

Abstract
MicroRNAs (miRNAs) are small noncoding RNAs that can function as either powerful tumor promoters or suppressors in numerous types of cancer. The ability of miRs to target multiple genes and biological signaling pathways has created intense interest in their potential clinical utility as predictive and diagnostic biomarkers, and as innovative therapeutic agents. Recently, accumulating preclinical studies have illustrated the feasibility of slowing tumor progression by either overexpressing tumor suppressive miRNAs, or by neutralizing the activities of oncogenic miRNAs in cell- and animal-based models of cancer. Here we highlight prominent miRNAs that may represent potential therapeutic targets in human malignancies, as well as review current technologies available for inactivating or restoring miRNA activity in clinical settings.
Keywords: Antisense oligonucleotides, Biomarkers, Chemotherapeutics, Locked nucleic acids, MetastamiR, Metastasis, microRNA, OncomiR
Review
Introduction
The war against cancer commenced in 1971 with the enactment of the National Cancer Act and continues unabated today against this most common killer of American men and women between the ages of 40-79 [1]. Indeed, the American Cancer Society estimates that 25% of all deaths in the United States can be directly attributed to cancer, which is anticipated to account for nearly 600,000 deaths and 1.7 million new invasive cancer cases in 2014. Despite this rather bleak outlook, the collective efforts of science and medicine have nevertheless dramatically reduced the annualized cancer death rate by 20% over the last two decades, thereby sparing the lives of more than 1.3 million Americans [1]. Building upon this success will require the development of new diagnostic and prognostic tests, as well as the creation of novel targeted chemotherapies derived from enhanced knowledge of the molecular mechanisms coupled to tumorigenesis and metastatic progression.
The central dogma of molecular biology states that the transfer of genetic information within cells transpires sequentially from DNA to RNA to proteins, whose coding sequences comprise a paltry 1.5-2% of the human genome [2, 3]. Although genetic and epigenetic aberrations that occur in components of the central dogma clearly elicit disease development in humans, recent findings also point to a prominent role for non-protein-coding regions of the genome in regulating cell and tissue homeostasis, as well as in contributing to the formation of human tumors. Included amongst the various classes of noncoding RNAs are members of the PIWI-interacting RNA (piRNA) family, the small nucleolar RNA (snoRNA) family, the large intragenic noncoding RNA (lincRNA) family, the long noncoding RNA (lncRNA) family, and the transcribed ultraconserved regions (T-UCR) family of the lncRNAs [3–5]. However, the best characterized and most extensively studied class of noncoding RNAs belong to the family of microRNAs (miRNAs), which play essential functions during embryogenesis and tissue development, and during cell differentiation, proliferation, and survival [4, 5]. As a group, miRNAs are small (17-27 nucleotides) noncoding RNAs that govern gene expression in a post-transcriptional manner by binding directly to the 3′UTRs of target mRNAs, thereby repressing their translation or inducing their degradation [6]. In doing so, miRNAs have been ascribed as being potent tumor suppressors in normal cells, and as being robust tumor promoters in developing and progressing carcinomas [7, 8]. Moreover, miRNAs are frequently located at fragile genome sites or regions that are frequently amplified or deleted in human cancers [9]. Indeed, emerging evidence indicates that miRNAs function as a molecular rheostats that serve in fine-tuning cell signaling pathways [10, 11], doing so by modulating the expression of large numbers of genes and, consequently, impacting the flux through essential regulatory nodes of vast signaling networks. These functional characteristics underlie the belief that targeting and manipulating either the expression or activity of miRNAs may provide novel inroads to treat human cancers, a supposition currently being evaluated in phase I clinical trials for MRX34 (a miR-34 mimetic) against late-stage hepatocellular carcinomas and a variety of lymphomas ([5]; ClinicalTrials.gov number, NCT01829971), and in phase II clinical trials for miR-122 antagonists against hepatitis ([12]; ClinicalTrials.gov number, NCT01200420). In the succeeding sections, we review recent findings pertaining to how miRNA expression transpires in normal cells, and to how these events become dysregulated in malignant cells, leading to their acquisition of metastatic and chemoresistant phenotypes. Finally, we discuss current therapeutic strategies aimed at targeting inappropriate miRNA expression and activity in developing and progressing human carcinomas.
miRNA biogenesis
miRNAs are transcribed in the nucleus by the actions of RNA polymerases II or III to produce a long primary miRNA transcript (pri-miRNA), which subsequently associates with the Drosha-microprocessor complex that houses the (i) RNase III enzyme, Drosha, (ii) DGCR8 (DiGeorge critical region 8), and (iii) RNA helicases p68 and p72 (see [13]). In addition, the KH-type splicing regulatory protein (KSRP) can also associate with the microprocessor complex to regulate the biogenesis of a subset of miRNAs operant in regulating cell differentiation, proliferation, and survival [14]. Endonucleolytic cleavage of pri-miRNAs by the microprocessor complex leads to formation of precursor miRNAs (pre-miRNA), which exist as hairpin loop structures of ~60-70 nucleotides in length that are exported from the nucleus by the actions of Exportin-5, with assistance from Ran-GTPases [15]. Upon gaining access to the cytoplasm, the pre-miRNA is further processed and cleaved by the Dicer/TRBP (Tar RNA-binding protein) complex to produce a mature ~21 nucleotide RNA duplex [16], which together with Argonaute (Ago2) proteins is loaded into the RNA-induced silencing complex (RISC). Interestingly, the orientation of mature miRNAs as they are loaded into RISC complexes is governed by their 5′-antisense ends, which possess enhanced flexibility and lower internal stabilities that facilitate miRNA unwinding and strand retention by active RISCs [17]. Finally, the extent of sequence complementarity between the 5′-seed region of the loaded miRNA with that of its target mRNA 3′UTR largely dictates whether individual mRNAs are inactivated via cleavage, translational repression, or deadenylation (Figure 1; [13]). As will be discussed below, recent studies clearly demonstrate the importance of oncogenic signaling systems to impinge upon multiple steps of the miRNA biogenesis pathway, resulting in the aberrant expression and activity of miRNAs during tumorigenesis [18, 19].
Figure 1.
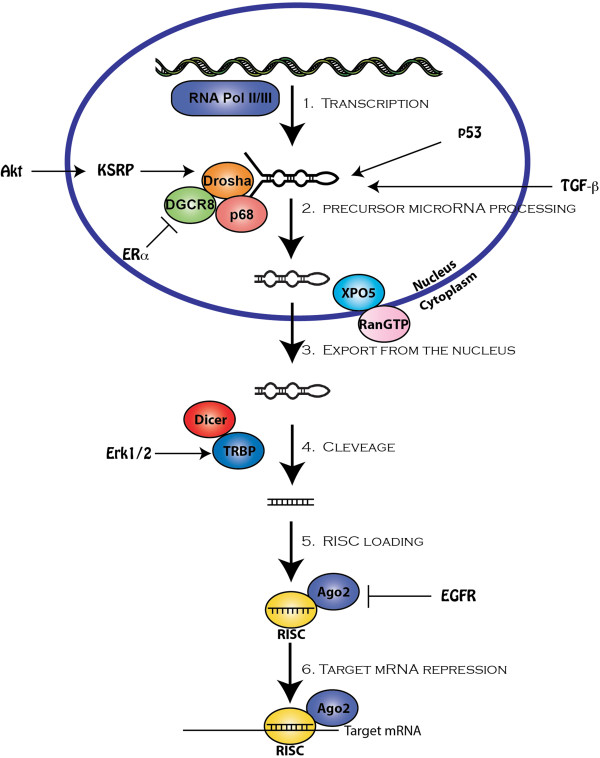
miRNA biogenesis. miRNAs are transcribed by RNA polymerase II or III (Pol II/III) to produce primary transcripts (pri-miRNAs), which are subsequently processed and cropped via the actions of the Drosha-DGCR8 complex, which together with the RNA helicases p68 and p72 generate the formation of precursor miRNAs (pre-miRNAs). Activation of the TGF-β, Akt/PI3K, and p53 signaling systems have all been shown to promote the processing of specific pri-miRNAs, while stimulation of the ER-α signaling system is capable of repressing pri-miRNA processing. Once processed, pre-miRNAs hairpins are exported from the nucleus by exportin-5 (XPO5)-RanGTPase complexes, and are subsequently cleaved by Dicer:TRBP complexes, thereby producing mature oligonucleotide duplexes. The rate at which Dicer cleaves pre-miRNAs is greatly enhanced by the phosphorylation of TRBP by Erk1/2. At the completion of pre-miRNA cleavage, Dicer serves with Argonaute (Ago) 2 in loading mature miRNAs into RNA-induced silencing complexes (RISCs), thereby silencing target mRNA expression through mRNA cleavage, translational repression, or deadenylation. Hypoxic conditions resulting in EGFR activation have been shown to induce the phosphorylation of Ago2, leading to diminished maturation reactions of select miRNAs.
Aberrant miRNA biogenesis in carcinomas
Although major progress has been achieved in terms of understanding the fundamental mechanisms whereby miRNAs are synthesized and processed, considerably less knowledge exists regarding the specific intracellular pathways and effector molecules coupled to the regulation of miRNA biogenesis. Recently, administration of either transforming growth factor-β (TGF-β) or bone morphogenetic protein-4 (BMP-4) to pulmonary artery smooth muscle cells (PASMCs) was observed to dramatically elevate their levels of mature miR-21 independent of any alterations in the transcription of pre-miR-21 [20], suggesting that these multifunctional cytokines drive the processing of miR-21, not its transcription. Accordingly, receptor-regulated Smad transcription factors for TGF-β (Smads 2 and 3) and BMP-4 (Smads 1, 5, and 8) were found to associate with Drosha complexes by interacting with the RNA helicase, p68 [20], a reaction dependent upon the presence the Smad-binding elements (SBEs) located in the stem regions of ~20 pre-miRNAs known to be responsive to TGF-β/BMP-4 stimulation [21]. Indeed, engineering SBE sequences into stem loop structures was sufficient to confer TGF-β/BMP-4-mediated processing of pre-miRNAs to yield their mature products (Figure 1; [21]). It should be noted that the ability of transcription factors to drive miRNA processing is not unique to TGF-β/BMP-regulated Smads, but is instead a widespread phenomenon as evidenced by the fact that ~45% of all pre-miRNAs harbor one or more consensus binding sites for transcription factors [22]. Indeed, similar to Smads, the tumor suppressor p53 has also been shown to interact with the Drosha complex through p68, resulting in enhanced Drosha processing of pre-miRNAs coupled to DNA damage responses and anticancer activities (e.g., miR-16-1, miR-143, miR-145; [23]). Additionally, hyperactivation of the Ras/ERK1/2, PI3K/Akt, and ATM/DNA damage pathways are commonplace in carcinoma cells and have recently been shown to promote the phosphorylation of TRBP, which enhances Dicer cleavage activity, and of KSRP, which enhances pri-miRNA processing reactions [24–27]. Thus, one mechanism whereby oncogenic signaling systems promote tumor development and metastatic progression transpires through upregulated miRNA processing and its associated inactivation of tumor suppressing genes and pathways.
In stark contrast to aforementioned mechanisms that underlie the increased processing of miRNAs, recent findings also observed significant reductions in miRNA processing in response to oncogenic signals. For instance, estradiol-mediated activation of estrogen receptor-α (ER-α) drives the association of this steroid receptor with p68, resulting in widespread inhibition of pri-miRNAs by Drosha complexes [28]. Likewise, hypoxic stress enables the epidermal growth factor receptor (EGFR) to phosphorylate Ago2 and prevent its binding to Dicer, thereby inhibiting the processing and maturation of tumor suppressive pre-miRNAs (e.g., miR-31, miR-192, and mir-193-5p; [29]). Future studies need to further elucidate the molecular mechanisms whereby oncogenic signaling pathways converge on the miRNA biogenesis network, particularly with respect to defining the sequence and specificity of these aberrant interactions, which may aid in discovering novel miRNA-based targeted therapies to alleviate carcinoma development and metastatic progression.
Regulation of miRNAs in carcinomas
The “Hallmarks of Cancer” were eloquently proposed by Hanahan and Weinberg to describe the essential features ingrained in the physiology of all malignant cells that drives their growth and dissemination. Indeed, included amongst the traits displayed by all carcinoma cells is their ability to (i) activate proliferative and replicative immortality programs, while simultaneously inactivating antigrowth programs; (ii) resist apoptotic programs; and (iii) stimulate angiogenic, invasive, and metastatic programs [30, 31]. Importantly, miRNAs play active roles in modulating all of these physiological processes in carcinomas, typically acting on multiple pathways and programs to elicit disease development. Identifying and understanding how miRNAs regulate the balance of and flux through these signaling systems may provide novel opportunities to design and implement miRNA-directed therapies to treat human malignancies. In the succeeding sections, we highlight a variety well-characterized and essential miRNAs operant in driving tumorigenesis based on their classification as oncomiRs (i.e., tumor promoting miRNAs), tumor suppressive miRNAs, or metastamiRs (i.e., metastasis promoting miRNAs) (Table 1). It should be noted that this discussion is by no means comprehensive, and as such, readers desiring more extensive summaries are directed to several recent reviews [5, 32, 33].
Table 1.
Identity and function of cancer-associated miRNAs
| microRNA | Disease setting | Targets | Hallmark (s) of cancer |
|---|---|---|---|
| oncomiRs | |||
| miR-17 ~ 92 Family | Lymphoma and some solid tumors [36] | PTEN [37, 38], Bim [7, 37], p21 [40, 41], TSP1 [42], CTGF [42] | Resisting cell death |
| Sustaining proliferative signaling, and inducing angiogenesis | |||
| miR-21 | Glioblastoma [44], breast, colon, lung, pancreas, prostate, chronic lymphocytic leukaemia, Diffuse Large B-Cell Lymphoma, acute myeloid leukaemia and others [45, 46] | PTEN, p63, PDCD4, and RECK [46] | Sustaining proliferative signaling, resisting cell death, activating invasion and metastasis |
| miR-155 | Lymphoma [50, 51], breast, colon, lung, pancreatic, and thyroid cancer [52] | RhoA [53], SOCS1 [54], FOXO3a [55] | Sustaining proliferative signaling, resisting cell death, activating invasion and metastasis |
| Tumor suppressor miRs | |||
| miR-15a ~ 16-1 | CLL [56], multiple myeloma, mantle cell lymphoma, and prostate cancers [57] | Bcl2, cyclin D1, and WNT3A [59] | Evading growth suppressors, Resisting cell death |
| let-7 | Lung [61], colon, breast, and ovarian cancers [62] | Ras [66], HMGA2 [67], c-Myc [65] | Evading growth suppressors |
| miR-29 Family | Lung cancer, melanoma and myeloid leukemia [75] | CDK6 [70, 71], Ppmid [68], osteonectin [72], Mcl1 [73], Bcl2 [74], DNMT3a [75], and extracellular matrix genes [76]. | Sustaining proliferative signaling, Enabling replicative immortality, Resisting cell death |
| miR-34 | Breast, lung, colon, kidney, bladder, pancreatic cancer, and melanoma [84] | Bcl2, cyclin D1, Cyclin E2, CDK4, CDK6, c-Myc , MET, N-Myc, and SIRT1 [78–83] | Resisting cell death, Evading tumor suppressors, Enabling replicative immortality. |
| metastamiRs | |||
| miR-200 Family | Breast cancer [91, 92] | Zeb1, Zeb2 [93], and Sec23a [95] | Activating invasion metastasis |
| miR-9 | Breast [96] and colon [98] cancer | E-cadherin [96], LIFR, Cyclin D1, and Ets1 [97, 98] | Activating invasion metastasis |
| miR-31 | Breast cancer [99] | ITGA5, RDX, RhoA and WAVE3 [100, 101] | Activating invasion metastasis |
| miR-10b | Breast [102, 103] and esophageal cancer [104] | HOXD10 [102] | Activating invasion metastasis |
| miR-181a | Breast cancer [108] | Bim [108], ATM [109] | Activating invasion metastasis |
oncomiRs
miR-17-92 cluster
The best characterized oncomiR is the polycistronic miR-17-92 (also known as oncomiR-1), whose expression from chromosome 13 is driven by c-Myc and results in the production of six mature miRNAs, namely miR-17-5p, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92a-1 [34]. Additionally, two miR-17-92 paralogs have been identified: (i) the miR-106a-363 cluster, which is located on the X chromosome and houses miR-106a, miR-18b, miR-20b, miR-19b-2, miR-92a-2, and miR-363, and (i) the miR106b-25 cluster, which is located on chromosome 7 and houses miR-106b, miR-93, and miR-25 [35]. Consistent with its designation as an oncomiR, the location of the miR-17-92 cluster on chromosome 13 is found within a genomic region that is frequently amplified in a variety of human tumors, including diffuse B-cell, follicular, and Burkit’s lymphomas, and lung carcinomas [36]. Initial studies demonstrated that overexpression of mir-17-92 in an Eμ-Myc transgenic mouse model of Burkit’s lymphoma was sufficient to accelerate the tumorigenic process by suppressing apoptosis [34]. Indeed, the anti-apoptotic effects of this oncomiR cluster in haematopoietic malignancies can be ascribed to its targeting of the Bim and PTEN tumor suppressors [37–39]. With respect to solid tumors, the miR-17-92 cluster has been shown to induce proliferation, as well as augment angiogenesis through its ability to target p21 [40, 41], thrombospondin-1 (TSP1), and connective tissue growth factor (CTGF; [42]). Along these lines, the miR-106b-25 cluster was observed to drive mammary tumorigenesis and epithelial-mesenchymal transition (EMT) programs by targeting the inhibitory Smad, Smad7, thereby enhancing oncogenic TGF-β signalling [43]. Although the exact physiological function and degree of functional redundancy possessed by individual miRNAs of the mir-17-92 cluster remains to be fully elucidated, recent evidence indicates that these miRNAs do in fact exhibit a combination of unique and overlapping functions that coalesce in regulating embryogenesis and tissue development, as well as in driving tumorigenesis [39]. Future studies clearly need to expand our understanding of which mRNAs are targeted by the miR-17-92 cluster and its paralogs, thereby providing potential inroads for the development of novel therapeutics capable of alleviating the oncogenic activities of these potent oncomiRs.
miR-21
Although miR-21 was originally identified as an oncomiR in glioblastomas [44], subsequent large-scale miRNA expression profiling analyses of 540 tumors spanning 6 distinct types of cancer (e.g., lung, breast, stomach, prostate, colon, and pancreas) demonstrated that miR-21 was upregulated in all tumor cohorts [45]. More recently, upregulated miR-21 expression has also been detected in lymphoid malignancies, including chronic lymphocytic leukemia, diffuse large B cell lymphoma, and acute myeloid leukemia (see [46]). Mechanistically, hyperactivation of the Ras/MAP kinase and NF-κB signaling systems in carcinomas elicits transcription of pri-miR-21 [47], whose processing to maturity is dramatically enhanced by the TGF-β signaling system [20]. Additionally, robust miR-21 expression has been observed to target a variety of essential tumor suppressors, including PTEN, p63, PDCD4, and RECK, which serve in promoting the proliferation, survival, and metastasis of carcinomas, as well as in driving their acquisition of chemoresistant phenotypes [46, 47]. Clinically, elevated expression of miR-21 is a negative predictor for disease-free survival in patients with cancers of the breast or lung [48, 49]. Collectively, these findings illustrate the profound ability of a single miRNA to target multiple oncogenic signaling nodes, resulting in global dysregulation of gene expression networks in carcinoma cells.
miR-155
miR-155 was first identified as an oncomiR in B cell lymphomas and chronic lymphocytic leukemias where it functions to accelerate Myc-mediated lymphomagenesis [50, 51]. More recently, miR-155 also appears to be prominently upregulated in solid tumors, including those of the breast, colon, lung, pancreas, and thyroid (see [52]). miR-155 transcription is induced by TGF-β, interferon-γ (IFNγ), and interleukin-6 (IL-6) and influences numerous cancer cell signaling pathways to promote tumorigenesis. For instance, by targeting the Rho GTPase, RhoA, miR-155 drives the dissolution of tight junctions that occur during EMT programs and breast cancer invasion stimulated by TGF-β [53]. Likewise, miR-155 also targets and downregulates the expression of the tumor suppressor SOCS1 (suppressor of cytokine signaling 1), leading to constitutive activation of the proto-oncogene STAT3 and its ability to induce inflammatory and proliferative phenotypes in mammary tumors [54]. Finally, miR-155 negates apoptotic signals and enhances cancer cell survival by repressing the expression of FOXO3a, thereby suppressing the expression of the pro-apoptotic proteins PUMA, Bim, FasL, and TRAIL [55].
Tumor suppressive miRNAs
miR-15a and miR-16-1 cluster
The miR-15a and miR-16-1 cluster of miRNAs were initially identified as the most frequently downregulated collection of miRNAs in chronic lymphocytic leukemias [56], an event that also occurs in a subset of multiple myelomas, mantle cell lymphomas, and prostate cancers [57]. Indeed, engineering human MEG-01 chronic myelogenous leukemia cells to overexpress miR-15a~16-1 dramatically suppressed their tumor forming ability when xenografted into nude mice [58]. Interestingly, genome-wide transcriptome profiling analyses indicate the miR-15a~16-1 cluster directly or indirectly regulates as much as 14% of the human genome, particularly for mRNAs housing AU-rich elements (AREs) [58]. These analyses also identified a signature of 60 genes whose expression is downregulated by the miR-15a~16-1 cluster, which preferentially targets genes operant in activating cell cycle progression and survival pathways [58]. Along these lines, intratumoral delivery of miR-15a and miR-16-1 to prostate tumor xenografts induced their regression via apoptosis programs that commenced following miRNA-directed downregulation of Bcl-2, cyclin D1, and WNT3A [59]. Collectively, these findings highlight the potent tumor suppressing activities mediated by restoration of the miR-15a~16-1 cluster.
Let-7 family
The let-7 family of miRNAs was originally discovered as negative regulators of cell cycle progression and inducers of terminal differentiation in C. elegans, and is comprised of 11 homologous miRNAs, including let-7a-1, let-7a-2, let-7a-3, let-7b, let-7c, let-7d, let-7e, let-7f-1, let-7f-2, let-7g, and let-7i [60]. Initial evidence linking the expression of let-7 family members to the process of tumor suppression was obtained in studies of lung carcinomas, which were determined to house significantly reduced expression of let-7 as compared to adjacent normal tumor tissues, an event correlated with diminished overall survival of lung cancer patients [61]. Subsequent studies have clearly established reduced let-7 expression to coincide with the development of cancers of the colon, breast, and ovary [62]. Indeed, overexpression of let-7 miRNAs in breast cancer tumor-initiating cells (TICs) dramatically reduced their ability to proliferate and form mammospheres, as well as enhanced their differentiation status. More importantly, these same let-7 manipulations significantly inhibited the growth and metastasis of breast cancer TICs in mice, doing so via let-7-mediated targeting of H-Ras, which negatively impacted TIC self-renewal, and HMGA2, which positively impacted TIC differentiation status [63]. Likewise, restoring let-7 expression in lung carcinoma cells reinstated their sensitivity to radiotherapy in vitro, while similarly enhanced or reduced expression of let-7 in C. elegans elicited sensitivity or resistance, respectively to radiation-induced cell death [64]. Functionally, oncogenic activation of c-Myc represses let-7 expression [65], while the converse scenario involving the elevated expression and activity of let-7 in human malignancies elicits downregulation of Ras family members [66], HMGA2 [67], and c-Myc [65], thereby suppressing the development, progression, and therapeutic resistance of human tumors.
miR-29 family
The miR-29 family contains three members, miR-29a, miR-29b, and miR-29c, all of which are transcriptionally induced by p53 [68] and repressed by c-Myc, NF-κB, and TGF-β signaling (see [69]). Functionally, miR-29 serves to reduce cell proliferation by targeting CDK6 [70, 71], to induce cell senescence or differentiation by targeting Ppmid [68] or osteonectin [72], respectively, and to stimulate cell apoptosis by targeting Mcl1 and Bcl2 [73, 74]. Additionally, miR-29 family members have also been observed to target the methyltransferase, DNMT3, leading to hypomethylated promoter regions of a variety of tumor suppressor genes in cancer of the lung, skin, and myeloid compartment [75]. Finally, expression levels of the miR-29 family are inversely correlated with the EMT status of carcinomas, presumably due to the ability of this miRNA family to target extracellular matrix (ECM) proteins operant in driving carcinoma cell migration and metastasis [76]. Thus, in addition to its role in countering malignant transformation, future studies also need to elucidate the extent to which expression of the miR-29 family also functions to suppress the metastatic progression of late-stage carcinomas.
miR-34 family
miR-34a and miR-34b/c represent the most highly upregulated miRNAs induced by the tumor suppressor, p53 [77, 78]. Once expressed, these miRNAs function to suppress the growth and metastasis of tumors by promoting apoptosis, cell cycle arrest, and senescence, doing so by targeting and downregulating the expression of oncogenic effectors, including Bcl2, cyclins D1 and E2, CDKs 4 and 6, c-Myc, MET, N-Myc, and SIRT1 [78–83]. Additionally, miR-34 expression is inactivated through epigenetic methylation of its promoter, a reaction that dominates over its transactivation by p53 and occurs in a variety of human malignancies, including those arising in the skin (63%), bladder (33%), lung (29%), breast (25%), kidney (21%), pancreas (16%), and colon (13%) [84]. Likewise, loss of miR-34 expression in prostate cancers has been linked to their acquisition of chemoresistant phenotypes [80]. Collectively, these findings suggest that developing and implementing novel measures capable of re-expressing miR-34 in human carcinomas may provide a unique approach to alleviate metastatic progression and disease recurrence.
MetastamiRs
The acquisition of metastatic phenotypes by developing carcinomas represents the greatest barrier to effective treatment and long-term survival of cancer patients, of which ~90% succumb to the presence of incurable primary and recurrent metastases [85, 86]. The intractability of metastases likely reflects the complex cascade and series of events necessary for carcinoma cells to disseminate and thrive beyond the confines of the primary tumor. Indeed, navigating the metastatic cascade requires carcinoma cells to (i) invade into and migrate through the supporting tumor stroma; (ii) intravasate into and traverse throughout the lymph and circulatory systems; and (iii) extravasate out of the circulation and ultimately infiltrate and colonize the secondary organ site [85–87]. Although a thorough understanding of the cellular and molecular mechanisms responsible for metastasis remain to be elucidated, recent findings have nevertheless identified a subclass of miRNAs whose expression is highly associated with the acquisition of metastatic phenotypes. Indeed, the importance miRNAs to drive metastasis is highlighted by the fact that diminished Dicer function elicited by miR-103/107 targeting was found to significantly enhance the metastatic activity of mammary tumors [88]. In the succeeding sections, we will discuss a variety of metastasis-related miRNAs, which are referred to as “metastamiRs” and endowed with either metastasis promoting or suppressing activities [89, 90].
miR-200 family
The miR-200 family is comprised of 5 related miRNAs, namely miR-200a, miR-200b, miR-200c, miR-141, and miR-429. Functionally, this family serves to maintain epithelial cell gene expression profiles, morphologies, and characteristics, thereby suppressing the acquisition of EMT and metastatic phenotypes [91, 92]. Mechanistically, miR-200 family members promote E-cadherin expression by repressing that of the master EMT transcription factors, Zeb1 and Zeb2, which function in preventing the production of E-cadherin transcripts [93]. Accordingly, enforced expression of miR-200 is sufficient to alleviate the ability of lung cancers to undergo EMT programs and, consequently, to engage in invasion and metastatic behaviors in mice [94]. Interestingly, recent studies have associated metastasis-promoting activities with the expression of miR-200 family members in the late-stages of metastatic colonization and outgrowth, doing so by downregulating the expression of Sec23a and preventing its secretion of the metastasis suppressing proteins, IGFBP4 (insulin-like growth factor-binding protein 4) and TINAGL1 (Axl receptor tyrosine kinase, tubulointerstitial nephritis antigen-like 1) [95]. Collectively, these findings indicate that the ability of miR-200 family members to either suppress (e.g., early-stage disease) or promote (e.g., late-stage disease) metastasis transpires in a context-dependent manner that may reflect alterations in the genetic landscape of developing carcinomas as they progress from early-stages to late-stages of the disease.
miR-9
In stark contrast to members of the miR-200 family and their indirect coupling to E-cadherin expression via Zeb1/2, the expression of miR-9 serves to promote invasive and metastatic phenotypes by directly downregulating the levels of E-cadherin transcripts in mammary carcinomas [96]. In addition to its ability to suppress the expression of E-cadherin, miR-9 also promotes carcinoma invasion and metastasis by targeting the leukemia inhibitory factor receptor (LIFR), leading to the inactivation of prometastatic signals mediated by the Hippo-YAP pathway [97]. Interestingly and reminiscent of the context-specific activities of miR-200 family members, Zheng et al. [98] recently observed the expression of miR-9 to be dramatically downregulated in gastric cancers, an event associated with their acquisition of proliferative, invasive, and metastatic phenotypes. Importantly, the tumorigenicity of gastric cancers was significantly suppressed by restoring miR-9 expression in miR-9-deficient gastric cancers, thereby reducing the expression of the cyclin D1 and Ets1 oncogenes [98]. Thus, future studies need to determine the contexts and genetic backgrounds that underlie the gain or loss of miR-9 expression in human malignancies, as well as how these events contribute to the growth and metastatic progression of the neoplastic lesions.
miR-31
The expression of miR-31 is inversely correlated with the metastatic capacity of more than 15 breast cancer cell lines [99], leading to its designation as a metastasis-suppressing miRNA. Accordingly, re-expression of miR-31 in human breast cancers that lack expression of this miRNA significantly impaired their metastatic dissemination in mice [99], doing so by inhibiting breast cancer migration and invasion, and by sensitizing these same cells to anoikis-mediated apoptosis [99]. Mechanistically, miR-31 suppresses metastasis by targeting the expression of α5 integrin (ITGA5), radixin (RDX), RhoA, and WAVE3 (WAS protein family member 3) [100, 101]. Clinically, abnormally low miR-31 expression levels in primary breast tumors is associated with increased metastasis and disease relapse [99, 101], indicating that miR-31 does indeed act as a potent inhibitor of the metastatic cascade.
miR-10b
The finding that miR-10b is differentially expressed and significantly higher in metastatic breast cancer cells as compared to their nonmetastatic counterparts suggest that this miRNA promotes the acquisition of metastatic phenotypes in developing carcinomas [102, 103]. Indeed, tumor specimens obtained from breast cancer patients demonstrated that elevated miR-10b expression was prevalent in patients harboring metastatic disease [102]. Moreover, abnormally elevated expression of miR-10b correlates with the appearance of high-grade malignancies in the liver, pancreas, and brain [104]. Consistent with its designation as a “metastamiR”, overexpression of miR-10b preferentially drives the metastasis of breast and esophageal cancers in mice without effecting the behaviors of the corresponding primary tumors [102, 105]. Mechanistically, miR-10b expression is upregulated by the EMT-promoting transcription factor, Twist, which results in miR-10b-mediated suppression of HOXD10 expression and, consequently, in the upregulated expression of the cell motility and ECM remodeling genes RhoC, uPAR (urokinase-type plasminogen activator receptor), α3 integrin, and MT1-MMP (membrane-type 1 MMP) [102]. Finally, systemic delivery of miR-10b antagomiRs potently inhibited the metastasis of breast cancers in mice, an event that occurred with little-to-no measurable toxicity [106], suggesting that inactivation of miR-10b expression and activity holds potential to prevent carcinoma metastasis.
miR-181a
miR-181a has been shown to function as either a tumor suppressor or a tumor promoter in a context-dependent manner (see [107]). For instance, we recently uncovered a novel role for miR-181a in promoting breast cancer metastasis and showed that the expression of this metastamiR correlates with the metastatic potential of breast cancers, particularly those classified as triple-negative (i.e., lack ER-α and progesterone receptor expression, and fail to amplify HER2) [108]. Functionally, upregulated miR-181a expression was observed to inhibit anoikis by targeting the pro-apoptotic protein, Bim, and to enhance EMT programs stimulated by TGF-β [108]. Likewise, miR-181a expression has been linked to the generation and expansion of cancer stem cells through its ability to target ATM [109]. Importantly, targeted inactivation of miR-181a reduced the metastatic outgrowth of breast cancer cells in mice [108], a finding consistent with the demonstration that high miR-181a levels correlated with a dramatic decline in the overall survival of breast cancer patients [108]. Along these lines, upregulated miR-181a expression in epithelial ovarian cancers enhanced TGF-β signaling and disease progression by targeting the inhibitory Smad, Smad7, leading to the acquisition EMT, motile, and chemoresistant phenotypes in caners of the ovary [110]. In stark contrast, loss of miR-181a expression suggestive of a tumor suppressive function has been detected in a variety of human cancer cell lines, including those derived from the lung and brain [107]. Although the mechanisms responsible for eliciting discrepant miR-181a expression profiles remains to be fully elucidated, recent findings implicate expression of p53 as a molecular determinant of miR-181a expression [107]. As such, future studies need to better understand the relationship between miR-181a expression and that of classical tumor suppressors and promoters before undertaking the therapeutic targeting of miR-181a to alleviate carcinoma metastasis.
Therapeutic strategies for targeting miRNAs
The small size, extreme stability, and potent biological activities exhibited by miRNA oligonucleotides suggests that measures capable of targeting and/or delivering miRNAs to developing carcinomas holds great promise to alleviate disease progression and improve overall survival rates of cancer patients. Accordingly, the pharmacologic delivery of miRNA oligonucleotides or viral-based miRNA expression constructs to tumors seeks limit their growth and dissemination via 3 general strategies: (i) inactivate the oncogenic activities of oncomiRs; (ii) reinstate the expression of tumor suppressive miRNAs; and (iii) administer chemotherapeutic agents capable of regulating miRNA transcription or processing.
Anti-oncomiR strategies
Antisense oligonucleotides
Antisense oligonucleotides inhibit target miRNAs by annealing to complementary sequences within mature miRNAs, thereby inducing their degradation or blocking their function. The inherent instability of single-stranded oligonucleotides has been circumvented by the incorporation of chemical modifications designed to increase the stability, binding affinity, and specificity of newly synthesized single-stranded oligonucleotides [111]. For instance, adding 2′-O-methyl modifications increases the stability of nucleotides [112], while adding sulphur atoms to form phosphorothiote bonds further increases nucleotide stability, but often at the expense decreasing binding affinity [113]. More recently, 2′-O-methyoxyethyl (2′-MOE) and 2′-Fluoro (2′-F) modifications have been shown to provide superior binding affinity as compared to 2′-O-methyl modifications [114, 115]. Thus, selective modification of both the 2′ sugar position and phosphodiester bonds is necessary to obtain optimal nucleotide stability and affinity. Indeed, single-stranded phosphorothioate-linked RNA analogues that contain 2′-O-methyl-modified cholesterol-conjugated nucleotides have been shown to be effective in specifically targeting miR-122 in the liver for 23 days following a single intravenous injection of the ‘miR-122 antagomiR’ [113].
Locked nucleic acid (LNA) constructs
Locked nucleic acid (LNA) constructs represent a novel class of nucleic acid analogs characterized by the presence of a “locked” ribose ring generated by the formation of a methylene bridge between the 2′-O atom and the 4′-C atom [116]. Interestingly, antisense compounds that house a “locked” configuration display several desirable features as compared to traditional “antagomiR” chemistry, including (i) increased binding affinity against complementary single stranded RNA molecules, (ii) enhanced discrimination against base mismatches, and (iii) improved efficiency of target miRNA knockdown and inactivation [116]. More recently, LNA chemistry served as the foundation during the synthesis of the anti-miR-122 agent developed by Santaris Pharma (SPC3649), which is highly effective in downregulating miR-122 levels in the liver in non-human primates (African Green Monkeys; [117]), and in treating chronic HCV infection in chimpanzees [118]. In both scenarios, administration of SPC3649 was demonstrated to possess low toxicity profiles, indicating that LNA molecules are well-tolerated in vivo and, consequently, may represent ideal agents designed to alleviate the tumor promoting activities of oncomiRs in cancer patients.
miRNA sponges and masks
miRNA sponges serve as competitive inhibitors by binding complementary miRNAs, thereby ‘soaking up’ available oncomiR reservoirs. Typically, miRNA sponges are retroviral vectors engineered to house multiple tandem miRNA binding sites whose expression is driven from a strong promoter, resulting in the supraphysiological expression of desired “antagomiRs”. At present, stable expression of miRNA sponges, particularly those that house fluorescent reporter genes within the vector backbone have proven to be valuable research tools to study the impact of miRNA loss-of-function scenarios in animal models of carcinoma development and metastatic progression. Unfortunately, the necessity of miRNA sponges to be stably expressed within target cells has greatly limited their overall therapeutic value due to issues related to the delivery of miRNA sponge retroviral particles specifically and efficiently to carcinoma cells, not normal host [119].
In stark contrast to the mechanism of action of miRNA sponges, miRNA masks antagonize miRNA function by binding directly to 3'-UTR sequences in target mRNAs, thereby protecting their integrity by preventing miRNA binding reactions [120]. Experimentally, this technique provides an innovative approach to determine the extent to which a given cellular phenotype is driven by distinct miRNA:mRNA pairs. Clinically, however, this technique is also limited by drug delivery and specificity issues, as well as by the fact that targeting and/or protecting a single mRNA via miRNA masks is unlikely to outperform the delivery of miRNA sponges, which will restore and/or protect the expression of hundreds of mRNAs in carcinoma cells.
miRNA overexpression strategies
“miRNA replacement therapy” has been proposed as a novel means to restoring tumor suppressing miRNAs at supraphysiological levels with developing and progressing carcinomas, doing so by engineering and delivering double-stranded 2′O-methyl phosphorothioate-containing miRNA mimics. Indeed, the growth of lung tumors in mice was severely compromised by the delivery of miRNA mimics for let-7b and miR-34 [121], as was that of prostate tumors following the administration of miRNA mimics for miR-15a and miR-16 [59]. Alternatively, the use of adenovirus-associated vectors (AVV), which do not integrate into the genome and are eliminated with minimal toxicity [122, 123], have also been employed to deliver and express tumor suppressing miRNAs in carcinomas. Indeed, systemic administration of AAV-miR-26a viral particles to mice bearing hepatocellular carcinomas resulted in a dramatic reduction in disease progression due to decreased cell proliferation and survival [124]. However, due to the extensive alterations that transpire within the miRNA processing system in malignant cells, it may prove to be especially challenging to achieve sustained therapeutic expression levels of tumor suppressing miRNAs in carcinomas, particularly over long periods of time and treatment durations.
Small-molecule inhibitors
Numerous small molecule inhibitors have been shown to elicit global alterations in miRNA expression profiles [5]. For instance, the administration of small molecule inhibitors against specific oncogenic effectors, transcription factors, or pathways can suppress the upregulated expression of oncomiRs. Alternatively, it may be feasible to develop novel small molecules capable of altering miRNA biogenesis, thereby impacting global miRNA expression profiles. Indeed, Gumireddy et al. [125] recently screened a luciferase-miR-21 reporter gene against chemical library that contained more than 1000 compounds, resulting in the identification of 2 small molecule inhibitors against miR-21 and that prevent its binding to 3′UTR seed sequences. Expanding similar miRNA-based reporter screens to larger and more complex chemical libraries, as well as to those comprised of FDA-approved drugs (i.e., drug repurposing) holds tremendous potential in isolating a variety of novel agents capable of inhibiting miRNA function both experimentally and clinically.
miRNAs as biomarkers
miRNAs are normally expressed in a developmental- and tissue-specific manner to maintain cell and tissue homeostasis [126]. However, disease development disrupts this cellular equilibrium and elicits aberrant miRNA expression in a disease-specific manner, particularly with respect to genetically distinct subtypes of breast cancer. In fact, miRNA signatures have recently been observed to possess more predictive power as compared to their larger and more extensive mRNA signature counterparts [127]. Along these lines, miRNA profiles are more effective in identifying tumors of unknown origin than their corresponding mRNA profiles [126]. Likewise, monitoring the presence of circulating miRNAs has recently been undertaken as a means to readily distinguish cancer patients from healthy individuals, and to predict overall and relapse-free survival rates with extreme accuracy [128]. Given the remarkable stability of miRNAs in the circulation, tissues, and other biological fluids, future studies clearly need to expand our understanding and repertoire of miRNA signatures and their specificity for diagnosing human malignancies, and for monitoring carcinoma development, metastatic progression, and recurrence.
Conclusions
The ability of miRNAs to function as tumor promoters or tumor suppressors is well-established, as is their role in regulating normal tissue homeostasis and disease development in humans. Moreover, the recent success of miRNA-based therapies to treat HCV, coupled with the apparent effectiveness of miRNA-targeted oligonucleotides to alleviate tumor development in preclinical models of cancer, suggests that miRNAs represent a feasible class of targets to treat human malignancies. Likewise, pharmacological targeting of miRNAs will impact a plethora of oncogenic signaling nodes and biological processes due to the extreme range of mRNAs governed by a single miRNA, thereby offering a unique therapeutic advantage over current single gene/molecule-based approaches to cancer. However, the broad spectrum of genes targeted by miRNA-based therapeutics might also present a number of challenges, particularly the potential for off-target miRNA activities that could lead to unwanted toxicities. As such, improving miRNA target prediction algorithms and garnering a greater understanding of miRNA signaling networks will aid significantly in the development of highly specific and safe miRNA-targeted therapies. Equally challenging are overcoming the difficulties associated with the administration and effective delivery miRNA cargos to tumor tissues. Although recent chemical modifications to RNA-based structures have greatly improved their stability and lessened their toxicities, the ability to target and deliver similarly modified miRNAs to tumors may nevertheless prove to be daunting undertaking. Thus, while the packaging of miRNAs within nanoparticles and liposomes may indeed improve their ultimate delivery to developing tumor microenvironments, it is clear that future studies need to elucidate the optimal chemistry profiles necessary to maximize oligonucleotide delivery to malignant tissues and cells, thereby alleviating the development, metastasis, and recurrence of human carcinomas.
Acknowledgements
We would also like to thank the members of the Schiemann laboratory for their helpful comments and suggestions. Research support was provided by the National Institutes of to W.P.S. (CA129359 and CA177069).
Abbreviations
- Ago
Argonaute
- BMP
Bone morphogenetic protein
- CDK
Cyclin-dependent protein kinase
- DNMT3
DNA (cystosine-5-)-methyltransferase 3
- HCV
Human cytomegalovirus
- EMT
Epithelial-mesenchymal transition
- HOXD10
Homeobox D10
- IFNγ
Interferon-γ
- IL-6
Interleukin-6
- ITGA3
α3 integrin
- ITGA5
α5 integrin
- HMGA2
High mobility group AT-hook 2
- LIFR
Leukemia inhibitory factor receptor
- lncRNA
Long noncoding RNA
- miRNA
MicroRNA
- MMP
Matrix metalloproteinase
- MT1-MMP
Membrane-type 1 MMP
- NF-κB
Nuclear factor-κB
- RDX
Radixin
- SBE
Smad-binding element
- TGF-β
Transforming growth factor-β
- TIC
Tumor-initiating cell
- Tsp1
Thrombospondin-1
- uPAR
Urokinase-type plasminogen activator receptor
- 3′UTR
3′-untranslated region
- WAVE3
WAS protein family member 3.
Footnotes
Competing interest
M.A.T is an employee of AstraZeneca.
Authors’ contribution
MAT and WPS organized, composed sections, and edited the final manuscript. MAT prepared the figure and table, which were edited by WPS. Both authors read and approved the final manuscript.
Contributor Information
Molly A Taylor, Email: molly.taylor@astrazeneca.com.
William P Schiemann, Email: william.schiemann@case.edu.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Li GW, Xie XS. Central dogma at the single-molecule level in living cells. Nature. 2011;475:308–315. doi: 10.1038/nature10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 4.Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov. 2013;12:847–865. doi: 10.1038/nrd4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 7.Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009;136:586–591. doi: 10.1016/j.cell.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicoloso MS, Spizzo R, Shimizu M, Rossi S, Calin GA. MicroRNAs–the micro steering wheel of tumour metastases. Nat Rev Cancer. 2009;9:293–302. doi: 10.1038/nrc2619. [DOI] [PubMed] [Google Scholar]
- 9.Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, Persson R, King BD, Kauppinen S, Levin AA, Hodges MR. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 13.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 14.Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A, Gherzi R, Rosenfeld MG. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature. 2009;459:1010–1014. doi: 10.1038/nature08025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- 17.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 18.Blahna MT, Hata A. Regulation of miRNA biogenesis as an integrated component of growth factor signaling. Curr Opin Cell Biol. 2013;25:233–240. doi: 10.1016/j.ceb.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun X, Jiao X, Pestell TG, Fan C, Qin S, Mirabelli E, Ren H, Pestell RG. Oncogene. 2013. MicroRNAs and cancer stem cells: the sword and the shield. [DOI] [PubMed] [Google Scholar]
- 20.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol Cell. 2010;39:373–384. doi: 10.1016/j.molcel.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piriyapongsa J, Jordan IK, Conley AB, Ronan T, Smalheiser NR. Transcription factor binding sites are highly enriched within microRNA precursor sequences. Biol Direct. 2011;6:61. doi: 10.1186/1745-6150-6-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Wan G, Berger FG, He X, Lu X. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol Cell. 2011;41:371–383. doi: 10.1016/j.molcel.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Briata P, Lin WJ, Giovarelli M, Pasero M, Chou CF, Trabucchi M, Rosenfeld MG, Chen CY, Gherzi R. PI3K/AKT signaling determines a dynamic switch between distinct KSRP functions favoring skeletal myogenesis. Cell Death Differ. 2012;19:478–487. doi: 10.1038/cdd.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paroo Z, Ye X, Chen S, Liu Q. Phosphorylation of the human microRNA-generating complex mediates MAPK/Erk signaling. Cell. 2009;139:112–122. doi: 10.1016/j.cell.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol. 2008;18:505–516. doi: 10.1016/j.tcb.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 28.Yamagata K, Fujiyama S, Ito S, Ueda T, Murata T, Naitou M, Takeyama K, Minami Y, O’Malley BW, Kato S. Maturation of microRNA is hormonally regulated by a nuclear receptor. Mol Cell. 2009;36:340–347. doi: 10.1016/j.molcel.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 29.Shen J, Xia W, Khotskaya YB, Huo L, Nakanishi K, Lim SO, Du Y, Wang Y, Chang WC, Chen CH, Hsu JL, Wu Y, Lam YC, James BP, Liu X, Liu CG, Patel DJ, Hung MC. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature. 2013;497:383–387. doi: 10.1038/nature12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 31.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Chou J, Shahi P, Werb Z. microRNA-mediated regulation of the tumor microenvironment. Cell Cycle. 2013;12(20):3262–3271. doi: 10.4161/cc.26087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yates LA, Norbury CJ, Gilbert RJ. The long and short of microRNA. Cell. 2013;153:516–519. doi: 10.1016/j.cell.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 34.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanzer A, Stadler PF. Molecular evolution of a microRNA cluster. J Mol Biol. 2004;339:327–335. doi: 10.1016/j.jmb.2004.03.065. [DOI] [PubMed] [Google Scholar]
- 36.Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, Yoshida Y, Seto M. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004;64:3087–3095. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- 37.Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koralov SB, Muljo SA, Galler GR, Krek A, Chakraborty T, Kanellopoulou C, Jensen K, Cobb BS, Merkenschlager M, Rajewsky N, Rajewsky K. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell. 2008;132:860–874. doi: 10.1016/j.cell.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 39.Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petrocca F, Visone R, Onelli MR, Shah MH, Nicoloso MS, de Martino I, Iliopoulos D, Pilozzi E, Liu CG, Negrini M, Cavazzini L, Volinia S, Alder H, Ruco LP, Baldassarre G, Croce CM, Vecchione A. E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell. 2008;13:272–286. doi: 10.1016/j.ccr.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 41.Ivanovska I, Ball AS, Diaz RL, Magnus JF, Kibukawa M, Schelter JM, Kobayashi SV, Lim L, Burchard J, Jackson AL, Linsley PS, Cleary MA. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol Cell Biol. 2008;28:2167–2174. doi: 10.1128/MCB.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38:1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith AL, Iwanaga R, Drasin DJ, Micalizzi DS, Vartuli RL, Tan AC, Ford HL. The miR-106b-25 cluster targets Smad7, activates TGF-beta signaling, and induces EMT and tumor initiating cell characteristics downstream of Six1 in human breast cancer. Oncogene. 2012;31:5162–5171. doi: 10.1038/onc.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 45.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jazbutyte V, Thum T. MicroRNA-21: from cancer to cardiovascular disease. Curr Drug Targets. 2010;11:926–935. doi: 10.2174/138945010791591403. [DOI] [PubMed] [Google Scholar]
- 47.Pan X, Wang ZX, Wang R. MicroRNA-21: a novel therapeutic target in human cancer. Cancer Biol Ther. 2010;10:1224–1232. doi: 10.4161/cbt.10.12.14252. [DOI] [PubMed] [Google Scholar]
- 48.Qian B, Katsaros D, Lu L, Preti M, Durando A, Arisio R, Mu L, Yu H. High miR-21 expression in breast cancer associated with poor disease-free survival in early stage disease and high TGF-b1. Breast Cancer Res Treat. 2009;117:131–140. doi: 10.1007/s10549-008-0219-7. [DOI] [PubMed] [Google Scholar]
- 49.Yang M, Shen H, Qiu C, Ni Y, Wang L, Dong W, Liao Y, Du J. High expression of miR-21 and miR-155 predicts recurrence and unfavourable survival in non-small cell lung cancer. Eur J Cancer. 2013;49:604–615. doi: 10.1016/j.ejca.2012.09.031. [DOI] [PubMed] [Google Scholar]
- 50.Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005;102:3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tam W, Hughes SH, Hayward WS, Besmer P. Avian bic, a gene isolated from a common retroviral site in avian leukosis virus-induced lymphomas that encodes a noncoding RNA, cooperates with c-myc in lymphomagenesis and erythroleukemogenesis. J Virol. 2002;76:4275–4286. doi: 10.1128/JVI.76.9.4275-4286.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tili E, Croce CM, Michaille JJ. miR-155: on the crosstalk between inflammation and cancer. Int Rev Immunol. 2009;28:264–284. doi: 10.1080/08830180903093796. [DOI] [PubMed] [Google Scholar]
- 53.Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, Cheng JQ. MicroRNA-155 is regulated by the transforming growth factor b/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol. 2008;28:6773–6784. doi: 10.1128/MCB.00941-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang S, Zhang HW, Lu MH, He XH, Li Y, Gu H, Liu MF, Wang ED. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 2010;70:3119–3127. doi: 10.1158/0008-5472.CAN-09-4250. [DOI] [PubMed] [Google Scholar]
- 55.Kong W, He L, Coppola M, Guo J, Esposito NN, Coppola D, Cheng JQ. MicroRNA-155 regulates cell survival, growth, and chemosensitivity by targeting FOXO3a in breast cancer. J Biol Chem. 2010;285:17869–17879. doi: 10.1074/jbc.M110.101055. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F, Croce CM. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dong JT, Boyd JC, Frierson HF., Jr Loss of heterozygosity at 13q14 and 13q21 in high grade, high stage prostate cancer. Prostate. 2001;49:166–171. doi: 10.1002/pros.1131. [DOI] [PubMed] [Google Scholar]
- 58.Calin GA, Cimmino A, Fabbri M, Ferracin M, Wojcik SE, Shimizu M, Taccioli C, Zanesi N, Garzon R, Aqeilan RI, Alder H, Volinia S, Rassenti L, Liu X, Liu CG, Kipps TJ, Negrini M, Croce CM. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–5171. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bonci D, Coppola V, Musumeci M, Addario A, Giuffrida R, Memeo L, D’Urso L, Pagliuca A, Biffoni M, Labbaye C, Bartucci M, Muto G, Peschle C, De Maria R. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008;14:1271–1277. doi: 10.1038/nm.1880. [DOI] [PubMed] [Google Scholar]
- 60.Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 61.Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, Mitsudomi T, Takahashi T. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- 62.Shi XB, Tepper CG, deVere White RW. Cancerous miRNAs and their regulation. Cell Cycle. 2008;7:1529–1538. doi: 10.4161/cc.7.11.5977. [DOI] [PubMed] [Google Scholar]
- 63.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, Song E. Let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 64.Weidhaas JB, Babar I, Nallur SM, Trang P, Roush S, Boehm M, Gillespie E, Slack FJ. MicroRNAs as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res. 2007;67:11111–11116. doi: 10.1158/0008-5472.CAN-07-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P, Petrelli NJ, Dunn SP, Krueger LJ. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007;67:9762–9770. doi: 10.1158/0008-5472.CAN-07-2462. [DOI] [PubMed] [Google Scholar]
- 66.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 67.Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–1030. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ugalde AP, Ramsay AJ, de la Rosa J, Varela I, Marino G, Cadinanos J, Lu J, Freije JM, Lopez-Otin C. Aging and chronic DNA damage response activate a regulatory pathway involving miR-29 and p53. EMBO J. 2011;30:2219–2232. doi: 10.1038/emboj.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y, Wu L, Wang Y, Zhang M, Li L, Zhu D, Li X, Gu H, Zhang CY, Zen K. Protective role of estrogen-induced miRNA-29 expression in carbon tetrachloride-induced mouse liver injury. J Biol Chem. 2012;287:14851–14862. doi: 10.1074/jbc.M111.314922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Garzon R, Heaphy CE, Havelange V, Fabbri M, Volinia S, Tsao T, Zanesi N, Kornblau SM, Marcucci G, Calin GA, Andreeff M, Croce CM. MicroRNA 29b functions in acute myeloid leukemia. Blood. 2009;114:5331–5341. doi: 10.1182/blood-2009-03-211938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, Schwind S, Pang J, Yu J, Muthusamy N, Havelange V, Volinia S, Blum W, Rush LJ, Perrotti D, Andreeff M, Bloomfield CD, Byrd JC, Chan K, Wu LC, Croce CM, Marcucci G. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–6418. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kapinas K, Kessler CB, Delany AM. miR-29 suppression of osteonectin in osteoblasts: regulation during differentiation and by canonical Wnt signaling. J Cell Biochem. 2009;108:216–224. doi: 10.1002/jcb.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mott JL, Kobayashi S, Bronk SF, Gores GJ. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26:6133–6140. doi: 10.1038/sj.onc.1210436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xiong Y, Fang JH, Yun JP, Yang J, Zhang Y, Jia WH, Zhuang SM. Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology. 2010;51:836–845. doi: 10.1002/hep.23380. [DOI] [PubMed] [Google Scholar]
- 75.Filkowski JN, Ilnytskyy Y, Tamminga J, Koturbash I, Golubov A, Bagnyukova T, Pogribny IP, Kovalchuk O. Hypomethylation and genome instability in the germline of exposed parents and their progeny is associated with altered miRNA expression. Carcinogenesis. 2010;31:1110–1115. doi: 10.1093/carcin/bgp300. [DOI] [PubMed] [Google Scholar]
- 76.Luna C, Li G, Qiu J, Epstein DL, Gonzalez P. Cross-talk between miR-29 and transforming growth factor-bs in trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2011;52:3567–3572. doi: 10.1167/iovs.10-6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 78.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, Chen C, Lowe SW, Cleary MA, Hannon GJ. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, MacDougald OA, Cho KR, Fearon ER. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–1307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 80.Fujita Y, Kojima K, Hamada N, Ohhashi R, Akao Y, Nozawa Y, Deguchi T, Ito M. Effects of miR-34a on cell growth and chemoresistance in prostate cancer PC3 cells. Biochem Biophys Res Commun. 2008;377:114–119. doi: 10.1016/j.bbrc.2008.09.086. [DOI] [PubMed] [Google Scholar]
- 81.Leucci E, Cocco M, Onnis A, De Falco G, van Cleef P, Bellan C, van Rijk A, Nyagol J, Byakika B, Lazzi S, Tosi P, van Krieken H, Leoncini L. MYC translocation-negative classical Burkitt lymphoma cases: an alternative pathogenetic mechanism involving miRNA deregulation. J Pathol. 2008;216:440–450. doi: 10.1002/path.2410. [DOI] [PubMed] [Google Scholar]
- 82.Wei JS, Song YK, Durinck S, Chen QR, Cheuk AT, Tsang P, Zhang Q, Thiele CJ, Slack A, Shohet J, Khan J. The MYCN oncogene is a direct target of miR-34a. Oncogene. 2008;27:5204–5213. doi: 10.1038/onc.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T, Korner H, Knyazev P, Diebold J, Hermeking H. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7:2591–2600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- 85.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 86.Vanharanta S, Massague J. Origins of metastatic traits. Cancer Cell. 2013;24:410–421. doi: 10.1016/j.ccr.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-b in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010;15:169–190. doi: 10.1007/s10911-010-9181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martello G, Rosato A, Ferrari F, Manfrin A, Cordenonsi M, Dupont S, Enzo E, Guzzardo V, Rondina M, Spruce T, Parenti AR, Daidone MG, Bicciato S, Piccolo S. A MicroRNA targeting dicer for metastasis control. Cell. 2010;141:1195–1207. doi: 10.1016/j.cell.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 89.Hurst DR, Edmonds MD, Welch DR. Metastamir: the field of metastasis-regulatory microRNA is spreading. Cancer Res. 2009;69:7495–7498. doi: 10.1158/0008-5472.CAN-09-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.White NM, Fatoohi E, Metias M, Jung K, Stephan C, Yousef GM. Metastamirs: a stepping stone towards improved cancer management. Nat Rev Clin Oncol. 2011;8:75–84. doi: 10.1038/nrclinonc.2010.173. [DOI] [PubMed] [Google Scholar]
- 91.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 94.Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, Thilaganathan N, Du L, Zhang Y, Pertsemlidis A, Kurie JM. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009;23:2140–2151. doi: 10.1101/gad.1820209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celia-Terrassa T, Mercatali L, Khan Z, Goodarzi H, Hua Y, Wei Y, Hu G, Garcia BA, Ragoussis J, Amadori D, Harris AL, Kang Y. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17:1101–1108. doi: 10.1038/nm.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, Westermann F, Speleman F, Vandesompele J, Weinberg RA. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen D, Sun Y, Wei Y, Zhang P, Rezaeian AH, Teruya-Feldstein J, Gupta S, Liang H, Lin HK, Hung MC, Ma L. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat Med. 2012;18:1511–1517. doi: 10.1038/nm.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zheng L, Qi T, Yang D, Qi M, Li D, Xiang X, Huang K, Tong Q. microRNA-9 suppresses the proliferation, invasion and metastasis of gastric cancer cells through targeting cyclin D1 and Ets1. PLoS One. 2013;8:e55719. doi: 10.1371/journal.pone.0055719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Valastyan S, Reinhardt F, Benaich N, Calogrias D, Szasz AM, Wang ZC, Brock JE, Richardson AL, Weinberg RA. A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell. 2009;137:1032–1046. doi: 10.1016/j.cell.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 100.Valastyan S, Benaich N, Chang A, Reinhardt F, Weinberg RA. Concomitant suppression of three target genes can explain the impact of a microRNA on metastasis. Genes Dev. 2009;23:2592–2597. doi: 10.1101/gad.1832709. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 101.Sossey-Alaoui K, Downs-Kelly E, Das M, Izem L, Tubbs R, Plow EF. WAVE3, an actin remodeling protein, is regulated by the metastasis suppressor microRNA, miR-31, during the invasion-metastasis cascade. Int J Cancer. 2011;129:1331–1343. doi: 10.1002/ijc.25793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 103.Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, Petrocca F, Martinvalet D, Song E, Lim B, Lieberman J. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One. 2009;4:e7181. doi: 10.1371/journal.pone.0007181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ma L. Role of miR-10b in breast cancer metastasis. Breast Cancer Res. 2010;12:210. doi: 10.1186/bcr2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tian Y, Luo A, Cai Y, Su Q, Ding F, Chen H, Liu Z. MicroRNA-10b promotes migration and invasion through KLF4 in human esophageal cancer cell lines. J Biol Chem. 2010;285:7986–7994. doi: 10.1074/jbc.M109.062877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ma L, Reinhardt F, Pan E, Soutschek J, Bhat B, Marcusson EG, Teruya-Feldstein J, Bell GW, Weinberg RA. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 2010;28:341–347. doi: 10.1038/nbt.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Seoudi AM, Lashine YA, Abdelaziz AI. MicroRNA-181a - a tale of discrepancies. Expert Rev Mol Med. 2012;14:e5. doi: 10.1017/S1462399411002122. [DOI] [PubMed] [Google Scholar]
- 108.Taylor MA, Sossey-Alaoui K, Thompson CL, Danielpour D, Schiemann WP. TGF-β upregulates miR-181a expression to promote breast cancer metastasis. J Clin Invest. 2013;123:150–163. doi: 10.1172/JCI64946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang Y, Yu Y, Tsuyada A, Ren X, Wu X, Stubblefield K, Rankin-Gee EK, Wang SE. Transforming growth factor-b regulates the sphere-initiating stem cell-like feature in breast cancer through miRNA-181 and ATM. Oncogene. 2011;30:1470–1480. doi: 10.1038/onc.2010.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Parikh A, Lee C, Peronne J, Marchini S, Baccarini A, Kolev V, Romualdi C, Fruscio R, Shah H, Wang F, Mullokandov G, Fishman D, D'Incalci M, Rahaman J, Kalir T, Redline RW, Brown BD, Narla G, Di Feo A. microRNA-181a has a critical role in ovarian cancer progression through the regulation of the epithelial-mesenchymal transition. Nat Commun. 2014;5:2977. doi: 10.1038/ncomms3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Weiler J, Hunziker J, Hall J. Anti-miRNA oligonucleotides (AMOs): ammunition to target miRNAs implicated in human disease? Gene Ther. 2006;13:496–502. doi: 10.1038/sj.gt.3302654. [DOI] [PubMed] [Google Scholar]
- 112.Cummins LL, Owens SR, Risen LM, Lesnik EA, Freier SM, McGee D, Guinosso CJ, Cook PD. Characterization of fully 2′-modified oligoribonucleotide hetero- and homoduplex hybridization and nuclease sensitivity. Nucleic Acids Res. 1995;23:2019–2024. doi: 10.1093/nar/23.11.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 114.Manoharan M. 2′-carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. Biochim Biophys Acta. 1999;1489:117–130. doi: 10.1016/s0167-4781(99)00138-4. [DOI] [PubMed] [Google Scholar]
- 115.Davis S, Lollo B, Freier S, Esau C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 2006;34:2294–2304. doi: 10.1093/nar/gkl183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vester B, Wengel J. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry. 2004;43:13233–13241. doi: 10.1021/bi0485732. [DOI] [PubMed] [Google Scholar]
- 117.Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S, Lindholm M, Hedtjarn M, Hansen HF, Berger U, Gullans S, Kearney P, Sarnow P, Straarup EM, Kauppinen S. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–899. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 118.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Orum H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Choi WY, Giraldez AJ, Schier AF. Target protectors reveal dampening and balancing of Nodal agonist and antagonist by miR-430. Science. 2007;318:271–274. doi: 10.1126/science.1147535. [DOI] [PubMed] [Google Scholar]
- 121.Trang P, Wiggins JF, Daige CL, Cho C, Omotola M, Brown D, Weidhaas JB, Bader AG, Slack FJ. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol Ther. 2011;19:1116–1122. doi: 10.1038/mt.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Aagaard L, Rossi JJ. RNAi therapeutics: principles, prospects and challenges. Adv Drug Deliv Rev. 2007;59:75–86. doi: 10.1016/j.addr.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Michelfelder S, Trepel M. Adeno-associated viral vectors and their redirection to cell-type specific receptors. Adv Genet. 2009;67:29–60. doi: 10.1016/S0065-2660(09)67002-4. [DOI] [PubMed] [Google Scholar]
- 124.Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P, Torbenson M, Clark KR, Mendell JR, Mendell JT. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gumireddy K, Young DD, Xiong X, Hogenesch JB, Huang Q, Deiters A. Small-molecule inhibitors of microrna miR-21 function. Angew Chem Int Ed Engl. 2008;47:7482–7484. doi: 10.1002/anie.200801555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 127.Liu A, Tetzlaff MT, Vanbelle P, Elder D, Feldman M, Tobias JW, Sepulveda AR, Xu X. MicroRNA expression profiling outperforms mRNA expression profiling in formalin-fixed paraffin-embedded tissues. Int J Clin Exp Pathol. 2009;2:519–527. [PMC free article] [PubMed] [Google Scholar]
- 128.Weiland M, Gao XH, Zhou L, Mi QS. Small RNAs have a large impact: circulating microRNAs as biomarkers for human diseases. RNA Biol. 2012;9:850–859. doi: 10.4161/rna.20378. [DOI] [PubMed] [Google Scholar]