

Abstract
Immune stimulation triggered by siRNAs is one of the major challenges in the development of safe RNAi-based therapeutics. Within an immunostimulatory siRNA sequence, this hurdle is commonly addressed by using ribose modifications (e.g. 2′-OMe or 2′-F) which results in decreased cytokine production. However, since immune stimulation by siRNAs is a sequence-dependent phenomenon, recognition of the nucleobases by the trigger receptor(s) is also likely. Here, we use the recently published crystal structures of Toll-like receptor 8 (TLR8) bound to small molecule agonists to generate computational models for ribonucleotide binding by this immune receptor. Our modeling suggested that modification of either the Watson-Crick or Hoogsteen face of adenosine would disrupt nucleotide/TLR8 interactions. We employed chemical synthesis to alter either the Watson-Crick or Hoogsteen face of adenosine and evaluated the effect of these modifications in an siRNA guide strand by measuring the immunostimulatory and RNA interference properties. For the siRNA guide strand tested, we found that modifying the Watson-Crick face is generally more effective at blocking TNFα production in human peripheral blood mononuclear cells (PBMCs) than modification at the Hoogsteen edge. We also observed that modifications near the 5′ end were more effective at blocking cytokine production than those placed at the 3′ end. This work advances our understanding of how chemical modifications can be used to optimize siRNA performance.
Keywords: TLR8, siRNA, immune stimulation, nucleobase modification
Introduction
One of the hurdles in the development of potent and safe RNA interference (RNAi) therapeutics is countering the immune response triggered by certain short interfering RNAs (siRNAs).[1–4] In humans, Toll-like receptors (TLRs) 7 and 8 have been implicated in the siRNA-induced immune response.[5] At this time, molecular recognition of RNA by TLR7 or TLR8 is poorly understood, however it has been shown that certain sequence motifs found in either the passenger or guide strands of siRNA duplexes activate these TLRs. For instance, adenosine-rich RNAs containing the sequences AUGU, AUAU, or UAUA, among others, are known to stimulate TLR8 in human immune cells.[6] In addition, recent crystal structures of human TLR8 bound to small molecule agonists identify binding sites for aromatic heterocycles at the dimer interface, suggesting where nucleobases within immunostimulatory RNA strands may interact with this receptor.[7] Indeed, site directed mutagenesis studies show that residues involved in small molecule agonist binding (e.g. hTLR8 D543) are also required for RNA binding.[7] In the absence of high-resolution structures of TLRs bound to RNA, models for the RNA-protein complex are valuable in guiding the design of new nucleoside analogs capable of blocking this interaction.
Here, we report the use of automated docking to evaluate binding modes for nucleoside bisphosphates in the small molecule agonist-binding site of TLR8. The docking process led to models for adenosine and uridine binding by TLR8 that are supported by the available site directed mutagenesis and RNA activation data. Furthermore, the model for adenosine binding suggested nucleobase modification strategies that would disrupt recognition of the nucleobase by TLR8. Indeed, using either a Hoogsteen or Watson-Crick (WC) face-modified adenosine analog reduced TNFα production in human peripheral blood mononuclear cells (PBMCs) when placed within an immunostimulatory siRNA. We also analyzed the gene knockdown activity of our modified siRNAs and identified a modified siRNA with similar RNAi activity to the unmodified siRNA, but with substantially reduced TNFα stimulation.
Results and Discussion
Models for nucleotide binding to TLR8
Recent crystal structures of human TLR8 bound to small molecule agonists provide a starting point for modeling RNA binding to this immune receptor.[7] Since agonist compounds are aromatic heterocycles (e.g. CLO97, Figure 1A), they share structural similarities to the nucleobases found in RNA. Furthermore, certain amino acid residues present in human TLR8 have been shown to be vital for both small molecule agonist and RNA recognition indicating overlap of the binding sites for these two TLR8 ligand classes.[8] As a result of this observation, we used the OpenEye suite of programs to generate models for how a nucleotide within an oligoribonucleotide activator could bind into the small molecule agonist binding site in human TLR8. This method would allow us to generate possible ligand conformations, dock them into the receptor-ligand binding site, and score the resulting binding poses.[9–10] We used the 3′,5′-bisphosphates of the four canonical ribonucleosides to account for possible roles a phosphodiester backbone might play in RNA binding and to easily identify poses that would not be compatible with oligonucleotide binding (e.g. 5′ or 3′ phosphate directed into the protein interior or buried in a protein pocket). We analyzed the top ten scoring poses assumed by the four 3′, 5′-bisphosphates (Figures 1B and 1C, Supplementary Figures 2–4, Supplementary Table 1) to assess overall binding plausibility. For both the adenosine and uridine compounds, multiple, similar binding poses were compatible with oligonucleotide binding (i.e. exterior phosphate positions) which also invoke roles for residues known to be involved in RNA recognition (i.e. D543, R429, and Y353)[7–8] (Figures 1B and 1C, Supplementary Figures 2 and 3, Supplementary Table 1). In addition, high scoring poses for these bisphosphates place the 5′ phosphate in a similar position within the binding pocket as the sulphate ion found in the TLR8-CLO97 crystal (Figure 1).[7] Less compelling binding models for guanosine and cytidine were generated by this approach, suggesting that these nucleotides present in immunostimulatory sequences either occupy different binding pockets than the small molecule agonists of TLR8 or that the receptor undergoes significant conformational changes to accommodate their binding (Supplementary Figure 4). Interestingly, our models for A and U binding suggest close approach of the nucleotide 2′-hydroxyl and 3′-phosphate to the side chain of R429 which is held in position through a H-bonding interaction to Y353 (Figure 1B, 1C). These two residues are known to be important for activation of TLR8 by RNA.[7–8] In addition, the nucleotide 2′-hydroxyl has been implicated in TLR8 activation in previous studies that focus on the effect of sugar modifications within an siRNA strand on immune stimulation.[11–16]
Figure 1.

(A) Complex of the imidazoloquinoline CLO97 bound to human Toll-like receptor 8 (TLR8) determined by x-ray crystallography.[7] The sulphate ion shown is from the crystallization liquor and is present near the CLO97 binding site in the crystallized complex. (B) Model of adenosine 3′, 5′-bisphosphate and (C) uridine 3′, 5′-bisphosphate bound to the agonist binding site of TLR8 (see Materials and Methods for docking procedure).
Immune stimulation by siRNAs bearing modified adenosines
Chemical modification of siRNAs is a useful tool for enhancing the potency of RNAi therapeutics[17–19], and has also been shown to abrogate immunostimulatory effects. Fucini et al. reported that ribose modifications, particularly 2′-F, placed on the adenosines of an siRNA significantly decreased cytokine release from human PBMCs while maintaining knockdown activity.[20] While ribose modifications are known to inhibit immune stimulation, the effects of siRNA base modifications on immune response remain largely unexplored.[21] Our model for adenosine recognition by TLR8 (Figure 1B) suggested that modification of either the WC or Hoogsteen edge of the purine could block this interaction. However, base modifications to an siRNA guide strand pose the risk of disrupting essential hydrogen bonding crucial for the formation of the A-form duplex needed to activate RNAi. Previously, our lab has developed replacements for adenosine in siRNAs that maintain base pairing with uridine while altering either the Hoogsteen (7-triazolo-8-aza-7-deazaadenosines)[22–23] or WC face (N2-alkyl-2-aminopurines)[24–25] (Scheme 1).
Scheme 1.
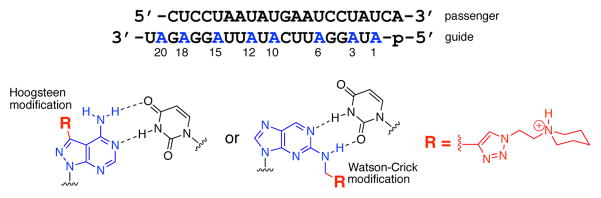
(Top) Sequence of the siRNA used in this study with guide strand adenosines replaced by nucleoside analogs highlighted in blue. (Bottom) Structures of adenosine analogs with Hoogsteen or Watson-Crick (WC) face-localized N-ethylpiperidine triazole modification.
While initial studies suggested that replacement of adenosines with N2-alkyl-2-aminopurines at specific positions was tolerated by the RNAi machinery and reduced immune stimulation, a systematic study of both types of modification at multiple positions in a single siRNA has not been reported.[21, 25]
To evaluate the effects of adenosine modification on immune stimulation and RNAi activity, we used a previously published siRNA sequence targeting the PIK3CB mRNA.[26] This siRNA has a guide strand rich in adenosines (8/21 positions) and stimulates cyokine production in human PBMCs when complexed with transfection agent (Figure 2A). Interestingly, higher levels of TNFα are stimulated by this siRNA formulation than IFNα, which is consistent with TLR8 activation (Figure 3A).[15, 27–29]
Figure 2.
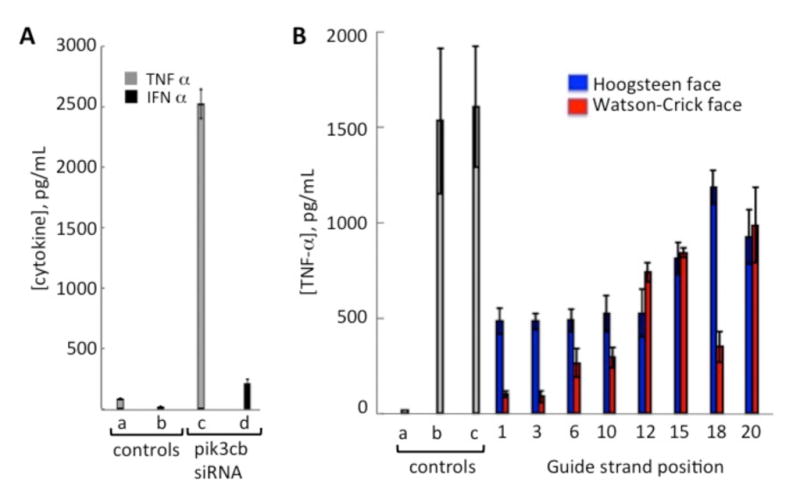
Cytokine production by human PBMCs treated with siRNAs. (A) TNFα and IFNα levels generated by transfection with the PIK3CB siRNA. (a) buffer only (b): PIK3CB without transfection agent (c) and (d): transfection with PIK3CB siRNA. (B) Comparison of TNFα levels resulting from transfection of human PBMCs with siRNAs bearing adenosine modifications at different positions on the PIK3CB guide strand. Controls: (a) buffer alone; (b) R-848 (10 μg/ml, positive control), (c) unmodified duplex, Blue bars: Hoogsteen face-localized modification (N-ethylpiperidine triazole linked through C7 of 7-deaza-8-azaadenosine); Red bars: Watson-Crick (WC) face localized modification (N-ethylpiperidine triazole linked through N2 of 2-aminopurine). SiRNAs were transfected at a concentration of 125 nM.
Figure 3.
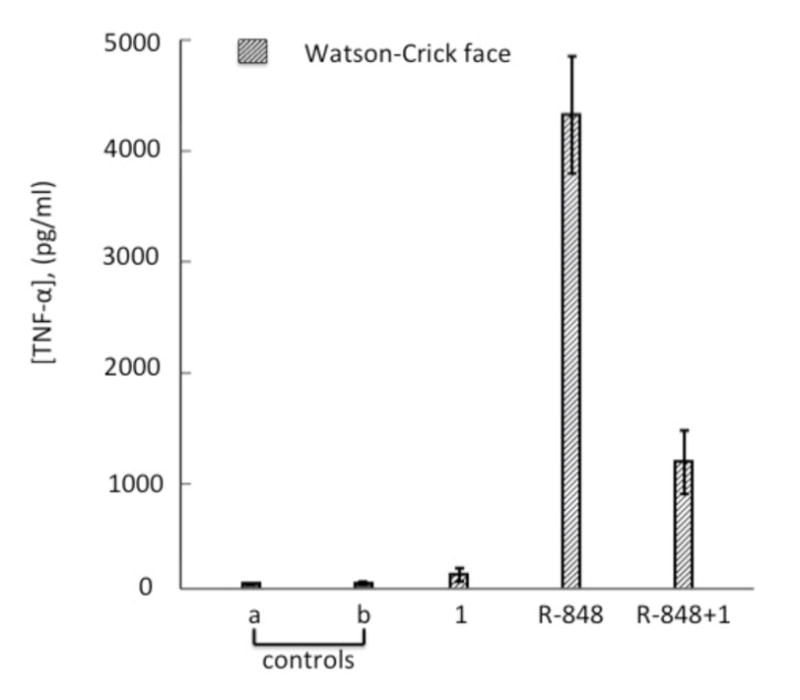
Antagonistic activity of the Watson-Crick (WC) face localized modification at guide position 1. Modified PIK3CB siRNA (125nM) was cotransfected with the agonist R-848 (1μg/mL) into human PBMCs. Controls: (a) buffer alone; (b) siRNA without transfection agent; Red bars: (1) WC face localized modification at guide position 1.
We used our previously described nucleoside analog phosphoramidites to install a reactive alkyne at different positions along the PIK3CB siRNA guide strand, then carried out copper-catalyzed azide/alkyne cycloaddition (CuAAC) reactions with an N-ethylpiperidine azide to introduce the triazole modifications shown in Scheme 1.[22, 24] This particular azide was chosen because the resulting triazoles are sterically demanding, but are only minimally destabilizing to RNA duplexes.[22, 25] To test the immunostimulatory properties of the modified siRNAs, we transfected the duplex oligonucleotides into human PBMCs and quantified TNFa production after 16–20 h of incubation (Figure 2B). We found that, for this sequence, modifying positions near the 5′ end of the siRNA (i.e. guide positions 1, 3, 6, and 10) with either nucleobase analog, position 12 with the Hoogsteen analog or position 18 on the WC face, resulted in a greater than three-fold reduction in cytokine production. Interestingly, modification at positions 1 or 3 with the WC-directed triazole resulted in more than a 15-fold reduction in TNFα production. These results are consistent with the observation that modification of a single nucleotide position is sufficient to block cytokine production caused by an immunostimulatory siRNA.[14, 16, 20]
We also performed a cytotoxicity assay using two WC-edge modified siRNAs that showed the lowest cytokine stimulation, in comparison with unmodified siRNA and untreated cells. We found minimal toxicity for all the tested siRNAs, similar to the untreated control, which shows that the decrease in cytokine production is due to the chemical modification and not cellular toxicity (Supplementary Figure 1).
Watson-Crick face modification generates an antagonist
Our model for adenosine binding to TLR8 suggested that modifying the Hoogsteen face would be highly disruptive since it would block interaction with D543, a residue known to be important for RNA recognition by TLR8 (Figure 1B). However, our results show that at the positions most sensitive to modification (i.e. positions 1 and 3), altering the WC edge provided a larger reduction in TNFα than modification at the Hoogsteen face. This led us to consider the possibility that our WC edge modification generated an RNA capable of binding the receptor but incapable of activation. Such a molecule could function as a TLR antagonist, preventing cytokine production due to other stimulatory molecules present, such as the passenger strand of the transfected siRNA. Others have shown that 2′-O-methylation of short RNA strands generates TLR antagonists,[16, 30–31] which explains how modification of only one strand in an siRNA can block immune stimulation caused by the other strand. To determine whether our nucleobase modification was functioning as an antagonist, we stimulated TNFα production in PBMCs using the small-molecule agonist R-848 in the presence and absence of the PIK3CB siRNA modified at guide strand position 1 on the WC edge. Indeed, the presence of this modified siRNA reduced R-848-mediated TNFα production, consistent with its function as a TLR8 antagonist (Figure 3).
RNA interference activity of siRNAs bearing adenosine modifications
To evaluate the RNAi performance of modified PIK3CB siRNAs, we tested their activity in HeLa cells using a dual-luciferase assay. We used the previously reported psiCHECK-2 vector[22], wherein the PIK3CB target sequence was inserted into the 3′-UTR of the Renilla luciferase gene. The knockdown activities of the WC face modified siRNAs were compared to the previously reported knockdown activities of the Hoogsteen face modified siRNA (Figure 4).[22]
Figure 4.
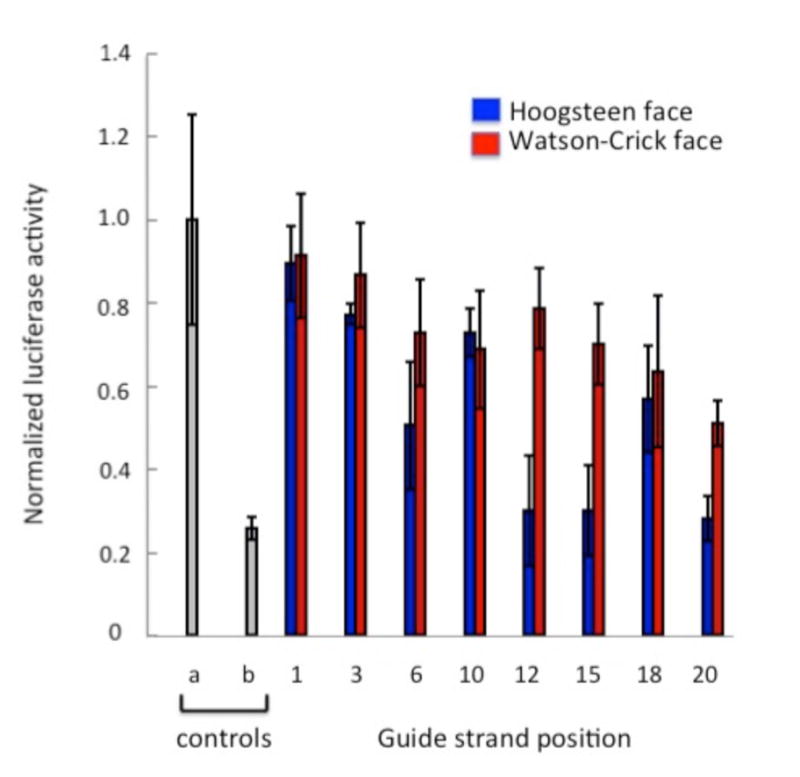
Knockdown activity of modified PIK3CB siRNAs. Activity is reported as the ratio of Renilla/firefly luciferase signals. Controls: (a) buffer alone; (b) unmodified duplex, Blue bars: Hoogsteen face localized modification; Red bars: Watson-Crick (WC) face localized modification. SiRNAs were transfected at a concentration of 30 pM in HeLa cells.
We found that modifications to the WC face at each of the eight guide strand adenosines inhibited RNAi activity (Figure 5). Previous RNAi studies with a different sequence had shown that position 2 of the guide strand is sensitive to this modification whereas modification at position 14 was tolerated.[25] Here, all eight positions tested showed significantly reduced RNAi activity, with position 20 as the most tolerant to modification (~ two-fold reduction in knockdown compared to the unmodified siRNA). On the other hand, modification on the Hoogsteen face at positions 12, 15, and 20 in this sequence retains RNAi activity (Figure 4).[22] Thus, for this sequence, the best strategy for maintaining RNAi activity while reducing immune stimulation using these nucleobase modifications, is to substitute position 12 with the Hoogsteen face modification. This modification at position 12 causes a three-fold decrease in TNFa production (Figure 2) while retaining native levels of knockdown activity (Figure 4).[22] While this modification strategy is not yet optimal because immune stimulation is not completely inhibited, our previous studies suggest that pairing this modification at guide stand position 12 with a modified passenger strand would likely reduce immune stimulation further.[21]
Conclusions
Molecular modeling with recently published crystal structures of Toll-like receptor 8 (TLR8) bound to small molecule agonists suggested that modification of either the Watson-Crick (WC) or Hoogsteen face of adenosine could disrupt nucleotide/TLR8 interactions. We found that modifying the WC face of adenosine is more effective at blocking TNFα production in PBMCs than modification to the Hoogsteen face. For the sequence tested, modifications near the 5′ end of an siRNA were more effective at blocking cytokine production than those placed near the 3′ end. This position dependence highlights the importance of specific nucleotide positions within an immunostimulatory oligonucleotide. This work advances our understanding of how chemical modifications can be used to optimize siRNA performance.
Experimental Section
Modeling nucleotide binding to TLR8
Nucleoside-3′,5′-bisphosphates were modeled into the CLO97 binding site in human TLR8 via conformer generation, receptor creation and molecular docking protocols using the OpenEye suite of programs.[9–10] Conformer Generation: Each of the four canonical ribonucleoside 3′, 5′-bisphosphates had conformers generated using OMEGA. Conformer generation was expanded to 5000 conformers from the default 200 conformers in order to account for more binding poses. Receptor Generation: Receptors were generated from the high-resolution structure of CLO97 bound to TLR8.[7] A receptor using CLO97 as the ligand was first used to study how adenosine-3′,5′-bisphosphate could bind into the receptor. The position of the exocyclic amine of CLO97 was used as a constraint in order to retain this interaction with D543, a residue known be necessary for both small molecule and RNA interactions (Supplementary Figure 4).[7–8] Using the best fit pose of the adenosine-3′,5′-bisphosphate shown in Figure 1B, a new receptor was generated constraining the 3′ phosphate, 3′ carbon and 2′ hydroxyl positions (Supplementary Figure 5). This was done to minimize binding poses that would not accommodate the nucleotide as part of an RNA strand. Molecular Docking: Using FRED, the nucleoside bisphosphates were docked and analyzed for potential ligand/protein interactions. Pose categories were generated when the nucleobase of a particular nucleoside would consistently overlay with the nucleobase position from its other poses (Supplementary Figures 1–3, Supplementary Tables 1). Therefore, binding modes represented in the top 10 poses that had nucleotide/protein interactions consistent with mutagenesis and available binding data as well as observed at high pose frequencies were regarded to be significant. The top 10 poses were ordered in rank of their computed chemguass4 scores via FRED.
Synthesis, purification and quantification of 21-mer RNA
The 7-ethynyl-8-aza-7-deazaadenosine and N2-propargyl-2-aminopurine ribonucleoside phosphoramidites were synthesized as previously described[22, 24]. The RNA 21-mer oligonucleotides incorporating these phosphoramidites were synthesized on an ABI 394 synthesizer (DNA/Peptide Core Facility, University of Utah, Salt Lake City) using 5′-DMTr protected β-cyanoethyl phosphoramidites (1.0 mmol scale). All the oligonucleotides were deprotected as previously described.[32] The RNA oligonucleotides containing ethynyl and propargyl modifications were gel purified and quantified as previously described[33]. MALDI mass spectrometry was used to confirm the identity of the RNAs.
Triazole formation with alkyne-modified PIK3CB siRNA guide strands
The reaction of the 7-ethynyl-8-aza-7-deazaadenosine modified siRNA with 1-(2-azidoethyl)-piperidine was performed as described previously.[22] Products were gel purified[33] and subjected to mass spectrometry analysis with mass values as previously reported.[22] A dry pellet of pure propargyl containing RNA (20 nmol) was dissolved in H2O (1 μL), then treated sequentially with tris-[1-(3-hydroxypropyl)-1H-[1,2,3]triazol-4-yl)methyl]amine (THPTA) ligand[34] (1 μL, 1 M in H2O), CuSO4 (1 μL, 100 mM in H2O), sodium ascorbate (1 μL, 1 M in H2O) and 1-(2-azidoethyl)-piperidine (1 μL of 50 mM) in Tris-HCl (0.5 M, pH 8.0). The resulting reaction mixture was incubated at room temperature for 6.5 h. The reaction mixtures were diluted to twice the original volume with PAGE loading buffer (80% formamide containing 10 mM EDTA). The 21-mer RNA product was gel purified and quantified as above. MALDI-MS or ESI-MS was used to confirm the identity of the reaction products for all PIK3CB guide strands modified with N-ethylpiperidine triazole linked through N2 of 2-aminopurine. ESI-MS was used for the guide strands modified at positions 1, 3, 6, 10, 12, 15, and 20. The calculated monoisotopic mass (positive mode) is equal for all the siRNAs containing this modification. [M+H]+ calculated: 7017.031; [M+Na]+ calculated: 7039.013; [M+2Na]+ calculated: 7060.995. Observed: position 1 = 7037.06; position 3 = 7016.07; position 6 = 7036.07; position 10 = 7059.01; position 12 = 7059.02; position 15 = 7039.06; position 20 = 7036.10. MALDI-MS was used to verify the identity of the position 18 modified guide strand. Calculated average mass (positive mode) [M+H]+: 7020.175. Observed: 7021.22.
siRNA duplex formation
Hybridization of the siRNA duplex was carried out by mixing equal amounts of pure modified guide and passenger strands, reaching a final concentration of 20 uM in Tris-HCl (10 mM) and KCl (50 mM, pH 7.5). The samples were heated for 5 minutes at 95°C, and were slowly cooled to room temperature for over 2–3 hours.
Cytokine assay
Peripheral blood mononuclear cells (PBMCs) (Sanguine Biosciences) were plated in a 96-well plate at 2.5–5 × 105 cells per well in RPMI medium (Cellgro) supplemented with penicillin/streptomycin (2X) and 10% fetal bovine serum. N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium methyl-sulfate (DOTAP) (Roche Applied Science) was used as a liposomal transfection agent. Formulation was carried out by mixing siRNAs and DOTAP at a working concentration of 10 μg DOTAP/μg of RNA, and incubated at room temperature for 15–40 minutes. Cultured PBMCs were treated with the formulated siRNAs and incubated for 16 – 20 h at 37 °C. Human TNFa in the cell culture supernatants was quantified in triplicate by using LEGEND MAX™ ELISA kit with pre-coated plates (BioLegend, Inc.), while human IFNa was measured in triplicate using VeriKine™ Human IFNa ELISA Kit (PBL InterferonSource). Small molecule agonist R-848 (Imgenex) was used as a positive control.
Cell culture and RNAi activity assay
HeLa cells (ATCC) were grown in Dulbecco’s modified Eagle’s medium (DMEM, GIBCO) supplemented with 1x Antibiotic-Antimycotic (Anti-Anti) (Gibco) and 10% fetal bovine serum, and incubated in 5% CO2 at 37°C. SiPORT NeoFX transfection reagent (Ambion) was used for the reverse transfection of HeLa cells. Cells were allowed to grow in flasks until reaching 80–90% confluence, then treated with Accutase (Innovative Cell Technologies) and diluted in fresh medium (DMEM, 10% FBS, 1x Anti-Anti) to a concentration of 1 × 105 cells mL−1. The (psiCHECK-2-PIK3CB) plasmid[22], which contains Renilla (hRluc) and Firefly (hluc+) luciferase reporter genes and the PIK3CB siRNA target sequence (5′-GCACATCTCCTAAUATGAATCCTATCAGAA-3′) inserted into the 3′ UTR of the Renilla luciferase gene was used for RNAi experiments. The Renilla luciferase functions as a reporter of siRNA activity, while the firefly luciferase functions as an internal control. SiRNA and plasmid transfections were carried out as previously described[21–22] using 20 ng of psiCHECK-2-PIK3CB per siRNA assayed. This assay was carried out in triplicate.
Supplementary Material
Acknowledgments
P.A.B. acknowledges the National Institutes of Health for financial support in the form of grant R01-GM080784.
Footnotes
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Supporting information for this article is given via a link at the end of the document.
References
- 1.Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. Nat Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 2.Semizarov D, Frost L, Sarthy A, Kroeger P, Halbert DN, Fesik SW. Proc Natl Acad Sci U S A. 2003;100:6347–6352. doi: 10.1073/pnas.1131959100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sioud M, Sorensen DR. Biochem Biophys Res Commun. 2003;312:1220–1225. doi: 10.1016/j.bbrc.2003.11.057. [DOI] [PubMed] [Google Scholar]
- 4.Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR. Nat Cell Biol. 2003;5:834–839. doi: 10.1038/ncb1038. [DOI] [PubMed] [Google Scholar]
- 5.Judge A, MacLachlan I. Hum Gene Ther. 2008;19:111–124. doi: 10.1089/hum.2007.179. [DOI] [PubMed] [Google Scholar]
- 6.Forsbach A, Nemorin JG, Montino C, Muller C, Samulowitz U, Vicari AP, Jurk M, Mutwiri GK, Krieg AM, Lipford GB, Vollmer J. J Immunol. 2008;180:3729–3738. doi: 10.4049/jimmunol.180.6.3729. [DOI] [PubMed] [Google Scholar]
- 7.Tanji H, Ohto U, Shibata T, Miyake K, Shimizu T. Science. 2013;339:1426–1429. doi: 10.1126/science.1229159. [DOI] [PubMed] [Google Scholar]
- 8.Colak E, Leslie A, Zausmer K, Khatamzas E, Kubarenko AV, Pichulik T, Klimosch SN, Mayer A, Siggs O, Hector A, Fischer R, Klesser B, Rautanen A, Frank M, Hill AV, Manoury B, Beutler B, Hartl D, Simmons A, Weber AN. J Immunol. 2014;192:5963–5973. doi: 10.4049/jimmunol.1303058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hawkins PC, Nicholls A. J Chem Inf Model. 2012;52:2919–2936. doi: 10.1021/ci300314k. [DOI] [PubMed] [Google Scholar]
- 10.Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT. J Chem Inf Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Judge AD, Bola G, Lee AC, MacLachlan I. Mol Ther. 2006;13:494–505. doi: 10.1016/j.ymthe.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Kariko K, Buckstein M, Ni H, Weissman D. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 13.Morrissey DV, Lockridge JA, Shaw L, Blanchard K, Jensen K, Breen W, Hartsough K, Machemer L, Radka S, Jadhav V, Vaish N, Zinnen S, Vargeese C, Bowman K, Shaffer CS, Jeffs LB, Judge A, MacLachlan I, Polisky B. Nat Biotechnol. 2005;23:1002–1007. doi: 10.1038/nbt1122. [DOI] [PubMed] [Google Scholar]
- 14.Sioud M. Eur J Immunol. 2006;36:1222–1230. doi: 10.1002/eji.200535708. [DOI] [PubMed] [Google Scholar]
- 15.Sioud M. Methods Mol Biol. 2009;487:41–59. doi: 10.1007/978-1-60327-547-7_2. [DOI] [PubMed] [Google Scholar]
- 16.Sioud M, Furset G, Cekaite L. Biochem Biophys Res Commun. 2007;361:122–126. doi: 10.1016/j.bbrc.2007.06.177. [DOI] [PubMed] [Google Scholar]
- 17.Chiu YL, Rana TM. RNA. 2003;9:1034–1048. doi: 10.1261/rna.5103703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haringsma HJ, Li JJ, Soriano F, Kenski DM, Flanagan WM, Willingham AT. Nucleic Acids Res. 2012;40:4125–4136. doi: 10.1093/nar/gkr1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kenski DM, Butora G, Willingham AT, Cooper AJ, Fu W, Qi N, Soriano F, Davies IW, Flanagan WM. Molecular therapy Nucleic acids. 2012;1:e5. doi: 10.1038/mtna.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fucini RV, Haringsma HJ, Deng P, Flanagan WM, Willingham AT. Nucleic Acid Ther. 2012;22:205–210. doi: 10.1089/nat.2011.0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peacock H, Fucini RV, Jayalath P, Ibarra-Soza JM, Haringsma HJ, Flanagan WM, Willingham A, Beal PA. J Am Chem Soc. 2011;133:9200–9203. doi: 10.1021/ja202492e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ibarra-Soza JM, Morris AA, Jayalath P, Peacock H, Conrad WE, Donald MB, Kurth MJ, Beal PA. Org Biomol Chem. 2012;10:6491–6497. doi: 10.1039/c2ob25647a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phelps KJ, Ibarra-Soza JM, Tran K, Fisher AJ, Beal PA. ACS Chem Biol. 2014;9:1780–1787. doi: 10.1021/cb500270x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peacock H, Maydanovych O, Beal PA. Org Lett. 2010;12:1044–1047. doi: 10.1021/ol100019r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peacock H, Fostvedt E, Beal PA. ACS Chem Biol. 2010;5:1115–1124. doi: 10.1021/cb100245u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L, Linsley PS. RNA. 2006;12:1179–1187. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gorden KB, Gorski KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, Tomai MA, Alkan SS, Vasilakos JP. J Immunol. 2005;174:1259–1268. doi: 10.4049/jimmunol.174.3.1259. [DOI] [PubMed] [Google Scholar]
- 28.Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, Noronha A, Manoharan M, Akira S, de Fougerolles A, Endres S, Hartmann G. Nat Med. 2005;11:263–270. doi: 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- 29.Philbin VJ, Dowling DJ, Gallington LC, Cortes G, Tan Z, Suter EE, Chi KW, Shuckett A, Stoler-Barak L, Tomai M, Miller RL, Mansfield K, Levy O. J Allergy Clin Immunol. 2012;130:195–204 e199. doi: 10.1016/j.jaci.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furset G, Floisand Y, Sioud M. Immunology. 2008;123:263–271. doi: 10.1111/j.1365-2567.2007.02695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robbins M, Judge A, Liang L, McClintock K, Yaworski E, MacLachlan I. Mol Ther. 2007;15:1663–1669. doi: 10.1038/sj.mt.6300240. [DOI] [PubMed] [Google Scholar]
- 32.Maydanovych O, Easterwood LM, Cui T, Veliz EA, Pokharel S, Beal PA. Methods Enzymol. 2007;424:369–386. doi: 10.1016/S0076-6879(07)24017-0. [DOI] [PubMed] [Google Scholar]
- 33.Peacock H, Kannan A, Beal PA, Burrows CJ. J Org Chem. 2011;76:7295–7300. doi: 10.1021/jo2012225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong V, Presolski SI, Ma C, Finn MG. Angew Chem Int Ed Engl. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.