 4′-methylated derivatives were more cytotoxic than corresponding demethylated compounds (397 vs. 399 and 392 vs. 393; Fig. 20).149
Figure 20.

Structures of compounds 387–399.
As shown in Figure 21, Kamal et al.150 also synthesized 4β-N-heteroaryl analogs (400–404) that exhibited better in vitro cytotoxic activity than ETO. Compound 402 with a fluoro substituent on the phenyl ring showed significant activity with GI50 values less than 0.010 μM against all six tested cancer cell lines, while the GI50 values of 401 without a fluoro group were 0.31, 0.02, 0.06, 0.03, 0.09, and 0.11 μM against DU145, HT29, MCF7, MCF7ADR, NCIH460, and U251 human cancer cell lines, respectively.
Figure 21.
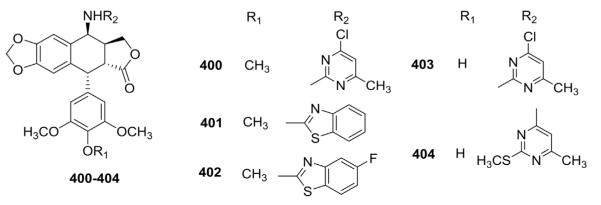
Structures of heteroarylamino analogs 400–404.
Current interest in dimeric analogs of lipophilic, neutral, DNA mono-intercalating agents as potential antitumor drugs prompted Kamal et al.151 to prepare bis-4β-amino-PPT dimers by linking the amino groups through various aryl spacers (405–418; Fig. 22). Most of the analogs exhibited promising in vitro cytotoxic activity against different human tumor cell lines. Interestingly, compared with ETO, 4′-methylated analogs showed superior topo II poisoning activity. Dimer 409 linked through a biphenyl spacer was the most active tested compound with GI50 values of 0.2, 0.2, and 0.6 μM against DU145, HT29, and MCF7, respectively.
Figure 22.

Structures of bis-N-linked dimers 405–418.
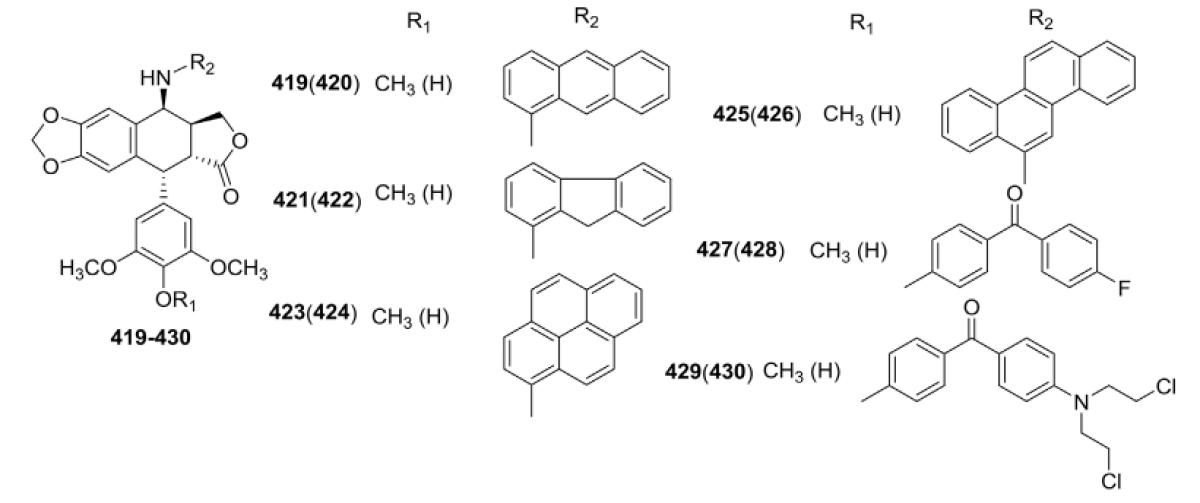
More recently, six series of 4-N-substituted PPT derivatives were synthesized by Kamal et al.152–157 and evaluated for cytotoxicity against selected human cancer cell lines or for DNA topo II inhibitory activity. The first new compounds were a series of 4β-N-polyarylamino congeners (Table XIII) reported in 2010.152 Cytotoxic activity was evaluated against several tumor cell lines (502713, HCT-15, HEP-2, IMR-32, A-549, DU-145, and PC-3). As shown in Table XIII, most of the new compounds exhibited significant cytotoxic activity compared with ETO. Selected compounds 423, 425, and 426 also caused similar inhibition of DNA topo II catalytic activity compared with ETO. Cell cycle studies with 425 revealed that these compounds exert apoptosis-inducing activity.
Table XIII.
Cytotoxicity Data for Polyaromatic Amino Analogs 419–430
| |||||||
|---|---|---|---|---|---|---|---|
| IC50 (μM) |
|||||||
| Compound | Colo502713 | HCT-15 | HEP-2 | IMR-32 | A549 | DU145 | PC-3 |
| ETO | 0.1 | 0.9 | 0.9 | 6.3 | 3.08 | 3.7 | 2.6 |
| 419 | 9.4 | 6.3 | 6.7 | 6.7 | 8.8 | 21 | 7.9 |
| 420 | – | 8.2 | 8.6 | 2.3 | 8.3 | 18 | – |
| 421 | 5.5 | 0.09 | 3 | 5.5 | 7.1 | 6.6 | 4.3 |
| 422 | 0.001 | 0.0001 | 1 | 10 | 1 | 2.7 | 0.5 |
| 423 | 0.002 | 0.0001 | – | – | 0.0009 | – | 0.0001 |
| 424 | 0.002 | 0.0001 | 0.0002 | 0.2 | 1.7 | 2.2 | – |
| 425 | 0.1 | 0.002 | 0.002 | 0.003 | 0.04 | 2.2 | 0.1 |
| 426 | 0.1 | 0.1 | 0.01 | 0.001 | 0.0003 | 0.0003 | 0.0003 |
| 427 | 4.1 | 6.2 | 7.1 | 19 | 17 | – | 6.7 |
| 428 | 1.4 | 0.08 | 1.3 | 1.3 | 0.2 | 0.3 | 6.0 |
| 429 | 0.1 | 0.9 | 0.9 | 6.3 | 3.08 | 3.7 | 2.6 |
| 430 | NTa | NT | NT | NT | NT | NT | NT |
NT, not tested.
In 2011, the same group designed and synthesized a series of benzothiazolo-4β-anilino-PPT derivatives through a one-pot iodination methodology.153 These compounds exhibited significant cytotoxic activity against Colo205, HT1080, and DWD cell lines, and moderate activity against Hop-62 (Table XIV). In particular, compound 432 [IC50 2.7 μM (Colo205), 2.3 μM (DWD)] was more potent than PPT (5.1 and 5.0 μM). All 16 compounds exhibited similar in vitro inhibition of topo II catalytic activity compared with m-AMSA.
Table XIV.
Cytotoxicity Data for Arylamino Analogs 431–446
| ||||
|---|---|---|---|---|
| IC50 (μM) |
||||
| Compound | Colo 205 | Hop62 | HT1080 | DWD |
| PPT | 5.0 | 13.1 | 8.1 | 6.1 |
| 431 | 8.1 | >80 | 8.2 | 22 |
| 432 | 2.7 | 30.1 | 9.7 | 2.3 |
| 434 | 8.3 | 8.6 | >80 | 6.5 |
| 437 | 7.0 | 15.2 | 9.5 | 5.2 |
| 441 | 7.1 | 60.3 | 9.1 | 7.3 |
| 442 | 8.3 | 10.1 | >80 | 9.1 |
As depicted in Table XV, Kamal et al.154 obtained a series of new 4β-acrylamido-PPT derivatives (447–461) synthesized by coupling substituted stilbene moieties with 4β-amino-PPT. These compounds showed significant cytotoxic activity with GI50 values ranging from <0.1 to 0.29 μM. Compounds 456–458 caused G2/M cell cycle arrest, with 457 being slightly more effective. Interestingly, compound 457 also caused both single-strand (30%) and double-strand (70%) DNA damage, eventually leading to cancer cell death. The study results suggested that these compounds exhibit dual topo I and topo II inhibitory activities.
Table XV.
Cytotoxicity Data for Acrylamido Analogs 447–461
| ||||||||
|---|---|---|---|---|---|---|---|---|
| GI50 (μM) |
||||||||
| Compound | Zr-75–1 | MCF7 | Gurav | DWD | Colo 205 | A549 | Hop62 | A2780 |
| ETO | 0.2 | 2.1 | 0.5 | 0.6 | 0.13 | 3.08 | 0.8 | 1.3 |
| 447 | 2.2 | – | 2.4 | – | – | −2 | 2.5 | – |
| 448 | – | 2.7 | – | – | 2.4 | – | – | – |
| 449 | 2.7 | – | 2.5 | – | – | – | – | – |
| 450 | 2.5 | 2.4 | 0.18 | 0.19 | – | 2.1 | 0.17 | 2.7 |
| 451 | 2.2 | – | 2.6 | 2.9 | 2.7 | – | 2.3 | 2.5 |
| 452 | 0.18 | 2.7 | 0.19 | <0.1 | 2.0 | – | 0.19 | 2.2 |
| 453 | 2.2 | – | 2.1 | 2.4 | 2.1 | – | 2.7 | 2.6 |
| 454 | – | 2.7 | 2.1 | <0.1 | – | 2.4 | 0.17 | 2.5 |
| 455 | 2.9 | 2.9 | 2.2 | 2.3 | 0.17 | – | – | 2.5 |
| 456 | 0.18 | <0.1 | 2.1 | 2.2 | – | 2.7 | 0.18 | <0.1 |
| 457 | 2.1 | <0.1 | 2.1 | 2.3 | – | 2.7 | 2.0 | 2.3 |
| 458 | 2.2 | <0.1 | 2.2 | 2.3 | – | 2.8 | 2.1 | 2.2 |
| 459 | 2.9 | – | – | – | – | 2.4 | 2.5 | – |
| 460 | – | – | – | – | – | 2.9 | 2.9 | – |
| 461 | 2.7 | – | 2.7 | 2.7 | – | 2.3 | 2.6 | – |
As outlined in Table XVI, a series of 4β-alkylamidochalcone as well as 4β-cinnamido linked PPTs were also synthesized by Kamal et al.155 and evaluated for cytotoxic activity against five human cancer cell lines (A-549, A375, MCF-7, HT-29, and ACHN). From the screening results, chalcone–PPT conjugates 462–477 showed moderate activity against different cancer cell lines (IC50 5.3–26.7 μM). The activity did not change with different lengths of the alkane chain spacer between the chalcone and PPT moieties. Comparatively, quinolino–chalcone linked PPTs 487–490 showed promising activity with IC50 values ranging from 2.2 to 15.4 μM. Finally, the cinnamido–PPT conjugates 478–486 exhibited IC50 values ranging from 2.1 to 9.5 μM against the A-549 cancer cell line, but generally were less active against the other tested cell lines. However, compounds 478 and 483 showed significant activity against A-549 (IC50 2.7 and 2.1 μM), as well as HT-29 (IC50 2.36 and 0.37 μM) and ACHN (IC50 3.91 and 2.18 μM) cancer cell lines. The IC50 values of ETO in the respective cell lines were 2.34, 1.81, and 7.61 μM. Flow cytometric analysis showed that compounds 478 and 483 arrested the cell cycle in the G2/M phase leading to caspase-3 dependent apoptotic cell death. Furthermore, Hoechst 33258 staining and DNA fragmentation also suggested that 478 and 483 induced cell death by apoptosis.
Table XVI.
Cytotoxicity Data for Alkylamido Chalcone and Cinnamido Analogs 462–490
| |||||
|---|---|---|---|---|---|
| GI50 (μM) |
|||||
| Compound | A549 | A375 | MCF-7 | HT-29 | ACHN |
| ETO | 2.34 | 1.39 | 0.68 | 1.81 | 7.61 |
| PPT | 3.75 | 2.62 | 1.18 | 0.78 | 2.12 |
| 462 | 2.15 | 18.4 | 24.8 | 15.3 | 16.5 |
| 463 | 14.4 | 20.1 | 17.5 | 11.9 | 13.7 |
| 464 | 19 | 13 | 12 | 16 | 10 |
| 465 | 10.7 | 10.8 | 18.1 | 8.75 | 11.9 |
| 466 | 13.6 | 9.8 | 10.2 | 12.1 | 19.5 |
| 467 | 5.9 | 11.4 | 15.8 | 11.7 | 9.3 |
| 468 | 14.7 | 11.5 | 8.3 | 21.1 | 20.8 |
| 469 | 16.7 | 10.3 | 12.6 | 6.47 | 13.4 |
| 470 | 19.1 | 25.8 | 24.2 | 12.4 | 10.18 |
| 471 | 9.1 | 13.8 | 21.8 | 8.1 | 13.8 |
| 472 | 8.3 | 9.3 | 16.2 | 16.9 | 16.1 |
| 473 | 9 | 5.3 | 6.7 | 8.41 | 13.2 |
| 474 | 10.7 | 7.8 | 14.9 | 9.54 | 8.51 |
| 475 | 26.4 | 12.2 | 19.1 | 21.6 | 26.7 |
| 476 | 16.8 | 18.2 | 24.4 | 15.6 | 9.1 |
| 477 | 11.7 | 11.5 | 15.4 | 5.98 | 11.1 |
| 478 | 2.7 | 13.4 | 19.7 | 2.36 | 391 |
| 479 | 6.8 | 9.5 | 14.6 | 7.1 | 18.6 |
| 480 | 8.8 | 5.7 | 13.7 | 11.4 | 7.54 |
| 481 | 9.5 | 17.8 | 11.2 | 19.2 | 14.3 |
| 482 | 7.7 | 19.1 | 15.7 | 17.6 | 17.8 |
| 483 | 2.1 | 18 | 19.2 | 0.37 | 2.18 |
| 484 | 8.2 | 14.5 | 10.1 | 18.1 | 12.8 |
| 485 | 8.4 | 13.2 | 20.1 | 21 | 17.7 |
| 486 | 7.9 | 14 | 17.7 | 8.34 | 13.1 |
| 487 | 15.4 | 14.5 | 13.8 | 12.3 | 10.8 |
| 488 | 13.4 | 7.7 | 11.2 | 7.75 | 15.7 |
| 489 | 10.6 | 10.3 | 8.6 | 11.8 | 10.7 |
| 490 | 7.7 | 6.8 | 2.2 | 8.9 | 9.46 |
A series of new 4β-sulfonamido and 4β-[(4′-sulfonamido)benzamide] derivatives of PPT (491–500 and 501–507) were synthesized and evaluated for cytotoxic activity against six human cancer cell lines (Table XVII).156 All compounds exhibited significant cytotoxic activity with GI50 values ranging from 0.04 to 2.90 μM. Compounds 492, 494, and 495 showed more potent cytotoxic activity against Colo-205 cells than ETO. Flow cytometric analysis showed that the three compounds caused G2/M cell cycle arrest, and especially 494 caused both single- and double-strand DNA breaks. Compound 494 inhibited topo IIα as observed from Western blot analysis, as well as activated caspase-3, p21, p16, and NF-kB, and down-regulated Bcl-2 protein. These findings suggested that 494 can induce apoptotic cell death, apart from acting as a topo IIα inhibitor.
Table XVII.
Cytotoxicity Data for Sulfonamido Analogs 491–507
| ||||||||
|---|---|---|---|---|---|---|---|---|
| GI50 (μM)a |
||||||||
| Compound | Zr-75–1 | MCF7 | KB | DWD | Colo 205 | A549 | Hop62 | A2780 |
| ETO | 0.20 | 2.11 | 0.31 | 0.62 | 0.13 | 3.08 | 0.80 | 1.31 |
| 491 | 2.10 | 0.09 | 2.30 | 2.00 | 2.10 | 0.16 | 2.40 | 1.91 |
| 492 | 0.08 | 0.12 | 0.06 | 0.17 | 0.04 | 0.04 | 0.06 | 0.08 |
| 493 | 2.50 | 0.11 | 2.10 | 0.15 | 0.15 | 0.14 | 2.00 | 0.16 |
| 494 | 0.18 | 0.13 | 0.04 | 0.05 | 0.04 | 0.12 | 0.04 | 0.06 |
| 495 | 0.06 | 0.18 | 0.04 | 0.04 | 0.04 | 0.15 | 0.04 | 0.05 |
| 496 | 0.17 | 0.15 | 0.17 | 0.14 | 0.11 | 0.12 | 0.16 | 0.15 |
| 497 | 2.30 | 2.61 | NTb | 2.21 | 2.71 | 2.00 | 2.01 | 2.11 |
| 498 | 2.31 | 2.21 | 2.31 | 2.91 | NT | 2.11 | NT | 2.40 |
| 499 | 2.11 | 0.14 | 2.11 | 2.61 | NT | NT | NT | 2.30 |
| 500 | 2.51 | 2.21 | NT | NT | NT | 2.71 | 2.21 | 2.311 |
| 501 | 2.90 | 2.71 | NT | 2.50 | NT | NT | 2.01 | 2.11 |
| 502 | 2.23 | 0.17 | NT | 2.80 | NT | NT | 2.91 | 2.11 |
| 503 | 2.81 | 2.32 | 2.00 | NT | 2.12 | 2.60 | 2.22 | 2.00 |
| 504 | NT | 2.90 | NT | 0.14 | 2.31 | 2.51 | 2.61 | 2.42 |
| 505 | 2.11 | 2.30 | 2.50 | 2.88 | 2.80 | NT | NT | 2.51 |
| 506 | 2.90 | NT | 2.01 | 2.10 | NT | 2.70 | 2.00 | NT |
| 507 | 2.12 | 0.16 | – | 2.02 | 2.51 | – | 2.71 | – |
50% Growth inhibition; values are means of three determinations.
NT, not tested.
More recently, Kamal et al.157 synthesized three series of heteroaromatic linked 4β-carboxamido-PPT derivatives (508–516, 517–525, and 526–529, Table XVIII) and evaluated their cytotoxicity against five human cancer cell lines (A-549, HeLa, MCF-7, HT-29,and ACHN). Among all compounds, analog 523 with a (3,4-dichlorophenyl)-1H-pyrazolecarboxamide exhibited significant activity against A549 (IC50 2.1 μM) and good activity against other cell lines, including HT-29 (7.1 μM), B-16 (9.3 μM) and HeLa (9.5 μM), whereas most of the other derivatives showed moderate to weak activity against the tested cancer cell lines. Flow cytometric analysis of 523-treated cells showed cell cycle arrest in the G2/M phase. Hoechst 33258 staining and DNA fragmentation assays revealed that 523 induced cell death by apoptosis. Further, activation of caspase-3 also suggested that 523 produced apoptotic cell death.
Table XVIII.
Cytotoxicity Data for Compounds 508–529
| IC50 (μM) |
|||||
|---|---|---|---|---|---|
| Compound | A549 | HT-29 | ACHN | B-16 | HELA |
| ETO | 2.8 | 1.81 | 7.61 | 1.39 | 1.52 |
| PPT | 3.6 | 1.02 | 2.3 | 2.5 | 2.8 |
| 508 | 13.6 | 19.5 | 17.5 | 16.3 | 5.9 |
| 509 | 17.9 | 22.6 | 26.4 | 16.8 | 7.5 |
| 510 | 24.7 | 27.9 | 28.5 | 20.3 | 5.26 |
| 511 | 38.1 | 32.1 | 30.3 | 22.5 | 11.2 |
| 512 | 23.1 | 38.4 | 23.1 | 12.7 | 13.3 |
| 513 | 19.8 | 25.1 | 34.2 | 23.4 | 18.4 |
| 514 | 31.07 | 11.2 | 16.8 | 22.7 | 12.6 |
| 515 | 12.7 | 14.1 | 16.4 | 27.5 | 21.2 |
| 516 | 20.6 | 19.9 | 15.6 | 23.5 | 16.6 |
| 517 | 28.9 | 26.5 | 30.2 | 16.3 | 7.5 |
| 518 | 19.4 | 20.6 | 23.5 | NA | 14.6 |
| 519 | 22.3 | 26.5 | 25.8 | 16.6 | 16.9 |
| 520 | 5.3 | 8.4 | 11.6 | 9.5 | 9.4 |
| 521 | 39.5 | 36.3 | 31.6 | 45.3 | 25.1 |
| 522 | 15.2 | 16.8 | 27.1 | 18.3 | 11.5 |
| 523 | 2.1 | 7.1 | 15.9 | 9.5 | 9.3 |
| 524 | 15.8 | 10.8 | 32.5 | 25.6 | 14.9 |
| 525 | 21.2 | 26.3 | 18.3 | 26.1 | 15.2 |
| 526 | 10.6 | 10.9 | 17.7 | 18.4 | 12.1 |
| 527 | 11.2 | 5.7 | 17.3 | 18.3 | 8.9 |
| 528 | 17.3 | 14.5 | 19.3 | 10.7 | 14.4 |
| 529 | 9.5 | 9.2 | 13.8 | 12.7 | 13.7 |
During the period of 2002–2009, Tian and co-worker’s laboratory158–160 reported the synthesis and biological evaluation of many 4β-N-substituted-5-fluorouracil-DEPPT derivatives. Most of these analogs showed more significant cytotoxic activity against certain tumor cell lines compared with ETO and 5-fluorouracil (5-FU). Also, compounds 530–536 derived from DEPPT were evaluated as inhibitors of stromelysin-1 as well as collagenase-1 (Fig. 23). Among them, compounds 530 and 531 demonstrated superior inhibitory activity against stromelysin-1 compared with ETO and 5-FU.
Figure 23.
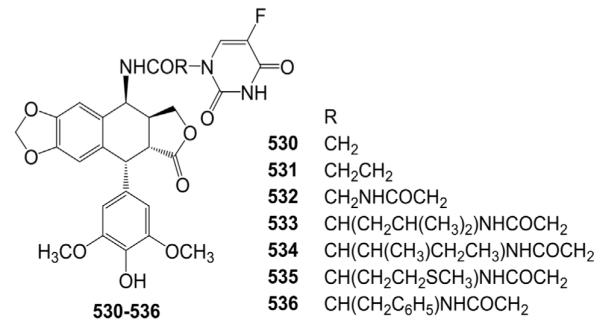
Structures of 5-FU–DEPPT analogs 530–536.
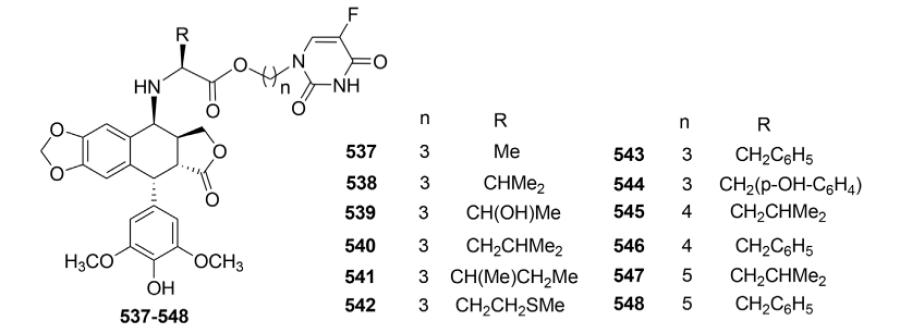
Furthermore, 12 novel conjugates were synthesized by coupling DEPPT with 5-FU-N1-alkyl amino acid esters (537–548).161 When evaluated against four tumor cell lines (HL-60, K562, AGS, and A-549), most of the compounds showed more potent inhibition than ETO (Table XIX). Also, the new analogs showed superior water solubility, with lower logP values than ETO (Table XIX). In addition, the DNA conformation changed from B- to C-form in the presence of 548, likely due to interaction of the compound with calf thymus DNA. Compound 548 was also relatively resistant to metabolism by human plasma.
Table XIX.
Biological Data for 5-FU–DEPPT Conjugates 537–548
| |||||
|---|---|---|---|---|---|
| Cytotoxic activity (IC50, μM)a |
|||||
| Compound | K562b | AGSc | HL-60b | A-549c | log P |
| ETO | >100 | 50.8 | 2.75 | 7.38 | 0.69 |
| 5-FU | >100 | >100 | 65.3 | 50.5 | – |
| 537 | 19.5 | 40.4 | 22.3 | 2.59 | −0.07 |
| 538 | 13.5 | 62.5 | 5.80 | 1.92 | 0.34 |
| 539 | 30.8 | 140.8 | 23.2 | 6.95 | −0.12 |
| 540 | 9.5 | 59.2 | 0.13 | 0.01 | 0.33 |
| 541 | 5.1 | 56.2 | 0.24 | 0.18 | 0.59 |
| 542 | 8.8 | 24.8 | 0.99 | 0.29 | 0.23 |
| 543 | 5.8 | 23.6 | 0.31 | 0.48 | 0.65 |
| 544 | 28.6 | 114.7 | 26.6 | 21.7 | 0.07 |
| 545 | 5.3 | 34.5 | 0.73 | <0.01 | 0.35 |
| 546 | 9.2 | 23.7 | 0.42 | 0.30 | 0.43 |
| 547 | 6.0 | 23.5 | 0.31 | 0.53 | 0.51 |
| 548 | 13.2 | 38.8 | 0.04 | <0.01 | 0.29 |
Average of triplicate experiments.
The microculture [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay method, 48 hr drug exposure.
the sulforhodamine B (SRB) colorimetric assay method, 72 hr drug exposure.
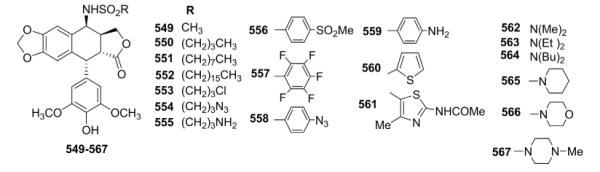
Guianvarch et al.162 synthesized a series of novel 4α-sulfonamide derivatives of DEPPT as depicted in Table XX. Their effects on human topo II and, in some cases, tubulin polymerization were evaluated. The alkyl side chain on the 4β-sulfonamide substituent had an important effect on the topo II inhibitory activity. Methyl sulfonamide analog 549 with the shortest side chain showed the strongest inhibitory activity that decreased sharply for butyl sulfonamide analog 550 with an increased chain length. Derivatives bearing an aromatic ring on the 4β-sulfonamide substituent displayed low topo II inhibition but comparable or slightly better cytotoxic potency (except 557 and 561) than ETO. Substitution of the 4β-sulfonamide group with six different amino (562–567) rather than alkyl or aryl substituents preserved high topo II inhibition and cytotoxic activity (except for 564). With T/C of 235 and 229% against P388 leukemia in vivo, the most active compounds contained morpholine (566) and piperazine (567) sulfonamides. However, these two compounds did not induce long-term survivors (LTS). Comparatively, ETO exhibited a T/C equal to 233%, with 6 of 42 LTS. Against an A-549 orthotopic model of lung carcinoma in vivo, 566 and 567 (T/C 155 and 159%, respectively) were more efficient than ETO (T/C 122–131%). The activity correlated with the inhibitory activity against topo II and marked accumulation of cells in the G2/M phase.
Table XX.
Biological Data for Sulfonamide Analogs 549–567
| |||
|---|---|---|---|
| Compound | Topo IIa (% linear DNA) | IC50 (μM)b | Cell cycle effectc |
| ETO | 50 | 0.83 | 80% (2.5 μM) |
| 549 | 50 | 0.07 | 77% (0.5 μM) |
| 550 | 10 | 0.033 | 75 (0.2 μM) |
| 551 | 35 | 0.25 | 76 (1 μM) |
| 552 | 0 | 5.5 | 73 (25 μM) |
| 553 | 14 | 0.045 | 66 (0.25 μM) |
| 554 | 32 | 0.035 | 65 (0.25 μM) |
| 555 | 45 | 2.5 | 75 (50 μM) |
| 556 | 10 | 0.55 | 75 (2.5 μM) |
| 557 | 0 | 1 | 52 (5 μM) |
| 558 | 9 | 0.21 | 69 (0.5 μM) |
| 559 | 0 | 0.34 | 75 (0.2 μM) |
| 560 | 9 | 0.17 | 71 (0.5 μM) |
| 561 | 7 | 3.6 | 68 (10 μM) |
| 562 | 44 | 0.037 | 66 (0.25 μM) |
| 563 | 24 | 0.083 | 86 (0.25 μM) |
| 564 | 0 | 0.51 | 60 (2.5 μM) |
| 565 | 34 | 0.12 | 77 (0.5 μM) |
| 566 | 31 | 0.1 | 76 (0.25 μM) |
| 567 | 45 | 0.048 | 84 (0.1 μM) |
Average of three independent experiments in the presence of drug at 50 μM.
IC50 is the concentration of drug required to reduce L1210 cell growth by 50%.
Percent of L1210 cells in the G2 + M phases at the indicated concentration.
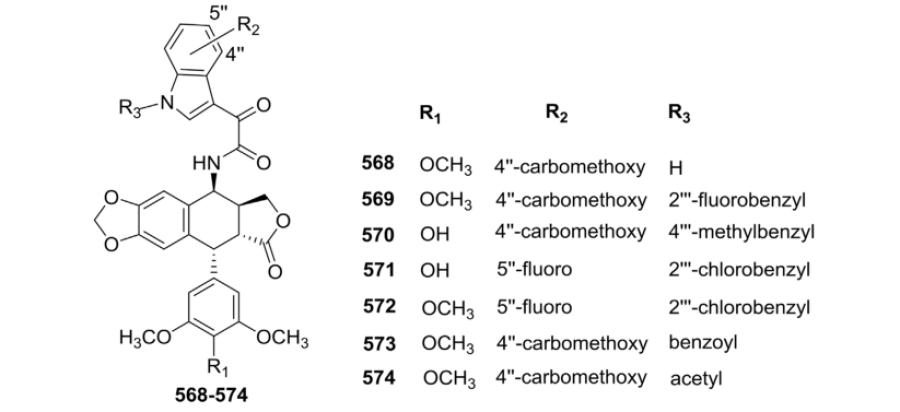
Guo et al.163 synthesized seven novel PPT and DEPPT derivatives (568–574, Table XXI) with 2-(1H-indol-2-yl)-2-oxoacetamides at the C-4β position and tested their cytotoxic activity against five human cancer cell lines, HeLa, KB, KBV, K562, and K562/AO2. Most of the compounds demonstrated improved in vitro antitumor activity and, most importantly, improvedanti-MDR activity compared with ETO. As shown in Table XXI, compounds 568, 571, and 572 exhibited stronger cytotoxic activity than ETO against HeLa cells, while derivatives 569–571, 573, and 574 were more effective than ETO against K562 and K562/AO2 cells.
Table XXI.
Cytotoxicity Data for Indole-Substituted Analogs 568–574
| |||||
|---|---|---|---|---|---|
| IC50 (μM) |
|||||
| Compound | HeLa | K562 | K562/AO2 | KB | KBV |
| ETO | 2.11 ± 1.17 | 0.9 ± 0.08 | 7.05 ± 5.08 | 3.36 ± 1.05 | 55.47 ± 4.36 |
| 568 | 1.05 | 0.99 ± 0.60 | 4.04 ± 0.97 | 3.99 ± 1.28 | 6.28 ± 2.05 |
| 569 | 5.237 | 0.29 ± 0.19 | 0.12 ± 00.6 | 2.49 ± 0.94 | 3.49 ± 1.05 |
| 570 | 3.805 | 0.23 ± 0.16 | 0.70 ± 0.02 | 0.88 ± 0.23 | 3.62 ± 2.17 |
| 571 | 0.15 | 0.40 ± 0.14 | 0.29 ± 0.26 | 1.00 ± 0.78 | 3.16 ± 1.46 |
| 572 | 0.73 | 1.22 ± 0.35 | 0.62 ± 0.28 | 4.99 ± 1.36 | 5.46 ± 3.65 |
| 573 | 10.67 | 0.31 ± 0.18 | 0.22 ± 0.20 | 1.03 ± 0.38 | 2.59 ± 0.28 |
| 574 | 11.11 | 0.46 ± 0.37 | 0.10 ± 0.01 | 1.62 ± 1.25 | 2.30 ± 2.17 |
Cytotoxicity against HeLa, KB, and KBV cell lines and against K562 and K562/AO2 cell lines measured by standard MTT and SRB assay methods, respectively.
Each value represents mean ± SD of three independent experiments.
Shang et al.164 synthesized ten new 4β-imidazolyl PPT and DEPPT analogs (575–584, Fig. 24). All compounds were evaluated for cytotoxicity against three human cancer cell lines. Compound 580 exhibited the highest cytotoxicity with IC50 values of 8.12 ± 1.03, 7.43 ± 0.62, and 4.16 ± 0.54 μM. The latter activity against the K562/ADM drug resistant cell line was particularly notable. In comparison, the IC50 values of ETO were 2.99 ± 0.87, 6.0 ± 1.84, and 76.55 ± 8.36 μM, respectively.
Figure 24.
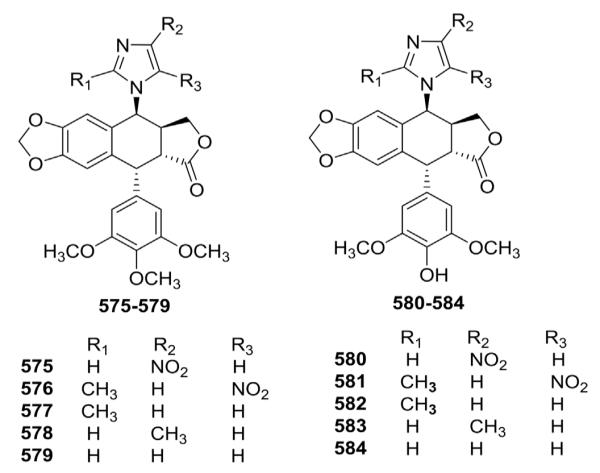
Structures of imidazolyl analogs 575–584.
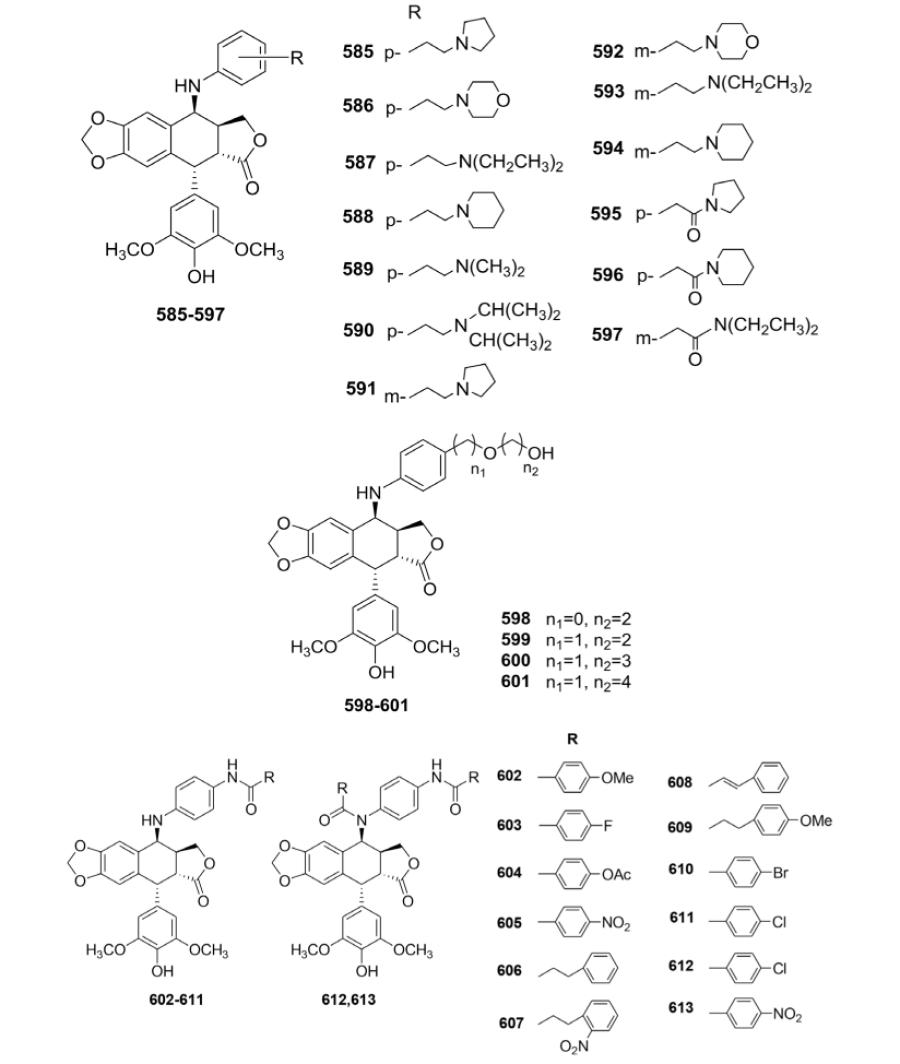
As outlined in Table XXII, Wang et al.165 synthesized three series of new 4β-anilino-DEPPT derivatives (585–613) for cytotoxicity evaluation against four human cancer cell lines, KB, KB/VCR, A549, and 95D. The IC50 values of 585–597 indicated that the alkylamino group attached to the 4β-anilino group was important to the in vitro cytotoxic activity. The ethyl pyrrolidine analog 585 exhibited the highest potency with IC50 values ranging from 0.036 to 1.57 μM against the tested cancer cell lines. Among compounds 598–601 with a hydrophilic alkoxy group on the end of the 4β-side chain, compound 598 with the shortest chain length showed the most potent cytotoxic activity. As the number of atoms between the benzene ring and the terminal hydroxy group increased (599–601), activity decreased (except for 599 against 95D cell line). Compared with ETO, compounds 602–613, which have amide moieties on the para-position of the anilino ring, displayed comparable or slightly weaker cell growth inhibition against KB and A549 cell lines, but superior cytotoxic potency against KB/VCR and 95D cell lines (except 612 and 613).
Table XXII.
Cytotoxicity Data for Anilino Analogs 585–613
| |||||
|---|---|---|---|---|---|
| IC50 (μM) |
|||||
| Compound | KB | KB/VCR | A549 | 95D | RFa |
| ETO | 4.61 | 83.4 | 2.56 | 20.2 | 18.09 |
| 585 | 0.21 | 1.57 | 0.24 | 0.036 | 7.84 |
| 586 | 0.78 | 1.27 | 1.94 | 11.5 | 1.63 |
| 587 | 0.45 | 1.53 | 0.40 | 9.84 | 3.4 |
| 588 | 4.21 | 19.1 | 13.4 | 17.2 | 4.54 |
| 589 | 2.42 | 3.95 | 19.0 | 17.1 | 1.63 |
| 590 | 0.44 | 2.20 | 0.49 | 10.0 | 5 |
| 591 | 0.39 | 1.85 | 3.91 | 1.17 | 4.74 |
| 592 | 0.48 | 2.09 | 5.10 | 14.8 | 4.35 |
| 593 | 0.77 | 3.87 | 7.49 | 14.6 | 5.03 |
| 594 | 0.43 | 1.78 | 2.75 | 7.40 | 4.14 |
| 595 | 1.52 | 2.20 | 15.9 | >50 | 1.45 |
| 596 | 0.40 | 2.33 | 5.20 | 25.8 | 5.83 |
| 597 | 0.42 | 27.2 | 4.78 | >50 | 64.76 |
| 598 | 2.88 | 6.51 | 4.54 | 4.13 | 2.26 |
| 599 | 3.12 | 7.39 | 5.11 | 0.55 | 2.37 |
| 600 | 6.00 | 5.08 | 7.45 | 12.04 | 0.85 |
| 601 | 6.60 | 9.55 | 8.5 | 20.30 | 1.45 |
| 602 | 6.76 | 9.28 | 3.41 | 0.44 | 1.37 |
| 603 | 2.91 | 5.31 | 1.56 | 1.08 | 1.82 |
| 604 | 1.91 | 8.48 | 2.68 | 0.42 | 4.44 |
| 605 | >50 | 13.7 | 11.0 | 3.41 | <0.27 |
| 606 | 6.88 | 3.70 | 3.31 | 1.15 | 0.54 |
| 607 | 6.69 | 5.30 | 8.43 | 0.41 | 0.79 |
| 608 | 6.53 | 2.80 | >50 | 1.25 | 0.43 |
| 609 | >50 | 3.08 | >50 | 1.23 | <0.06 |
| 610 | 8.75 | 5.72 | 18.9 | 1.66 | 0.65 |
| 611 | 22.1 | 13.06 | 19.5 | 6.56 | 0.59 |
| 612 | >50 | >50 | >50 | 15.7 | NTb |
| 613 | >50 | 14.21 | >50 | 19.6 | <0.28 |
Resistance factor calculated as ratio of IC50 against MDR cells and IC50 against corresponding drug-sensitive parental cells.
NT, not tested.
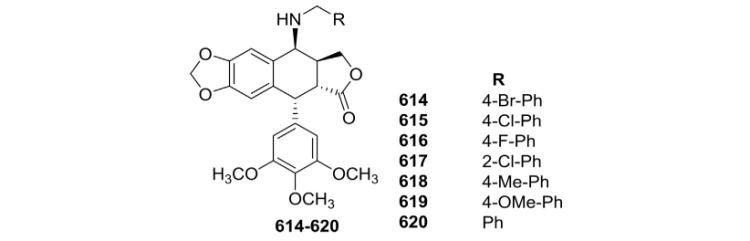
As depicted in Table XXIII, seven 4β-benzylamino PPT derivatives (614–620) were also synthesized by Wang et al.166 The seven compounds displayed strong cytotoxic activity against A549, HCT-116, and HepG2 cancer cell lines. Among them, compound 615 possessed the highest cytotoxicity with an average IC50 value of 3.8 μM. The new compounds were generally more potent than ETO against the three tested cancer cell lines, indicating that PPT derivatives with a benzylamino structural modification possess potent cytotoxic activity.
Table XXIII.
Cytotoxicity Data for Benzylamino Analogs 614–620
| ||||
|---|---|---|---|---|
| IC50 (μM) |
||||
| Compound | A549 | HCT-116 | HepG2 | Average |
| ETO | 21.4 | 13.5 | 4.9 | 13.3 |
| 614 | 4.9 | 6.3 | 5.8 | 5.7 |
| 615 | 2.9 | 5.6 | 3.0 | 3.8 |
| 616 | 4.0 | 5.1 | 5.0 | 4.7 |
| 617 | 5.5 | 6.4 | 6.3 | 6.1 |
| 618 | 3.2 | 5.2 | 5.8 | 4.7 |
| 619 | 9.5 | 8.7 | 8.1 | 8.8 |
| 620 | 4.8 | 6.1 | 5.1 | 5.3 |
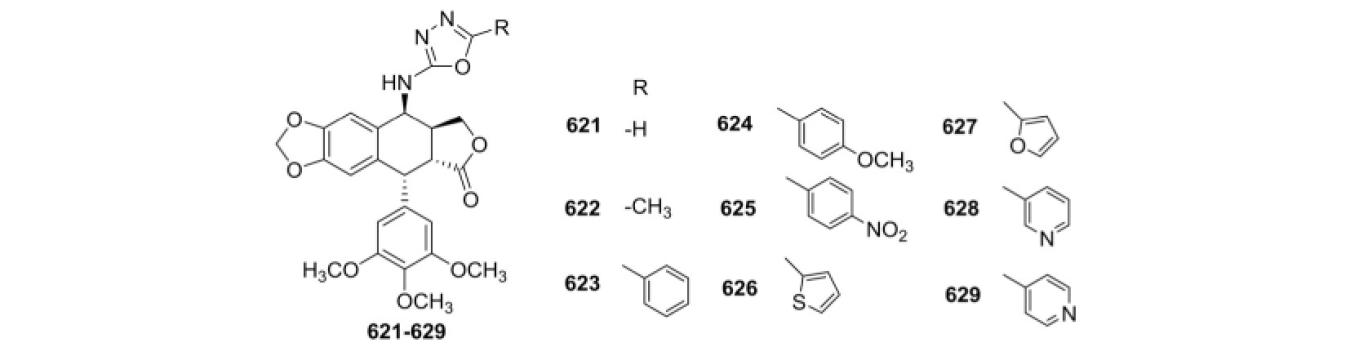
Ren et al.167 introduced 2-amino-1,3,4-oxadiazole moieties into the C-4 position to give nine novel 4β-N-substituted PPT derivatives (621–629) and evaluated their cytotoxicity against DU-145, SGC-7901, A549, SH-SY5Y, HepG2, and HeLa cell lines (Table XXIV). These derivatives displayed much lower cytotoxicity toward the tested normal cell lines (L929 and Vero) than PPT. Among them, compound 622 exhibited the highest potency against HepG2 and HeLa with IC50 values of 1.29 and 2.68 μM, respectively. It also showed much better selectivity between the tumor and normal cells than ETO and PPT. Moreover, in MOA studies, 622 inhibited gene and protein expressions of DNA topo IIb, suspended the cell cycle at S-phase, and eventually caused apoptosis of the tumor cells.
Table XXIV.
Cytotoxicity Data for Oxadiazole-Amino Analogs 621–629
| ||||||||
|---|---|---|---|---|---|---|---|---|
| IC50 (μM) |
||||||||
| Compound | DU-145 | SGC-7901 | A549 | SH-SY5Y | HepG2 | HeLa | L929 | Vero |
| ETO | 64.26 | 12.09 | 10.12 | 5.37 | 5.48 | 96.26 | 1.00 | 31.14 |
| PPT | >100 | 2.95 | 13.62 | 2.54 | 2.27 | 7.58 | 1.37 | 1.78 |
| 621 | 11.34 | >100 | >100 | 25.81 | 11.85 | >100 | 43.29 | 15.37 |
| 622 | >100 | 38.11 | >100 | 14.66 | 1.51 | 2.68 | 19.62 | 5.49 |
| 623 | >100 | >100 | NTa | 26.88 | 74.20 | 85.34 | >100 | >100 |
| 624 | 76.12 | >100 | 53.66 | 7.39 | 27.95 | 17.66 | 18.15 | 7.43 |
| 625 | >100 | >100 | NT | 16.58 | >100 | >100 | >100 | NT |
| 626 | >100 | 56.58 | >100 | 20.02 | 38.82 | 42.53 | 70.85 | >100 |
| 627 | 35.42 | NT | 69.22 | 29.48 | 38.81 | 58.85 | >100 | >100 |
| 628 | >100 | >100 | >100 | 22.47 | 72.77 | >100 | 54.56 | >100 |
| 629 | >100 | >100 | >100 | 41.53 | 96.56 | >100 | 20.47 | >100 |
NT, not tested.
Zhao et al.168 reported 12 new aroylthiourea derivatives (630–641, Fig. 25) of 4β-amino-DEPPT. Compound 641 was the most potent (IC50 4.4, 5.0, and 9.5 μM), although most of the derivatives displayed significant cytotoxic activity against HepG2, A549, and HCT-116 cancer cell lines. MOA studies indicated inhibition of the catalytic activity of DNA topo II, which caused HCT-116 cell cycle arrest at the G2/M phase.
Figure 25.
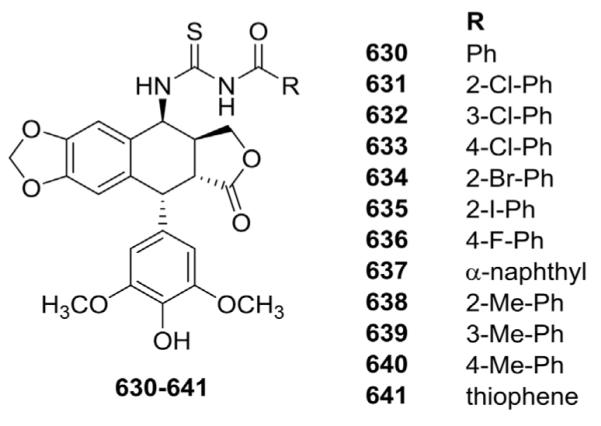
Structures of aroylthiourea-amino analogs 630–641.
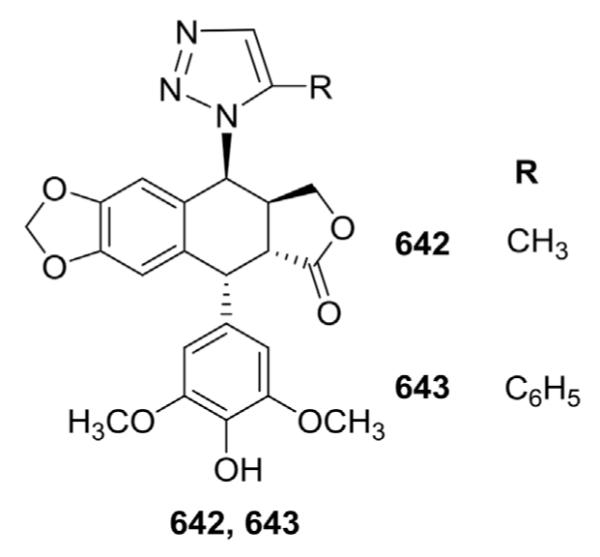
A 1,2,3-triazole ring is widely present in various drugs and has also been incorporated by several research groups into PPT analogs. In 1999, Tao et al.169 reported two novel 4β-[(5-substituted)-1,2,3-triazol-1-yl]-DEPPT derivatives (642 and 643, Fig. 26). In cytotoxicity testing against L1210 cells, compound 642 (ID50 0.13 μM, 5-methyl) and ETO (ID50 0.15 μM) exhibited almost equivalent activity, while compound 643 (ID50 0.0030 μM, 5-phenyl) was 50-fold more active. Subsequently, Cao et al.170 also reported a brief synthesis of 4-(1,2,3-triazol-1-yl)-PPT derivatives as a new class of antitumor compounds.
Figure 26.
Structures of triazole analogs 642–643.
In 2008, Reddy’s group171,172 synthesized a library of 4β-[(4-substituted)-1,2,3-triazol-1-yl]-PPTs and DEPPTs using click chemistry protocols. The library focused on aniline-based (644–653, Table XXV), phenol-based (654–667, Table XXVI)-, and thiophenol-based (668–673, Table XXVI) 1,2,3-triazole derivatives. Many compounds showed promising antitumor activity, and certain analogs with aromatic substituents on the triazole moiety displayed excellent cytotoxicity. PPT analog 648 with a 3-nitroaniline moiety showed significant cytotoxicity against DU-145, PC-3, SF-295, HCT-15, and 502713 cell lines with highest potency against the HCT-15 cell line (IC50 0.04 μM). PPT analogs 658 and 659 with 2-chloro- and 4-chlorophenoxy groups also showed significant cytotoxicity against the seven tested cancer cell lines with highest potency against HT-29 (IC50 0.34 μM) and HCT-15 (IC50 0.31 μM) cell lines, respectively.
Table XXV.
Cytotoxicity Data for Triazole Analogs 644–653
| |||||||
|---|---|---|---|---|---|---|---|
| IC50 (μM) |
|||||||
| Prostate |
CNS SF-295 |
Colon |
Liver HEP-2 |
Lung A-549 |
|||
| Compound | DU-145 | PC-3 | HCT-15 | 502713 | |||
| PPT | 2.97 | 18.9 | 5.69 | 1.00 | 3.88 | 1.42 | 7.63 |
| ETO | 3.85 | 17.4 | 13.3 | 1.48 | 2.42 | 2.15 | 5.62 |
| 644 | 1.06 | 1.09 | 1.94 | 0.36 | 0.78 | 9.14 | 7.88 |
| 645 | 1.19 | 1.53 | 0.96 | 0.14 | 0.62 | 1.50 | 5.17 |
| 646 | 0.93 | 1.18 | 0.84 | 0.34 | 0.71 | 1.37 | 4.99 |
| 647 | 4.16 | 1.37 | 3.11 | 4.22 | 0.98 | 1.22 | 8.63 |
| 648 | 0.65 | 0.74 | 0.68 | 0.04 | 0.56 | 3.65 | 5.78 |
| 649 | 4.37 | 6.63 | 4.20 | 1.55 | 1.19 | 4.10 | 7.43 |
| 650 | 1.12 | 0.88 | 0.78 | 0.14 | 0.61 | 4.64 | 6.10 |
| 651 | 1.09 | 1.03 | 0.70 | 0.40 | 0.68 | 2.96 | 7.46 |
| 652 | 3.57 | 3.45 | 1.00 | 1.43 | 1.88 | 1.26 | 9.24 |
| 653 | 2.75 | 4.49 | 6.03 | 1.69 | 1.34 | 3.27 | 7.41 |
Table XXVI.
Cytotoxicity Data for Triazole Analogs 654–673
| |||||||
|---|---|---|---|---|---|---|---|
| IC50 (μM) |
|||||||
| Compound | HT-29 | HCT-15 | 502713 | HOP-62 | A549 | MCF-7 | SF-295 |
| ETO | 5.31 | 7.07 | 3.88 | 4.80 | 7.63 | 2.51 | 5.69 |
| 654 | 9.65 | 1.91 | 0.96 | NTa | 1.0 | 0.90 | 0.93 |
| 655 | 0.34 | 0.53 | 0.32 | 0.50 | 1.42 | 1.60 | 3.79 |
| 656 | 0.53 | 1.46 | 0.72 | 4.18 | 1.67 | 1.84 | 8.86 |
| 657 | 8.25 | 5.11 | 20.8 | NT | 8.11 | 4.84 | 3.65 |
| 658 | 0.34 | 0.62 | 0.41 | 0.69 | 0.64 | 0.79 | 1.0 |
| 659 | 0.35 | 0.31 | 0.43 | 0.57 | 0.56 | 1.6 | 1.67 |
| 660 | 21.8 | 23.4 | 44.2 | 50.0 | 48.1 | NT | 50.6 |
| 661 | 1.46 | 3.22 | 4.07 | 4.32 | 4.12 | 3.68 | 1.21 |
| 662 | 4.72 | 4.33 | NT | NT | 5.06 | 4.22 | 9.15 |
| 663 | 3.37 | 3.31 | 7.56 | 6.5 | 4.26 | 4.74 | 16.6 |
| 664 | 2.86 | 4.83 | 22.0 | 7.87 | 6.22 | 4.79 | 1.22 |
| 665 | 2.88 | 3.53 | 4.69 | 10.7 | 5.3 | 4.74 | 4.64 |
| 666 | 8.87 | 4.79 | 41.6 | NT | 19.8 | 4.29 | 2.92 |
| 667 | 9.23 | 6.67 | 5.17 | 28.6 | 29.6 | 5.18 | 1.45 |
| 668 | 0.37 | 0.65 | 0.46 | 0.81 | 0.7 | 1.91 | 5.44 |
| 669 | 22.7 | 21.8 | 28.5 | 32.9 | 29.2 | 23.1 | 49.5 |
| 670 | 24.4 | 27.5 | 37.0 | 50.0 | 32.2 | 17.5 | 30.2 |
| 671 | 2.08 | 3.31 | 4.76 | 4.84 | 5.06 | 4.95 | 9.56 |
| 672 | 1.38 | 3.68 | 4.37 | 5.0 | 6.22 | 4.64 | 3.89 |
| 673 | 1.51 | 3.39 | 3.74 | 5.0 | 4.59 | 3.7 | NT |
NT, not tested.
In the same year, Reddy and co-workers173 also generated a series of carbohydrate-based 1,2,3-triazole derivatives by condensing 1-O-propargyl monosaccharides with 4β-azido-PPT and 4β-azido-4′-O-demethyl-PPT (Table XXVII). The new compounds were screened for cytotoxicity against six human cancer cell lines, DU-145 (prostate), PC-3 (prostate), A-549 (lung), HOP-62 (lung), HCT-15 (colon), and SF-295 (CNS), but these generally were not as potent as ETO. Compound 675, in which the sugar moiety was per-acylated, was less potent than the corresponding free glycoside (674). The presence of a hydroxy moiety on ring E was essential to the activity; for example, compounds containing dimethoxy substitution (679–682) were more active than those having trimethoxy substitution (674–678). Thus, compound 679, which contained dimethoxy rather than trimethoxy substitution on ring E and a glucose moiety on the triazolyl ring, exhibited the best activity among the nine tested compounds.
Table XXVII.
Cytotoxicity Data for Triazole Analogs 674–682
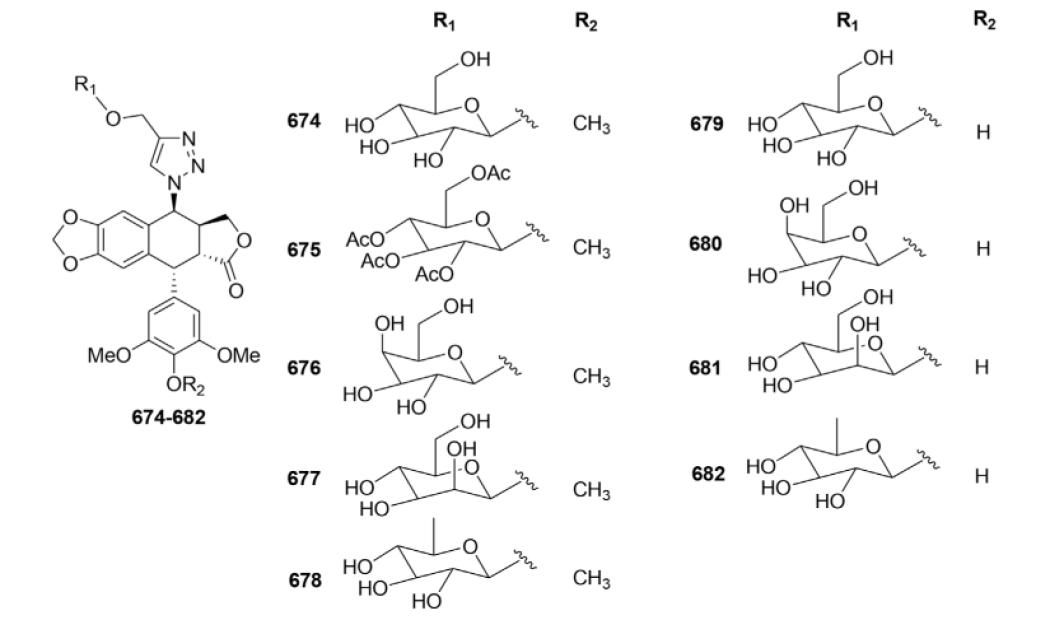
| ||||||
|---|---|---|---|---|---|---|
| IC50 (μM) |
||||||
| Compound | DU-145 | PC-3 | A549 | HCT-15 | HOP-62 | SF-295 |
| ETO | 2.97 | 100 | 7.63 | 1 | 4.8 | 5.69 |
| 674 | 25.8 | 127 | 125 | 15 | NTa | 26.8 |
| 675 | 42 | 221 | 556 | NT | 142 | 35.1 |
| 676 | 237 | 316 | 59.9 | 16 | 178 | 14.5 |
| 677 | 18.6 | 94.9 | 51.8 | 237 | 121 | 53.4 |
| 678 | 26.5 | 268 | 114 | NT | NT | 88 |
| 679 | 3.11 | 34.6 | 3 | 0.93 | 8.55 | 2.01 |
| 680 | 4.95 | 24.3 | 6.88 | 1 | 5.27 | 16.1 |
| 681 | 2.73 | 8.62 | 7.33 | 3.57 | 5.96 | 2.51 |
| 682 | 5.04 | 20.8 | 7.04 | 2.14 | 16.2 | 8.29 |
NT, not tested.
Information on docking studies prompted Reddy et al.174 to synthesize additional 4β-[(4-alkyl)-1,2,3-triazol-1-yl] derivatives of PPT and DEPPT using click chemistry (Table XXVIII). Most of the derivatives exhibited better cytotoxicity than ETO. In particular, three compounds, 683, 691, and 692, showed notable potency against PC-3 (IC50 0.03, 0.06, 0.06 μM, respectively) and HEP-2 (IC50 0.06, 0.06, 0.05 μM, respectively) cell lines. Compounds 691 and 692 also displayed excellent IC50 values (0.01 and 0.04 μM, respectively) against MCF-7. The cytotoxic activity generally decreased as the alkyl chain length increased, so that analogs with ethyl (683, 692) or hydroxymethyl (691) groups on the triazole moiety were the most potent. Moreover, DNA fragmentation and flow cytometric results revealed that these derivatives induced dose-dependent apoptosis. Docking experiments showed a good correlation between their calculated interaction energies with observed IC50 values and topo II inhibitory effects of all compounds.
Table XXVIII.
Cytotoxicity Data for Alkyltriazole Analogs 683–700
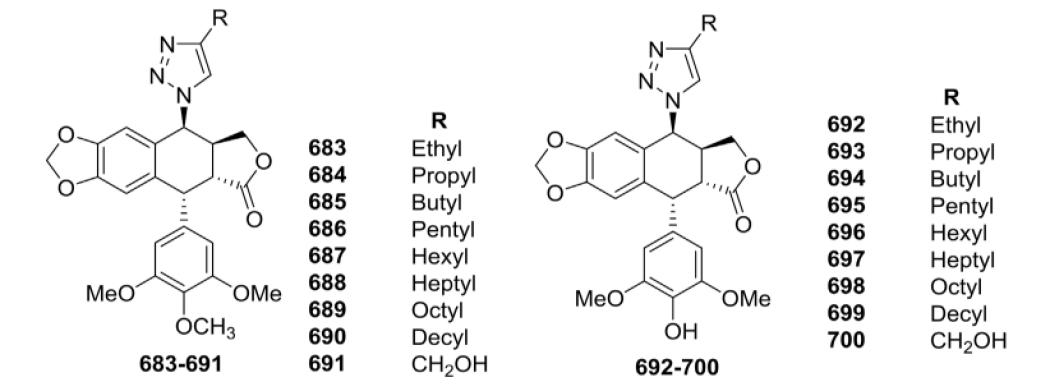
| ||||||
|---|---|---|---|---|---|---|
| IC50 (μM) |
||||||
| Compound | SF-295 | A549 | PC-3 | Hep-2 | HCT-15 | MCF-7 |
| ETO | 13.5 | 5.62 | 17.5 | 2.15 | 7.15 | 19 |
| 683 | 1.8 | 35 | 0.03 | 0.06 | 0.4 | 0.4 |
| 684 | 3.6 | 39 | 5.1 | 1.6 | 0.03 | 1.4 |
| 685 | 8.1 | 82 | 6.6 | 9.6 | 11 | 9.8 |
| 686 | 24 | 39 | 8.4 | 7.7 | 11 | 9.5 |
| 687 | 10 | 35 | 6.4 | 10 | 27 | 8.6 |
| 688 | 25 | 36 | 23 | 9.8 | 45 | 6.4 |
| 689 | 14 | >100 | 17 | 18 | >100 | 9.3 |
| 690 | 6.2 | >100 | 5.5 | 5.8 | 29 | 5.7 |
| 691 | 2.1 | >100 | 0.06 | 0.06 | 15 | 0.01 |
| 692 | 4.8 | 17 | 0.06 | 0.05 | 1.9 | 0.04 |
| 693 | 2.3 | 27 | 0.2 | 2.9 | 0.8 | 5.7 |
| 694 | 21 | 35 | 12 | 17 | 34 | 20 |
| 695 | 4.3 | 26 | 4.7 | 21 | 2.1 | 14 |
| 696 | 18 | 26 | 8.7 | >100 | 11 | 19 |
| 697 | 10 | 45 | 5.8 | 6 | 15 | 13 |
| 698 | 5.7 | >100 | 5.1 | 3.8 | 13 | 11 |
| 699 | 13 | 30 | 19 | 19 | 15 | 11 |
| 700 | 15 | 18 | 8.2 | 6.7 | 18 | 0.6 |
In addition, Chen’s group175 synthesized and evaluated nine different 4β-[(4-substituted)-1,2,3-triazol-1-yl]-PPT derivatives (701–709, Fig. 27) for cytotoxicity against human cancer cell lines (HeLa, K562, K562/A02). Most of the compounds demonstrated significant cytotoxic activity. Some derivatives exhibited high cytotoxicity toward the drug resistant K562/A02 leukemic cell line, whereas ETO was not active. Among them, compound 707 showed excellent potency, with IC50 values of 0.082, 0.053, and 0.059 μM against HeLa, K562, and K562/A02, respectively, more than 40 times more cytotoxic than ETO.
Figure 27.
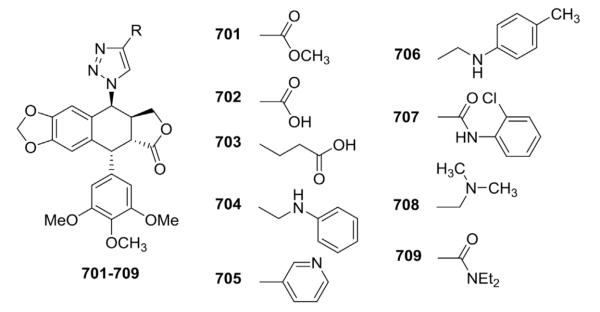
Structures of compounds 701–709.
The same synthetic route was also used by Chen et al.176 to prepare several additional series of 4β-[(4-substituted)-1,2,3-triazol-1-yl]-PPT congeners (710–734, Table XXIX). When evaluated against four tumor cell lines (HepG2, MKN-45, NCI-H1993, and B16), seven compounds (717–721, 728, and 730) were notably more potent compared with ETO. Among phenyoxymethyl and phenylthiomethyl substituted compounds (710–723), analogs with an electron-donating group (e.g., 712, 714, 717, 719, and 720) at the meta or para position of the aromatic ring tended to be more active than those with an electron-withdrawing group (e.g., 710, 711, 715, and 716). Methoxy groups at both meta and para positions of the phenyl ring were also tolerated, for example, compound 720 displayed significant activity with an average IC50 value of 1.58 μM. The two methyl benzamide substituted compounds 724 and 725 were less active. Significantly, the presence of a fluorine atom on the phenyl moiety (e.g., 730) was optimal for chalcone-containing analogs (726–734).
Table XXIX.
Cytotoxicity Data for Triazole Analogs 710–734
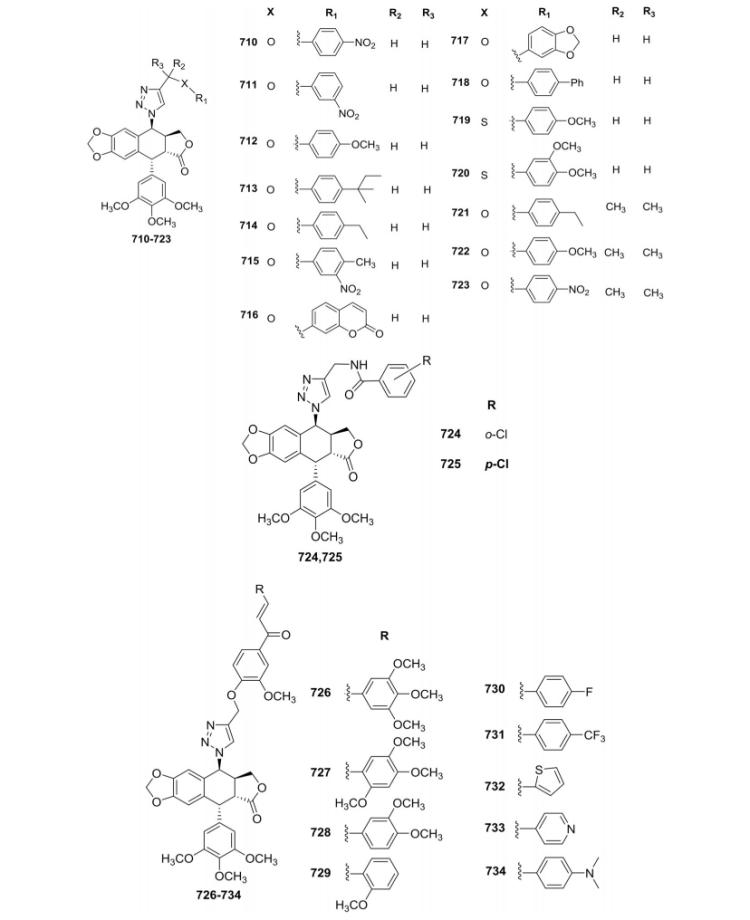
| ||||
|---|---|---|---|---|
| IC50 at 48 hr (μM)a |
||||
| Compound | HepG2 | MKN-45 | NCI-H1993 | B16 |
| ETO | >10.00 | 6.34 ± 0.89 | 7.93 ± 0.77 | >10.00 |
| 710 | 3.87 ± 0.23 | 6.10 ± 0.98 | 59.60 ± 2.34 | 30.00 ± 1.87 |
| 711 | 1.05 ± 0.04 | 0.99 ± 0.01 | 54.50 ± 4.78 | 10.00 ± 1.89 |
| 712 | 0.98 ± 0.04 | 1.05 ± 0.06 | 10.70 ± 1.26 | 2.85 ± 0.67 |
| 713 | 19.00 ± 1.89 | 7.62 ± 0.87 | 64.00 ± 4.76 | 66.60 ± 4.56 |
| 714 | 4.18 ± 0.56 | 4.26 ± 0.65 | 8.25 ± 0.89 | >80.00 |
| 715 | 3.80 ± 0.19 | 3.11 ± 0.22 | 4.13 ± 0.34 | 4.53 ± 0.55 |
| 716 | 7.78 ± 0.77 | 1.45 ± 0.13 | 6.92 ± 0.55 | 4.35 ± 0.65 |
| 717 | 0.25 ± 0.01 | 0.93 ± 0.04 | 0.85 ± 0.05 | 2.93 ± 0.32 |
| 718 | 0.15 ± 0.01 | 0.22 ± 0.01 | 0.24 ± 0.03 | 0.54 ± 0.09 |
| 719 | 0.26 ± 0.02 | 0.13 ± 0.02 | 0.49 ± 0.05 | 2.52 ± 0.33 |
| 720 | 0.31 ± 0.08 | 0.44 ± 0.04 | 1.45 ± 0.12 | 0.90 ± 0.35 |
| 721 | 0.18 ± 0.01 | 0.31 ± 0.02 | 0.29 ± 0.05 | 0.57 ± 0.01 |
| 722 | 1.72 ± 0.09 | 1.72 ± 0.09 | 3.00 ± 0.14 | 2.50 ± 0.34 |
| 723 | 3.14 ± 0.19 | 3.14 ± 0.78 | 2.04 ± 0.43 | 3.35 ± 0.12 |
| 724 | 8.55 ± 0.98 | 4.20 ± 0.98 | 7.25 ± 0.98 | 8.25 ± 0.65 |
| 725 | 4.85 ± 0.56 | 3.10 ± 0.81 | 3.90 ± 0.56 | 4.10 ± 0.45 |
| 726 | 2.80 ± 0.09 | 1.05 ± 0.05 | 1.63 ± 0.07 | 2.48 ± 0.32 |
| 727 | >80.00 | >10.00 | >10.00 | >80.00 |
| 728 | 1.61 ± 0.23 | 1.01 ± 0.05 | 2.14 ± 0.07 | 1.56 ± 0.09 |
| 729 | 9.65 ± 0.98 | 6.80 ± 0.66 | >10.00 | >20.00 |
| 730 | 0.81 ± 0.04 | 0.69 ± 0.04 | 0.85 ± 0.11 | 0.53 ± 0.01 |
| 731 | 2.39 ± 0.11 | 2.34 ± 0.22 | 8.20 ± 0.99 | 3.45 ± 0.45 |
| 732 | 2.20 ± 0.15 | 2.33 ± 0.19 | 4.37 ± 0.76 | 2.18 ± 0.88 |
| 733 | 11.95 ± 1.01 | >10.00 | >10.00 | >10.00 |
| 734 | 2.26 ± 0.44 | 0.91 ± 0.04 | 2.04 ± 0.17 | 2.93 ± 0.22 |
Cytotoxicity results are expressed as IC50 values, the compound concentration producing a 50% cell growth inhibition and represent the mean ± SD of three independent experiments.
5. PPT DERIVATIVES SPIN-LABELED AT C-4 POSITION
Our prior review on PPT46 included rationale for introduction of a stable nitroxide radical into PPT derivatives. Free radical compounds of this type have also been introduced into many antitumor drugs, such as thiotepa, 5-fluorouracil, nitrosourea, rubomycin and 6-mercaptopurine,177 camptothecin,178 rotenone,179 and glycyrrhetinic acid.180 The resulting new compounds often showed superior pharmacological properties compared with the parent compounds. Nitroxyl radicals can serve as transporter vehicles through cell membranes, regulate levels of oxidized cytochrome P-450 that have been brought down by lethal doses of cytostatic agents, and generally improve antitumor and antioxidant properties of drugs.
In view of the above advantages, our laboratory introduced stable radicals as spin labels at different positions of the PPT skeleton, particularly the C- or D-ring, over the past 27 years.181–194 Such analogs frequently exhibited significant antitumor activity against several mouse transplantable tumors together with remarkably decreased toxicity.
Both antitumor and antioxidant activities of D-ring spin-labeled PPT derivative GP–1 (735) and two congeners GP-1-OH (736) and GP-1-H (737) were determined (Fig. 28).187 The former testing used mice with transplanted S180 and HepA tumors. The rank order of potency was GP-1 > GP-OH > GP-1-H, which was attributed to the influence of partition coefficients and ionization constants on the compounds’ properties. Moreover, the related GP-11 (738, Fig. 28) was reported as a low immunosuppressive antitumor agent;188 it increased the mitotic index and arrested cells at the G2/M rather than S phase. In addition, the 4β-amino DEPPT-modified compound, GP-7 (739, Fig. 28), which is spin-labeled on the C-ring, showed lower toxicity but equivalent activity both in vivo (mouse solid tumors S180 and HePA) and in vitro (mouse leukemia L1210 and human stomach carcinoma SGC-7901) in comparison with ETO.189 Additional spin-labeled PPT derivatives included 4β-amido190 and 4β-ester191 compounds. These modifications resulted in substantially increased activity relative to ETO, as exemplified in three spin-labeled 4β-amido derivatives (740–742, Fig. 28).190 Compounds 740 and 741 were as or more potent than ETO against human nasopharyngeal carcinoma KB, lung cancer A-549, stomach carcinoma SGC-7901, and mouse leukemia L1210 and P388 cells.191
Figure 28.

Structures of spin-labeled compounds 735–742.
Most spin-labeled PPT esters (743–754, Table XXX) displayed greater cytotoxicity than ETO against P-388 and A-549 cell lines.191 Compound 750 showed the highest potency with IC50 values less than 0.01 μM against P-388 and 0.13 μM against A-549. The cytotoxic and antioxidative activities of 4α-substituted compounds (747–754) generally were superior to those of 4α-substituted compounds (743–746).191 The studies also found a correlation between cytotoxicity against tumor cells and antioxidant activity, suggesting that these compounds may act on tumor cells through an antioxidative mechanism. No distinct correlation was found between cytotoxic activity and the size or degree of saturation of the ring system in the nitroxide moiety.
Table XXX.
Biological Data for Spin-Labeled Compounds743–754
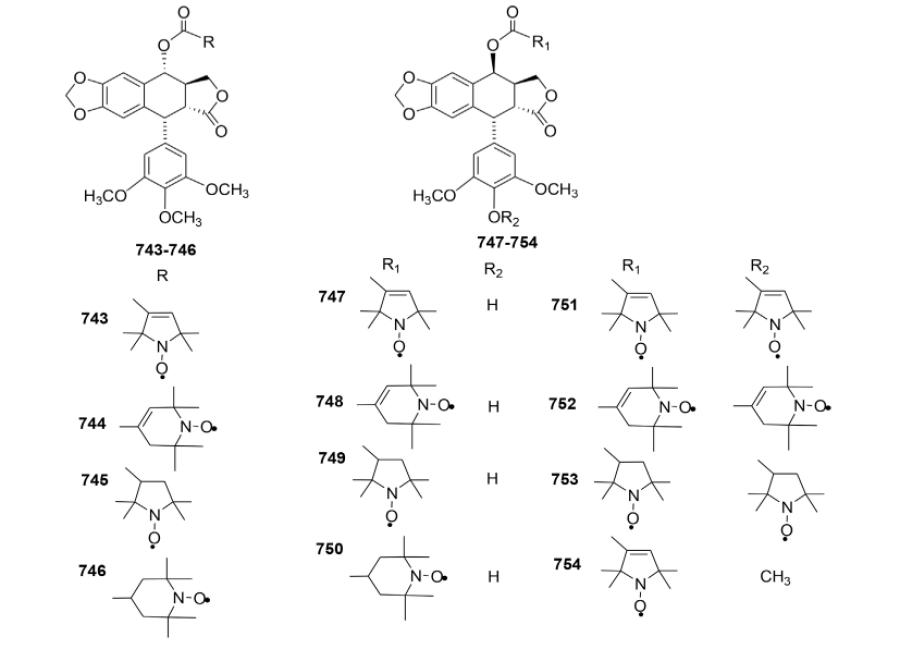
| |||||
|---|---|---|---|---|---|
| Cytotoxic activity (IC50, μM) |
Antioxidative activity |
||||
| Compound | P388 | A549 | Liver | Kidney | Heart |
| ETO | 1.18 | 7.14 | 46.38 | 64.63 | 82.73 |
| 743 | 0.82 | 0.85 | 7.92 | 8.80 | 8.76 |
| 744 | 0.18 | 8.42 | 13.55 | 10.35 | 10.47 |
| 745 | 0.01 | 6.43 | 15.41 | 15.42 | 13.39 |
| 746 | <0.01 | 6.86 | 10.59 | 10.96 | 10.88 |
| 747 | 0.02 | 0.30 | 8.98 | 7.82 | 3.8 |
| 748 | 0.09 | 0.46 | 4.19 | 5.10 | 4.84 |
| 749 | 0.07 | 0.90 | 5.50 | 5.54 | 9.07 |
| 750 | <0.01 | 0.13 | 5.26 | 4.96 | 5.54 |
| 751 | 0.34 | >10 | 4.26 | 3.04 | 5.50 |
| 752 | 0.03 | 0.28 | 2.93 | 5.47 | 2.93 |
| 753 | 0.24 | 8.19 | 5.54 | 5.41 | 6.20 |
| 754 | 0.04 | 0.42 | 7.32 | 7.64 | 7.56 |
Because of their good water solubility, L-amino acids are often used as carriers for drugs. Therefore, we linked N-(1-oxyl-2,2,6,6-tetramethyl-4-piperidinyloxycarbonyl)-amino acids with PPT to give five novel spin-labeled amino acid-linked PPT derivatives (755–759, Table XXXI).192,193 All five compounds showed significant activity against K562, L-1210, and HL-60 cell lines, with compounds 757–759 exhibiting the highest cytotoxicity against HL-60 cells (IC50 0.108, 0.552, and 0.171 μM, respectively).
Table XXXI.
Cytotoxicity Data for Spin-Labeled Compounds 755–759
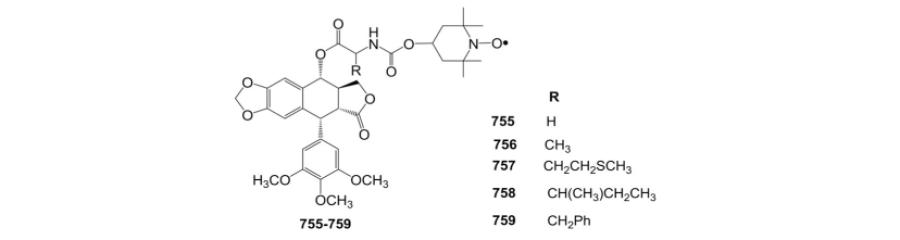
| |||||
|---|---|---|---|---|---|
| IC50 (μM)a |
|||||
| Compound | K562 | HL-60 | SPCA-1 | Lewis | L-1210 |
| ETO | 3.29 × 10−2 | 4.44 × 10−2 | 3.2 × 10−2 | 7.2 × 10−2 | 3.49 × 10−2 |
| 755 | 1.08 × 10−3 | 1.96 × 10−3 | 0.248 | 0.190 | 1.57 × 10−2 |
| 756 | 1.52 × 10−2 | 1.52 × 10−2 | 5.7 × 10−2 | 0.155 | 8.64 × 10−3 |
| 757 | 9.64 × 10−3 | 1.08 × 10−4 | 9.7 × 10−2 | 6.6 × 10−2 | 5.37 × 10−3 |
| 758 | 1.67 × 10−2 | 5.52 × 10−4 | 0.127 | 9.3 × 10−2 | 1.50 × 10−2 |
| 759 | 1.48 × 10−2 | 1.71 × 10−4 | 7.6 × 10−2 | 7.3 × 10−2 | 1.63 × 10−4 |
IC50, concentration of drug that affords 50% reduction in cell number using the MTT method with drug exposure for 48 hr.
As outlined in Table XXXII, we subsequently prepared similar spin-labeled derivatives linked through a 4β-amide rather than 4α-ester bond by reaction of 4β-amino-DEPPT with N-(1-oxyl-2,2,6,6-tetramethyl-4-piperidinyloxycarbonyl)-amino acids.194 Compounds 760–766 were tested for in vitro cytotoxicity against three tumor cell lines as well as for antioxidative activity in tissues of SD rats. Most of the new compounds showed superior or comparable activity to ETO against A-549, HL-60, and RPMI-8226. Notably, compounds 762–764 and 766 exhibited excellent inhibitory activity against RPMI-8226 with IC50 values ranging from 0.06 to 0.09 μM. Furthermore, all target compounds displayed three- to sixfold more potent antioxidative activity (IC50 5.89–9.63 μM) in homogenate tissues of rat liver, heart, and kidney compared with ETO. Thus, introduction of l-amino acids with a stable nitroxyl radical into PPT could lead to improved biological activity.
Table XXXII.
Biological Data for Spin-Labeled Compounds 760–766
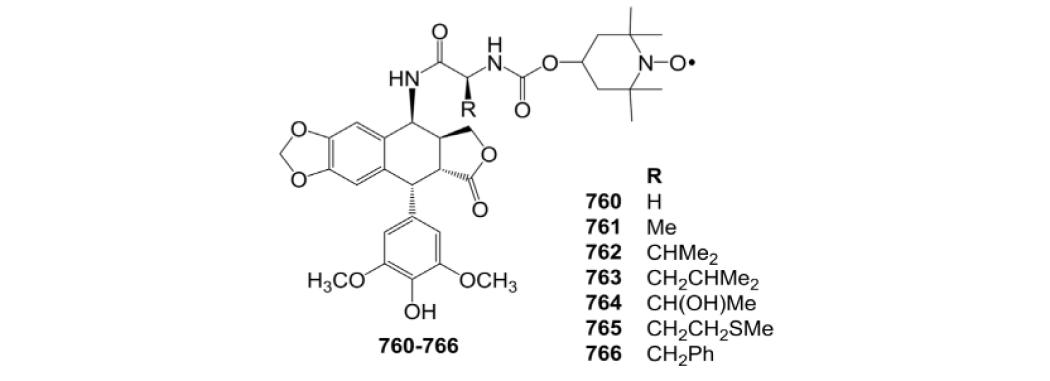
| ||||||
|---|---|---|---|---|---|---|
| Cytotoxic activity (IC50, μM) |
Antioxidative activity |
|||||
| Compound | A-549 | HL-60 | RPMI-8226 | Liver | Kidney | Heart |
| ETO | 0.29 | 0.42 | 0.14 | 43.89 | 23.77 | 40.67 |
| 760 | 0.21 | 0.32 | 0.33 | 7.47 | 8.9 | 7.19 |
| 761 | 0.12 | 0.22 | 0.26 | 7.64 | 7.68 | 6.39 |
| 762 | 0.15 | 0.24 | 0.061 | 8.69 | 8.88 | 8.04 |
| 763 | 0.19 | 0.16 | 0.089 | 8.85 | 8.47 | – |
| 764 | 0.21 | 0.21 | 0.078 | 7.12 | 7.45 | 8.79 |
| 765 | 0.42 | 0.67 | 0.48 | 6.85 | 7.55 | 5.89 |
| 766 | 0.21 | 0.21 | 0.090 | 6.55 | 9.63 | 7.39 |
Overall, novel spin-labeled PPTs provide a promising direction in antitumor chemotherapy, not only because they exhibit superior activity, but also because they can be monitored by electron spin resonance in pharmacological experiments.46 As a whole, the introduction of a stable nitroxyl radical into the PPT molecule led to potentiated antitumor and antioxidative effects, proving that the design and synthesis of these compounds should be beneficial.
6. CONCLUSION
In summary, the successful history of PPT and related lignans confirms the importance of natural products as a major source of lead compounds for drug research. Since PPT’s isolation in 1880 but unsuccessful clinical usage, the dynamics of PPT research has remained very high, resulting in hundreds of publications every year. The tremendous efforts in PPT research have mainly focused on the semisynthetic construction of novel PPT derivatives, prodrugs, and new forms of administration. These efforts have resulted in PPT-derived drugs, including approval for clinical use of ETO and teniposide, as well as many other drug candidates, such as NK611, TOP53, and GL-331, with C-4 modifications. However, the development of safe, economical, and site-specific anticancer drugs still faces challenges. Undoubtedly, continued studies on PPT analogs and their interaction with topo II will expand our understanding of the detailed MOA, which in turn will suggest new synthetic directions toward more effective anticancer drugs. The expectations and value of PPT as a lead compound go beyond the development of anticancer agents, based on the numerous biological activities continually being discovered for it and its derivatives. In conclusion, although the conversion of PPT-derived compounds to clinically effective drugs has been slow and unpredictable, the application of rational drug design technologies should promote the process at a faster pace.
ACKNOWLEDGMENTS
The authors are indebted to their excellent graduate students and research associates, whose names are given in the text and references, for their invaluable experimental and theoretical contributions to the work carried out on PPT’s chemistry and medicinal chemistry in our laboratory. They thank their collaborators who have contributed to this research in many ways and are cited in the accompanying references. This work was supported financially by the National Natural Science Foundation of China (30800720, 31371975), the Fundamental Research Funds for the Central Universities (lzujbky-2013-69), and the Young Scholars Science Foundation of Lanzhou Jiaotong University (2011011). Partial support was also supplied by NIH grant CA177584-01 from the National Cancer Institute awarded to K.H.L. Thanks are also due to the support of Taiwan Department of Health Cancer Research Center of Excellence (DOH-100-TD-C-111-005).
Contract grant sponsor: National Natural Science Foundation of China; Contract grant numbers: 30800720, 31371975; Contract grant sponsor: Fundamental Research Funds for the Central Universities; Contract grant number: lzujbky-2013-69; Contract grant sponsor: Young Scholars Science Foundation of Lanzhou Jiaotong University; Contract grant number: 2011011; Contract grant sponsor: NIH; Contract grant number: CA177584-01.
Biography
Ying-Qian Liu received his B.S. in chemistry from Yebei Normal University in 2002, and his Ph.D. degree in Bioorganic Chemistry from Lanzhou University in 2007, he worked as postdoctoral scholar in School of Life Sciences from Lanzhou University in 2008–2013, and worked as a visiting scholar at the University of North Carolina at Chapel Hill in 2011–2012. He is currently an associate professor of school of pharmacy in Lanzhou University. His research interests include the design and synthesis of bioactive compounds as potential anticancer or pesticide agents, development of novel methodologies in organic chemistry, as well as drug mechanism and pharmacokinetics evaluation. He has published over 40 research articles, been applied over 10 patents, and received several projects.
Jing Tian received her B.S. degree in Pharmacy from Shandong University in 2011, and her M.S. degree in Medicinal Chemistry from Lanzhou University in 2013. Her research interests include the isolation, structural elucidation, and structural modification of plant-derived anti-cancer and anti-diabetic natural products.
Keduo Qian received her B.S. degree in Pharmacy from Beijing University in 2004 and her Ph.D. degree in Pharmaceutical Sciences from the University of North Carolina at Chapel Hill (UNCCH) in 2008. She became Research Assistant Professor in the Division of Chemical Biology and Medicinal Chemistry, UNC-CH in 2009. Her research interests include the design and synthesis of bioactive compounds, development of analytical methods, as well as drug metabolism and pharmacokinetics (DMPK) evaluation.
Xiao-Bo Zhao received his B.S. degree in School of Pharmacy from Lanzhou University in 2012. He is currently a M.S. student in Medicinal Chemistry at Lanzhou University. His research interests include the design and synthesis of plant-derived anti-cancer agents, and development of novel methodologies in medicinal chemistry.
Susan L. Morris-Natschke received her B.S. in Chemistry from the University of Maryland-College Park (1975) and her Ph.D. in Organic Chemistry from the University of North Carolina-Chapel Hill (1982). She is currently Research Professor in the Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, UNC-CH, where she has been on the faculty since 1983. Her interests include scientific writing and editing, as well as the synthesis and structure-activity relationships of bioactive natural products.
Liu Yang received her B.S. in Chemistry from Yebei Normal University in 2002, and her M.S. degree in Analytical Chemistry from Shanxi University in 2005. She is currently a lecturer of Environmental and Municipal Engineering School in Lanzhou Jiaotong University. Her research interests include development of new methodologies in environmental chemistry, and drug pharmacokinetics evaluation. She has published over 20 research articles, and received several projects.
Xiang Nan received his B.S. degree in School of Pharmacy from Lanzhou University in 2011. He is currently a M.S. student in Medicinal Chemistry at Lanzhou University. His research interests include the design and synthesis of plant-derived pesticide agents, development of novel methodologies in organic chemistry.
Xuan Tian received his B.S. in Chemistry from Lanzhou University in 1969, and his M.S. degree in Organic Chemistry from Lanzhou University in 1983. He is a professor of School of State Key Laboratory of Applied Organic Chemistry in Lanzhou University. His research interests include the isolation, structural elucidation, and structural modification of bioactive compounds. He has published over 80 research articles, and received several projects.
Kuo-Hsiung Lee received his B.S. in Pharmacy from Kaohsiung Medical University, Taiwan (1961), M.S. in Pharmaceutical Chemistry from Kyoto University, Japan (1965), and Ph.D. in Medicinal Chemistry from University of Minnesota, Minneapolis (1968), and was a postdoctoral scholar in organic chemistry at UCLA (1968–70). Dr. Lee joined the faculty of the UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill as Assistant Professor in 1970, where he is currently Kenan Distinguished Professor and Director of the Natural Products Research Laboratories. Dr. Lee combines the fields of natural products and synthetic medicinal chemistry research to design and discover bioactive natural products and their analogs as clinical trials drug candidates against cancer, AIDS, and other diseases. His productive, internationally known research has resulted in more than 800 research articles and over 100 patents.
REFERENCES
- 1.Ayres DC, Loike JD. Lignans, chemical, biological and clinical properties. Cambridge University Press; Cambridge: 1990. Chapters 3 and 4. [Google Scholar]
- 2.Jardine I. Podophyllotoxin, in anticancer agents based on natural products models. Academic Press, Inc.; New York: 1980. pp. 319–351. [Google Scholar]
- 3.King J. Discovery of podophyllin. Coll J M Sci. 1857;2:557–559. [Google Scholar]
- 4.Podwyssotzki V. The active constituent of podophyllin. Pharm J Trans. 1881;12:217–218. [Google Scholar]
- 5.Podwyssotzki V. On the active constituents of podophyllin. Am J Pharm. 1882;12:102–115. [Google Scholar]
- 6.Hartwell JL, Schrecker AW. Chemistry of podophyllum. Fortshr Chem Org Naturst. 1958;15:83–166. doi: 10.1007/978-3-7091-7162-2_3. [DOI] [PubMed] [Google Scholar]
- 7.Kelly MG, Hartwell JL. The biological effects and the chemical composition of podophyllin: A review. J Natl Cancer Inst. 1954;14:967–1010. [PubMed] [Google Scholar]
- 8.MacRae DW, Towers GH. Biological activity of lignans. Phytochemistry. 1984;23:1207–1220. [Google Scholar]
- 9.Kaplan IW. Condylomata acuminata. N Orleans Med Surg J. 1942;94:388–390. [Google Scholar]
- 10.King ML, Sullivan MM. The similarity of the effect of podophyllin and colchicine and their use in the treatment of Condylomata acuminata. Science. 1946;104:244–245. doi: 10.1126/science.104.2698.244. [DOI] [PubMed] [Google Scholar]
- 11.Sullivan BJ, Wechsler HJ. The cytological effects of podophyllin. Science. 1947;105:433. doi: 10.1126/science.105.2730.433. [DOI] [PubMed] [Google Scholar]
- 12.Stähelin H, von Wartburg A. From podophyllotoxin glucoside to etoposide. Prog Drug Res. 1989;33:169–266. doi: 10.1007/978-3-0348-9146-2_8. [DOI] [PubMed] [Google Scholar]
- 13.Canetta R, Hilgard P, Florentine S, Bedogni P, Lenaz L. Current development of podophyllotoxins. Cancer Chemother Pharmacol. 1982;7:93–98. doi: 10.1007/BF00254528. [DOI] [PubMed] [Google Scholar]
- 14.Kuhn M, von Wartburg A. Concerning a new glycoside preparation method. Synthesis of epipodophyllotoxin-β-D-glucopyranoside. Helv Chim Acta. 1968;51:1631–1641. doi: 10.1002/hlca.19680510719. [DOI] [PubMed] [Google Scholar]
- 15.Kuhn M, von Wartburg A. On a new glycoside synthesis process. Glycosides of 4′-demethylepipodophyllotoxin. Helv Chim Acta. 1969;52:948–955. doi: 10.1002/hlca.19690520411. [DOI] [PubMed] [Google Scholar]
- 16.Keller-Juslén C, Kuhn M, Stähelin H, von Wartburg A. Synthesis and antimitotic activity of glycosidic lignan derivatives related to podophyllotoxin. J Med Chem. 1971;14:936–940. doi: 10.1021/jm00292a012. [DOI] [PubMed] [Google Scholar]
- 17.Joel S. The clinical pharmacology of etoposide: An update. Cancer Treatment Rev. 1996;22:179–221. doi: 10.1016/s0305-7372(96)90002-x. [DOI] [PubMed] [Google Scholar]
- 18.Stähelin HF, von Wartburg A. The chemical and biological route from podophyllotoxin glucoside to etoposide: Ninth Cain Memorial Award Lecture. Cancer Res. 1991;51:5–15. [PubMed] [Google Scholar]
- 19.Ross W, Rowe T, Glisson B, Yalowich J, Liu L. Role of topoisomerase II in mediating epipodophyllotoxin-induced DNA cleavage. Cancer Res. 1984;44:5857–5860. [PubMed] [Google Scholar]
- 20.Long BH, Brattain MG. The activity of etoposide and teniposide against human lung tumor in vitro: Cytotoxicity and DNA breakage. In: Issell BF, Muggia FM, Carter SA, editors. Etoposide: Current Status and New Developments. Academic Press; New York: 1984. p. 223. [Google Scholar]
- 21.Budman DR. Early studies of etoposide phosphate, a water-soluble prodrug. Semin Oncol. 1996;23:8–14. [PubMed] [Google Scholar]
- 22.Greco FA, Hainsworth JD. Clinical studies with etoposide phosphate. Semin Oncol. 1996;23:45–50. [PubMed] [Google Scholar]
- 23.Long BH, Minocha A. Inhibition of topoisomerase II by VP-16 (etoposide), VM-26 (teniposide) and structure congeners as explanation for in vivo DNA breakage and cytotoxicity. Proc Am Assoc Cancer Res. 1983;24:1271–1277. [Google Scholar]
- 24.Hande KR. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur J Cancer. 1998;34:1514–1521. doi: 10.1016/s0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- 25.Long BH, Musial ST, Brattain MG. Comparison of cytotoxicity and DNA breakage activity of congeners of podophyllotoxin including VP16–213 and VM26: A quantitative structure-activity relationship. Biochemistry. 1984;23:1183–1188. doi: 10.1021/bi00301a024. [DOI] [PubMed] [Google Scholar]
- 26.Saito H, Yoshikawa H, Nishimura Y, Kondo S, Takeuchi T, Umezawa H. Studies on lignan lactone antitumor agents. II. Synthesis of N-alkylamino- and 2,6-dideoxy-2-aminoglycosidic lignan variants related to podophyllotoxin. Chem Pharm Bull. 1986;34:3741–3746. doi: 10.1248/cpb.34.3741. [DOI] [PubMed] [Google Scholar]
- 27.Pagani O, Zucchetti M, Sessa C, de Jong J, D’Incalci M, De Fusco M, Kaeser-Fröhlich A, Hanauske A, Cavalli F. Clinical and pharmacokinetic study of oral NK611, a new podophyllotoxin derivative. Cancer Chemother Pharmacol. 1996;38:541–547. doi: 10.1007/s002800050524. [DOI] [PubMed] [Google Scholar]
- 28.Lee KH. Novel antitumor agents from higher plants. Med Res Rev. 1999;19:569–596. doi: 10.1002/(sici)1098-1128(199911)19:6<569::aid-med7>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 29.Lee KH, Beers SA, Mori M, Wang ZQ, Kuo YH, Li L, Liu SY, Chang JY, Han FS, Cheng YC. Antitumor agents. 111. New 4-hydroxylated and 4-halogenated anilino derivatives of 4′-demethylepipodophyllotoxin as potent inhibitors of human DNA topoisomerase II. J Med Chem. 1990;33:1364–1368. doi: 10.1021/jm00167a013. [DOI] [PubMed] [Google Scholar]
- 30.Wang ZQ, Kuo YH, Schnur D, Bowen JP, Liu SY, Han FS, Chang JY, Cheng YC, Lee KH. Anti-tumor agents. 113. New 4β-arylamino derivatives of 4′-O-demethyl epipodophyllotoxin and related compounds as potent inhibitors of human DNA topoisomerase II. J Med Chem. 1990;33:2660–2666. doi: 10.1021/jm00171a050. [DOI] [PubMed] [Google Scholar]
- 31.Terada T, Fujimoto K, Nomura M, Yamashita J, Wierzba K, Yamazaki R, Shibata J, Sugimoto Y, Yamada Y, Kobunai T. Antitumor agents. 3. Synthesis and biological activity of 4β-alkyl derivatives containing hydroxy, amino, and amido groups of 4′-O-demethyl-4-desoxypodophyllotoxin as antitumor agents. J Med Chem. 1993;36:1689–1699. doi: 10.1021/jm00064a002. [DOI] [PubMed] [Google Scholar]
- 32.Cragg GM, Newman DJ. A Tale of two tumor targets: Topoisomerase I and tubulin. The Wall and Wani contribution to cancer chemotherapy. J Nat Prod. 2004;67:232–244. doi: 10.1021/np030420c. [DOI] [PubMed] [Google Scholar]
- 33.Barret JM, Etiévant C, Baudouin C, Skov K, Charvéron M, Hill BT. F11782, a novel catalytic inhibitor of topoisomerases I and II, induces atypical, yet cytotoxic DNA double-strand breaks in CHO-K1 cells. Anticancer Res. 2002;22:187–192. [PubMed] [Google Scholar]
- 34.Cho SJ, Tropsha A, Suffness M, Cheng YC, Lee KH. Antitumor agents. 163. Three-dimensional quantitative structure-activity relationship study of 4′-O-demethyl-epipodophyllotoxin analogs using the modified CoMFA/q2-GRS approach. J Med Chem. 1996;39:1383–1395. doi: 10.1021/jm9503052. [DOI] [PubMed] [Google Scholar]
- 35.Imbert TF. Discovery of podophyllotoxins. Biochimie. 1998;80:207–222. doi: 10.1016/s0300-9084(98)80004-7. [DOI] [PubMed] [Google Scholar]
- 36.Bohlin L, Rosen B. Podophyllotoxin derivatives: Drug discovery and development. Drug Discov Today. 1996;1:343–351. [Google Scholar]
- 37.Hande KR. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur J Cancer. 1998;34:1514–1521. doi: 10.1016/s0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- 38.Ward RS. Synthesis of podophyllotoxin and related compounds. Synthesis. 1992;1:719–730. [Google Scholar]
- 39.Damayanthi Y, Lown JW. Podophyllotoxins: Current status and recent developments. Curr Med Chem. 1998;5:205–252. [PubMed] [Google Scholar]
- 40.Gordaliza M, Castro MA, del Corral JM, Feliciano AS. Antitumor properties of podophyllotoxin and related compounds. Curr Pharm Des. 2000;6:1811–1839. doi: 10.2174/1381612003398582. [DOI] [PubMed] [Google Scholar]
- 41.Sackett DL. Podophyllotoxin, steganacin and combretastatin: Natural products that bind at the colchicine site of tubulin. Pharmacol Ther. 1993;59:163–228. doi: 10.1016/0163-7258(93)90044-e. [DOI] [PubMed] [Google Scholar]
- 42.Gordaliza M, García PA, del Corral JM, Castro MA, Gomez-Zurita MA. Podophyllotoxin: Distribution, sources, applications and new cytotoxic derivatives. Toxicon. 2004;44:441–459. doi: 10.1016/j.toxicon.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 43.Desbène S, Giorgi-Renault S. Drugs that inhibit tubulin polymerization: The particular case of podophyllotoxin and analogues. Curr Med Chem Anticancer Agents. 2002;2:71–90. doi: 10.2174/1568011023354353. [DOI] [PubMed] [Google Scholar]
- 44.You Y. Podophyllotoxin derivatives: Current synthetic approaches for new anticancer agents. Curr Pharm Des. 2005;11:1695–1717. doi: 10.2174/1381612053764724. [DOI] [PubMed] [Google Scholar]
- 45.Xu H, Lv M, Tian X. A review on hemisynthesis, biosynthesis, biological activities, mode of action, and structure-activity relationship of podophyllotoxins: 2003–2007. Curr Med Chem. 2009;16:327–349. doi: 10.2174/092986709787002682. [DOI] [PubMed] [Google Scholar]
- 46.Liu YQ, Yang L, Tian X. Podophyllotoxin: Current perspectives. Curr Bioact Compd. 2007;3:37–66. [Google Scholar]
- 47.Lee KH. Current developments in the discovery and design of new drug candidates from plant natural product leads. J Nat Prod. 2004;67:273–283. doi: 10.1021/np030373o. [DOI] [PubMed] [Google Scholar]
- 48.Lee KH, Xiao Z. Antitumor agents 277. Podophyllotoxins and analogs. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. Taylor & Francis; New York: 2011. pp. 95–122. [Google Scholar]
- 49.Lv M, Xu H. Recent advances in semisynthesis, biosynthesis, biological activities, mode of action, and structure-activity relationship of podophyllotoxins: An update (2008–2010) Mini Rev Med Chem. 2011;11:901–909. doi: 10.2174/138955711796575461. [DOI] [PubMed] [Google Scholar]
- 50.Liu CJ, Hou SS. Current research status of podophyllotoxin lignans. Nat Prod Res Dev. 1997;9:81–89. [Google Scholar]
- 51.Pujol MD, Romero M, Sánchez I. Synthesis and biological activity of new class of dioxygenated anticancer agents. Anticancer Agents Med Chem. 2005;5:215–237. doi: 10.2174/1568011053765930. [DOI] [PubMed] [Google Scholar]
- 52.Bedows E, Hatfield GM. An investigation of the antiviral activity of Podophyllum peltatum. J Nat Prod. 1982;45:725–729. doi: 10.1021/np50024a015. [DOI] [PubMed] [Google Scholar]
- 53.Markkanen T, Makinen ML, Maunuksela E, Himanen P. Podophyllotoxin lignans under experimental antiviral research. Drug Exp Clin Res. 1981;7:711–718. [Google Scholar]
- 54.Gordaliza M, Castro MA, Garcia-Gracalos MD, Ruiz P, Miguel del Corral JM, Feliciano AS. Antineoplastic and antiviral activities of podophyllotoxin. Arch Pharm (Weinheim) 1994;327:175–179. doi: 10.1002/ardp.19943270309. [DOI] [PubMed] [Google Scholar]
- 55.Hammonds TR, Denyer SP, Jackson DE, Irving WL. Studies to show that with podophyllotoxin the early replicative stages of herpes simplex virus type 1 depend upon functional cytoplasmic microtubules. J Med Microbiol. 1996;45:167–172. doi: 10.1099/00222615-45-3-167. [DOI] [PubMed] [Google Scholar]
- 56.Canel C, Moraes RM, Dayan FE, Ferreira D. Podophyllotoxin. Phytochemistry. 2000;54:115–120. doi: 10.1016/s0031-9422(00)00094-7. [DOI] [PubMed] [Google Scholar]
- 57.Charlton JL. Antiviral activity of lignans. J Nat Prod. 1998;61:1447–1451. doi: 10.1021/np980136z. [DOI] [PubMed] [Google Scholar]
- 58.Hara H, Fujihashi T, Sakata T, Kaji A, Kaji H. Tetrahydronaphthalene lignan compounds as potent anti-HIV type 1 agents. AIDS Res Hum Retroviruses. 1997;13:695–705. doi: 10.1089/aid.1997.13.695. [DOI] [PubMed] [Google Scholar]
- 59.Markos AR. The successful treatment of Molluscum contagiosum with podophyllotoxin (0.5%) self-application. Int J STD AIDS. 2001;12:833–834. [PubMed] [Google Scholar]
- 60.Mukherjee AK, Basu S, Sarkar N, Ghosh AC. Advances in cancer therapy with plant based natural products. Curr Med Chem. 2001;8:1467–1486. doi: 10.2174/0929867013372094. [DOI] [PubMed] [Google Scholar]
- 61.Hermanns-Lê T, Arrese JE, Goffin V, Piérard GE. Podophyllotoxin-induced acantholysis and cytolysis in a skin equivalent model. Eur J Morphol. 1998;36:183–187. doi: 10.1076/ejom.36.3.183.4768. [DOI] [PubMed] [Google Scholar]
- 62.Liu YQ, Xiao H, Gao R, Tian X. Synthesis and insecticidal activities of novel derivatives of podophyllotoxin: Part XII. Pest Biochem Physiol. 2008;91:116–121. doi: 10.1080/14786410701371520. [DOI] [PubMed] [Google Scholar]
- 63.Liu YQ, Zhao CY, Yang L, Zhao YL, Liu TT. Evaluation of insecticidal activity of podophyllotoxin derivatives against Brontispa longissima. Pest Biochem Physiol. 2011;99:39–44. [Google Scholar]
- 64.Liu YQ, Zhao YL, Yang L, Zhou XW, Feng G. Design, semisynthesis and insecticidal activity of novel podophyllotoxin derivatives against Brontispa longissima in vivo. Pest Biochem Physiol. 2012;102:11–18. [Google Scholar]
- 65.Oliva A, Moraes RM, Watson SB, Duke SO, Dayan FE. Aryltetralin lignans inhibit plant growth by affecting the formation of mitotic microtubular organizing centers. Pest Biochem Physiol. 2002;72:45–54. [Google Scholar]
- 66.Garcia ES, Azambuja P. Lignoids in insects: Chemical probes for the study of ecdysis, excretion and Trypanosoma cruzi-triatomine interactions. Toxicon. 2004;44:431–440. doi: 10.1016/j.toxicon.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 67.Botta B, Delle Monache G, Misiti D, Vitali A, Zappia G. Aryltetralin lignans: Chemistry, pharmacology and biotransformations. Curr Med Chem. 2001;8:1363–1381. doi: 10.2174/0929867013372292. [DOI] [PubMed] [Google Scholar]
- 68.AIDS International CPH 82. BioDrugs. 2003;17:373–374. doi: 10.2165/00063030-200317050-00007. [DOI] [PubMed] [Google Scholar]
- 69.Pugh N, Khan IA, Moraes RM, Pasco DS. Podophyllotoxin lignans enhance IL-1β but suppress TNF-α mRNA expression in LPS-treated monocytes. Immunopharmacol Immunotoxicol. 2001;23:83–95. doi: 10.1081/iph-100102570. [DOI] [PubMed] [Google Scholar]
- 70.Gordaliza M, Faircloth GT, Castro MA, Miguel del Corral JM, López-Vázquez ML, San Feliciano A. Immunosuppressive cyclolignans. J Med Chem. 1996;39:2865–2868. doi: 10.1021/jm960023h. [DOI] [PubMed] [Google Scholar]
- 71.Gordaliza M, Castro MA, del Corral JMM, López-Vázquez ML, San Feliciano A, Faircloth GT. In vivo immunosuppressive activity of some cyclolignans. Bioorg Med Chem Lett. 1997;21:2781–2786. [Google Scholar]
- 72.Loike JD, Horwitz SB. Effects of podophyllotoxin and VP-16–213 on microtubule assembly in vitro and nucleoside transport in HeLa cells. Biochemistry. 1976;15:5435–5443. doi: 10.1021/bi00670a003. [DOI] [PubMed] [Google Scholar]
- 73.Gordaliza M, del Corral JM, Castro MA, López-Vázquez ML, San Feliciano A, García-Grávalos MD, Carpy A. Synthesis and evaluation of pyrazolignans. A new class of cytotoxic agents. Bioorg Med Chem. 1995;3:1203–1210. doi: 10.1016/0968-0896(95)00091-t. [DOI] [PubMed] [Google Scholar]
- 74.Schönbrunn E, Phlippen W, Trinczek B, Sack S, Eschenburg S, Mandelkow EM, Mandelkow E. Crystallization of a macromolecular ring assembly of tubulin liganded with the anti-mitotic drug podophyllotoxin. J Struct Biol. 1999;128:211–215. doi: 10.1006/jsbi.1999.4183. [DOI] [PubMed] [Google Scholar]
- 75.Cowan CR, Cande WZ. Meiotic telomere clustering is inhibited by colchicine but does not require cytoplasmic microtubules. J Cell Sci. 2002;115:3747–3756. doi: 10.1242/jcs.00055. [DOI] [PubMed] [Google Scholar]
- 76.Tseng CJ, Wang YJ, Liang YC, Jeng JH, Lee WS, Lin JK, Chen CH, Liu IC, Ho YS. Microtubule damaging agents induce apoptosis in HL 60 cells and G2/M cell cycle arrest in HT 29 cells. Toxicol. 2002;175:123–142. doi: 10.1016/s0300-483x(02)00073-2. [DOI] [PubMed] [Google Scholar]
- 77.López-Pérez JL, del Olmo E, de Pascual Teresa B, Merino M, Barajas M, San Feliciano A. A role for dipole moment in the activity of cyclolignans. J Mol Struct (Theochem) 2000;504:51–57. [Google Scholar]
- 78.van Maanen JM, Retèl J, de Vries J, Pinedo HM. Mechanism of action of antitumor drug etoposide: A review. J Natl Cancer Inst. 1988;80:1526–1533. doi: 10.1093/jnci/80.19.1526. [DOI] [PubMed] [Google Scholar]
- 79.Champoux JJ. DNA topoisomerases: Structure, function, and mechanism. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 80.Burden DA, Osheroff N. Mechanism of action of eukaryotic topoisomerase II and drug targeted to the enzyme. Biochim Biophys Acta. 1998;1400:139–154. doi: 10.1016/s0167-4781(98)00132-8. [DOI] [PubMed] [Google Scholar]
- 81.Berger JM, Gambin SJ, Harrison SC, Wang JC. Structure and mechanism of DNA topoisomerase II. Nature. 1996;379:225–232. doi: 10.1038/379225a0. [DOI] [PubMed] [Google Scholar]
- 82.Haim N, Nemec J, Roman J, Sinha BK. Peroxidative free radical formation and O-demethylation of etoposide (VP-16) and teniposide (VM-26) Biochem Biophys Res Commun. 1986;135:215–220. doi: 10.1016/0006-291x(86)90965-4. [DOI] [PubMed] [Google Scholar]
- 83.Haim N, Nemec J, Roman J, Sinha BK. Peroxidase-catalyzed metabolism of etoposide (VP-16–213) and covalent binding of reactive intermediates to cellular macromolecules. Cancer Res. 1987;47:5835–5840. [PubMed] [Google Scholar]
- 84.Sinha BK. Free radicals in anticancer drug pharmacology. Chem Biol Interact. 1989;69:293–317. doi: 10.1016/0009-2797(89)90117-8. [DOI] [PubMed] [Google Scholar]
- 85.Lin S, Huang HC, Chen LL, Lee CC, Huang TS. GL331 induces down-regulation of cyclin D1 expression via enhanced proteolysis and repressed transcription. Mol Pharmacol. 2001;60:768–775. [PubMed] [Google Scholar]
- 86.Tseng CJ, Wang YJ, Liang YC, Jeng JH, Lee WS, Lin JK, Chen CH, Liu IC, Ho YS. Microtubule damaging agents induce apoptosis in HL 60 cells and G2/M cell cycle arrest in HT 29 cells. Toxicology. 2002;175:123–142. doi: 10.1016/s0300-483x(02)00073-2. [DOI] [PubMed] [Google Scholar]
- 87.Boldt S, Weidle UH, Kolch W. The role of MAPK pathways in the action of chemotherapeutic drugs. Carcinogenesis. 2002;23:1831–1838. doi: 10.1093/carcin/23.11.1831. [DOI] [PubMed] [Google Scholar]
- 88.Qi SN, Yoshida A, Wang ZR, Ueda T. GP7 can induce apoptotic DNA fragmentation of human leukemia cells through caspase-3-dependent and -independent pathways. Int J Mol Med. 2004;13:163–167. [PubMed] [Google Scholar]
- 89.Qi SN, Jing YX, Dong GX, Chen Y, Yoshida A, Ueda T. GP7 induces internucleosomal DNA fragmentation independent of caspase activation and DNA fragmentation factor in NB4 cells. Oncol Rep. 2007;18:273–277. [PubMed] [Google Scholar]
- 90.Suh SJ, Kim JR, Jin UH, Choi HS, Chang YC, Lee YC, Kim SH, Lee IS, Moon TC, Chang HW, Kim CH. Deoxypodophyllotoxin, flavolignan, from Anthriscus sylvestris Hoffm inhibits migration and MMP-9 via MAPK pathways in TNF-α-induced HASMC. Vasc Pharmacol. 2009;51:13–20. doi: 10.1016/j.vph.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 91.Yong Y, Shin SY, Lee YH, Lim Y. Antitumor activity of deoxypodophyllotoxin isolated from Anthriscus sylvestris: Induction of G2/M cell cycle arrest and caspase-dependent apoptosis. Bioorg Med Chem Lett. 2009;19:4367–4371. doi: 10.1016/j.bmcl.2009.05.093. [DOI] [PubMed] [Google Scholar]
- 92.Shin SY, Yong Y, Kim CG, Lee YH, Lim Y. Deoxypodophyllotoxin induces G2/M cell cycle arrest and apoptosis in HeLa cells. Cancer Lett. 2010;287:231–239. doi: 10.1016/j.canlet.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 93.Chen YQ, Xie X. Podophyllotoxin induces CREB phosphorylation and CRE-driven gene expression via PKA but not MAPKs. Mol Cells. 2010;29:41–50. doi: 10.1007/s10059-010-0015-1. [DOI] [PubMed] [Google Scholar]
- 94.Tadafumi T, Katsuhiko F, Makoto N. Antitumor agents II: Regio- and stereospecific synthesis of 1β-alkyl-1-desoxypodophyllotoxin derivatives and biological activity. Chem Pharm Bull. 1993;41:907–912. doi: 10.1248/cpb.41.907. [DOI] [PubMed] [Google Scholar]
- 95.Tadafumi T, Katsuhiko F, Makoto TOP-53. Drugs Future. 1996;21:1136–1139. [Google Scholar]
- 96.Roulland E, Magiatis P, Arimondo P, Bertounesque E, Monneret C. Hemi-synthesis and biological activity of new analogues of podophyllotoxin. Bioorg Med Chem. 2002;10:3463–3471. doi: 10.1016/s0968-0896(02)00255-9. [DOI] [PubMed] [Google Scholar]
- 97.Chen ZX, Ma WY, Zhang CN. Stereoselective synthesis and antitumor activities of new podophyllotoxin derivatives: 4β-Cyano-4-deoxy-4′-demethylepipodophyllotoxin and 4β-carboxyl-4-deoxy-4′-demethylepipodophyllotoxin. Chin Chem Lett. 2000;11:505–508. [Google Scholar]
- 98.Chen ZX, Ma WY, Zhang CN. Synthesis and antitumor activity of 4-β-alkylthiocarbonyl-4-deoxy-4′-demethylepipodophyllotoxin. Chin J Med Chem. 2000;10:84–87. [Google Scholar]
- 99.Chen ZX, Ma WY, Chen XH, Wang JD, Zhang CN. Synthesis and antitumor activity of 4-β-substituted carbamoyl-4-deoxy-4′-demethylepipodophyllotoxin. Chin J Pharm. 2000;30:265–267. [Google Scholar]
- 100.Abad A, López-Pérez JL, del Olmo E, Garcia-Fernández LF, Francesch A, Trigili C, Barasoain I, Andreu JM, Diaz JF, San Feliciano A. Synthesis and antimitotic and tubulin interaction profiles of novel pinacol derivatives of podophyllotoxins. J Med Chem. 2012;55:6724–67377. doi: 10.1021/jm2017573. [DOI] [PubMed] [Google Scholar]
- 101.Showalterk HD, Winters RT, Sercel AD, Michel A. Facile synthesis of thioglucose analogues of the anticancer agent etoposide. Tetrahedron Lett. 1991;32:2849–2852. [Google Scholar]
- 102.Wang ZG, Ma WY, Zhang CN. Stereocontrolled synthesis of 4-sulfanyl-4-deoxy-4-demethylepipodophyllotoxin. Acta Chim Sin. 1992;50:696–701. [Google Scholar]
- 103.Wang ZG, Yin SF, Zhuang W, Li BS, Ma WY, Zhang CN. Synthesis and antitumor activities of 4β-alkylthio-4-deoxy-4′-demethylepipodophyllotoxin and its derivatives. Acta Pharm Sin. 1992;50:696–701. [Google Scholar]
- 104.Wang ZG, Yin SF, Zhuang W, Li BS, Ma WY, Zhang CN. Synthesis and antitumor activities of 4β-(2-aminoethylthio)-4-deoxy-4′-demethylepipodophyllotoxin and its derivatives. Chin J Pharm. 1992;23:159–167. [Google Scholar]
- 105.Lu KK, Liu FM, Chen YZ. Synthesis and antitumor activity of podophyllotoxin analogues. J Chin Pharm Sci. 1998;7:182–185. [Google Scholar]
- 106.Sanmartin C, Plano D, Sharma AK, Palop JA. Selenium compounds, apoptosis and other types of cell death: An overview for cancer chemotherapy. Int J Mol Sci. 2012;13:9649–9672. doi: 10.3390/ijms13089649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang ZG, Ma WY, Zhang CN. Synthesis and antitumor activities of 4-alkylselenyl-4-deoxy-4′-demethylepipodophyllotoxin. Acta Chim Sin. 1993;51:933–936. [PubMed] [Google Scholar]
- 108.Miao RD, Han Y, An LZ, Yang JB, Wang Q. Seleno-podophyllotoxin derivatives induce hepatoma SMMC-7721 cell apoptosis through Bax pathway. Cell Biol Int. 2008;32:217–223. doi: 10.1016/j.cellbi.2007.08.034. [DOI] [PubMed] [Google Scholar]
- 109.Zhang YZ, Cao CN. Cytotoxicity determination and synthesis of selenoyl-4-deoxy-4′-demethylepipodophyllotoxin. J Qinghai Norm Univ. 2004;3:76–79. [Google Scholar]
- 110.Terada T, Fujimoto K, Nomura M, Yamashita J, Kobunai T, Takeda S, Wierzba K, Yamada Y, Yamaguchi H. Antitumor agents. I. DNA topoisomerase II inhibitory activity and the structural relationship of podophyllotoxin derivatives as antitumor agents. Chem Pharm Bull. 1992;40:2720–2727. doi: 10.1248/cpb.40.2720. [DOI] [PubMed] [Google Scholar]
- 111.Zhuang W, Ma WY, Zhang CN. Synthesis and anticancer activity of 4β-ether-4′-demethylepipodophyllotoxin derivatives. Chin J Pharm. 1991;22:446–448. [Google Scholar]
- 112.Yin SF, Ma WY, Zhang CN. Synthesis and antitumor activity of 4,4′-O-bis(2-hydroxy-3-substituted-amino)propyl-4′-demethylepipodophyllotoxins. Chin J Pharm. 1993;24:486–488. [Google Scholar]
- 113.Bathini Y, Scholte A, Kelly S, Micetich RG, Daneshtalab M. New podophyllotoxin derivatives as potential anticancer agents: Synthesis and cytotoxicity of 4β-O-propenylpodophyllotoxin ethers. Syn Commun. 1999;29:379–385. [Google Scholar]
- 114.Levy RK, Hall IH, Lee KH. Antitumor Agents LXII: Synthesis and biological evaluation of podophyllotoxin esters and related derivatives. J Pharm Sci. 1983;72:1158–1161. doi: 10.1002/jps.2600721012. [DOI] [PubMed] [Google Scholar]
- 115.Gupta RS, Chenchaiah PC. Synthesis and biological activities of the C-4 esters of 4′-demethylepipodophyllotoxin. Anticancer Drug Des. 1987;2:13–23. [PubMed] [Google Scholar]
- 116.López-Pérez JL, del Olmo E, de Pascual-Teresa B, Abad A, San Feliciano A. Synthesis and cytotoxicity of hydrophobic esters of podophyllotoxins. Bioorg Med Chem Lett. 2004;14:1283–1286. doi: 10.1016/j.bmcl.2003.12.039. [DOI] [PubMed] [Google Scholar]
- 117.Duca M, Guianvarch D, Meresse P, Bertounesque E, Dauzonne D, Kraus-Berthier L, Thirot S, Léonce S, Pierré A, Pfeiffer B, Renard P, Arimondo PB, Monneret C. Synthesis and biological study of a new series of 4′-demethylepipodophyllotoxin derivatives. J Med Chem. 2005;48:593–603. doi: 10.1021/jm0495733. [DOI] [PubMed] [Google Scholar]
- 118.Kamal A, Kumar BA, Suresh P, Juvekar A, Zingde S. Synthesis of 4β-carbamoyl epipodophyllotoxins as potential antitumour agents. Bioorg Med Chem. 2011;19:2975–2979. doi: 10.1016/j.bmc.2011.03.030. [DOI] [PubMed] [Google Scholar]
- 119.Mustafa J, Khan SI, Ma G, Walker LA, Khan IA. Synthesis and anticancer activities of fatty acid analogs of podophyllotoxin. Lipids. 2004;39:167–172. doi: 10.1007/s11745-004-1215-5. [DOI] [PubMed] [Google Scholar]
- 120.Mustafa J, Khan SI, Ma G, Walker LA, Khan IA. Synthesis and in vitro cytotoxic activity of N-, F-, and S-ether derivatives of podophyllotoxin fatty acid adducts. Lipids. 2005;40:375–381. doi: 10.1007/s11745-006-1397-x. [DOI] [PubMed] [Google Scholar]
- 121.Liu YQ, Wei DF, Zhao YL, Cheng WD, Lu Y, Ma YQ, Li X, Han C, Wei YX, Cao HM, Zhao CY. Synthesis and biological evaluation of a series of podophyllotoxin derivatives as a class of potent antitubulin agents. Bioorg Med Chem. 2012;20:6285–6295. doi: 10.1016/j.bmc.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 122.Yu PF, Chen H, Wang J, He CX, Cao B, Li M, Yang N, Lei ZY, Cheng MS. Design, synthesis and cytotoxicity of novel podophyllotoxin derivatives. Chem Pharm Bull. 2008;56:831–834. doi: 10.1248/cpb.56.831. [DOI] [PubMed] [Google Scholar]
- 123.Chen H, Wang J, Zhang J, Wang Y, Cao B, Bai S, Yu PF, Bi W, Xie W. L1EPO, a novel podophyllotoxin derivative overcomes P-glycoprotein-mediated multidrug resistance in K562/A02 cell line. Biol Pharm Bull. 2009;32:609–613. doi: 10.1248/bpb.32.609. [DOI] [PubMed] [Google Scholar]
- 124.Gupta R, Al-said NH, Oreski B, Lown JW. Design, synthesis and topoisomerase II inhibition activity of 4′-demethylepipodophyllotoxin-lexitropsin conjugates. Anticancer Drug Des. 1996;11:325–328. [PubMed] [Google Scholar]
- 125.Derry WB, Pamidi CC, Gupta RS. Synthesis and biological activity of novel thymidine derivatives of podophyllotoxin and 4′-demethylepipodophyllotoxin. Anticancer Drug Des. 1993;8:203–221. [PubMed] [Google Scholar]
- 126.Lee NJ, Jeong IC, Cho MY, Jeon CW, Yun BC, Kim YO, Kim SH, Chung I. Synthesis and in vitro antitumor activity of phthalimide polymers containing podophyllotoxin. Eur Polym J. 2006;42:3352–3359. [Google Scholar]
- 127.Passarella D, Giardini A, Peretto B, Fontana G, Sacchetti A, Silvani A, Ronchi C, Cappelletti G, Cartelli D, Borlake J, Danieli B. Inhibitors of tubulin polymerization: Synthesis and biological evaluation of hybrids of vindoline, anhydrovinblastine and vinorelbine with thiocolchicine, podophyllotoxin and baccatin III. Bioorg Med Chem. 2008;16:6269–6285. doi: 10.1016/j.bmc.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 128.Passarella D, Peretto B, Yepes RB, Cappelletti G, Cartelli D, Ronchi C, Snaith J, Fontana G, Danieli B, Borlak J. Synthesis and biological evaluation of novel thiocolchicine–podophyllotoxin conjugates. Eur J Med Chem. 2010;45:219–226. doi: 10.1016/j.ejmech.2009.09.047. [DOI] [PubMed] [Google Scholar]
- 129.Cappelletti G, Cartelli D, Peretto B, Ventura M, Riccioli M, Colombo F, Snaith JS, Borrelli S, Passarella D. Tubulin-guided dynamic combinatorial library of thiocolchicine-podophyllotoxin conjugates. Tetrahedron. 2011;67:7354–7357. [Google Scholar]
- 130.Danieli B, Giardini A, Lesma G, Passarella D, Peretto B, Sacchetti A, Silvani A, Pratesi G, Zunino F. Thiocolchicine-podophyllotoxin conjugates: Dynamic libraries based on disulfide exchange reaction. J Org Chem. 2006;71:2848–2853. doi: 10.1021/jo052677g. [DOI] [PubMed] [Google Scholar]
- 131.Liu YQ, Liu YQ, Tian X, Yang L. Synthesis of novel derivatives of bispodophyllotoxin as potential antineoplastic agents—part XVI. Nat Prod Res. 2008;22:285–291. doi: 10.1080/14786410701757298. [DOI] [PubMed] [Google Scholar]
- 132.Allevi P, Anastasia M, Ciuffreda P. The first synthesis of the N-glucosyl analogue of the antitumor agent etoposide. Tetrahedron Lett. 1993;34:7313–7316. [Google Scholar]
- 133.KH Lee, Imakura Y, Haruna M, Beers SA, Thurston LS, Dai HJ, Chen CH, Cheng YC. Antitumor agents. 107: New cytotoxic 4-alkylamino analogues of 4′-demethyl-epipodophyllotoxin as inhibitors of human DNA topoisomerase II. J Nat Prod. 1989;52:606–613. doi: 10.1021/np50063a021. [DOI] [PubMed] [Google Scholar]
- 134.Zhu XK, Guan J, Tachibana Y, Bastow KF, Cho SJ, Cheng HH, Cheng YC, Gurwith M, Lee KH. Antitumor agents. 194. Synthesis and biological evaluations of 4β-mono-, -di-, and -trisubstituted aniline-4′-demethyl-podophyllotoxin and related compounds with improved pharmacological profiles. J Med Chem. 1999;42:2441–2446. doi: 10.1021/jm990055f. [DOI] [PubMed] [Google Scholar]
- 135.Zhou XM, Wang ZQ, Chang JY, Chen HX, Cheng YC, Lee KH. Antitumor agents. 120. New 4-substituted benzylamine and benzyl ether derivatives of 4′-O-demethylepipodophyllotoxin as potent inhibitors of human DNA topoisomerase II. J Med Chem. 1991;34:3346–3350. doi: 10.1021/jm00116a001. [DOI] [PubMed] [Google Scholar]
- 136.Zhou XM, Wang ZQ, Chen HX, Cheng YC, Lee KH. Antitumor agents. 125. New 4β-benzoylamino derivatives of 4′-O-demethyl-4-desoxypodophyllotoxin and 4β-benzoyl derivatives of 4′-O-demethylpodophyllotoxin as potent inhibitors of human DNA topoisomerase II. Pharm Res. 1993;10:214–219. doi: 10.1023/a:1018978525533. [DOI] [PubMed] [Google Scholar]
- 137.Zhang YL, Guo X, Cheng YC, Lee KH. Antitumor agents. 148. Synthesis and biological evaluation of novel 4β-amino derivatives of etoposide with better pharmacological profiles. J Med Chem. 1994;37:446–452. doi: 10.1021/jm00030a003. [DOI] [PubMed] [Google Scholar]
- 138.Xiao Z, Bastow KF, Vance JR, Lee KH. Antitumor agents. Part 227: Studies on novel 4′-O-demethyl-epipodophyllotoxins as antitumor agents targeting topoisomerase II. Bioorg Med Chem. 2004;12:3339–3344. doi: 10.1016/j.bmc.2004.03.067. [DOI] [PubMed] [Google Scholar]
- 139.Xiao Z, Bastow KF, Vance JR, Sidwell RS, Wang HK, Chen MS, Shi Q, Lee KH. Antitumor agents. 234. Design, synthesis, and biological evaluation of novel 4β-[(4″-benzamido)-amino]-4′-O-demethyl-epipodophyllotoxin derivatives. J Med Chem. 2004;47:5140–5148. doi: 10.1021/jm030609l. [DOI] [PubMed] [Google Scholar]
- 140.Bastow KF, Wang HK, Cheng YC, Lee KH. Antitumor agents–CLXXIII. Synthesis and evaluation of camptothecin-4β-amino-4′-O-demethylepipodophyllotoxin conjugates as inhibitors of mammalian DNA topoisomerases and as cytotoxic agents. Bioorg Med Chem. 1997;5:1481–1488. doi: 10.1016/s0968-0896(97)00102-8. [DOI] [PubMed] [Google Scholar]
- 141.Shi Q, Wang HK, Bastow KF, Tachibana Y, Chen K, Lee FK, Lee KH. Antitumor agents. 210. Synthesis and evaluation of taxoid-epipodophyllotoxin conjugates as novel cytotoxic agents. Bioorg Med Chem. 2001;9:2999–3004. doi: 10.1016/s0968-0896(01)00206-1. [DOI] [PubMed] [Google Scholar]
- 142.Wang ZG, Yin SF, Ma WY, Li BS, Zhang CN. Synthesis and antitumor activities of 4-acylamino-4-deoxy-4′-demethylepipodophyllotoxin analogues. Acta Pharm Sin. 1993;28:422–427. [PubMed] [Google Scholar]
- 143.Tian X, Yang MG, Chen YZ. Synthesis, antitumor activity and QSAR study of derivatives of 4β-halogenated arylacylamido-4-deoxy-4′-demethylepipodophyllotoxin. J Lanzhou Univ. 1998;34:114–119. [Google Scholar]
- 144.Hansen HF, Jensen RB, Willumsen AM, Nørskov-Lauritsen N, Ebbesen P, Nielsen PE, Buchardt O. New compounds related to podophyllotoxin and congeners: Synthesis, structure elucidation and biological testing. Acta Chim Scand. 1993;47:1190–1200. doi: 10.3891/acta.chem.scand.47-1190. [DOI] [PubMed] [Google Scholar]
- 145.Kamal A, Gayatri NL, Rao NV. Facile and improved synthesis of 4β-aminopodophyllotoxin congeners. Bioorg Med Chem Lett. 1998;8:3097–3100. doi: 10.1016/s0960-894x(98)00570-8. [DOI] [PubMed] [Google Scholar]
- 146.He Y, Ma WY, Chen XH, Zhang CN. Synthesis and activities of 4-deoxy-4β-arylmethylenesulfonylamino-4′-demethylpodophyllotoxins. Acta Pharm Sin. 2001;36:105–107. [PubMed] [Google Scholar]
- 147.Ma WY, Li Y, Zhang CN. Synthesis and activities of 4-deoxy-4β-sulfonylamino-4′-demethylepipodophyllotoxins. Chin J Pharm. 1993;24:198–201. [Google Scholar]
- 148.Kamal A, Gayatri NL, Reddy DR, Mohan Reddy PS, Arifuddin M, Dastidar SG, Kondapi AK, Rajkumar M. Synthesis and biological evaluation of new 4β-anilino- and 4β-imido-substituted podophyllotoxin congeners. Bioorg Med Chem. 2005;13:6218–6225. doi: 10.1016/j.bmc.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 149.Kamal A, Ashwini Kumar B, Arifuddin M, Dastidar SG. Synthesis of 4β-amido and 4β-sulphonamido analogues of podophyllotoxin as potent antitumor agents. Bioorg Med Chem. 2003;11:5135–5142. doi: 10.1016/j.bmc.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 150.Kamal A, Kumar BA, Arifuddin M, Dastidar SG. Synthesis and biological activity of new 4β-N-heteroaryl analogues of podophyllotoxin. Lett Drug Des Dis. 2006;3:205–213. [Google Scholar]
- 151.Kamal A, Laxman E, Khanna GB, Reddy PS, Rehana T, Arifuddin M, Neelima K, Kondapi AK, Dastidar SG. Design, synthesis, biological evaluation and QSAR studies of novel bisepipodophyllotoxins as cytotoxic agents. Bioorg Med Chem. 2004;12:4197–4209. doi: 10.1016/j.bmc.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 152.Kamal A, Kumar BA, Suresh P, Agrawal SK, Chashoo G, Singh SK, Saxena AK. Synthesis of 4β-N-polyaromatic substituted podophyllotoxins: DNA topoisomerase inhibition, anticancer and apoptosis-inducing activities. Bioorg Med Chem. 2010;18:8493–8500. doi: 10.1016/j.bmc.2010.10.042. [DOI] [PubMed] [Google Scholar]
- 153.Kamal A, Kumar BA, Suresh P, Shankaraiah N, Kumar MS. An efficient one-pot synthesis of benzothiazolo-4β-anilino-podophyllotoxin congeners: DNA topoisomerase-II inhibition and anti-cancer activity. Bioorg Med Chem Lett. 2011;21:350–353. doi: 10.1016/j.bmcl.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 154.Kamal A, Suresh P, Janaki Ramaiah M, Mallareddy A, Kumar BA, Raju P, Vinay Gopal J, Pushpavalli SN, Lavanya A, Sarma P, Pai-Bhadra M. Synthesis and biological evaluation of 4β-acrylamidopodophyllotoxin congeners as DNA damaging agents. Bioorg Med Chem. 2011;19:4589–4600. doi: 10.1016/j.bmc.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 155.Kamal A, Mallareddy A, Suresh P, Lakshma Nayak V, Shetti RV, Sankara Rao N, Tamboli JR, Shaik TB, Vishnuvardhan MV, Ramakrishna S. Synthesis and anticancer activity of 4β-alkylamidochalcone and 4β-cinnamido linked podophyllotoxins as apoptotic inducing agents. Eur J Med Chem. 2012;47:530–545. doi: 10.1016/j.ejmech.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 156.Kamal A, Suresh P, Ramaiah MJ, Mallareddy A, Imthiajali S, Pushpavalli SN, Lavanya A, Pai-Bhadra M. Synthesis and biological evaluation of 4β-sulphonamido and 4β-[(4′-sulphonamido)benzamide]podophyllotoxins as DNA topoisomerase-IIα and apoptosis inducing agents. Bioorg Med Chem. 2012;20:2054–2066. doi: 10.1016/j.bmc.2012.01.039. [DOI] [PubMed] [Google Scholar]
- 157.Kamal A, Tamboli JR, Vishnuvardhan MV, Adil SF, Nayak VL, Ramakrishna S. Synthesis and anticancer activity of heteroaromatic linked 4β-amido podophyllotoxins as apoptotic inducing agents. Bioorg Med Chem Lett. 2013;23:273–280. doi: 10.1016/j.bmcl.2012.10.099. [DOI] [PubMed] [Google Scholar]
- 158.Zhang FM, Tian X. Synthesis and anticancer activity of novel derivatives of 4′-demethylepipodophyllotoxin. Acta Chim Sin. 2002;60:720–724. [Google Scholar]
- 159.Liu YQ, Yang H, Tian X. Design, synthesis and biological evaluation of novel etoposide analogue as cytotoxic agents. Chin J Chem. 2006;24:785–790. [Google Scholar]
- 160.Chen SW, Tian X, Tu YQ. Synthesis and cytotoxic activity of novel derivatives of 4′-demethylepipodophyllotoxin. Bioorg Med Chem Lett. 2004;14:5063–5066. doi: 10.1016/j.bmcl.2004.07.094. [DOI] [PubMed] [Google Scholar]
- 161.Chen SW, Xiang R, Liu J, Tian X. Synthesis and biological evaluation of novel conjugates of podophyllotoxin and 5-FU as antineoplastic agents. Bioorg Med Chem. 2009;17:3111–3117. doi: 10.1016/j.bmc.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 162.Guianvarch D, Duca M, Boukarim C, Kraus-Berthier L, Léonce S, Pierré A, Pfeiffer B, Renard P, Arimondo PB, Monneret C, Dauzonne D. Synthesis and biological activity of sulfonamide derivatives of epipodophyllotoxin. J Med Chem. 2004;47:2365–2374. doi: 10.1021/jm031117b. [DOI] [PubMed] [Google Scholar]
- 163.Guo YE, Chen H, Zuo S, Liu DL, Lu YL, Lv JJ, Wen SP, Zhang TC. Synthesis and antitumor activity of novel podophyllotoxin derivatives against multidrug-resistant cancer cells. J Asian Nat Prod Res. 2011;13:417–424. doi: 10.1080/10286020.2011.568941. [DOI] [PubMed] [Google Scholar]
- 164.Shang H, Chen H, Zhao DM, Tang XW, Liu YF, Pan L, Cheng MS. Synthesis and biological evaluation of 4α/4β-imidazolylpodophyllotoxin analogues as antitumor agents. Arch Pharm Chem Life Sci. 2012;345:43–48. doi: 10.1002/ardp.201100094. [DOI] [PubMed] [Google Scholar]
- 165.Wang L, Yang FY, Yang XC, Guan XH, Hu CQ, Liu T, He QJ, Yang B, Hu YZ. Synthesis and biological evaluation of new 4β-anilino-4′-O-demethyl-4-desoxypodophyllotoxin derivatives as potential antitumor agents. Eur J Med Chem. 2011;46:285–296. doi: 10.1016/j.ejmech.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 166.Wang CN, Wu ZH, Zhao Y, Ni CY, Zhao XD, Zhu L. Synthesis and cytotoxicity evaluation of novel podophyllotoxin derivatives. Arch Pharm Chem Life Sci. 2011;344:735–740. doi: 10.1002/ardp.201100095. [DOI] [PubMed] [Google Scholar]
- 167.Ren J, Wu L, Xin WQ, Chen X, Hu K. Synthesis and biological evaluation of novel 4β-(1,3,4-oxadiazole-2-amino)-podophyllotoxin derivatives. Bioorg Med Chem Lett. 2012;22:4778–4782. doi: 10.1016/j.bmcl.2012.05.059. [DOI] [PubMed] [Google Scholar]
- 168.Zhao Y, Ge CW, Wu ZH, Wang CN, Fang JH, Zhu L. Synthesis and evaluation of aroylthiourea derivatives of 4β-amino-4′-O-demethyl-4-desoxypodophyllotoxin as novel topoisomerase II inhibitors. Eur J Med Chem. 2011;46:901–906. doi: 10.1016/j.ejmech.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 169.Tao L, Wang YG, Ma C, Zheng B, Chen YZ. Synthesis of 4β-(1,2,3-triazol-1-yl) podophyllotoxins as potential antitumor drugs. Syn Commun. 1999;29:2053–2059. [Google Scholar]
- 170.Cao CN, Chen YY, Wang YG, Chen YZ. Synthesis and antitumor activity of 4-(1,2,3-triazol-1-yl) podophyllotoxins. Chin Chem Lett. 1999;10:1003–1006. [Google Scholar]
- 171.Bhat BA, Reddy PB, Agrawal SK, Saxena AK, Kumar HM, Qazi GN. Studies on novel 4β-[(4-substituted)-1,2,3-triazol-1-yl] podophyllotoxins as potential anticancer agents. Eur J Med Chem. 2008;43:2067–2072. doi: 10.1016/j.ejmech.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 172.Reddy PB, Agrawal SK, Singh S, Bhat BA, Saxena AK, Kumar HM, Qazi GN. Synthesis and biological evaluation of 4β-[(4-substituted)-1,2,3-triazol-1-yl] podophyllotoxins as potential anticancer agents. Chem Biodiver. 2008;5:1792–1802. doi: 10.1002/cbdv.200890168. [DOI] [PubMed] [Google Scholar]
- 173.Reddy PB, Paul DV, Agrawal SK, Saxena AK, Kumar M, Qazi GN. Design, synthesis, and biological testing of 4β-[(4-substituted)-1,2,3-triazol-1-yl]podophyllotoxin analogues as antitumor agents. Arch Pharm (Weinheim) 2008;341:126–131. doi: 10.1002/ardp.200700116. [DOI] [PubMed] [Google Scholar]
- 174.Reddy DM, Srinivas J, Chashoo G, Saxena AK, Sampath Kumar HM. 4β-[(4-Alkyl)-1,2,3-triazol-1-yl]podophyllotoxins as anticancer compounds: Design, synthesis and biological evaluation. Eur J Med Chem. 2011;46:1983–1991. doi: 10.1016/j.ejmech.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 175.Chen H, Zuo S, Wang X, Tang X, Zhao M, Lu Y, Chen L, Liu J, Liu Y, Liu D, Zhang S, Li T. Synthesis of 4β-triazole-podophyllotoxin derivatives by azide-alkyne cycloaddition and biological evaluation as potential antitumor agents. Eur J Med Chem. 2011;46:4709–4714. doi: 10.1016/j.ejmech.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 176.Chen J, Ma L, Zhang R, Tang J, Lai H, Wang J, Wang G, Xu Q, Chen T, Peng F, Qiu J, Liang X, Cao D, Ran Y, Peng A, Wei Y, Chen L. Semi-synthesis and biological evaluation of 1,2,3-triazole-based podophyllotoxin congeners as potent antitumor agents inducing apoptosis in HepG2 cells. Arch Pharm (Weinheim) 2012;345:945–956. doi: 10.1002/ardp.201100438. [DOI] [PubMed] [Google Scholar]
- 177.Wang YG, Tian X, Chen YZ. Recent advances in spin labeled anticancer drugs. Acta Pharm Sin. 1988;23:792–798. [PubMed] [Google Scholar]
- 178.Liu YQ, Tian X, Yang L, Zhan ZC. First synthesis of novel spin-labeled derivatives of camptothecin as potential antineoplastic agents. Eur J Med Chem. 2008;43:2610–2614. doi: 10.1016/j.ejmech.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 179.Liu YQ, Ohkoshi E, Li LH, Yang L, Lee KH. Design, synthesis and cytotoxic activity of novel spin-labeled rotenone derivatives. Bioorg Med Chem Lett. 2012;22:920–923. doi: 10.1016/j.bmcl.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 180.Liu Y, Qian K, Wang CY, Chen CH, Yang XM, Lee KH. Synthesis and biological evaluation of novel spin labeled 18β-glycyrrhetinic acid derivatives. Bioorg Med Chem Lett. 2012;22:7530–7533. doi: 10.1016/j.bmcl.2012.10.041. [DOI] [PubMed] [Google Scholar]
- 181.Chen YZ, Zhang CJ, Tian X. Spin-labeled antitumor derivatives of podophyllotoxin. Sci Sin B. 1987;30:1070–1079. [PubMed] [Google Scholar]
- 182.Tian X, Yan ZQ, Li JX, Chen YZ. Anticancer drugs (V)—Synthesis of etoposide derivatives. Chem Res Chin Univ. 1995;11:79–83. [Google Scholar]
- 183.Tian X, Yang MG, Chen YZ. Anticancer drugs (VI)—Synthesis and activity of spin labeled derivatives of 4-amino-4-deoxy-4′-demethylepipodophyllotoxin. Chem Res Chin Univ. 1996;12:304–308. [Google Scholar]
- 184.Lu KK, Wang YG, Chen YZ. Synthesis of novel spin labeled analogues of podophyllotoxin glycoside. Syn Commun. 1997;27:1963–1968. [Google Scholar]
- 185.Wang YG, Pan JL, Shi JF, Chen YZ. New spin labeled analogues of podophyllotoxin as potential antitumor agents. Life Sci. 1997;61:537–542. doi: 10.1016/s0024-3205(97)00413-x. [DOI] [PubMed] [Google Scholar]
- 186.Pan JL, Wang YG, Shi JF, Chen YZ. Synthesis of new spin labeled derivatives of podophyllotoxin as potential anticancer agents. Chin Chem Lett. 1997;8:207–208. [Google Scholar]
- 187.Tian X, Zhang FM, Li WG. Antitumor and antioxidant activity of spin labeled derivatives of podophyllotoxin (GP-1) and congeners. Life Sci. 2002;70:2433–2443. doi: 10.1016/s0024-3205(02)01482-0. [DOI] [PubMed] [Google Scholar]
- 188.Wang JZ, Tian X, Tsumura H, Shimura K, Ito H. Antitumor activity of a new low immunosuppressive derivative of podophyllotoxin (GP-11) and its mechanisms. Anticancer Drug Des. 1993;8:193–202. [PubMed] [Google Scholar]
- 189.Chen YZ, Wang YG, Li JX, Tian X, Jia ZP, Zhang PY. Anticancer drugs. II. Synthesis and biological evaluation of spin labeled derivatives of podophyllotoxin. Life Sci. 1989;45:2569–2575. doi: 10.1016/0024-3205(89)90241-5. [DOI] [PubMed] [Google Scholar]
- 190.Tian X, Wang YG, Yang MG, Chen YZ. Synthesis and antitumor activity of spin labeled derivatives of podophyllotoxin. Life Sci. 1997;60:511–517. doi: 10.1016/s0024-3205(96)00689-3. [DOI] [PubMed] [Google Scholar]
- 191.Jin Y, Chen SW, Tian X. Synthesis and biological evaluation of new spin-labeled derivatives of podophyllotoxin. Bioorg Med Chem. 2006;14:3062–3068. doi: 10.1016/j.bmc.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 192.Liu YQ, Tian X. Synthesis of novel spin-labeled derivatives of podophyllotoxin as potential anti-neoplastic agents. Syn Commun. 2005;35:2479–2758. [Google Scholar]
- 193.Yang L, Liu YQ, Tan H, Li WG, Tian X. Design, synthesis, and biological evaluation of novel spin-labeled derivatives of podophyllotoxin as potential antineoplastic agents. Part XII. Nat Prod Res. 2007;21:998–1008. doi: 10.1080/14786410701371546. [DOI] [PubMed] [Google Scholar]
- 194.Zhang JQ, Zhang ZW, Hui L, Chen SW, Tian X. Novel semisynthetic spin-labeled derivatives of podophyllotoxin with cytotoxic and antioxidative activity. Bioorg Med Chem Lett. 2010;20:983–986. doi: 10.1016/j.bmcl.2009.12.048. [DOI] [PubMed] [Google Scholar]