

Abstract
The aim of the present study was the evaluation and automation of the radiosynthesis of [11C]harmine for clinical trials. The following parameters have been investigated: amount of base, precursor concentration, solvent, reaction temperature and time. The optimum reaction conditions were determined to be 2–3 mg/mL precursor activated with 1 eq. 5 M NaOH in DMSO, 80 °C reaction temperature and 2 min reaction time. Under these conditions 6.1±1 GBq (51.0±11% based on [11C]CH3I, corrected for decay) of [11C]harmine (n=72) were obtained. The specific activity was 101.32±28.2 GBq/µmol (at EOS). All quality control parameters were in accordance with the standards for parenteral human application. Due to its reliability and high yields, this fully-automated synthesis method can be used as routine set-up.
Keywords: [11C]harmine, PET, MAO-A, Carbon-11, Monoamine oxidase, Radiosynthesis
Highlights
-
•
Preparation of [11C]harmine on a commercially available synthesizer for the routine application.
-
•
High reliability: only 4 out of 72 failed syntheses; 5% due to technical problems.
-
•
High yields: 6.1±1 GBq overall yield (EOS).
-
•
High specific activities: 101.32±28.2 GBq/µmol.
1. Introduction
Monoamine oxidases (MAO) are primarily located in the outer mitochondrial membrane of catecholaminergic neurons where they are responsible for the degradation of monoamine neurotransmitters (Weyler et al., 1990). Two subtypes of the enzyme, MAO-A and MAO-B, have been identified on the basis of their substrate selectivity, biochemical properties and gene products (Johnston, 1968; Shih et al., 1999). MAO-A preferentially oxidizes serotonin, norepinephrine and epinephrine and is therefore an attractive target in the study of psychiatric and neurological diseases. Highly elevated levels of MAO-A in the human brain are associated with major depression disorder (Meyer et al., 2006), whereas inhibition of MAO-A is useful in the treatment of depression and anxiety disorders (Livingston and Livingston, 1996). For the in vivo measurement of the MAO-A density the positron emission tomography (PET) radioligands [11C]clorgyline, [11C]clorgyline-D2 (Fowler et al., 2001), [11C]befloxatone (Dollé et al., 2003) and [11C]harmine (Bacher et al., 2011; Bergström et al., 1997a, 1997b; Ginovart et al., 2006; Meyer et al., 2009; Sacher et al., 2010) have been used since several years. The β-carboline derivative [11C]harmine is a selective and reversible inhibitor of MAO-A with high brain uptake and high affinity (Ki=5 nM) (Kim et al., 1997).
The aim of the present study was the implementation and automation of the radiosynthesis of [11C]harmine into our clinical setup in order to investigate seasonal depression in a clinical trial. We slightly modified the synthesis described by Bergström et al. (1997a) and conducted additional evaluation steps.
2. Experimental
2.1. Materials
All chemicals and solvents were obtained from commercial sources with analytical grade and used without further purification. Iodine (sublimated grade for analysis; ACS, Pharm. Eur.) was purchased from Merck (Darmstadt, Germany; product number: 1.04761.0100). The Ni catalyst (Shimalilte Ni reduced, 80/100 mesh) was purchased from Shimadzu (Kyoto, Japan). The precursor harmol (7-hydroxy-1-methyl-9H-pyrido[3,4-b]indole; GMP grade) and the reference compound harmine (7-methoxy-1-methyl-9H-[3,4-b]indole) were obtained from ABX (ABX-Advanced Biochemical Compounds, Radeberg, Germany). Harmaline (4,9-dihydro-7-methoxy-1-methyl-3H-pyrido[3,4-b]indole), the reference compound for a potential radioactive by-product, dimethyl sulfoxide (DMSO) (anhydrous, ≥99.9%), N,N-dimethylformamide (DMF) (puriss., ≥99.8%) and acetonitrile were obtained from Sigma Aldrich (Vienna, Austria). Ammonium formate, ammonium acetate, acetic acid, sodium hydroxide (NaOH) and ethanol (absolute) were purchased from Merck (Darmstadt, Germany). 0.9% Saline solution was purchased from B. Braun (Melsungen, Germany). 3% Saline solution was obtained from a local pharmacy (Landesapotheke Salzburg, Austria). 125 mM phosphate buffer was prepared by dissolving 0.224 g sodium dihydrogenphosphate-monohydrate and 1.935 g disodiumhydrogenphosphate-dihydrate (both from Merck, Darmstadt, Germany) in 100 mL sterile water. Sterile water was purchased from B. Braun (Melsungen, Germany). C18plus SepPak® cartridges for solid phase extraction (SPE) were purchased from Waters (Waters® Associates Milford, MA, USA). Low-protein binding Millex GS® 0.22 µm sterile filters were purchased from Millipore® (Bedford, MA, USA). Semipreparative high performance liquid chromatography (HPLC) column (Supelcosil™ LC-ABZ+; 5 µm, 250 mm×10 mm; Nr. 59179) was purchased from Supelco (Bellfonte, PA, USA). Analytical HPLC-column (Prodigy 10 µm ODS (3); 250 mm×4.6 mm; Nr. 00G-4244-E0) was obtained from Phenomenex (Aschaffenburg, Germany).
2.2. Instrumentation
[11C]CO2 was produced at a GE PET trace cyclotron (General Electric Medical System, Uppsala, Sweden) via the 14N(p,α)11C nuclear reaction by irradiation of a gas target (Aluminium) filled with N2 (+1% O2) (Air Liquide, Vienna, Austria). Typical beam currents were 50–56 µA and the irradiation was stopped as soon as the desired activity level was reached (approx. 50–65 GBq [11C]CO2, calculated by cyclotron operating software; corresponding to 20–30 min irradiation time). The production of [11C]CH4, [11C]CH3I and [11C]harmine including semi-preparative HPLC were performed on a TRACERlab™ FX C Pro synthesis module (GE Healthcare, Uppsala, Sweden). Analytical HPLC was performed using a Merck-Hitachi LaChrom system consisting of a L-7100 pump, a Merck-Hitachi LaChrom L7400 UV-detector (operated at 254 nm) and a lead shielded NaI-radiodetector (Raytest Isotopenmessgeräte GmbH, Straubenhardt, Germany). The osmolality was measured using a Wescor osmometer Vapro® 5600 (Sanova Medical Systems, Vienna, Austria) and pH was measured using a WTW inoLab 740 pH metre (WTW, Weilheim, Germany).
2.3. Radiosynthesis of [11C]harmine
2.3.1. Manually operated small scale syntheses
[11C]CH3I was produced in the TRACERlab™ FX C Pro synthesis module using the gas phase conversion described by Larsen et al. (1997) and trapped in 1 mL DMSO and DMF, respectively. The reaction of [11C]harmine was started by adding a portion of the trapped [11C]CH3I to the precursor solution (final precursor concentration: 0.5–3 mg/mL), dissolved in DMSO/DMF and activated with 0–6 equivalent (eq.) of 5 M NaOH. The reaction was conducted in a 0.5 mL Wheaton-Vial. A reaction scheme is shown in Fig. 1. Different reaction temperatures (25–100 °C) were evaluated. After 0.5–2 min reaction time, the reaction mixture was quenched with water and the radiochemical incorporation yield (RCIY) was determined using analytical radio-HPLC (mobile phase: 0.1 M ammonium formate (pH 4)/acetonitrile 75/25 v/v; flow: 1.5 mL/min). Chromatograms were registered using an UV-detector (254 nm) and a NaI radioactivity detector in series.
Fig. 1.
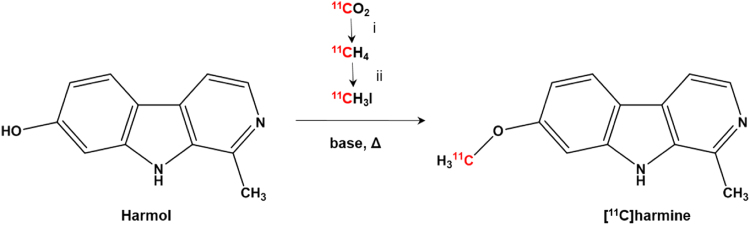
Radiosynthesis of [11C]harmine (i: H2/Ni, 400 °C; ii: I2, 734 °C).
2.3.2. Fully-automated large scale syntheses
2.3.2.1. Preparation of the synthesis module
A scheme of the synthesis module is presented in Fig. 2. All parts prior to HPLC-purification (reactor, HPLC injector, tubing) were rinsed with water and acetone and then dried with a stream of helium. Parts after preparative HPLC (i.e. SPE tubing, dilution flask, product collection vial, product outlet tubing) were cleaned using ethanol and physiological saline (0.9%).
Fig. 2.

Scheme of the commercial 11C-synthesizer used for the radiosynthesis and purification of [11C]harmine (SPE: solid phase extraction; PCV: product collection vial). For details in set-up and processing refer to Section 2.3.2.
Vessels were filled as followed:
Vial 2: 0.5 mL of HPLC eluent (0.1 M ammonium acetate and acetonitrile (60/40; v/v)) for quenching of the reaction and dilution prior to HPLC injection;
Vial 4: 5 mL of physiological saline (0.9%) for final rinsing of the SPE cartridge and dilution of the eluate;
Vial 5: 1.5 mL of ethanol for elution of the SPE cartridge;
Vial 6: 10 mL of sterile water for washing the SPE cartridge;
Vials 1 & 3 remained empty and were not used for the synthesis;
Bulb: 60 mL of sterile water for dilution of the lipophilic HPLC fraction after peak cutting.
A C18 SPE cartridge was preconditioned using 5 mL of ethanol and 10 mL of sterile water, dried and connected to its designated position. The product collection vial (PCV) was filled with 4 mL of physiological saline (0.9%), 1 mL of 3% saline and 1 mL of phosphate buffer (125 mM). The reactor was filled with 1–1.5 mg harmol and 1 eq. of 5 M NaOH dissolved in 0.5 mL of DMSO. Subsequently the reactor was placed into the heating block.
2.3.2.2. Synthesis of [11C]harmine
The fully automated preparation of [11C]harmine was performed on the prepared radiosynthesizer. [11C]CH3I was bubbled through the precursor solution at room temperature. The reactor was sealed and the reaction mixture heated to 80 °C and kept at this temperature for 2 min. After cooling down to room temperature (<1 min), the reaction mixture was quenched by addition of 0.5 mL of HPLC-eluent (through V2) and subsequently transferred to the 5 mL injection loop of the HPLC-system by passing the fluid detector.
2.3.2.3. Purification and formulation of [11C]harmine
Injection of the crude reaction mixture to the semi-preparative RP-HPLC column was controlled by an automated fluid detector. Semi-preparative HPLC was performed with 0.1 M ammonium acetate and acetonitrile (60/40; v/v) as a mobile phase with an isocratic flow of 8 mL/min. Chromatograms (Fig. 3) were registered using an UV-detector (254 nm) and a NaI radioactivity detector in series. The [11C]harmine fraction was cut into a bulb (containing 60 mL of sterile water) through V14. The resulting solution was then pushed through a C18 SPE cartridge. After washing with 10 mL of water (Vial 6) the purified product was completely eluted with 1.5 mL of ethanol (Vial 5) and 5 mL of 0.9% saline (Vial 4). Formulation was done with a further 4 mL of physiological saline (0.9%), 1 mL of saline solution (3%) and 1 mL of phosphate buffer (125 mM) in the product collection vial. This final solution was transferred to a shielded laminar air-flow hot cell and, there, sterile-filtered (0.22 µm) into a sterile 25 mL vial containing another 5 mL of saline solution 0.9%. Hence, the final total volume was 17.5 mL (containing 8.5% ethanol).
Fig. 3.

Semi-preparative chromatogram of the reaction solution of [11C]harmine (top: UV cannel; bottom: radioactivity channel).
2.3.2.4. Quality control of [11C]harmine
Chemical and radiochemical impurities were detected and quantified using analytical radio-HPLC (for conditions see Section 2.3.1), according to the monograph in the European Pharmacopoeia (2008). The whole HPLC analysis was completed within 7 min, the retention time of the precursor (harmol) was 3.1–3.3 min (kʹ=0.4–0.5), harmaline was eluted with a retention time of 5.2–5.5 min (kʹ=1.3–1.5) and the product [11C]harmine was eluted at 5.5–6.4 min (kʹ=1.5–1.9). The chemical identity of [11C]harmine was determined by co-injection of the unlabelled reference compound harmine. For calculation of radiochemical purity, the portion of [11C]harmine in relation to total radioactivity was determined (threshold ≥95%). Sample chromatograms are given in Fig. 4.
Fig. 4.

Typical chromatogram of the purified and formulated [11C]harmine using analytical HPLC (top: UV cannel; bottom: radioactivity channel).
Osmolality and pH were determined to assure safe administration using standard methods. Radionuclidic purity was assessed by recording of the corresponding gamma spectrum and additional measurement of the physical half-life.
Testing of sterility and concentration of bacterial endotoxins was performed using standard protocols at the Department of Infection Diseases and Tropical Medicine (Medical University of Vienna, Austria).
2.4. Statistical analysis
All values are given by arithmetic means±standard deviation.
To determine significant differences a two-tailed t-test with α=0.95 was performed using the statistics add-on in Microsoft® Excel. A value of P<0.05 was considered as significant.
3. Results and discussion
3.1. Small scale syntheses
Following parameters were investigated: amount of 5 M NaOH, precursor concentration, solvent, reaction temperature and time. The sample size was ≥2.
Using 1 mg/mL precursor in DMSO and 2 min reaction time, the added amount of 5 M NaOH had a high impact on the RCIY. The highest yields could be achieved using 1 eq. of 5 M NaOH: 73.55±18.7% at 25 °C, 86.05±5.1% at 50 °C and 84.86±2.2% at 80 °C. Using 6 eq. of 5 M NaOH resulted in a significantly lower yield: 14.57±2.0% at 25 °C, 20.44±4.9% at 50 °C and 29.65±0.0% at 80 °C. Almost no conversion could be observed conducting the synthesis without 5 M NaOH (Fig. 5A).
Fig. 5.

Dependence pf the radiochemical incorporation yield (RCIY) of [11C]harmine (n≥2) on (A) amount of 5 M NaOH (@ 1 mg/mL precursor, 2 min), (B) precursor concentration (@ 80 °C, 2 min), (C) reaction temperature (@ 2 mg/mL precursor, 2 min) and (D) reaction time (@ 2 mg/mL precursor, 25 °C). If not visible, error bars are within the margin of the symbols.
A clear trend was observed between the RCIY and the precursor concentration (experiments conducted in DMSO at 80° and for 2 min). RCIY increased significantly with increasing precursor amount. The highest RCIY was obtained using 3 mg/mL precursor (84.09±2.0%); 72.37±1.2% using 2 mg/mL, 62.40±1.1% using 1 mg/mL and 56.57±0.2% using 0.5 mg/mL (Fig. 5B).
The reaction temperature had no significant impact on the RCIY but a greater variance could be observed at 25 °C (Fig. 5C; experiments conducted in DMSO with 2 mg/mL precursor and for 2 min). This temperature was favourable for the formation of the by-product [11C]harmaline reducing the radiochemical purity of the product.
Regarding the reaction time, 74.32±19.2% RCIY were already achieved after 0.5 min; 57.35±23.8 after 1 min and 68.24±22.7 after 2 min (Fig. 5D; experiments conducted in DMSO with 2 mg/mL precursor at 25 °C). There was no significant difference in the RCIYs between the reaction times.
Changing the solvent from DMSO to DMF resulted in a significantly lower RCIY. Using 1 mg/mL precursor, 80 °C reaction temperature and 2 min reaction time the RCIY was 24.63±13.7% in DMF and 56.57±0.2% in DMSO.
Hence, the optimum reaction conditions were determined and summarized in Table 1.
Table 1.
Optimum reaction conditions and outcome for large scale preparations of [11C]harmine (n=72).
| Reaction temperature (°C) | 80 |
| Reaction time (min) | 2 |
| Amount of precursor (mg/mL) | 2–3 |
| Solvent | DMSO |
| Yield (GBq)a | 6.1±1 |
| Yield (%)b | 51.0±11 |
| Specific activity (GBq/µmol)a | 101.3±28 |
| Radiochemical purity (%) | 100±0 |
At end of synthesis (EOS).
Based on [11C]CH3I, corrected for decay.
3.2. Fully-automated large scale syntheses
The fully-automated synthesis and purification of [11C]harmine was performed successfully and with high reliability within 35 min after the end of bombardment (EOB). In 72 runs 6.1±1 GBq (51.0±11% based on [11C]CH3I trapped on the [11C]CH3I-Trap, corrected for decay) of [11C]harmine were obtained under the optimum reaction conditions (Table 1) as a sterile solution ready for application. 4 Syntheses failed due to technical problems. Typical loss during sterile filtration was <10% of the product activity. The radiochemical purity was in every synthesis 100% as determined by radio-HPLC. The precursor concentration in the final product ranged from 0 to 2.66 µg/mL (0.34±1.5 µg/mL) and the specific activity was 101.32±28.2 GBq/µmol (0.75±0.3 µg/mL harmine) at the end of synthesis (EOS). Sample chromatograms are shown in Fig. 4. Osmolality was 292±11 mosmol/kg and pH was 7.5±0.1. Concentration of endotoxins was found to be below 1.0 EU/mL and all samples passed the test for sterility. Concluding, all quality control parameters were in accordance with the standards for parenteral human application. The quality control (except tests for endotoxins and sterility) was completed within 7 min.
Compared to the radiochemical yields (72.5±3.6% based on [11C]CH3I, corrected for decay) in the synthesis described by Bergström et al. (1997a), in our set-up, we obtained lower yields (51.0±11%) with the same parameters (DMSO, 1 eq. 5 M NaOH and 80 °C) but using a lower precursor concentration (2 mg/mL compared to 3.3–6.6 mg/mL) and shorter reaction time (2 min compared to 5 min). We also achieved higher specific activities (101.32±28.2 GBq/µmol compared to 18.0–87.3 GBq/µmol) which is of advantage for clinical trials.
4. Conclusion
We established a rapid, reproducible and reliable preparation and purification method on a commercially available synthesis module. The optimum reaction conditions were 2–3 mg/mL precursor activated with 1 eq. 5 M NaOH in DMSO, 80 °C reaction temperature and 2 min reaction time. Due to its reliability and high yields (6.1±1 GBq), the presented method can be used as routine set-up.
Acknowledgement
The authors thank Daniela Haeusler for her support in the radiosyntheses and Thomas Zenz and Andreas Krcal for their skilful help with the cyclotron and synthesis module. This research was partly supported by a grant of the Austrian Science Fund (FWF P24359) to Dietmar Winkler and Rupert Lanzenberger.
References
- Bacher I., Houle S., Xu X., Zawertailo L., Soliman L., Wilson A.A., Selby J., George T.P., Sacher J., Miler L., Kish S.J., Rusjan P., Meyer J.H. Monoamine oxidase a binding in the prefrontal and anterior cingulate cortices during acute withdrawal from heavy cigarette smoking. Arch. Gen. Psychiatry. 2011;68:817–826. doi: 10.1001/archgenpsychiatry.2011.82. [DOI] [PubMed] [Google Scholar]
- Bergström M., Westerberg G., Kihlberg T., Langström B. Synthesis of some 11C-labelled MAO-A inhibitors and their in vivo uptake kinetics in rhesus monkey brain. Nucl. Med. Biol. 1997;24:381–388. doi: 10.1016/s0969-8051(97)80003-0. [DOI] [PubMed] [Google Scholar]
- Bergström M., Westerberg G., Langström B. 11C-harmine as a tracer for monoamine oxidase A (MAO-A): in vitro and in vivo studies. Nucl. Med. Biol. 1997;24:287–293. doi: 10.1016/s0969-8051(97)00013-9. [DOI] [PubMed] [Google Scholar]
- Dollé F., Valette H., Bramoulle Y., Guenther I., Fuseau C., Coulon C., Lartizien C., Jegham S., Curet O., Pinquier J.L., George P., Bottlaender M. Synthesis and in vivo imaging properties of [11C]befloxatone: a novel highly potent positron emission tomography ligand for monoamine oxidase-A. Bioorg. Med. Chem. Lett. 2003;13:1771–1775. doi: 10.1016/s0960-894x(03)00215-4. [DOI] [PubMed] [Google Scholar]
- Fowler J.S., Ding Y.-S., Logan J., MacGregor R.R., Shea C., Garze V., Gimi R., Volkow N.D., Wang G.-J., Schlyer D., Ferrieri R., Gatley S.J., Alexoff D., Carter P., King P., Pappas N., Arnett C.D. Species differences in [11C]clorgyline binding in brain. Nucl. Med. Biol. 2001;28:779–785. doi: 10.1016/s0969-8051(01)00245-1. [DOI] [PubMed] [Google Scholar]
- Ginovart N., Meyer J.H., Boovariwala A., Hussey D., Rabiner E.A., Houle S., Wilson A.A. Positron emission tomography quantification of [11C]harmine binding to monoamine oxidase-A in the human brain. J. Cereb. Blood Flow Metab. 2006;26:330–344. doi: 10.1038/sj.jcbfm.9600197. [DOI] [PubMed] [Google Scholar]
- Johnston J.P. Some observations upon a new inhibitor of monoamine oxidase in brain tissue. Biochem. Pharmacol. 1968;17:1285–1297. doi: 10.1016/0006-2952(68)90066-x. [DOI] [PubMed] [Google Scholar]
- Kim H., Sablin S.O., Ramsay R.R. Inhibition of monoamine oxidase A by beta-carboline derivatives. Arch. Biochem. Biophys. 1997;337:137–142. doi: 10.1006/abbi.1996.9771. [DOI] [PubMed] [Google Scholar]
- Larsen P., Ulin J., Dahlstrom K., Jensen M. Synthesis of [C-11]iodomethane by iodination of [C-11]methane. Appl. Radiat. Isot. 1997;48:153–157. [Google Scholar]
- Livingston M.G., Livingston H.M. Monoamine oxidase inhibitors. An update on drug interactions. Drug Saf. 1996;14:219–227. doi: 10.2165/00002018-199614040-00002. [DOI] [PubMed] [Google Scholar]
- Meyer J.H., Ginovart N., Boovariwala A., Sagrati S., Hussey D., Garcia A., Young T., Praschak-Rieder N., Wilson A.A., Houle S. Elevated monoamine oxidase a levels in the brain: an explanation for the monoamine imbalance of major depression. Arch. Gen. Psychiatry. 2006;63:1209–1216. doi: 10.1001/archpsyc.63.11.1209. [DOI] [PubMed] [Google Scholar]
- Meyer J.H., Wilson A.A., Sagrati S., Miler L., Rusjan P., Bloomfield P.M., Clark M., Sacher J., Voineskos A.N., Houle S. Brain monoamine oxidase A binding in major depressive disorder. Arch. Gen. Psychiatry. 2009;66:1304–1312. doi: 10.1001/archgenpsychiatry.2009.156. [DOI] [PubMed] [Google Scholar]
- Sacher J., Wilson A.A., Houle S., Rusjan P., Hassan S., Bloomfield P.M., Stewart D.E., Meyer J.H. Elevated brain monoamine oxidase A binding in the early postpartum period. Arch. Gen. Psychiatry. 2010;67:468–474. doi: 10.1001/archgenpsychiatry.2010.32. [DOI] [PubMed] [Google Scholar]
- Shih J.C., Chen K., Ridgill M.P. Monoamine oxidase from gene to behaviour. Ann. Rev. Neurosci. 1999;22:197–217. doi: 10.1146/annurev.neuro.22.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyler W., Hsu Y.-P.P., Breakefield X.O. Biochemistry and genetics of monoamine oxidase. Pharmacol. Ther. 1990;47:391–417. doi: 10.1016/0163-7258(90)90064-9. [DOI] [PubMed] [Google Scholar]