

Abstract
Objectives
Here we aimed to clarify the role of CXCR2 in the macrophage migration inhibitory factor (MIF)-mediated effects after myocardial ischemia and reperfusion (I/R). As a pleiotropic chemokine-like cytokine, MIF has been identified to activate multiple receptors including CD74 and CXCR2. In models of myocardial infarction (MI), MIF exerts both pro-inflammatory effects and protective effects in cardiomyocytes. Likewise, CXCR2 displays opposing effects in resident versus circulating cells.
Approach and results
Using bone marrow (BM) transplantation, we generated chimeric mice with CXCR2−/− BM-derived inflammatory cells and wild-type resident cells (wt/CXCR2−/−), with CXCR2−/− cardiomyocytes and wild-type BM-derived cells (CXCR2−/−/wt) and wild-type controls reconstituted with wild-type BM (wt/wt). All groups were treated with anti-MIF or isotype control antibody before they underwent myocardial I/R. Blocking MIF increased infarction size and impaired cardiac function in wt/wt and wt/CXCR2−/− mice but ameliorated functional parameters in CXCR2−/−/wt mice, as analyzed by echocardiography and Langendorff perfusion. Neutrophil infiltration and angiogenesis were unaltered by MIF blockade or CXCR2 deficiency. Monocyte infiltration was blunted in wt/CXCR2−/− mice and reduced by MIF blockade in wt/wt and CXCR2−/−/wt mice. Moreover, MIF blockade attenuated collagen content in all groups in a CXCR2-independent manner.
Conclusions
The compartmentalized and opposing effects of MIF after myocardial I/R are largely mediated by CXCR2. Whereas MIF confers protective effects improving myocardial healing and function through CXCR2 in resident cells, complementing paracrine effects through CD74/AMPK, it exerts detrimental effects on CXCR2-bearing inflammatory cells, increasing monocyte infiltration and impairing heart function. These dichotomous findings should be considered, when developing novel therapeutic strategies to treat MI.
Keywords: Macrophage migration inhibitory factor, CXCR2, neoangiogenesis, myocardial ischemia/reperfusion, myocardial infarction
Introduction
Macrophage migration inhibitory factor (MIF) was one of the first cytokines to be identified almost 40 years ago1. Today, MIF is known as a structurally unique, pleiotropic inflammatory cytokine with chemokine-like properties that functions as a potent mediator of several inflammatory conditions2–4. However, the role of MIF in cardiovascular diseases, especially in myocardial infarction (MI), has not yet been definitively clarified.
MIF is up-regulated in endothelial cells and smooth muscle cells of atherosclerotic arteries, where it contributes to macrophage accumulation and plaque formation5, 6. It appears that MIF acts sequentially by first triggering monocyte arrest through the CXCR axis and then promoting monocyte transmigration through the intermediate production of CCL27, 8. Being predominantly expressed in vulnerable plaques and inducing collagen-degrading matrix metalloproteinases9, 10, MIF has been implicated in plaque destabilization.
In the context of MI, the levels of MIF were found to be significantly elevated in patients with reversible myocardial ischemia11. A model of acute MI revealed that both MIF mRNA and MIF protein are constitutively expressed at low levels in myocytes of normal and sham-operated rats but rapidly up-regulated by surviving cardiomyocytes in the infarcted versus the non-infarcted regions, thereby increasing macrophage infiltration one day after acute MI12. Moreover, it seems that cardiomyocytes can secrete MIF through a protein kinase C-dependent pathway in response to reactive oxygen species and hypoxia in the myocardium13, 14. Of note, MIF released by cardiomyocytes in a model of myocardial injury induced by 15 mins of ischemia followed by reperfusion (I/R) injury exerts cardioprotective effects via the CD74/AMPK/JNK axes15, 16, and the protective effect can be enhanced by S-nitrosylation of MIF17. CD74 is the cell surface form of the major histocompatibility complex class II-associated invariant chain (Ii) and was identified to bind MIF by high-affinity interaction and to mediate MIF-induced ERK1/ERK2 phosphorylation and cell proliferation18, 19. However, some of the cell types targeted by MIF (e.g. neutrophils or the cell lines HEK293 and HeLa) do not express CD74 at the surface7, raising the need to further clarify the molecular action and functional receptors of MIF in inflammation. Despite the beneficial role of MIF in cardiomyocytes, it was indeed shown that MIF deficiency protected the heart from prolonged, severe I/R injury by suppressing inflammatory responses20. Thus, the exact role of MIF in healing after MI and the receptors involved therein are thus far from being completely understood.
Recent data have provided evidence that MIF is a non-cognate ligand of the CXC chemokine receptors CXCR2 and CXCR4, thus extending the range of binding partners for MIF7. MIF harbors ELR- and N-loop-like motives typically required for CXCR2 activation21, 22 and has chemoattractant activity towards monocytes and neutrophils through CXCR2, both crucial mechanisms in inflammatory pathologies like atherosclerosis7. Although MIF can bind to CXCR2 or CD74 individually, binding to a CXCR2/CD74 complex appears to further enhance G-protein-coupled receptor (GPCR) activation and atherogenic functions7. In addition, MIF can also bind the CXCL12 receptor CXCR4, thereby promoting T-cell and endothelial progenitor cell recruitment7, 23, enhancing angiogenesis24, 25 and inducing phosphorylation of Akt and mitogen-activated protein kinase (MAPK) p42/4426.
Notably, CXCR2 has been found to exert opposing effects on myocardial viability during I/R with the recruitment of damaging inflammatory cells prevailing over direct myocardial protection promoted in resident cells27, suggesting that CXCR2 may prominently contribute to mediating the effects of MIF in myocardial I/R injury. To shed light on the precise mechanisms enacted by MIF during the myocardial healing response after MI, we aimed to dissect and elucidate the compartment-specific role of CXCR2 in mediating the effects of endogenous MIF in a mouse model of I/R.
Materials and Methods
See Supplementary File.
Results
The effect of anti-MIF-antibody on heart parameters one week after I/R
To examine the role of the MIF-Cxcr2 pathway in healing following I/R, we obtained chimeric mice with wild-type resident cardiomyocytes and Cxcr2−/− BM-derived inflammatory cells, mice with Cxcr2−/− cardiomyocytes and wild-type inflammatory cells and wild-type controls (Table 1). Each group received either anti-MIF or isotype control antibody. The high mortality of Cxcr2−/− mice after irradiation (50%) reduced the number of mice in these groups (4 per group).
Table 1.
Treatment groups of bone marrow chimeras
| Mouse | Bone marrow | Anti-MIF antibody (100 μg/week) | Appellation | Cells resident/recruited | CXCR2 |
|---|---|---|---|---|---|
| wild-type | wild-type | − | wt/wt BM | cardiomyocytes | + |
| + | inflammatory cells | + | |||
| wild-type | CXCR2−/− | − | wt/CXCR2−/− BM | cardiomyocytes | + |
| + | inflammatory cells | − | |||
| CXCR2−/− | wild-type | − | CXCR2−/−/wt BM | cardiomyocytes | − |
| + | inflammatory cells | + |
One week after I/R, both wt/wt BM and wt/Cxcr2−/− BM mice treated with anti-MIF antibody showed a significant increase in the size of the infarcted area as compared to isotype control, whereas the Cxcr2−/−/wt BM mice displayed no effect with anti-MIF (Figure 1). In contrast to a previous report27, Cxcr2 deficiency in BM cells itself only slightly but not significantly reduced the size of the infarcted area after one week. In addition, heart function was evaluated by echocardiography before and after myocardial I/R. Before MI, no significant differences were observed among the groups. One week after I/R, anti-MIF antibody treatment significantly decreased the ejection fraction in wt/wt BM and wt/Cxcr2−/− BM mice, as compared to the untreated mice (Table 2, Figure 2A). By contrast, treatment with anti-MIF antibody led to a significant increase in ejection fraction in the Cxcr2−/−/wt BM chimera mice as compared to isotype control (Table 2, Figure 2A). Analysis of cardiac output showed good compensation in all groups (Table 2). Heart rate and weight did not differ among BM chimeras (Table 2).
Figure 1. The effect of anti-MIF antibody on infarction size after I/R.
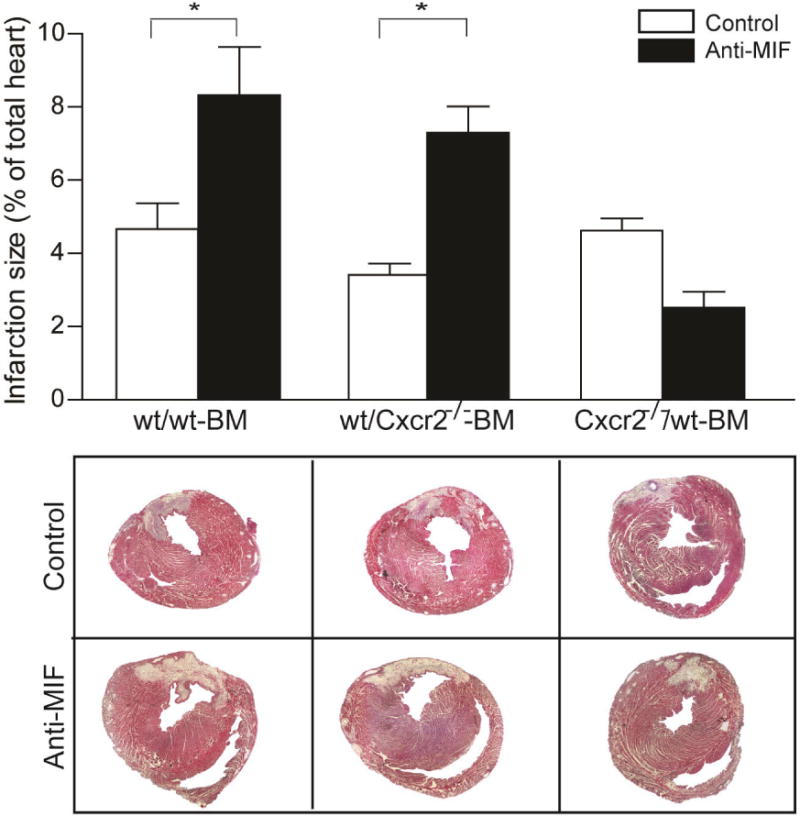
Infarction size was analyzed in wt/wt BM, wt/Cxcr2−/− BM, Cxcr2−/−/wt BM treated with anti-MIF antibody (dark bars) or isotype control (open bars) one week after I/R. Both wt/wt BM and wt/Cxcr2−/− BM mice treated with anti-MIF antibody showed a significant increase in the size of the infarcted area as compared to isotype control, whereas the Cxcr2−/−/wt BM mice displayed no effect after anti-MIF treatment (*p<0.05, n=4–6).
Table 2.
Echocardiography and functional parameters
| wt/wt BM | wt/CXCR2 BM | CXCR2/wt BM | ||||
|---|---|---|---|---|---|---|
| Control | Anti-MIF | Control | Anti-MIF | Control | Anti-MIF | |
| Ejection fraction (%) | 56.5±2.5 | 41.8±2.7 | 61.2±4.5 | 46.0±3.5 | 41.6±2.2 | 54.6±4.8 |
|
Cardiac output (ml/min) |
10.9±1.5 | 10.8±0.9 | 10.5±1.7 | 9.6±0.7 | 9.5±0.5 | 9.7±0.9 |
|
Heart rate (bpm) |
388±11.8 | 413±14.5 | 377±15.0 | 425±25.7 | 434±19.0 | 407±5.0 |
|
Heart weight (mg) |
89.4±8.8 | 84.5±9.7 | 88.4±6.3 | 83.8±2.6 | 88.3±20.2 | 93.7±6.7 |
Figure 2. The effect of anti-MIF-antibody on functional parameters after I/R.
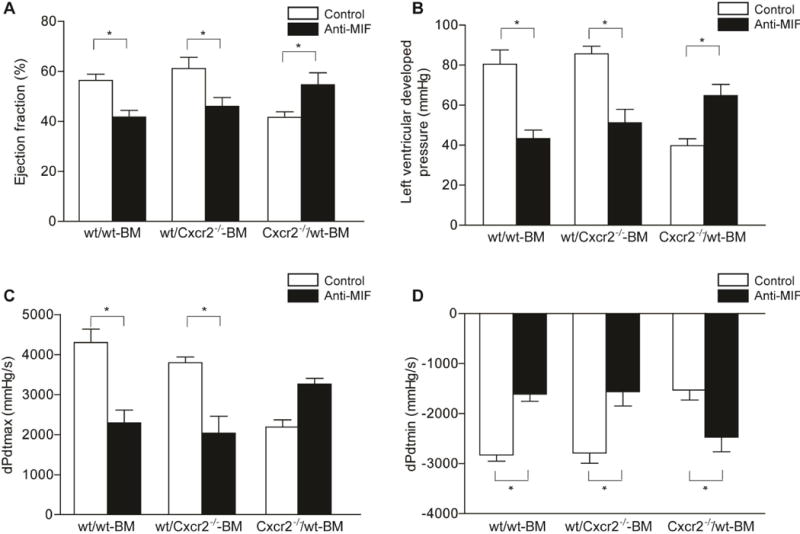
Ejection fraction was analyzed by echocardiography (A, *p<0.05, n=4–6). One week after I/R, anti-MIF antibody treatment (dark bars) significantly decreased the ejection fraction in wt/wt BM and wt/Cxcr2−/− BM mice, as compared to the isotype treated mice (open bars), while treatment with anti-MIF antibody led to a significant increase in ejection fraction in the Cxcr2−/−/wt BM chimera mice as compared to isotype control. Similar results are observed after analysis of cardiac function by Langendorff perfusion, as shown by left-ventricular developed pressure (LVDP) (B, *p<0.05, n=4–6), maximal − dP/dtmax (C, *p<0.05, n=4–6) and minimal − dP/dtmin (D, *p<0.05, n=4–6) pressure changes in the left ventricle.
The analysis of cardiac function by Langendorff perfusion one week after myocardial I/R revealed corresponding results. Whereas treatment with anti-MIF antibody decreased left ventricular developed pressure (LVDP) in wt/wt BM and wt/Cxcr2−/− BM mice as compared to isotype control, it significantly improved and restored the impaired LVDP in Cxcr2−/−/wt BM mice (Figure 2B). The maximal (dP/dtmax) and minimal (dP/dtmin) pressure change in left ventricle measurements confirmed these results. Treatment with anti-MIF antibody impaired contraction (dP/dtmax, Figure 2C) and relaxation (dP/dtmin, Figure 2D) in wt/wt BM mice and wt/CXCR2−/− BM mice but preserved myocardial function in Cxcr2−/−/wt BM chimeras.
These data demonstrate that in the presence of wild-type cardiomyocytes, blocking MIF aggravates myocardial healing and function, thus highlighting that the protective effect of MIF is mediated by a Cxcr2-dependent pathway in resident cardiomyocytes. In contrast, neutralizing MIF in the presence of Cxcr2-deficient cardiomyocytes only blocked the detrimental effects of MIF mediated by Cxcr2 on BM-derived cells.
Histological and immunohistochemical analysis of infarcted area
We next analyzed the recruitment of inflammatory cells, neo-angiogenesis and collagen synthesis in the infarcted area one day after myocardial I/R. One day after MI, neutrophil infiltration did not significantly differ between the groups (Figure 3A, Table 3), suggesting an involvement of receptor axes other than MIF/Cxcr2−/−. In contrast, monocyte infiltration into the infarcted area was reduced by treatment with anti-MIF antibody in wt/wt BM and Cxcr2−/−/wt BM mice compared with isotype control (Figure 3B, Table 3). In wt/Cxcr2−/− BM mice, monocyte infiltration was substantially impaired and not further attenuated by treatment with anti-MIF antibody (Figure 3B, Table 3), suggesting that the recruitment of monocytes towards the area of the infarction is strictly dependent on Cxcr2 in our model. Using CD14/CCR2 double staining, we could now show that most of the infiltrated monocytes are CCR2-positive inflammatory monocytes. This indicates that MIF promotes the recruitment of inflammatory CCR2+ monocytes into the heart tissue (Figure 3C, Table 3), which dominate the early phase of healing after infarction and exhibit phagocytic, proteolytic, and inflammatory features28 Of note, it has already been shown that MIF can recruit inflammatory monocytes in a CCL2-dependent manner29, indicating a functional interaction between CXC and CC receptors in the myocardial recruitment of inflammatory monocytes.
Figure 3. The analysis of anti-MIF-antibody effect on inflammatory reaction after I/R.
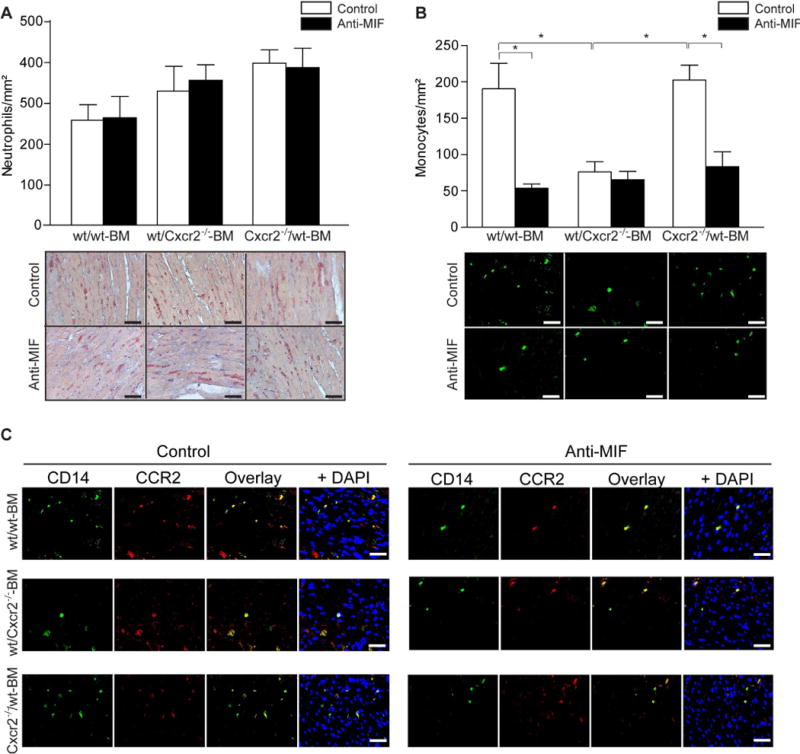
The infarcted area of chimeric wt/wt BM, wt/Cxcr2−/− BM and Cxcr2−/−/wt BM mice treated with anti-MIF (dark bars) or isotype control (open bars) antibody were analyzed one day after myocardial I/R. Neutrophil infiltration (specific esterase – red) did not significantly differ between the groups (A, n=4–6, Scale bars 50 μm), while monocyte infiltration (CD14 – green) was reduced by treatment with anti-MIF antibody in wt/wt BM and Cxcr2−/−/wt BM mice as compared to isotype control. In wt/Cxcr2−/− BM mice the monocyte recruitment was significantly impaired and anti-MIF antibody treatment did not further reduce the monocyte infiltration (B, *p<0.05, n=4–6, Scale bars 50 μm). The most infiltrated monocytes are CCR2 positive inflammatory monocytes, as shown by CD14 (green)/CCR2 (red) double staining (C, Scale bars 50 μm).
Table 3.
Histological and immunohistochemical analysis of infarcted area.
| wt/wt BM | wt/CXCR2 BM | CXCR2/wt BM | |||||
|---|---|---|---|---|---|---|---|
| Control | Anti-MIF | Control | Anti-MIF | Control | Anti-MIF | ||
| Neutrophils/mm2 | 250±37 | 256±50 | 319±59 | 345±37 | 386±31 | 375±46 | |
| Monocytes/mm2 | 191±35 | 54±6 | 78±10 | 66±11 | 202±20 | 83±20 | |
|
CCR2+ monocytes (% of CD14+ monocytes) |
86±5 | 72±14 | 84±7 | 73±11 | 86±10 | 83±9 | |
| Vessels/mm2 | 44±16 | 55±10 | 43±9 | 52±13 | 46±12 | 38±18 | |
|
Collagen (% infarcted area) |
15.1±1.4 | 7.3±0.4 | 13.5±0.5 | 10.3±0.5 | 14.2±0.5 | 11.6±0.3 | |
| Myofibroblasts/mm2 | 80±14 | 88±10 | 78±8 | 91±11 | 88±13 | 86±25 | |
Analysis of neo-angiogenesis in the infarcted area revealed no significant differences in the level of CD31-positive capillaries between the groups one week after MI (Figure 4A, Table 3), indicating that it was independent of Cxcr2 and MIF. Similarly, Langendorff measurements failed to reveal changes in coronary flow in any of the groups (Figure 4B).
Figure 4. The analysis of angiogenesis in the infarcted area after anti-MIF-antibody after I/R.
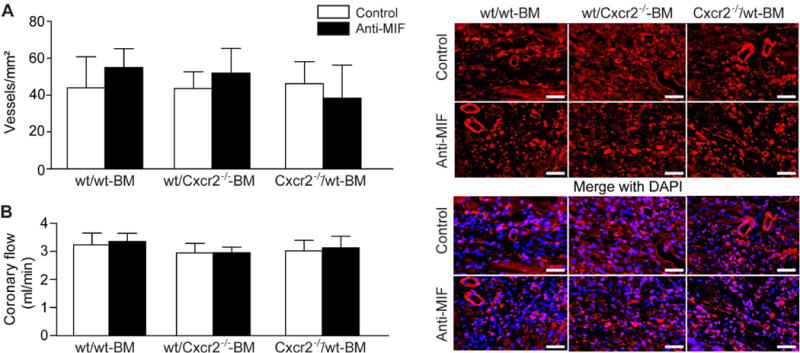
Analysis of neo-angiogenesis in the infarcted area (A) revealed no significant differences in the level of CD31-positive capillaries (red rings, right panel) between the groups one week after MI (n=4–6, Scale bars 50 μm). Similarly, Langendorff measurements failed to reveal changes in coronary flow in any of the groups (B, n=4–6).
Notably, collagen content was reduced in all three groups by treatment with anti-MIF antibody but not altered by Cxcr2 deficiency in BM or resident cells, demonstrating that collagen deposition one week after myocardial I/R was regulated by MIF, but not by Cxcr2 (Figure 5A, Table 3). The number of myofibroblasts did not differ between the groups (Figure 5B, Table 3), implying that MIF rather affects their function in collagen synthesis. Hence, protective effects of MIF are in part attributable to increased collagen deposition, whereas its deleterious effects are due to a stimulation of myocardial monocyte infiltration.
Figure 5. The effect of anti-MIF-antibody treatment on fibrosis after I/R.
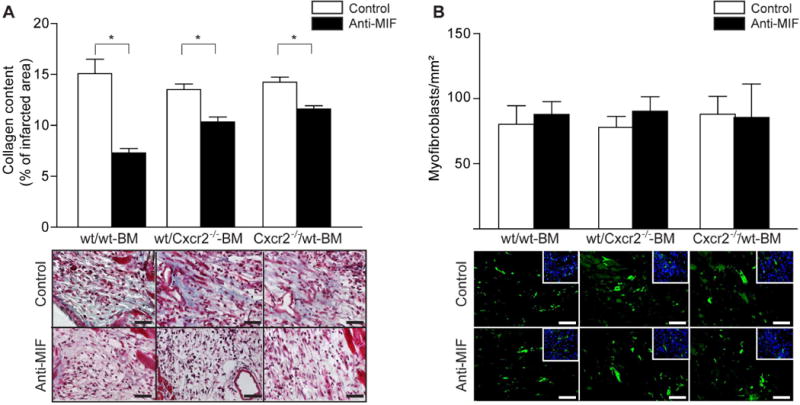
Notably, collagen content (blue) was reduced in all three groups by treatment with anti-MIF antibody but not altered by Cxcr2 deficiency in BM or resident cells (A, *p<0.05, n=4–6, Scale bars 50 μm), while the number of myofibroblasts (green) did not differ between the groups (B, n=4–6, Scale bars 50 μm).
Discussion
Our data establish a double-edged role of endogenous MIF in the myocardial healing response after I/R injury. To analyze the MIF-Cxcr2 pathway, chimeric mice were obtained by reconstitution of wild-type mice with wild-type or Cxcr2−/− BM, and of Cxcr2−/− mice with wild-type BM. We demonstrate that MIF exerts opposing effects on resident cardiac cells versus BM-derived inflammatory cells mediated through Cxcr2. Whereas blocking endogenous MIF acting through Cxcr2 on cardiomyocytes impaired heart function and increased infarction size, blocking endogenous MIF acting through Cxcr2 on circulating inflammatory cells protected heart function and decreased infarction size Notably, the latter effect was not related to an infiltration with neutrophils, which did not require Cxcr2 or MIF, but due to the control of monocyte infiltration in the infarcted area mediated by MIF and Cxcr2. Since we did not observe significant differences between the infarction size in isotype-treated wt/wt BM and Cxcr2−/−/wt BM, it is conceivable that Cxcr2−/− mice harbor compensatory mechanisms, which may substitute for CXCR2 functions, e.g. the CXCR4/CXCL12 axis, CCL2, TNFα or HIF30. The oxidoreductive function of MIF has been elegantly shown to provide an anti-oxidative and cardioprotective capacity in myocardial infarction31 and could therefore be another compensatory MIF-based protective mechanism in myocardial ischemia/reperfusion injury.
It has previously been shown that MIF released by cardiomyocytes during I/R injury exerts cardioprotective effects by activating AMPK (JNK) through CD7415. We observed a reduction in the functional parameters of the heart (ejection fraction, LVED, contraction and relaxation) and increased infarction size after anti-MIF antibody treatment in wt/wt BM and wt/Cxcr2−/− BM mice. MIF blockade improved ejection fraction, LVDP, contraction and relaxation without influencing infarction size in Cxcr2−/−/wt BM mice, which do not express Cxcr2 on cardiomyocytes and therefore show impaired heart function following I/R injury, as compared to controls. It thus appears that protective effects of endogenous MIF secreted by cardiomyocytes can prevent severe functional loss in heart tissue in wt/CXCR2−/− BM mice with Cxcr2-deficient circulating cells. Considering what is known about the CD74/AMPK-mediated MIF protection15, these results are surprising. Hence, we can assume that CD74 and CXCR2 are both required for the myocardial protection in ischemia injury. Although MIF is able to exert his biological effects by binding to CD74 and CXCR2 individually, it was shown that MIF binding to the CXCR2/CD74 complex enhances G-protein-coupled receptor activation and downstream cardiovascular effects7. Accordingly, predictably the deletion of one or both of the receptors would attenuate the protective effect in cardiomyocytes exposed to ischemia. Therefore, we suppose that the deletion of both receptors similar to that of Cxcr2 alone would decrease the protective MIF effect. However, as Cxcr2/Cd74-DKO mice are not available, we can currently not verify this notion and thus cannot exclude the compensation of this effect by other MIF-independent protective mechanisms in cardiomyocytes. Nevertheless, the CXCR2/CD74 complex should play a crucial role in mediating the protective effects of MIF in cardiomyocytes after MI.
Notably, deficiency in Cxcr2 in cardiomyocytes and in BM-derived inflammatory cells caused only moderate alterations in the size of the infarcted area one week after I/R, which contrasts previous findings in bone marrow chimeras after one day, as reported by Tarzami et al27. Since one day after MI the infarction size is largely defined by cardiomyocyte death, this discrepancy may be explained by adaptive remodeling of the infarction area following the inflammatory reaction over the course of one week after I/R injury.
In general, MIF blockade reduced the number of inflammatory cells, namely monocytes, infiltrating the site of injury. Mononuclear cells infiltrating the site of inflammation release cytokines and chemokines, which further enhances the recruitment and activation of these pro-inflammatory cells8, 32. It has been shown that MIF can induce monocyte arrest through CXCR27, and accordingly myocardial monocyte infiltration was also dependent on CXCR2 in our study. Studies in animal models characterized by impaired monocyte infiltration have consistently shown preserved heart function following experimental induction of MI33–35. Since we performed our analysis one day after I/R, most of the recruited monocytes should be Gr-1high (also known as Gr-1+CCR2+CX3CR1lo) monocytes, which dominate the early phase and exhibit phagocytic, proteolytic, and inflammatory features28. Reducing inflammatory monocytes following MI seems to beneficially promote cardiac remodeling30. Since CXCR2 is knocked out in inflammatory cells in the wt/Cxcr2−/− BM mice, and since the number of infiltrating monocytes was not further reduced by MIF blockade in these mice, we can assume that MIF is able to control the recruitment of inflammatory monocytes. Indeed, MIF has been shown to recruit monocyte/macrophage in a CCL2-dependent manner, as CCL2 deficiency or anti-CCL2 antibody treatment significantly inhibited MIF-induced monocyte adhesion and transmigration in mice29. This reveals the importance of CXCR in the myocardial recruitment of inflammatory monocytes.
Surprisingly, infiltration of the infarcted myocardial tissue with neutrophils was unaffected by MIF blockade or Cxcr2 deficiency. Neutrophils have been found to express CXCR2 but not CD743. The moderate chemotactic activity towards MIF in neutrophils has been related to a lack of CD74 in these cells7. Since the function of MIF depends on the CXCR2/CD74 complex7, the absence of CD74 might explain why neutrophil infilitration was unaltered in our myocardial I/R model. Furthermore, this is consistent with findings showing that early neutrophil infiltration following I/R injury was greatly diminished by Ccr1 deficiency and is hence rather dependent on the presence of the CCL5 receptor CCR136.
Unexpectedly, our evaluation of neoangiogenesis in the infarcted tissue one week after myocardial I/R injury failed to revealed changes in coronary flow or CD31-positive capillaries in any of the experimental groups. This is surprising, given that MIF and CXCR2 have been implicated in angiogenic responses5 under hypoxic conditions by initiating tube formation and differentiation towards endothelial cell phenotypes or stimulating endothelial proliferation and capillary-like structure formation, respectively23, 24, 37. Our data support the notion that the MIF-CXCR2 pathway plays a minimal role in angiogenesis after MI, and rather indicate the involvement of other factors such as vascular endothelial growth factor or the CXCR4 ligand stromal-cell derived factor-1, acting through receptors other than CXCR225.
Another unexpected effect of MIF blockade was the CXCR2-independent reduction of collagen content in the infarcted area. Until recently, it was believed that reduced collagen content in the infarcted area might facilitate cardiac function. Our results rather indicate that increased collagen content in the infracted area can lead to a more stable myocardial scar and improved heart function30, 38. Therefore, we assume that different collagen subtypes can be synthesized in response to MIF and, together with other factors, influence myocardial function. Accordingly, we have shown that MIF can favor the differentiation of smooth muscle-actin (SMA)-positive cells in vivo24. Moreover, MIF in conjunction with CD74 can antagonize myocardial hypertrophy and fibrosis39 and experimental liver fibrosis40. Thus, MIF may contribute to fibroblast differentiation towards SMA-positive myofibroblasts, which is a crucial event in myocardial healing and scar formation30. Because the number of myofibroblasts did not differ in our groups, MIF seems to exert a direct effect on myofibroblast function and collagen synthesis. However, the mechanism underlying MIF-dependent fibrosis and scar formation following MI remain to be clarified in detail.
In conclusion, this study provides novel insights into the mechanisms of MIF and its functional receptor CXCR2 in myocardial regeneration and scar formation. Without affecting neutrophil infiltration or angiogenesis, MIF protects cardiac tissue via CXCR2. This benefit of MIF is counteracted by the CXCR2-dependent recruitment of monocytes associated with an impaired heart function. Our findings should be taken into account, when developing tailored therapeutic strategies for improved remodeling and preservation of heart function after MI.
Supplementary Material
Significance.
Myocardial infarction remains the most common cause of death in the western countries, despite the extensive research in the last decades. Developing new therapeutic strategies to prevent and treat myocardial damage after an ischemic insult represents a priority for the cardiovascular scientific community. To this end, more detailed and advanced knowledge about the mechanisms of cardiac cell protection and regeneration is urgently required.
Macrophage migration inhibitory factor (MIF) was found to be significantly elevated in patients with myocardial ischemia and also appears to protect the heart, thus possibly serving as a suitable therapeutic concept. However, the underlying mechanisms and its implications for cardioprotection are not completely elucidated.
This study provides novel insights into the mechanisms, by which MIF exerts its effects after myocardial infarction. Whereas MIF protects cardiac tissue via its receptor CXCR2 on resident cells, it exerts detrimental effects on CXCR2-bearing inflammatory cells, associated with an impaired heart function. These findings are of particular importance when developing novel therapeutic strategies to preserve the heart function after myocardial infarction.
Acknowledgments
We want to thank Susanna Kintig, Christiane Herold, Manfred Dewor, and Birgitt Lennartz for excellent technical assistance.
Source of funding
This study was supported by the Deutsche Forschungsgemeinschaft (Forschergruppe 809, TP01 and TP04; and IRTG1508, project P13 to J.B., C.W., and I.K.), and the Interdisciplinary Centre for Clinical Research IZKF Aachen (junior research group to E.L., project K5 to J.B. and junior career funding to I.K.) within the faculty of Medicine at RWTH Aachen University.
Abbreviations
- MIF
Macrophage migration inhibitory factor
- I/R
ischemia and reperfusion
- BM
bone marrow
- wt
wild-type
- MI
myocardial infarction
- GPCR
G-protein-coupled receptor
- MAPK
mitogen-activated protein kinase
- SMA
smooth muscle-actin
Footnotes
Disclosures
None
References
- 1.Bloom BR, Bennett B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science. 1966;153:80–82. doi: 10.1126/science.153.3731.80. [DOI] [PubMed] [Google Scholar]
- 2.Bucala R, Lolis E. Macrophage migration inhibitory factor: a critical component of autoimmune inflammatory diseases. Drug news & perspectives. 2005;18:417–426. doi: 10.1358/dnp.2005.18.7.939345. [DOI] [PubMed] [Google Scholar]
- 3.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noels H, Bernhagen J, Weber C. Macrophage migration inhibitory factor: a noncanonical chemokine important in atherosclerosis. Trends in cardiovascular medicine. 2009;19:76–86. doi: 10.1016/j.tcm.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Asare Y, Schmitt M, Bernhagen J. The vascular biology of macrophage migration inhibitory factor (MIF). Expression and effects in inflammation, atherogenesis and angiogenesis. Thrombosis and haemostasis. 2013;109:391–398. doi: 10.1160/TH12-11-0831. [DOI] [PubMed] [Google Scholar]
- 6.Lin SG, Yu XY, Chen YX, Huang XR, Metz C, Bucala R, Lau CP, Lan HY. De novo expression of macrophage migration inhibitory factor in atherogenesis in rabbits. Circ Res. 2000;87:1202–1208. doi: 10.1161/01.res.87.12.1202. [DOI] [PubMed] [Google Scholar]
- 7.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–596. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 8.Zernecke A, Bernhagen J, Weber C. Macrophage migration inhibitory factor in cardiovascular disease. Circulation. 2008;117:1594–1602. doi: 10.1161/CIRCULATIONAHA.107.729125. [DOI] [PubMed] [Google Scholar]
- 9.Burger-Kentischer A, Goebel H, Seiler R, Fraedrich G, Schaefer HE, Dimmeler S, Kleemann R, Bernhagen J, Ihling C. Expression of macrophage migration inhibitory factor in different stages of human atherosclerosis. Circulation. 2002;105:1561–1566. doi: 10.1161/01.cir.0000012942.49244.82. [DOI] [PubMed] [Google Scholar]
- 10.Schober A, Bernhagen J, Thiele M, Zeiffer U, Knarren S, Roller M, Bucala R, Weber C. Stabilization of atherosclerotic plaques by blockade of macrophage migration inhibitory factor after vascular injury in apolipoprotein E-deficient mice. Circulation. 2004;109:380–385. doi: 10.1161/01.CIR.0000109201.72441.09. [DOI] [PubMed] [Google Scholar]
- 11.Yu CM, Lau CP, Lai KW, Huang XR, Chen WH, Lan HY. Elevation of plasma level of macrophage migration inhibitory factor in patients with acute myocardial infarction. Am J Cardiol. 2001;88:774–777. doi: 10.1016/s0002-9149(01)01850-1. [DOI] [PubMed] [Google Scholar]
- 12.Yu CM, Lai KW, Chen YX, Huang XR, Lan HY. Expression of macrophage migration inhibitory factor in acute ischemic myocardial injury. J Histochem Cytochem. 2003;51:625–631. doi: 10.1177/002215540305100508. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi M, Nishihira J, Shimpo M, Mizue Y, Ueno S, Mano H, Kobayashi E, Ikeda U, Shimada K. Macrophage migration inhibitory factor as a redox-sensitive cytokine in cardiac myocytes. Cardiovasc Res. 2001;52:438–445. doi: 10.1016/s0008-6363(01)00408-4. [DOI] [PubMed] [Google Scholar]
- 14.Fukuzawa J, Nishihira J, Hasebe N, Haneda T, Osaki J, Saito T, Nomura T, Fujino T, Wakamiya N, Kikuchi K. Contribution of macrophage migration inhibitory factor to extracellular signal-regulated kinase activation by oxidative stress in cardiomyocytes. J Biol Chem. 2002;277:24889–24895. doi: 10.1074/jbc.M112054200. [DOI] [PubMed] [Google Scholar]
- 15.Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, Young LH. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451:578–582. doi: 10.1038/nature06504. [DOI] [PubMed] [Google Scholar]
- 16.Qi D, Hu X, Wu X, Merk M, Leng L, Bucala R, Young LH. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J Clin Invest. 2009;119:3807–3816. doi: 10.1172/JCI39738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luedike P, Hendgen-Cotta UB, Sobierajski J, Totzeck M, Reeh M, Dewor M, Lue H, Krisp C, Wolters D, Kelm M, Bernhagen J, Rassaf T. Cardioprotection through S-nitros(yl)ation of macrophage migration inhibitory factor. Circulation. 2012;125:1880–1889. doi: 10.1161/CIRCULATIONAHA.111.069104. [DOI] [PubMed] [Google Scholar]
- 18.Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197:1467–1476. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchell RA, Metz CN, Peng T, Bucala R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999;274:18100–18106. doi: 10.1074/jbc.274.25.18100. [DOI] [PubMed] [Google Scholar]
- 20.Gao XM, Liu Y, White D, Su Y, Drew BG, Bruce CR, Kiriazis H, Xu Q, Jennings N, Bobik A, Febbraio MA, Kingwell BA, Bucala R, Fingerle-Rowson G, Dart AM, Morand EF, Du XJ. Deletion of macrophage migration inhibitory factor protects the heart from severe ischemia-reperfusion injury: a predominant role of anti-inflammation. Journal of molecular and cellular cardiology. 2011;50:991–999. doi: 10.1016/j.yjmcc.2010.12.022. [DOI] [PubMed] [Google Scholar]
- 21.Kraemer S, Lue H, Zernecke A, Kapurniotu A, Andreetto E, Frank R, Lennartz B, Weber C, Bernhagen J. MIF-chemokine receptor interactions in atherogenesis are dependent on an N-loop-based 2-site binding mechanism. Faseb J. 2011;25:894–906. doi: 10.1096/fj.10-168559. [DOI] [PubMed] [Google Scholar]
- 22.Weber C, Kraemer S, Drechsler M, Lue H, Koenen RR, Kapurniotu A, Zernecke A, Bernhagen J. Structural determinants of MIF functions in CXCR2-mediated inflammatory and atherogenic leukocyte recruitment. Proc Natl Acad Sci U S A. 2008;105:16278–16283. doi: 10.1073/pnas.0804017105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simons D, Grieb G, Hristov M, Pallua N, Weber C, Bernhagen J, Steffens G. Hypoxia-induced endothelial secretion of macrophage migration inhibitory factor and role in endothelial progenitor cell recruitment. Journal of cellular and molecular medicine. 2011;15:668–678. doi: 10.1111/j.1582-4934.2010.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanzler I, Tuchscheerer N, Steffens G, Simsekyilmaz S, Konschalla S, Kroh A, Simons D, Asare Y, Schober A, Bucala R, Weber C, Bernhagen J, Liehn EA. Differential roles of angiogenic chemokines in endothelial progenitor cell-induced angiogenesis. Basic Res Cardiol. 2013;108:310. doi: 10.1007/s00395-012-0310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liehn EA, Tuchscheerer N, Kanzler I, Drechsler M, Fraemohs L, Schuh A, Koenen RR, Zander S, Soehnlein O, Hristov M, Grigorescu G, Urs AO, Leabu M, Bucur I, Merx MW, Zernecke A, Ehling J, Gremse F, Lammers T, Kiessling F, Bernhagen J, Schober A, Weber C. Double-edged role of the CXCL12/CXCR4 axis in experimental myocardial infarction. J Am Coll Cardiol. 2011;58:2415–2423. doi: 10.1016/j.jacc.2011.08.033. [DOI] [PubMed] [Google Scholar]
- 26.Tarnowski M, Grymula K, Liu R, Tarnowska J, Drukala J, Ratajczak J, Mitchell RA, Ratajczak MZ, Kucia M. Macrophage migration inhibitory factor is secreted by rhabdomyosarcoma cells, modulates tumor metastasis by binding to CXCR4 and CXCR7 receptors and inhibits recruitment of cancer-associated fibroblasts. Mol Cancer Res. 2010;8:1328–1343. doi: 10.1158/1541-7786.MCR-10-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tarzami ST, Miao W, Mani K, Lopez L, Factor SM, Berman JW, Kitsis RN. Opposing effects mediated by the chemokine receptor CXCR2 on myocardial ischemia-reperfusion injury: recruitment of potentially damaging neutrophils and direct myocardial protection. Circulation. 2003;108:2387–2392. doi: 10.1161/01.CIR.0000093192.72099.9A. [DOI] [PubMed] [Google Scholar]
- 28.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gregory JL, Morand EF, McKeown SJ, Ralph JA, Hall P, Yang YH, McColl SR, Hickey MJ. Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J Immunol. 2006;177:8072–8079. doi: 10.4049/jimmunol.177.11.8072. [DOI] [PubMed] [Google Scholar]
- 30.Liehn EA, Postea O, Curaj A, Marx N. Repair after myocardial infarction, between fantasy and reality: the role of chemokines. J Am Coll Cardiol. 2011;58:2357–2362. doi: 10.1016/j.jacc.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 31.Koga K, Kenessey A, Powell SR, Sison CP, Miller EJ, Ojamaa K. Macrophage migration inhibitory factor provides cardioprotection during ischemia/reperfusion by reducing oxidative stress. Antioxidants & redox signaling. 2011;14:1191–1202. doi: 10.1089/ars.2010.3163. [DOI] [PubMed] [Google Scholar]
- 32.Weber C, Schober A, Zernecke A. Chemokines: key regulators of mononuclear cell recruitment in atherosclerotic vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:1997–2008. doi: 10.1161/01.ATV.0000142812.03840.6f. [DOI] [PubMed] [Google Scholar]
- 33.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 34.Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker T, Kraemer D, Taffet G, Rollins BJ, Entman ML. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007;115:584–592. doi: 10.1161/CIRCULATIONAHA.106.646091. [DOI] [PubMed] [Google Scholar]
- 35.Liehn EA, Piccinini AM, Koenen RR, Soehnlein O, Adage T, Fatu R, Curaj A, Popescu A, Zernecke A, Kungl AJ, Weber C. A new monocyte chemotactic protein-1/chemokine CC motif ligand-2 competitor limiting neointima formation and myocardial ischemia/reperfusion injury in mice. J Am Coll Cardiol. 2010;56:1847–1857. doi: 10.1016/j.jacc.2010.04.066. [DOI] [PubMed] [Google Scholar]
- 36.Liehn EA, Merx MW, Postea O, Becher S, Djalali-Talab Y, Shagdarsuren E, Kelm M, Zernecke A, Weber C. Ccr1 deficiency reduces inflammatory remodelling and preserves left ventricular function after myocardial infarction. Journal of cellular and molecular medicine. 2008;12:496–506. doi: 10.1111/j.1582-4934.2007.00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hristov M, Zernecke A, Liehn EA, Weber C. Regulation of endothelial progenitor cell homing after arterial injury. Thrombosis and haemostasis. 2007;98:274–277. [PubMed] [Google Scholar]
- 38.Schuh A, Liehn EA, Sasse A, Hristov M, Sobota R, Kelm M, Merx MW, Weber C. Transplantation of endothelial progenitor cells improves neovascularization and left ventricular function after myocardial infarction in a rat model. Basic Res Cardiol. 2008;103:69–77. doi: 10.1007/s00395-007-0685-9. [DOI] [PubMed] [Google Scholar]
- 39.Koga K, Kenessey A, Ojamaa K. Macrophage migration inhibitory factor antagonizes pressure overload-induced cardiac hypertrophy. American journal of physiology. 2013;304:H282–293. doi: 10.1152/ajpheart.00595.2012. [DOI] [PubMed] [Google Scholar]
- 40.Heinrichs D, Knauel M, Offermanns C, Berres ML, Nellen A, Leng L, Schmitz P, Bucala R, Trautwein C, Weber C, Bernhagen J, Wasmuth HE. Macrophage migration inhibitory factor (MIF) exerts antifibrotic effects in experimental liver fibrosis via CD74. Proc Natl Acad Sci U S A. 2011;108:17444–17449. doi: 10.1073/pnas.1107023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.