

Abstract

Phospholipase D is a ubiquitous protein in eukaryotes that hydrolyzes phospholipids to generate the signaling lipid phosphatidic acid (PtdOH). PldA, a Pseudomonas aeruginosa PLD, is a secreted protein that targets bacterial and eukaryotic cells. Here we have characterized the in vitro factors that modulate enzymatic activity of PldA, including divalent cations and phosphoinositides. We have identified several similarities between the eukaryotic-like PldA and the human PLD isoforms, as well as several properties in which the enzymes diverge. Notable differences include the substrate preference and transphosphatidylation efficiency for PldA. These findings offer new insights into potential regulatory mechanisms of PldA and its role in pathogenesis.
Phospholipase D, or PLD, is a ubiquitous eukaryotic protein that produces the signaling lipid phosphatidic acid (PtdOH). Roles for human PLD (hPLD) in a variety of signaling processes include cytoskeleton rearrangement, vesicular trafficking, endocytosis, and cell survival.1–3 The involvement of hPLD in bacterial infections has long been recognized, but details on its function are lacking. PLD expressed in human leukocytes has been proposed to function in antimicrobial mechanism such as phagocytosis, degranulation, respiratory burst, and chemo-taxis.4–7 However, host PLD can be manipulated to promote internalization and intracellular survival of bacterial pathogens also.8,9 Several intracellular pathogens are known to secrete one or more phospholipase D enzymes to promote cell internalization, intracellular survival, or in vivo infectivity.10–17
Bacterial PLDs produced by human pathogens generally have poor sequence homology to the host PLD, but one exception is PldA produced by Pseudomonas aeruginosa. Higher order eukaryotic PLDs are generally larger than prokaryotic PLDs and contain one or more regulatory domains, such as phox and pleckstrin homology lipid binding domains or a C2 calcium binding domain. PldA is comparatively large for a bacterial PLD at 122 kDa, but based on primary sequence analysis does not appear to contain these regulatory domains.
PldA is a secreted effector of the H2 Type VI secretion system (H2-T6SS) of P. aeruginosa, an opportunistic pathogen that is a major cause of hospital acquired infections. Cystic fibrosis patients are particularly disposed to developing chronic infections that ultimately lead to respiratory failure. PldA has been associated with mediating intra- and interspecies bacterial competition18 promoting chronic infection,19, and targeting eukaryotic cells to promote cell invasion by P. aeruginosa.20 Given the sequence similarity of PldA to hPLD isoforms and that PldA targets eukaryotic cells, it is of interest to determine similarities and differences between the enzymes. Although enzymatic activity of PldA has been directly confirmed,19 an extensive analysis of catalytic activity and in vitro regulation has not previously been performed.
Here we are reporting a detailed in vitro characterization of PldA identifying its substrate specificities, transphosphatidylation capabilities, divalent cation sensitivity, and lipid and protein activation. We were also able to promote uptake of exogenous PldA into A549 cells to study its direct effects on host cellular lipid composition. Understanding the enzymology and regulation of PldA provides a framework from which to investigate its mechanistic roles during infection.
EXPERIMENTAL PROCEDURES
Materials
Human lung adenocarcinoma epithelial cells (A549) were purchased from ATCC (Manassas, VA). Lipids were obtained from Avanti Polar Lipids (Alabaster, AL) unless otherwise stated. Phosphatidylcholine, l-α-dipalmitoyl, [cholinemethyl-3H] (3H-DPPC) was purchased from PerkinElmer Life Science (Boston, MA). PldA polyclonal antibody was generated by the Vanderbilt Protein and Antibody Resource. All organic solvents were HPLC grade from EMD Millipore, used without further purification.
Cell Culture
A549 cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37 °C in 5% CO2.
Recombinant Expression and Purification of PldA
The PldA gene, provided by Michael Vasil (University of Colorado), was amplified by PCR. The amplified PCR product and pET32b vector were digested with BamHI and HindIII, then ligated using T4 DNA ligase to generate plasmid pET32b.PldA. PldA is expressed as a fusion protein with an N-terminal thioredoxin fusion protein and His6 tag. The PldA K169E mutant construct was generated using QuikChange II mutagenesis kit following the manufacturer's protocol. BL21 CodonPlus (DE3) RIPL cells were transformed with pET32b.PldA. Bacteria were grown at 37 °C to OD600 of 0.6 in the presence of 100 μg/mL ampicillin and 50 μg/mL chloramphenicol. Protein expression was induced at 16 °C with 0.1 mM IPTG for 12–16 h. Cells were lysed by treating with 0.5 mg/mL lysozyme for 20 min and sonication in wash buffer (8 mM Na2PO4, 2 mM KH2PO4 (pH 7.5), 500 mM NaCl) in the presence of protease inhibitor cocktail on ice. PldA was purified using HiTrap chelating column. Elution fractions were combined and further purified by size exclusion chromatography using a HiLoad 16/600 Superdex 200 column. Fractions were concentrated and stored at –20 °C. PldA K169E mutant was expressed and purified the same as wild type PldA. Protein concentrations were determined using Bradford reagent. Protein purity was determined by SDS-PAGE. To generate tagless wild type and K169 PldA, 10 mU thrombin/μg PldA was incubated with purified PldA fusion protein overnight at 4 °C. The reaction was quenched with 1 mM PMSF. Cleaved PldA was purified using a HiTrap chelating column followed by ultra-filtration to remove thrombin, the cleaved tag, and imidazole.
Mass Spectrometry Assay
General Protocol
Purified PldA (4 nM) was incubated with liposomes as indicated for each experiment at 37 °C for 10–30 min in reaction buffer (50 mM HEPES (pH 7.5), 100 mM KCl). Lipids in chloroform were dried under a nitrogen stream in a glass test tube and resuspended in reaction buffer using bath sonication. PldA, lipid, and cation solutions were combined on ice and reactions initiated by incubating at 37 °C. Reactions were quenched with 2:1 acidified methanol (0.1 N HCl/CH3OH, 1:1)/chloroform. Lipids were extracted in the organic layer and analyzed by mass spectrometry as previously described.21 Briefly, to the organic layer, a cocktail of 24 odd-carbon phospholipid standards of 200 ng each and for transphosphatidylation experiments 100 ng of 32:0 phosphatidylmethanol standard was added (detailed list in Supporting Information Table 1) and solvent was evaporated under vacuum. The resulting lipid film was dissolved in 100 μL of isopropanol (IPA)/hexane/100 mM NH4COOH(aq) 58:40:2. MS analysis of hydrolysis or transphosphatidylation products was performed on an Applied Biosystems/MDX SCIEX 4000QTRAP hybrid triple quadrupole/linear ion trap instrument, connected to a Shimadzu HPLC system. Phospholipid classes were separated on a Phenomenex Luna Silica column (2 × 250 mm, 5 μm particle size). Gradient elution was used for class and species separation consisting of IPA/hexane/100 mM NH4COOH(aq) 58:40:2 (mobile phase A) and IPA/hexane/100 mM NH4COOH(aq) 50:40:10 (mobile phase B) and the following program: 0 to 5 min, B = 50%; 5 to 30 min, B = 50 to 100%; 30 to 40 min, B = 100%; 40 to 41 min, B = 100 to 50%; and 41 to 50 min, B = 50%. Flow rate was 0.3 mL/min, and spectra were acquired in negative ionization mode, using turbo spray at 450 °C, and –3500 V ion voltage. Identification of the lipid species was based on LC–MS/MS data using chromatographic resolution of phospholipid classes and mass spectral fragmentation patterns. Quantitation of phospholipid and phosphatidylalcohol molecular species was achieved by applying algorithms using calculated slopes from standard curves developed for each molecular species. At least three independent experiments were performed in triplicate.
Substrate Profile Analysis
PldA was incubated with 25 μM of each lipid and 3 mM MgCl2. For dioleoylphosphatidylethanolamine (DOPE) and 18:0p/18:1 PE conditions, liposomes were composed of PE and 18:1 sphingomyelin (SM) in a 10:1 ratio.
Substrate Fatty Acyl Chain Analysis
Lipids of various acyl chain compositions (16:0/16:0, 16:0/18:1, 18:1/18:1, 18:0/ 18:0, and 18:0/20:4) were combined to generate liposomes composed of a single lipid class. 18:0/20:4 PG was not assayed. PldA was incubated with 6 μM of each lipid (30 μM total PC or PG and 24 μM total PG) and 3 mM MgCl2.
Divalent Cation Regulation
PldA was incubated with 50 μM 10:1 DOPE/SM, DOPG, or DOPC liposomes in the presence of 0.1–10 mM MgCl2 or CaCl2.
Transphosphatidylation Reaction Analysis
PldA was incubated with 50 μM 10:1 DOPE/SM, DOPG, or DOPC liposomes and 0.3% methanol, ethanol, n-butanol, ethanolamine, or glycerol, or 50 mM choline chloride and 3 mM MgCl2. Quantitation of phosphatidylmethanol, phosphatidylethanol, and phosphatidylbutanol produced was calculated based on offine calibration using 36:2 phosphatidylbutanol standard curves.
Phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) Regulation
PldA was incubated with 50 μM 10:1 DOPE/SM or DOPG liposomes in the absence and presence of 5 mol % porcine brain PI(4,5)P2 and 3 mM MgCl2.
Exogenous PLD Assay
Phospholipase D activity was measured using the modified exogenous substrate PLD assay.22 In brief, 6 nM purified PldA was incubated with liposomes containing 50 μM DOPC, 2.5 μCi 3H-DPPC, and any indicated added lipids for 10–30 min at 37 °C. Final reaction conditions contained 50 mM HEPES (pH 7.5), 100 mM KCl, and 3 mM MgCl2. The reaction was quenched with addition of 10% trichloroacetic acid and bovine serum albumin on ice. Protein and lipid were removed by centrifugation. Supernatant was collected and free 3H-choline was measured by scintillation counting. At least three independent experiments were performed in triplicate.
Liposome Binding Assay
The liposome binding assay was adapted from the procedure of Buser and McLaughlin.23 Lipids in chloroform and 3.2 μCi [3H]-DPPC were dried under a stream of nitrogen. Lipids were suspended in 50 mM HEPES (pH 7.5), and 176 mM sucrose buffer by vortexing vigorously and allowed to hydrate for 1 h at room temperature. Vesicles were subjected to five cycles of freezing in liquid N2 and thawing in a warm water bath after which, vesicles were extruded through two 0.1 μm polycarbonate filters. Vesicles were diluted 1:5 in an isotonic buffer (50 mM HEPES (pH 7.5), 100 mM KCl), pelleted by centrifugation at 100 000g for 1 h at room temperature and resuspended in isotonic buffer. Final lipid concentrations were calculated based on [3H]DPPC radioactivity. PldA (15 nM) was added to vesicles in a 1 mL reaction and incubated at room temperature for 5 min. Samples containing PldA without liposomes were used as controls to determine background. The final reaction conditions were 15 nM PldA, 50 mM HEPES (pH 7.5), 100 mM KCl, 3 mM MgCl2, and 100 μM lipid. Lipid-bound protein was isolated by centrifugation at 15 000g for 20 min at 4 °C. The supernatant was removed and the lipid and protein resuspended in 1× SDS loading buffer. For Western blot analysis, protein was transferred to a nitrocellulose membrane. The membrane was blocked with TBST containing 5% nonfat milk and incubated with anti-PldA rabbit polyclonal antibody. After washing, blots were probed with HRP conjugated rabbit secondary antibody and developed using ECL reagent.
Exogenous PldA Internalization Assay
To compare internalization of tagged and tagless PldA, in a 12-well plate, A549 cells were seeded at 1.5 × 105 cells/well in DMEM, 10% FBS, and 1% penicillin/streptomycin 18 h prior to treatment. Two μM PldA, PldA K169E, tagless PldA, or tagless PldA K169E was incubated with A549 cells in DMEM and 0.05% FBS containing medium for 3.5 h at 37 °C in 5% CO2. Control cells were vehicle treated. Cells were washed three times with PBS and lysed on the plate with 100 μg/mL digitonin in PBS with protease inhibitor cocktail at room temperature for 10 min. Cytosol was collected, and cell membranes scraped in PBS with protease inhibitor to collect membrane fraction. PldA was detected by Western blotting as described earlier using a rabbit anti-PldA polyclonal antibody and HRP conjugated antirabbit secondary antibody. β-Actin (Sigma) was used as a loading control. CAD, carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (Abcam), and Na+/K+ ATPase (Cell Signaling) were used as cytosolic and membrane protein markers.
To measure PldA internalization over time, A549 cells were seeded at 1.5 × 105 cells/well in DMEM, 10% FBS, and 1% penicillin/streptomycin 18 h prior to treatment in a 12-well plate. Two μM wild type PldA or K169E PldA was incubated with A549 cells in DMEM and 0.05% FBS containing medium for 30 min to 4 h at 37 °C in 5% CO2. Control cells were vehicle treated. Cells were washed three times with PBS and lysed on the plate with 1× SDS loading buffer with protease inhibitor cocktail. Cells were scraped and lysates analyzed by Western blotting. PldA was detected by Western blotting as described earlier using a rabbit anti-PldA polyclonal antibody and HRP conjugated anti-rabbit secondary antibody. β-Actin was used as a loading control.
To measure the effects of PldA on Akt, cells were treated as described above with wild type or K169E PldA treated cells for 30 min or 4 h. Cells were washed three times with PBS and lysed on the plate with 1× SDS loading buffer with protease inhibitor cocktail. Cells were scraped and lysates analyzed by Western blotting. PldA was detected as described earlier. Akt was probed using anti-phospho Akt S473 and anti-pan Akt antibodies (Cell Signaling).
For mass spectrometric analysis, in a 12-well plate, A549 cells were seeded at 1.5 × 105 cells/well in DMEM, 10% FBS, and 1% penicillin/streptomycin medium 18 h prior to treatment. Two μM PldA or PldA K169E was incubated with A549 cells for 3.5 h at 37 °C in 5% CO2. Cells were washed three times with PBS and lysed in 800 μL 0.1 N HCl:methanol (1:1). Lipids were extracted with 400 μL chloroform. LC–MS analysis was performed as previously published.21
Confocal Microscopy
A549 cells were seeded at 1.5 × 104 cells/well in a 35 mm glass bottom plate (MatTek) in DMEM, 10% FBS, and 1% penicillin/streptomycin 18 h prior to treatment. PldA and PldA K169E were labeled using the Alexa Fluor 488 Protein labeling kit according to the manufacturer's protocol to generate WT-488 or K169E-488. 1 μM WT-488 or K169E-488 was incubated with A549 cells for 4 h at 37 °C in 5% CO2. Extracellular PldA was removed by washing three times with PBS followed by addition of 10% FBS in DMEM. Cells were imaged using Zeiss LSM 510 inverted microscope at 5 and 7 h postincubation using a 63× 1.4 N.A. (Plan-Apochromat) oil immersion objective. Acquisition settings were held constant unless noted otherwise.
RESULTS
Expression, Optimization and Purification of Recombinant PldA
The PldA gene sequence was cloned into the pET32b vector to produce a PldA fusion protein with N-terminal thioredoxin and polyhistidine tags. Optimal protein expression required incubations at temperature below 18 °C. Lowering the induction temperature enhanced the stability of the enzyme by reducing accumulation of protein fragments. The Trx-His6-PldA fusion protein was isolated using immobilized metal affinity chromatography (IMAC). Protein was concentrated and further purified using size exclusion chromatography (SEC). Protein yields after IMAC and SEC were 8.1 and 6.4 mg/L, respectively. For select experiments, a catalytically dead mutant was used as a negative control. Site directed mutagenesis was used to generate mutant PldA K169E to render PldA catalytically inactive. For some experiments, tagless PldA was required. The thrombin cleavage site of the PldA fusion protein was utilized to remove the thioredoxin and polyhistidine tags and generate tagless wild type PldA and tagless PldA K169E constructs. SDS-PAGE analysis after purification for wild type PldA, PldA K169E, tagless PldA, and tagless PldA K169E is shown in Figure 1A. All experiments were formed with the wild type Trx-His6-PldA fusion protein, hereafter referred to as PldA, unless otherwise stated.
Figure 1.
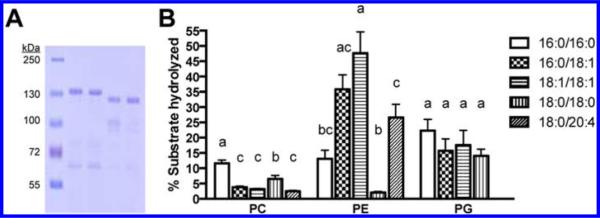
(A) SDS-PAGE analysis of purified recombinant PldA. Lane 1, molecular weight ladder; lane 2, wild type PldA; lane 3, PldA K169E; lane 4, tagless wild type PldA; lane 5, tagless PldA K169E. (B) PldA displays variable substrate hydrolysis based on phospholipid fatty acyl chain composition. 18:0/20:4 PG was not assayed. Mean values are shown ± SEM (n = 9). Statistical analysis performed using one-way ANOVA with Tukey's post hoc test. Groups differing (p < 0.05) have different letters.
PldA Has a Wide Substrate Profile in Vitro
The ability of diverse glycerophospholipids to serve as PldA substrates was assessed based on various chemical lipid features, including headgroup composition, fatty acyl chain unsaturation, and acyl or ether linkage to the glycerol backbone. The ability of PldA to hydrolyze the major glycerophospholipids, phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), phosphatidylinositol (PI), and phosphatidylserine (PS) at a single concentration was determined. Due to the inherent instability of PE in aqueous solutions, DOPE and 18:1 sphingomyelin (SM) in a 10:1 ratio was used to generate stable lipid solutions. All further experiments with DOPE shown also include 18:1 SM. PldA utilizes all of these lipids as substrates, except PI, with variable hydrolysis rates (Table 1). The order of substrate preference was DOPE >> DOPG > DOPS > DOPC. Lysolipids were also observed to be substrates for PldA, but the order of preference does not mirror that of the diacyl lipids (LPE >> LPC > LPG > LPS). Most lysolipids were also hydrolyzed to a lesser extent than their diacyl counterpart, except LPC. The hydrolysis rate of 18:1 LPC was significantly higher (8.6-fold) than hydrolysis of DOPC. PldA was not found to have any detectable sphingomyelinase activity. No hydrolysis of 18:1 SM was observed under any conditions including 10:1 PE/SM liposomes.
Table 1.
Specific Activity of Recombinant PldA with Various Lipidsa
| substrate | specific activity (nmol/min/mg) |
|---|---|
| DOPC | 15.3 ± 3.6 |
| DOPG | 344.1 ± 22.0 |
| DOPE | 2871.9 ± 218.1 |
| DOPS | 229.3 ± 25.8 |
| DOPI | NDb |
| 18:1 LPC | 130.4 ± 17.9 |
| 18:1 LPG | 57.3 ± 2.6 |
| 18:1 LPE | 1345.6 ± 94.9 |
| 18:1 LPS | 9.0 ± 5.9 |
| 18:1 LPI | NDb |
| 18:0p/18:1 PC | 46.3 ± 5.6 |
| 16:0e/18:1 PC | 12.3 ± 2.8 |
| 18:0p/18:1 PE | 1714.6 ± 79.2 |
| 18:1 SM | NDb |
Values are given as mean ± SEM (n = 7–9).
ND: not detected.
In order to further evaluate substrate specificity, alkenyl, or plasmalogen, and alkyl ether phospholipids were assessed as potential substrates. The hydrolysis rate of 18:0p/18:1 plasmenylethanolamine and 18:0p/18:1 plasmenylcholine was 50% and 300% that of the corresponding diacyl-linked lipids, DOPE and DOPC, respectively. To improve solubility of 18:0p/18:1 plasmenylethanolamine in solution, 18:1 SM was added in a 10:1 ratio of PE to SM. The alkyl-linked lipid 16:0e/18:1 PC also displayed a similar hydrolysis rate relative to DOPC. In vitro PldA can hydrolyze acyl-linked phospholipids as well as ether-linked lipids commonly found in eukaryotic cells.
We next examined whether PldA has preference for substrates of particular fatty acyl compositions. The effects of fatty acid unsaturation on PldA lipid hydrolysis were evaluated for three lipid substrate classes (Figure 1B). PldA was incubated with mixed liposomes containing a single lipid class, such as PC, but a variety of fatty acyl moiety combination, such as 18:1/18:1, 16:0/18:1, 16:0/16:0, and so forth in equal proportions. In terms of PC hydrolysis, lipids with two saturated acyl chains were preferentially hydrolyzed over species containing one or more unsaturated acyl chains. This trend is opposite that of PE substrates, in which lipids containing unsaturated acyl chains were preferentially hydrolyzed over disaturated PE lipids. As for PG substrates, little preference in hydrolysis was observed based on fatty acid unsaturation.
PldA Has Low Transphosphatidylation Activity
PLD hydrolyzes phospholipids to generate phosphatidic acid; however, many enzymes are capable of using primary alcohols as a nucleophile to produce a transphosphatidylation product. The ability of PldA to perform transphosphatidylation using a series of primary alcohols was evaluated (Figure 2). PldA was capable of using methanol (MeOH), ethanol (EtOH), and n-butanol (BuOH) to produce transphosphatidylation products. The order of transphosphatidylation rates correlated with the order of hydrolysis rates in the absence of primary alcohols for the lipid classes (DOPE > DOPG > DOPC) (Table 1). Interestingly, the trends for transphosphatidylation rates for the alcohols varied for each substrate. When using DOPC as a substrate, transphosphatidylation rates with BuOH were slightly lower than with MeOH or EtOH, although not significant. But when DOPG served as the substrate, transphosphatidylation rates with BuOH were significantly higher than with MeOH or EtOH. And finally, when DOPE served as the substrate, transphosphatidylation rates were significantly higher with MeOH than other two alcohols.
Figure 2.

PldA transphosphatidylation and hydrolysis activity in the presence of primary alcohols. (A) PldA transphosphatidylation and (B) hydrolysis activity in the presence of methanol, ethanol, or n-butanol. (C) Effects of free phospholipid headgroups on PldA hydrolysis activity. Substrates are shown above each panel. Mean values are shown ± SEM (n = 9). Statistical analysis performed using one-way ANOVA with Tukey's post hoc test. *p < 0.01.
The effects of MeOH, EtOH, and BuOH on PldA substrate hydrolysis are summarized in Figure 2B. Changes in hydrolysis rates of DOPC and DOPG in the presence of MeOH, EtOH, and BuOH trended with phosphatidylalcohol production. A major difference was the stimulation of DOPC hydrolysis by MeOH and EtOH, but an inhibition of DOPG hydrolysis by MeOH and EtOH. In the case of DOPE hydrolysis, the addition of any of the alcohols resulted in a slight increase in hydrolysis rates that was not significant. Most interestingly, in many cases, these primary alcohols were ineffective at inhibiting PtdOH production, especially PE, which is the major substrate of PldA. Observed transphosphatidylation rates were low relative to hydrolysis rates. Transphosphatidylation rates for PG were only 1.5–1.8% of the hydrolysis rates in the presence of the alcohols, and transphosphatidylation rates using PC and PE were only 2–6% of the hydrolysis rates. Butanol is a commonly used tool for inhibiting hPLD mediated PtdOH production, but 0.3% alcohol was ineffective inhibiting in vitro PldA activity.
Because of the transphosphatidylation efficiency of some bacterial PLDs, many have been studied for use as biocatalysts for industrial production of phospholipids, such as PG, PE, and PS, from PC. Transphosphatidylation activity of PldA with physiologically relevant headgroups, like choline, ethanolamine, and glycerol was also investigated for PldA (Figure 2C). The order of inhibition of PtdOH formation correlated to the order of substrate preference (ethanolamine > glycerol > choline). In the presence of 0.3% ethanolamine, hydrolysis of both DOPG and DOPC was significantly inhibited. The addition of 0.3% glycerol produced a modest decrease in DOPC hydrolysis, while DOPE hydrolysis was not altered. The addition of 50 mM choline chloride had no impact on DOPG or DOPE hydrolysis. Glycerol was the only headgroup used by PldA under these conditions to generate a transphosphatidylation product and only from DOPE substrate with a transphosphatidylation rate of 114.2 ± 10.5 nmol/min/mg, which is only 5% of the hydrolysis reaction (Table 1).
Divalent Cations Stimulate PldA Activity
For some PLD enzymes, activity is stimulated by or dependent on divalent cations, such as magnesium and calcium.24–26 PldA activity as a function of Ca2+ and Mg2+ concentrations was measured. Robust hydrolysis of DOPE did not require either of these ions and only a modest 1.6-fold increase in activity at 10 mM MgCl2 was observed (Figure 3A). Increasing concentrations of CaCl2 produced a modest, but significant decrease in DOPE hydrolysis at 5 mM and 10 mM. In contrast, a large concentration dependent stimulation of DOPC and DOPG hydrolysis was observed with maximal activity observed above 2.5 mM MgCl2 and CaCl2 (Figure 3B, C). DOPC activity did require Mg2+ and Ca2+; however, PG hydrolysis was barely detectable without the ions present. Interestingly, for DOPG, Mg2+ and Ca2+ produced similar changes in the absolute specific activity at comparable concentrations; however, for DOPC, Mg2+ and Ca2+ produced similar patterns of stimulation but Mg2+ was more effective at stimulating higher specific activity at comparable cation concentrations.
Figure 3.
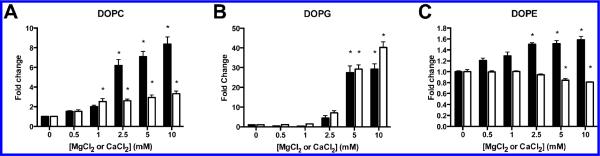
Divalent cations stimulate PldA activity in vitro in a concentration dependent manner. MgCl2 (black bar) and CaCl2 (white bar). Substrates are shown above each panel. Mean values are shown ± SEM (n = 9–12). Statistical analysis performed using one-way ANOVA with Tukey's post hoc test. *Significantly different from control, p < 0.01.
Phosphoinositides Stimulate PldA Activity in Vitro
Eukaryotic PLDs are regulated through a variety of protein–protein and protein–lipid interactions. Since PldA is a secreted protein and due to the significant sequence homology of PldA to the hPLD isoforms, some of these interactions for hPLD were investigated as regulators of PldA. hPLD activity is stimulated in vitro and in vivo by protein kinase C and small GTPases, such as Arf and Cdc42.27–31 Stimulation of PldA activity in vitro was not observed for these small GTPases or protein kinase C (data not shown).
Phosphatidylinositol-4,5-bisphosphate, PI(4,5)P2, is a potent lipid stimulator of human PLD1 and PLD2 activity. Here PI(4,5)P2 stimulation of PldA was also examined. Based on sequence analysis, the PI(4,5)P2 binding site located in the catalytic domain of hPLD is partially conserved within PldA (Figure 4A). As shown in Figure 4B, the addition of 5 mol % PI(4,5)P2 to DOPC liposomes resulted in an 8-fold stimulation of PC hydrolysis. To determine if this stimulation is specific for DOPC, hydrolysis of DOPG and DOPE liposomes in the absence and presence of 5 mol % PI(4,5)P2 was compared (Figure 4B). The addition of PI(4,5)P2 to DOPG liposomes resulted in a modest 1.2-fold increase in PldA activity, while PI(4,5)P2 caused a 20% reduction in DOPE hydrolysis. PI(4,5)P2 stimulation of PC hydrolysis by PldA was also concentration dependent as seen in Figure 4C.
Figure 4.

Phosphoinositides stimulate PldA activity and promote lipid binding in vitro. (A) Alignment of putative PI(4,5)P2 binding domain of PldA and PI(4,5)P2 binding domain of human PLD1 and PLD2. (B) Brain PI(4,5)P2 stimulates PC substrate hydrolysis by PldA. Statistical analysis performed using Student's t test. *p < 0.01. (C) Brain PI(4,5)P2 stimulation of DOPC hydrolysis is concentration dependent. Statistical analysis performed using one-way ANOVA with Tukey's post hoc test. Groups differing (p < 0.01) have different letters. (D) Phosphoinositides stimulate DOPC hydrolysis. All phosphoinositides were 36:2, except for brain PI(4)P. Statistical analysis performed using one-way ANOVA with Tukey's post hoc test. Groups differing (p < 0.01) have different letters. (E) Brain PI(4,5)P2 increases DOPC liposome binding of PldA. All experiments performed in triplicate. Shown are representative data. All mean values are shown ± SEM (n = 3).
We also investigated the specificity of the PI(4,5)P2 stimulation by assaying the effects of other phosphoinositides on PldA activity. PldA hydrolysis of DOPC liposomes containing 5 mol % PI, phosphoinositides, or diacylglycerol pyrophosphate (DGPP) was measured. PI did not stimulate PC hydrolysis; however, all phosphoinositides stimulated hydrolysis to varying extents (Figure 4D). The order of potency for phosphoinositides was PIP3 > PIP2 > PIP. The difference in stimulation was not dependent on the location of phosphate on the inositol headgroup, but rather the overall number of charges. DGPP also stimulated PldA activity to a similar extent as monophosphorylated phosphoinositides.
One potential mechanism by which PI(4,5)P2 can increase PLD activity is to increase lipid binding affinity. To examine this possibility, protein binding of PldA to liposomes comprised 100 mol % DOPC or 95 mol % DOPC/5 mol % PI(4,5)P2 was assessed. Based on the Western blotting results in Figure 4E, PI(4,5)P2 caused a significant increase in binding to DOPC liposomes.
Thioredoxin Tagged PldA Internalizes into A549 Cells
The H2-T6SS of P. aeruginosa and PldA have been shown to be involved in eukaryotic infections and to promote epithelial cell internalization of P. aeruginosa.19,20,32,33 Although translocation of PldA into eukaryotic cells has been demonstrated, its catalytic activity within human cells has not been characterized. Overexpression of PldA by transfection within a variety of cell lines resulted in modest and transient changes in PtdOH despite appreciable levels of protein expression (data not shown). Compensatory mechanisms or protein inactivation over the course of a 6–72 h transfection may explain the lack of observed changes in lipid composition.
Instead, we explored the effects of introducing exogenous PldA and determining if it was capable of internalizing into human cells and eliciting changes in lipid composition. PldA used throughout the in vitro studies was generated as a fusion protein with thioredoxin, a protein that has been shown to translocate across the plasma membrane of human cells.34,35 To examine whether the thioredoxin tag promotes PldA internalization, the thioredoxin tags of PldA and PldA K169E were removed utilizing thrombin, and internalization of tagless PldA and thioredoxin-PldA fusion proteins were analyzed by Western blotting (Figure 5A). Based on Western blot analysis of cell cytosolic and membrane fractions, PldA levels are significantly higher than PldA K169E levels after several hours of incubation. Tagless PldA and PldA K169E were detected in the cell membrane and cytosolic fractions at levels comparable to the PldA K169E fusion protein, suggesting that the thioredoxin tag promotes a significantly higher level of internalization of PldA. The large discrepancy in protein levels of PldA and PldA K169E fusion proteins led us to investigate whether the loss of catalytic activity for PldA K169E was affecting internalization or stability of PldA K169E. To answer this question, internalization of PldA and PldA K169E over time was determined by Western blotting (shown in Figure 5B) and quantitated (shown in Figure 5C). At early time points, (30 min), wild type and K169E PldA proteins are present at equal levels within A549 cells; however, by 3 h postincubation, protein levels for PldA are still increasing while those for PldA K169E have significantly declined, suggesting that catalytic activity is important for stability after internalization, but not for internalization itself.
Figure 5.

Recombinant PldA internalizes into A549 cells. (A) PldA fusion protein translocates into A549 cells. (B) Stability of catalytically dead PldA (K169E) decreases over time, while wild type PldA accumulates. Statistical analysis performed using unpaired t test. *p < 0.05. Shown is a representative experiment. (C) Quantitation of WT and K169E PldA levels in experiment in panel (B) normalized to β-actin and protein levels at 30 min. Results are combined from three independent experiments (n = 3). (D) Changes in localization of PldA in A549 cells are activity dependent. Fluorescence and DIC overlay images of A549 cells treated with PldA.
To confirm internalization of exogenous recombinant PldA into A549 cells, PldA and PldA K169E were covalently labeled with Alexa Fluor 488 and imaged using confocal microscopy (Figure 5D). Based on confocal imaging, both catalytically competent fluorophore-labeled (WT-488) and incompetent (K169E-488) PldA were localized to the perinuclear space within A549 cells 5 h post-treatment; however, over time differences are observed. At 5 and 7 h postincubation, WT-488 fluorescence intensity is comparably robust. Using the same acquisition settings, K169E-488 fluorescence intensity was much lower at 7 h compared to 5 h post-K169E-488 treatment or 7 h post-WT-488 treatment. This is consistent with results shown in Figure 5B. Both wild type and mutant PldA initially localize to perinuclear vesicles, but over time wild type PldA vesicles disperse toward the plasma membrane. Localization of mutant PldA however remained constant over time.
Internalized PldA Induces Changes in A549 Cellular Lipids
Once internalization was confirmed, we evaluated the effects of PldA on A549 cellular lipids. The lipid composition of A549 cells treated with PldA or PldA K169E was determined by LC–MS analysis (Figure 6). A complete list and species distribution of phospholipids and sphingolipids detected and quantitated is included in Supporting Information Table 2. The primary PtdOH species generated (32:1, 34:2, 34:1, and 36:2 PtdOH) correlate nicely with significant reductions in the corresponding species of PE (32:1, 34:2, 34:1, and 36:2) and PG (34:1 and 36:2). In vitro, PldA was observed to hydrolyze plasmalogen PE. In A549 cells, the 36:1p PE species was the only plasmalogen whose levels were reduced with wild type PldA treatment, although the change was not significant. The only PC species whose level decreased with PldA treatment relative to PldA K169E treatment was dipalmitoyl PC (DPPC) although the change was also not statistically significant. It is interesting to note here that DPPC was observed to be the preferred PC substrate in vitro as well (Figure 1B).
Figure 6.

Changes to (A) PtdOH, (B) PE, (C) PG, (D) PC, (E) BMP, and (F) SM lipids of A549 cells associated with PldA treatment. Fold change was normalized to PldA K169E treated samples. All mean values are shown ± SEM (n = 4). Statistical analysis performed using unpaired t test. *p < 0.01.
Bis(monoacylglycero)phosphate, or BMP, was the only lipid class that significantly decreased with PldA treatment that was not a substrate. BMP is specifically and exclusively localized to the late endosome or lysosome within cells and is important for intraluminal vesicle formation of late endosomes, sphingolipid metabolism, and cholesterol trafficking.36,37 BMP is a highly stable lipid and PldA likely does not directly hydrolyze BMP; instead the significant loss of PG, the starting material for BMP de novo synthesis, most likely limited biosynthesis of BMP. The decline in BMP with wild type PldA treatment suggests that PldA can alter the lipid membranes of endosomal vesicles. Changes to BMP would be expected to have an impact on sphingolipid metabolism, as BMP is an activator of many sphingolipid metabolizing enzymes. Although we could not analyze most sphingolipids using our LC–MS method, sphingomyelin was quantifiable. Treatment of A549 cells with wild type PldA caused an increase in SM levels, although not significant, suggesting a reduction in lipid turnover or an increase in SM synthesis. The decrease in BMP, which normally stimulates acid sphingomyelinase hydrolysis of sphingomyelin, suggests that the increase is due to a decline in SM turnover. As noted earlier, with confocal microscopy, PldA localized puncta appears to transition from the perinuclear region closer to the cell periphery. This effect is dependent on catalytic activity. The synthesis of PtdOH, a lipid that promotes vesicular trafficking in human cells, and/or the modulation of endosomal lipids such as BMP may mediate these changes and may promote stability of PldA. Based on confocal imaging and mass spectrometry, internalized PldA is capable of altering vesicle trafficking and lipid signaling within A549 cells.
Previous accounts demonstrated a link between PldA and human Akt that served to mediate cell internalization of P. aeruginosa.20 Herein, we assessed whether PldA internalized from the medium also induces Akt activation in A549 cells. Initial efforts focused at the 3.5 h time point, as that was where the most profound difference in wild type and mutant PldA levels were observed (Figure 7A). No significant difference was observed between WT and K169E treated cells. However, at earlier time points (30 min to 1 h) post-treatment, a reproducible and quantified increase in Akt phosphorylation was observed with WT treated cells, while K169E treated cells resembled control, or vehicle, treated cells (Figure 7B). Independent of the mechanism for internalization, either the T6SS20 or thioredoxin induced exogenous uptake, PldA stimulates Akt activation.
Figure 7.
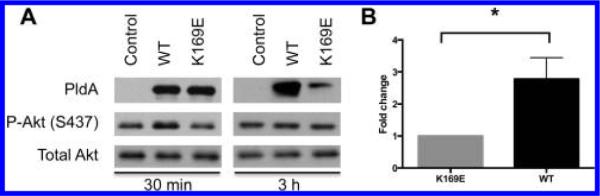
PldA induces a transient stimulation of Akt in A549 cells upon exogenous treatment. (A) Wild type PldA stimulates Akt phosphor-ylation 30 min after exogenous addition and uptake, but not after 3 h. Representative experiment shown. (B) Quantification of difference in Akt phosphorylation between WT and K169E treated cells after 30 min using densitometry. Fold change was normalized to PldA K169E treated samples. Mean values are shown ± SEM (n = 3). Statistical analysis performed using one-sample t test. *p < 0.05.
DISCUSSION
PldA is a secreted effector of the H2-T6SS of Pseudomonas aeruginosa that targets bacterial and eukaryotic cells, however little characterization of its catalytic activity has been performed. Our findings demonstrate that PldA hydrolyzes a wide variety of phospholipids in vitro, however a limited and specific set of products is generated in lung epithelial cells. PldA activity is stimulated in vitro by divalent cations and phosphoinositides; both may be factors in regulating PldA activity in human cells. Unlike many HKD-containing PLDs, transphosphatidylation by PldA is highly inefficient. We also observed that exogenous PldA translocated into epithelial cells induces changes in endosomal lipids and alters vesicular trafficking.
Despite the protein sequence similarity between PldA and eukaryotic PLD family members,19,20 the substrate preference of PldA is noticeably different from that of the human isoforms. Human PLD1 and PLD2 utilize PC as the primary substrate in vitro and in vivo, while PldA hydrolyzes PE and PG more readily than PC in a reconstitution assay and within A549 cells. The preference for PE instead of PC as the primary substrate correlates with the observed sensitization and disruption of bacterial membranes, which generally contain little to no PC.
In A549 cells treated with PldA, increased production of only four species of PtdOH (32:1, 34:2, 34:1, 36:2) was detected. Interestingly, these PtdOH species are the same characteristic signaling species of PtdOH produced by the human isoforms in cells.38,39 This selectivity is not based on species abundance. The PE species that were PldA substrates constituted only 17% of the total cellular PE, while the five most abundant PE species, all of which contain PUFAs, constituted 55% of total PE (Supporting Information Table 2). By mimicking host PLD signaling lipids, PldA may be able to induce host signaling pathways, such as vesicle trafficking (Figure 5D) or Akt signaling.20,40
The transphosphatidylation reaction is a commonly observed property of HKD-containing PLDs. Although PldA can perform this reaction, the hydrolysis rates are significantly higher than the transphosphatidylation rates. n-Butanol is commonly used to inhibit cellular PtdOH production by hPLD isoforms. In most cases assayed here, MeOH, EtOH, and BuOH were poor inhibitors of the production of PtdOH, suggesting that these primary alcohols would likely be ineffective tools to study and modulate PldA activity in cells.
Divalent cations often stimulate catalytic activity of PLDs.24,25,41 PldA does not display an absolute requirement of divalent cations for catalytic activity; however, robust stimulation does occur in a concentration dependent manner. Although the concentrations of free intracellular Mg2+ (0.5–1 mM)42 and Ca2+(0.3–0.5 μM)43 in human cells are below the levels observed for PldA stimulation in this study, the effects of the cations may be primarily due to increased PldA-lipid interactions rather than direct modulation of PldA. If divalent cations were required for catalysis, all substrates would be expected to be equally effected, but divalent cations were required only for the hydrolysis of the anionic substrate, PG. Stimulation of PC hydrolysis by the anionic lipid, PI(4,5)P2 is also divalent cation dependent (data not shown).
In eukaryotic cells, phosphoinositides commonly serve as unique docking sites for proteins and help to delineate various intracellular membrane compartments, primarily those involved in vesicular trafficking.44 PLD1 in a variety of cell lines has a basal localization at perinuclear vesicles that is dependent on phosphoinositide binding45,46 and upon stimulation PLD1 mediates vesicular trafficking that is dependent on catalytic activity.45 Both PldA lipid binding and activity are increased in the presence of phosphoinositides, as they are for human PLD. Phosphoinositides may modulate PldA activity and localization once the enzyme is translocated into the host cell cytosol. In this study, exogenous PldA was taken up into A549 cells and was associated with both the membrane and cytosolic fractions. Localization of cytosolic PldA to intracellular vesicles may be mediated by phosphoinositides.
Internalized active, but not mutant, PldA induced changes in vesicular trafficking in A549 cells. Many pathogens hijack host vesicular trafficking during infection to promote bacterial survival.47 Based on these findings, one can hypothesize that PldA is able to mimic host PLD mediated vesicular trafficking as well. The nature of these intracellular vesicles colocalized with PldA and the effects of PldA on various components of vesicular trafficking networks is the focus of future studies. The gonococcal PLD, NgPLD, was observed to directly bind and stimulate host Akt signaling. Previous findings suggest PldA also interacts with Akt and stimulates its phosphorylation and activation. Here, we reproduce the earlier findings with PldA using exogenous PldA that is capable of internalizing into A549 cells. The ability to reproduce these earlier findings of Akt activation support the notion that other observed effects in A549 cells are relevant despite the difference in the method of delivery of PldA into cells.
Understanding of the parallels between the human isoforms of PLD and PldA contributes to predictions of its mechanism of activity as a virulence factor. In this work, we have identified, for the first time, the substrate specificity of PldA in eukaryotic cells, as well as the products produced, which closely match those of the host PLD. Several similarities between hPLD isoforms and PldA suggest that PldA may mimic some of the functions of the host PLD, such as vesicular trafficking or Akt activation. Of particular interest for future studies is the putative role of phosphoinositides in regulating PldA localization and activity in eukaryotic cells. Further investigation is needed to confirm the physiological relevance of these in vitro regulators of PldA activity and to better understand the function of PldA during P. aeruginosa infection. Given the central importance of this bacterial virulence factor, such studies are likely to be illuminating.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Sarah Scott and Pavlina Ivanova for assistance with mass spectrometric studies. Confocal microscopy experiments were performed in part through the use of the VUMC Cell Imaging Shared Resource (supported by NIH Grants CA68485, DK20593, DK58404, DK59637, and EY08126).
Funding
This research was funded in part by the Vanderbilt Institute of Chemical Biology and the Bixler-Johnson-Mayes Endowed Chair in Pharmacology (H.A.B.). C.S. was supported by National Institutes of Health Training Grant 5T32GM7628 and National Science Foundation Graduate Fellowship DGE-0946822.
ABBREVIATIONS
- PLD
phospholipase D
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PG
phosphatidylglycerol
- PS
phosphatidylserine
- PtdOH
phosphatidic acid
- PI
phosphatidylinositol
- LPE
lysophosphatidylethanolamine
- LPG
lysophosphatidylglycerol
- LPC
lysophosphatidylcholine
- LPS
lysophosphatidylserine
- LPI
lysophosphatidylinositol
- DO
dioleoyl
- DP
dipalmitoyl
- H2-T6SS
H2-Type Six Secretion System
- LC–MS
liquid chromatography–mass spectrometry
- IMAC
immobilized affinity chromatography
- SEC
size exclusion chromatography
- MeOH
methanol
- EtOH
ethanol
- BuOH
n-butanol
- PI(4,5)P2
phosphatidylinositol-4,5-bisphosphate
- hPLD
human PLD
- FBS
fetal bovine serum
- PBS
phosphate buffered saline
- PUFA
polyunsaturated fatty acid
- SM
sphingomyelin
- BMP
bis(monoacylglycero)phosphate
- PIP3
phosphatidylinositol-3,4,5-triphosphate
- PIP
phosphatidylinositol phosphate
- DAG
diacylglycerol
- TBST
Tris buffered saline with 0.1% Tween 20
- HRP
horseradish peroxidase
- SEM
standard error of the mean
Footnotes
Supporting Information
Lipid internal standards used in LC–MS analysis (Table 1); species distribution of phospholipids and sphingolipids detected in A549 cells after treatment with wildtype (WT) or mutant (K169E) PldA. (Table 2). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Roth MG. Molecular mechanisms of PLD function in membrane traffic. Traffic. 2008;9:1233–1239. doi: 10.1111/j.1600-0854.2008.00742.x. [DOI] [PubMed] [Google Scholar]
- 2.Gomez-Cambronero J. Phosphatidic acid, phospholipase D and tumorigenesis. Adv. Biol. Regul. 2013;54:197–206. doi: 10.1016/j.jbior.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris AJ. Regulation of phospholipase D activity, membrane targeting and intracellular trafficking by phosphoinositides. Biochem. Soc. Symp. 2007:247–257. doi: 10.1042/BSS0740247. [DOI] [PubMed] [Google Scholar]
- 4.Gomez-Cambronero J, Di Fulvio M, Knapek K. Understanding phospholipase D (PLD) using leukocytes: PLD involvement in cell adhesion and chemotaxis. J. Leukocyte Biol. 2007;82:272–281. doi: 10.1189/jlb.0107033. [DOI] [PubMed] [Google Scholar]
- 5.Kusner DJ, Adams J. ATP-Induced killing of virulent Mycobacterium tuberculosis within human macrophages requires phospholipase D. J. Immunol. 2000;164:379–388. doi: 10.4049/jimmunol.164.1.379. [DOI] [PubMed] [Google Scholar]
- 6.Kusner DJ, Barton JA. ATP stimulates human macrophages to kill intracellular virulent Mycobacterium tuberculosis via calcium-dependent phagosome-lysosome fusion. J. Immunol. 2001;167:3308–3315. doi: 10.4049/jimmunol.167.6.3308. [DOI] [PubMed] [Google Scholar]
- 7.Fairbairn IP, Stober CB, Kumararatne DS, Lammas DA. ATP-mediated killing of intracellular kycobacteria by kacrophages is a P2X7-dependent process inducing bacterial death by phagosome-lysosome fusion. J. Immunol. 2001;167:3300–3307. doi: 10.4049/jimmunol.167.6.3300. [DOI] [PubMed] [Google Scholar]
- 8.Lam GY, Cemma M, Muise AM, Higgins DE, Brumell JH. Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenes during the early stages of macrophage infection. Autophagy. 2013;9:985–995. doi: 10.4161/auto.24406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han X, Yu R, Ji L, Zhen D, Tao S, Li S, Sun Y, Huang L, Feng Z, Li X, Han G, Schmidt M, Han L. InlB-mediated Listeria monocytogenes internalization requires a balanced phospholipase D activity maintained through phospho-cofilin. Mol. Microbiol. 2011;81:860–880. doi: 10.1111/j.1365-2958.2011.07726.x. [DOI] [PubMed] [Google Scholar]
- 10.Viner R, Chetrit D, Ehrlich M, Segal G. Identification of two Legionella pneumophila effectors that manipulate host phospholipids biosynthesis. PLoS Pathog. 2012;8:e1002988. doi: 10.1371/journal.ppat.1002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, Luo Z-Q. Comprehensive identification of protein substrates of the dot/Icm type IV transporter of Legionella pneumophila. PLoS One. 2011;6:e17638. doi: 10.1371/journal.pone.0017638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darville T, Welter-Stahl L, Cruz C, Sater AA, Andrews CW, Ojcius DM. Effect of the purinergic receptor P2X7 on chlamydia infection in cervical epithelial cells and vaginally infected mice. J. Immunol. 2007;179:3707–3714. doi: 10.4049/jimmunol.179.6.3707. [DOI] [PubMed] [Google Scholar]
- 13.Benagiano M, Munari F, Ciervo A, Amedei A, Paccani SR, Mancini F, Ferrari M, Della Bella C, Ulivi C, D'Elios S, Baldari CT, Prisco D, de Bernard M, D'Elios MM. Chlamydophila pneumoniae phospholipase D (CpPLD) drives Th17 inflammation in human atherosclerosis. Proc. Natl. Acad. Sci. U. S. A. 2012;109:1222–1227. doi: 10.1073/pnas.1111833109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renesto P, Dehoux P, Gouin E, Touqui L, Cossart P, Raoult D. Identification and characterization of a phospholipase D–superfamily gene in Rickettsiae. J. Infect. Dis. 2003;188:1276–1283. doi: 10.1086/379080. [DOI] [PubMed] [Google Scholar]
- 15.Edwards JL, Apicella MA. Neisseria gonorrhoeae PLD directly interacts with Akt kinase upon infection of primary, human, cervical epithelial cells. Cell. Microbiol. 2006;8:1253–1271. doi: 10.1111/j.1462-5822.2006.00707.x. [DOI] [PubMed] [Google Scholar]
- 16.Edwards JL, Entz DD, Apicella MA. Gonococcal phospholipase D modulates the expression and function of complement receptor 3 in primary cervical epithelial cells. Infect. Immun. 2003;71:6381–6391. doi: 10.1128/IAI.71.11.6381-6391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacobs AC, Hood I, Boyd KL, Olson PD, Morrison JM, Carson S, Sayood K, Iwen PC, Skaar EP, Dunman PM. Inactivation of phospholipase D diminishes Acinetobacter baumannii pathogenesis. Infect. Immun. 2010;78:1952–1962. doi: 10.1128/IAI.00889-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russell AB, LeRoux M, Hathazi K, Agnello DM, Ishikawa T, Wiggins PA, Wai SN, Mougous JD. Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature. 2013;496:508–512. doi: 10.1038/nature12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilderman PJ, Vasil AI, Johnson Z, Vasil ML. Genetic and biochemical analyses of a eukaryotic-like phospholipase D of Pseudomonas aeruginosa suggest horizontal acquisition and a role for persistence in a chronic pulmonary infection model. Mol. Microbiol. 2001;39:291–304. doi: 10.1046/j.1365-2958.2001.02282.x. [DOI] [PubMed] [Google Scholar]
- 20.Jiang F, Waterfield NR, Yang J, Yang G, Jin Q. A Pseudomonas aeruginosa type VI secretion phospholipase D effector targets both prokaryotic and eukaryotic cells. Cell Host Microbe. 2014;15:600–610. doi: 10.1016/j.chom.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 21.Ivanova PT, Milne SB, Byrne MO, Xiang Y, Brown HA. Glycerophospholipid identification and quantitation by electrospray ionization mass spectrometry. In: Brown HA, editor. Methods in Enzymology. Academic Press; New York: 2007. pp. 21–57. [DOI] [PubMed] [Google Scholar]
- 22.Brown HA, Henage LG, Preininger AM, Xiang Y, Exton JH. Biochemical analysis of phospholipase D. In: Brown HA, editor. Methods in Enzymology. Academic Press; New York: 2007. pp. 49–87. [DOI] [PubMed] [Google Scholar]
- 23.Buser C, McLaughlin S. Ultracentrifugation technique for measuring the binding of peptides and proteins to sucrose-loaded phospholipid vesicles. In: Bar-Sagi D, editor. Transmembrane Signaling Protocols. Humana Press; New York: 1998. pp. 267–281. [DOI] [PubMed] [Google Scholar]
- 24.Chalifa V, Möhn H, Liscovitch M. A neutral phospholipase D activity from rat brain synaptic plasma membranes. Identification and partial characterization. J. Biol. Chem. 1990;265:17512–17519. [PubMed] [Google Scholar]
- 25.Okamura S, Yamashita S. Purification and characterization of phosphatidylcholine phospholipase D from pig lung. J. Biol. Chem. 1994;269:31207–31213. [PubMed] [Google Scholar]
- 26.Wang X. Multiple forms of phospholipase D in plants: The gene family, catalytic and regulatory properties, and cellular functions. Prog. Lipid Res. 2000;39:109–149. doi: 10.1016/s0163-7827(00)00002-3. [DOI] [PubMed] [Google Scholar]
- 27.Henage LG, Exton JH, Brown HA. Kinetic analysis of a mammalian phospholipase D: Allosteric modulation by monomeric GTPases, protein kinase c, and polyphosphoinositides. J. Biol. Chem. 2006;281:3408–3417. doi: 10.1074/jbc.M508800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cockcroft S, Thomas GMH, Fensome A, Geny B, Cunningham E, Gout I, Hiles I, Totty NF, Truong O, Hsuan JJ. Phospholipase D: A downstream effector of ARF in granulocytes. Science. 1994;263:523–526. doi: 10.1126/science.8290961. [DOI] [PubMed] [Google Scholar]
- 29.Brown HA, Gutowski S, Moomaw CR, Slaughter C, Sternwels PC. ADP-ribosylation factor, a small GTP-dependent regulatory protein, stimulates phospholipase D activity. Cell. 1993;75:1137–1144. doi: 10.1016/0092-8674(93)90323-i. [DOI] [PubMed] [Google Scholar]
- 30.Sung T-C, Altshuller YM, Morris AJ, Frohman MA. Molecular analysis of mammalian phospholipase D2. J. Biol. Chem. 1999;274:494–502. doi: 10.1074/jbc.274.1.494. [DOI] [PubMed] [Google Scholar]
- 31.Kook S, Exton JH. Identification of interaction sites of protein kinase Cα on phospholipase D1. Cell. Signalling. 2005;17:1433–1432. doi: 10.1016/j.cellsig.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 32.Sana TG, Hachani A, Bucior I, Soscia C, Garvis S, Termine E, Engel J, Filloux A, Bleves S. The second type VI secretion system of Pseudomonas aeruginosa strain PAO1 is regulated by quorum sensing and fur and modulates internalization in epithelial cells. J. Biol. Chem. 2012;287:27095–27105. doi: 10.1074/jbc.M112.376368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lesic B, Starkey M, He J, Hazan R, Rahme LG. Quorum sensing differentially regulates Pseudomonas aeruginosa type VI secretion locus I and homologous loci II and III, which are required for pathogenesis. Microbiology. 2009;155:2845–2855. doi: 10.1099/mic.0.029082-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kondo N, Ishii Y, Kwon YW, Tanito M, Sakakura-Nishiyama J, Mochizuki M, Maeda M, Suzuki S, Kojima M, Kim YC, Son A, Nakamura H, Yodoi J. Lipid raft-mediated uptake of cysteine-modified thioredoxin-1: apoptosis enhancement by inhibiting the endogenous thioredoxin-1. Antioxid. Redox Signaling. 2007;9:1439–1448. doi: 10.1089/ars.2007.1665. [DOI] [PubMed] [Google Scholar]
- 35.Das KC, Lewis-Molock Y, White CW. Elevation of manganese superoxide dismutase gene expression by thioredoxin. Am. J. Respir. Cell Mol. Biol. 1997;17:713–726. doi: 10.1165/ajrcmb.17.6.2809. [DOI] [PubMed] [Google Scholar]
- 36.Hullin-Matsuda F, Luquain-Costaz C, Bouvier J, Delton-Vandenbroucke I. Bis(monoacylglycero)phosphate, a peculiar phospholipid to control the fate of cholesterol: Implications in pathology. Prostaglandins, Leukotrienes Essent. Fatty Acids. 2009;81:313–324. doi: 10.1016/j.plefa.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 37.Hullin-Matsuda F, Kawasaki K, Delton-Vandenbroucke I, Xu Y, Nishijima M, Lagarde M, Schlame M, Kobayashi T. De novo biosynthesis of the late endosome lipid, bis(monoacylglycero)-phosphate. J. Lipid Res. 2007;48:1997–2008. doi: 10.1194/jlr.M700154-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Pettitt TR, McDermott M, Saqib KM, Shimwell N, Wakelam MJ. Phospholipase D1b and D2a generate structurally identical phosphatidic acid species in mammalian cells. Biochem. J. 2001;360:707–715. doi: 10.1042/0264-6021:3600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pettitt TR, Martin A, Horton T, Liossis C, Lord JM, Wakelam MJO. Diacylglycerol and phosphatidate generated by phospholipases C and D, respectively, have distinct fatty acid compositions and functions: phospholipase D-derived diacylglycerol does not activate protein kinase C in porcine aortic endothelial cells. J. Biol. Chem. 1997;272:17354–17359. doi: 10.1074/jbc.272.28.17354. [DOI] [PubMed] [Google Scholar]
- 40.Bruntz RC, Taylor HE, Lindsley CW, Brown HA. Phospholipase D2 mediates survival signaling through direct regulation of Akt in glioblastoma cells. J. Biol. Chem. 2014;289:600–616. doi: 10.1074/jbc.M113.532978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang X, Waksman M, Ely Y, Liscovitch M. Characterization and regulation of yeast Ca2+-dependent phosphatidylethanolamine-phospholipase D activity. Eur. J. Biochem. 2002;269:3821–3830. doi: 10.1046/j.1432-1033.2002.03073.x. [DOI] [PubMed] [Google Scholar]
- 42.Günther T. Concentration, compartmentation and metabolic function of intracellular free Mg2+. Magnesium Res. 2006;19:225–36. [PubMed] [Google Scholar]
- 43.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 44.Kutateladze TG. Translation of the phosphoinositide code by PI effectors. Nat. Chem. Biol. 2010;6:507–513. doi: 10.1038/nchembio.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du G, Altshuller YM, Vitale N, Huang P, Chasserot-Golaz S, Morris AJ, Bader MF, Frohman MA. Regulation of phospholipase D1 subcellular cycling through coordination of multiple membrane association motifs. J. Cell. Biol. 2003;162:305–315. doi: 10.1083/jcb.200302033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hodgkin MN, Masson MR, Powner D, Saqib KM, Ponting CP, Wakelam MJO. Phospholipase D regulation and localisation is dependent upon a phosphatidylinositol 4,5-bisphosphate-specific PH domain. Curr. Biol. 2000;10:43–46. doi: 10.1016/s0960-9822(99)00264-x. [DOI] [PubMed] [Google Scholar]
- 47.Baxt LA, Garza-Mayers AC, Goldberg MB. Bacterial subversion of host innate immune pathways. Science. 2013;340:697–701. doi: 10.1126/science.1235771. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.