

Abstract
Myelinated axons have a distinct protein architecture essential for action potential propagation, neuronal communication, and maintaining cognitive function. Damage to myelinated axons, associated with cerebral hypoperfusion, contributes to age-related cognitive decline. We sought to determine early alterations in the protein architecture of myelinated axons and potential mechanisms after hypoperfusion. Using a mouse model of hypoperfusion, we assessed changes in proteins critical to the maintenance of paranodes, nodes of Ranvier, axon–glial integrity, axons, and myelin by confocal laser scanning microscopy. As early as 3 d after hypoperfusion, the paranodal septate-like junctions were damaged. This was marked by a progressive reduction of paranodal Neurofascin signal and a loss of septate-like junctions. Concurrent with paranodal disruption, there was a significant increase in nodal length, identified by Nav1.6 staining, with hypoperfusion. Disruption of axon–glial integrity was also determined after hypoperfusion by changes in the spatial distribution of myelin-associated glycoprotein staining. These nodal/paranodal changes were more pronounced after 1 month of hypoperfusion. In contrast, the nodal anchoring proteins AnkyrinG and Neurofascin 186 were unchanged and there were no overt changes in axonal and myelin integrity with hypoperfusion. A microarray analysis of white matter samples indicated that there were significant alterations in 129 genes. Subsequent analysis indicated alterations in biological pathways, including inflammatory responses, cytokine-cytokine receptor interactions, blood vessel development, and cell proliferation processes. Our results demonstrate that hypoperfusion leads to a rapid disruption of key proteins critical to the stability of the axon–glial connection that is mediated by a diversity of molecular events.
Introduction
The integrity of the brain's white matter, comprised mainly of myelinated axons, is critical in regulating efficient neuronal communication and maintaining cognitive function (Nave, 2010). Myelination of axons, by oligodendrocytes, determines the localization of key proteins along the axon and segregates the axonal membrane into defined regions: the node of Ranvier, paranode, juxtaparanode, and internode (Rios et al., 2003; Susuki and Rasband, 2008). In the adult brain, the nodes of Ranvier located at defined points along the axolemma are critical for action potential propagation. They are comprised of voltage-gated sodium channels that are clustered at a high density within a diffusion barrier of septate-like junctions. These paranodal septate-like junctions, consisting of glial Neurofascin (Nfasc)155, axonal contactin, and contactin-associated protein (Caspr), are critical to the maintenance of axon–glial junctions at the paranodes. A dynamic relationship exists between axons and glia, in addition to highly coordinated signaling with vascular components (endothelial cells, pericytes) that comprise the oligovascular niche (Arai and Lo, 2009), which, if disrupted, could impede neuronal communication and cognitive abilities.
With advancing age, there is a general decline in white matter integrity associated with cognitive decline (Bartzokis et al., 2004; Bastin et al., 2009). Although there is evidence that the protein architecture of myelinated axons may be altered at nodal regions in the aging brain (Lasiene et al., 2009), as yet the underlying molecular changes that occur early are not known. Cerebral hypoperfusion is suggested to contribute to the development of white matter changes in the aging brain (Fernando et al., 2006). In response to severe hypoperfusion as a result of stroke, axon–glial integrity is impaired, as demonstrated by a loss of myelin-associated glycoprotein (MAG) (Waxman, 2006), a protein important in the maintenance of the axon–glial connection (Quarles, 2007; Schnaar and Lopez, 2009). Glutamate excitotoxicity has also been shown to cause paranodal myelin splitting and retraction (Fu et al., 2009). Disruption of the protein architecture of myelinated axons is often associated with demyelinating diseases. In multiple sclerosis, paranodal disruption of Nfasc155 and Caspr leads to dispersion of Nav1.6 and Nfasc186 proteins along the axon (Howell et al., 2006; Waxman, 2006) and impaired nerve function (Quarles, 2007).
Using a mouse model of chronic cerebral hypoperfusion (Shibata et al., 2004), which mimics the modest, sustained reductions in blood flow of the aging brain, we previously demonstrated that hypoperfusion results in damage to white matter at the cellular level (Coltman et al., 2011) and in vivo (Holland et al., 2011). We hypothesized that hypoperfusion would cause alterations in axon–glial integrity associated with a disruption in key proteins responsible for anchoring nodal proteins and paranodal junctions. To test this, we undertook a systematic analysis of the integrity of key proteins at the paranodal and nodal regions in response to hypoperfusion. To define the complex alterations in gene expression and biological pathways that may contribute to axon–glial damage, microarray analysis was conducted in white matter-enriched tissue. Our results indicate that diverse molecular pathways are altered with hypoperfusion.
Materials and Methods
Animals.
Male C57BL/6J mice (25–30 g, ∼3–4 months old) were obtained from Charles River. All procedures were authorized under the Home Office approved project license number 60/3722 title “Pathophysiology of Alzheimer's disease: link to cerebrovascular disease” held by Dr. K. Horsburgh. This license was approved by the University of Edinburgh's Ethical Review Committee on 4 June 2007 and the Home Office on 30 July 2007, is valid until 30 July 2012, and adhered to regulations specified in the Animals (Scientific Procedures) Act (1986).
Chronic cerebral hypoperfusion.
Chronic cerebral hypoperfusion was induced by applying microcoils (0.18 mm internal diameter; Sawane Spring Co.) to both common carotid arteries as previously described (Shibata et al., 2004; Coltman et al., 2011) under isoflurane anesthesia. There was a period of 30 min between the insertion of each coil. Sham-operated animals underwent an identical surgical approach except that microcoils were not applied to the common carotid arteries. The temperature of the mice was maintained between 36.5°C and 37.5°C. The recovery of the mice was monitored closely, and the weight of the mice, whether they were eating and drinking, and any signs of overt neurological dysfunction (e.g., circling, rolling, hunching) were recorded.
Tissue preparation and immunohistochemistry.
At either 3 d or 1 month after surgery, mice were deeply anesthetized with 5% isoflurane and transcardially perfused with 20 ml of 0.9% heparinized PBS and then 20 ml of 4% paraformaldehyde in 0.1% phosphate buffer. After perfusion, brains were removed and postfixed either in 4% PFA overnight (for nodal and internodal structures) or for 30 min (for Nfasc186 immunolabeling or with subsequent Bouins fixation for Nfasc155 immunolabeling). Brains were cut along the midline and floating sagittal sections (50 μm thickness) were produced with a vibrating blade microtome (Hydrax V50; Zeiss). Sections were collected and stored in cryoprotective medium (30% glycerol/30% ethylene glycol in phosphate buffer) and stored at −20°C until use. Adjacent sections were stained with hematoxylin and eosin (H&E) stain to determine the presence of ischemic neuronal perikaryal damage. Any animals exhibiting ischemic damage were excluded from the study.
The antibodies used in this study were as follows: Nav1.6 (1:200, AB5580; Millipore), MAG (1:100, L20, sc-9543; Santa Cruz Biotechnology), myelin basic protein (MBP; 1:100, MAB386; Millipore), SMI312R (1:100; Covance), Caspr (1:100, clone K65/35; UC Davies NIH NeuroMab), Nfasc155 (NFF3, 1:1000) (Tait et al., 2000), Nfasc186 (MNF2, 1:200) (Tait et al., 2000), Neurofascin common epitope (1:1000, AB15188; Millipore), AnkyrinG (H-215, sc-28561; Santa Cruz Biotechnology), human/mouse active caspase-3 (1:100, AF835; R & D Systems). Secondary Cy2, 3, and 5 antibodies were purchased from Jackson ImmunoResearch Laboratories. All antibodies were incubated overnight at 4°C in PBS, pH 7.4, with 0.1% Triton X-100.
Analysis of immunohistochemistry.
Immunolabeled 50 μm vibratome sections were analyzed using a laser scanning confocal microscope (Zeiss Axioskope LSM 510). All images were acquired with a 63× oil-immersion objective (numerical aperture, 1.4), a pinhole of 1 Airy unit, and a 1024 × 1024 pixel resolution. The corresponding Nyquist settings, 3.1× zoom and 0.13 μm z-steps, were used to allow image deconvolution (Huygens Professional Deconvolution Software; SVI) for nodal counts, length measurements, and colocalization assessment. All length measurements and counts were performed in an image stack spanning 10 μm using ImageJ MBI 1.43 s. The observer was always blinded to the surgical procedure.
Sodium channel cluster (Nav1.6) length was assessed after 3 d hypoperfusion (n = 9 sham; n = 8 hypoperfusion) and 1 month hypoperfusion (n = 10 sham; n = 6 hypoperfusion). The first 40 nodes in the acquired 10 μm confocal stack that were not directly in contact with the border of the stack were analyzed to exclude only partially imaged nodes of Ranvier. The regions of the corpus callosum, internal capsule, and optic tract in sagittal sections were imaged at the stereotactic co-ordinates, lateral 2.40 ± 0.1 mm, according to Franklin and Paxinos (1997). The identity of nodes of Ranvier was confirmed by Caspr double-labeling. AnkyrinG and Nfasc186 cluster length was analyzed accordingly. AnkyrinG was assessed after 3 d (n = 9 sham; n = 8 hypoperfusion) and 1 month (n = 9 sham; n = 10 hypoperfusion) in the corpus callosum. Neurofascin186 was assessed after 3 d (n = 6 sham; n = 6 hypoperfusion) and 1 month (n = 6 sham; n = 4 hypoperfusion) in the corpus callosum. Different cohorts needed to be used due to differences in fixation methods. All animals with ischemic neuronal perikaryal damage or infarcts confirmed by H&E staining were excluded from analysis (12%).
Colocalization of paranodal proteins was assessed using Caspr and Neurofascin/Nfasc155 double-immunolabeling. The tissue was postfixed using Bouins fixative for Nfasc155 (Sigma Aldrich). The corpus callosum of animals after 3 d of hypoperfusion (n = 5 sham; n = 5 hypoperfusion) and 1 month hypoperfusion (n = 6 sham; n = 4 hypoperfusion) was imaged. To identify paranodal pairs, paired Caspr-positive rectangular clusters (paranodes), separated by a small void of labeling (indicating the node of Ranvier), were analyzed regarding their Nfasc155 colabeling. If both labels (Caspr and Nfasc155) were completely overlapping in a paranodal pair, the node was defined as intact. If Nfasc155 labeling was absent on one or more sides of a Caspr-positive paranodal pair, the paranode was defined as disrupted. Overall, after 3 d of hypoperfusion, 129 paranodal pairs in the sham group and 138 in the hypoperfused group and after 1 month 185 paranodal pairs in the sham group and 129 in the hypoperfused group were analyzed. For Caspr and Neurofascin, common epitope labeling colocalization analysis was performed (Rickman et al., 2007) using ImageJ 1.43 (NIH) and Manders' coefficients determined.
Fluorescent intensity of MAG, MBP, and SMI312R immunohistochemistry was measured on single optical confocal sections, spanning 47 × 47 μm, of the corpus callosum, internal capsule, and optic tract after 3 d of hypoperfusion (n = 10 sham; n = 8 hypoperfusion) and 1 month of hypoperfusion (n = 10 sham; n = 7 hypoperfusion) using ImageJ 1.43 (NIH). Activated caspase-3 immunolabeling was assessed using a Leica DMR fluorescence microscope. All activated caspase-3-positive cells in a 50 μm thick sagittal section in the entire corpus callosum, optic tract, and internal capsule were counted.
Number of nodes of Ranvier.
The number of the nodes of Ranvier was determined on the same sections as the voltage-gated sodium channel cluster measurements. A 47 × 47 × 5 μm3 confocal stack per region was analyzed. All nodes included in the first optical section and/or contacting the left or upper edge of the stack were excluded; all nodes contacting the last and/or right and lower edge of the stack were included.
TEM of optic nerves.
Animals were transcardially perfused with 5% glutaraldehyde/4% paraformaldehyde after 1 month hypoperfusion. Optic nerves were removed, samples were fixed in 3% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.3, for 2 h, then washed in three 10 min changes of 0.1 m sodium cacodylate. Specimens were then postfixed in 1% osmium tetroxide in 0.1 m sodium cacodylate. After dehydration, samples were embedded in Araldite resin. Ultrathin sections (60 nm thick) were cut on a Reichert OMU4 ultramicrotome (Leica Microsystems), stained in uranyl acetate and lead citrate, then viewed in a Philips CM120 transmission electron microscope (FEI). Images were taken with an Orius CCD camera (Gatan). Paranodes in longitudinal sections of optic nerves were analyzed. The presence or absence of paranodal septate-like junctions were assessed in 37 paranodes of sham and 35 paranodes of hypoperfused animals (n = 3 sham; n = 3 hypoperfusion). The micrographs were assessed by two observers who were blinded to the surgical procedure. The optic nerve was studied for the ease of identification of sufficient numbers of nodal and paranodal structures, which is precluded in other regions, such as the corpus callosum and internal capsule, and is known to be damaged in response to hypoperfusion (Coltman et al., 2011).
White matter micropunch.
Seventy-two hours after hypoperfusion or sham surgery, mice (n = 12/group) were deeply anesthetized with isoflurane and perfused with 0.9% PBS. Brains were excised and placed in ice-cold PBS. Brains were subsequently sectioned with a vibratome to obtain two 1 mm coronal sections containing the genu and body of the corpus callosum. Sections were placed on glass slides and flash frozen on dry ice. White matter microsamples were collected via micropunch from subthawed sections using a 0.5 mm blunt-end needle attached to an air-filled syringe under a dissecting microscope. Micropunches of the corpus callosum were collected and aspirated into microcentrifuge tubes and stored at −80°C until RNA extraction.
RNA extraction.
RNA was extracted from white matter micropunches using an RNeasy Lipid Tissue Mini Kit (Qiagen) according to the manufacturer's instructions. RNA concentrations were determined using a Nanodrop (Thermo Scientific).
Microarray analysis.
Microarray RNA was pooled from 12 animals per cohort in groups of three, resulting in four pooled samples per cohort. RNA quality was confirmed using an RNA BioAnalyzer 2100 (Agilent). cDNA preparation, the microarray assay, and primary analysis was performed in the Sir Henry Wellcome Functional Genomics Facility at the University of Glasgow using standard Affymetrix protocols. Briefly, samples were hybridized to GeneChip Mouse Exon 1.0 ST arrays (Affymetrix) representing >44,000 genes using the fluidic station 400. Raw data were obtained and quality control was assessed using the GeneChip Command Console Software and the Expression console (Affymetrix).
Raw data were then normalized using GC-content by the Robust Multichip Average method within Genomics Suite (Partek) using a selection of probes corresponding to the core annotation level, resulting in 23,332 transcripts. Transcripts lacking gene symbol annotation were filtered out and only transcripts with full annotations (16,711) were statistically analyzed.
The log2 transformation of normalized signal intensities of annotated transcripts were analyzed with Student's t test. To control for false positives, a p value cutoff of 0.001 corresponding to a false discovery rate <0.11 (Benjamini and Hochberg, 1995) was used. Significantly altered genes were uploaded to the WebGestalt Gene Set Analysis Toolkit version 2 (http://bioinfo.vanderbilt.edu/webgestalt, Vanderbilt University) for enrichment analysis.
Results
Paranodal breakdown with hypoperfusion
To test whether chronic cerebral hypoperfusion would cause alterations in proteins essential for the integrity of the paranodal junctions, we analyzed two key proteins (Caspr and Nfasc155) that are contained within the paranodal septate-like junctions. Caspr is located at the axonal paranode, and Nfasc155 is contained at the opposing paranodal loops of the myelin sheath. These proteins function as a diffusion barrier for the nodal Nav1.6 sodium channels and are essential for the integrity of nodes of Ranvier. Sections were double-immunolabeled with the axonal Caspr and the glial Nfasc155 proteins and the number of disrupted paranodes was assessed in the corpus callosum (Fig. 1A). In response to hypoperfusion, there was a significant reduction in the number of paranodes labeled with both Nfasc155 and Caspr, due to a marked reduction of Nfasc155 immunolabeling. After 3 d of hypoperfusion, 21% of the paranodal pairs were affected compared with 9% in the sham group; after 1 month, 55% displayed a loss of the glial Nfasc155 protein compared with 7% in the sham group (Fig. 1D). To confirm that there was a loss of colocalization of Caspr and NFasc155, sections were double-immunolabeled with the axonal Caspr and panNeurofascin (labeling glial Nfasc155 and axonal Nfasc186), and colocalization was determined within the paranodes (Fig. 1B). A significant reduction in colocalization of Caspr and panNfasc within the paranodes was determined at both 3 d and 1 month after hypoperfusion (Fig. 1E). These data suggest a progressive breakdown of the paranodal composition with hypoperfusion.
Figure 1.
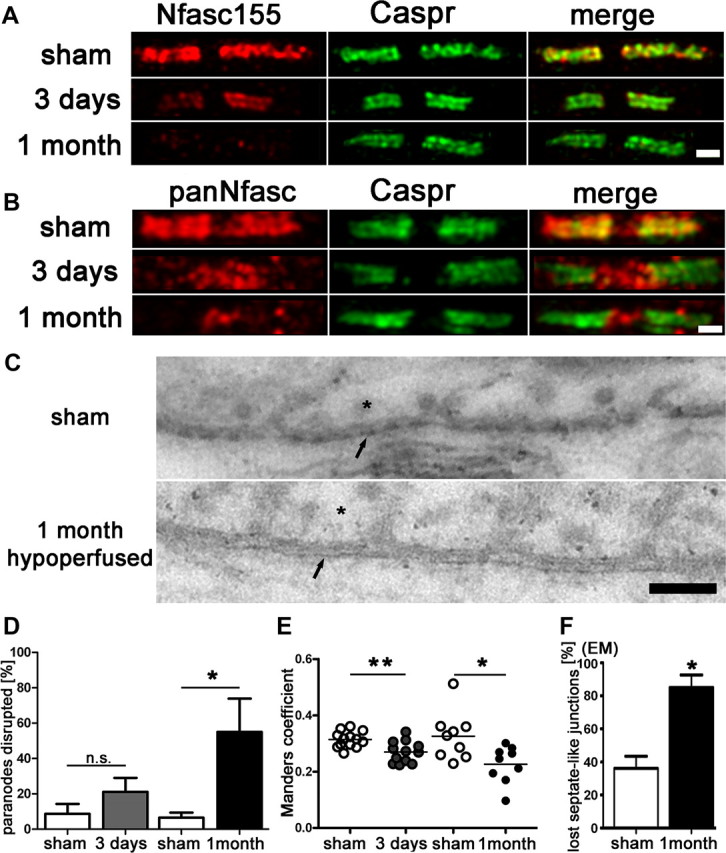
Paranodal disruption occurs early in response to hypoperfusion in the corpus callosum. A, B, Colocalization of axonal Caspr and glial Neurofascin protein indicates intact septate-like junctions at the paranodes in the sham group. In response to hypoperfusion, at both 3 d and 1 month, there is a selective loss of Neurofascin colocalization with Caspr, which is indicative of a disruption of the paranodes. Scale bar, 1 μm. C, Electron micrographs show paranodal disruption in response to hypoperfusion in the optic nerve. Arrows indicate axonal membrane at the paranodal region; asterisks indicate paranodal loop. Disruption of septate-like junctions is indicated by loss of transverse bands. Scale bars, 0.1 μm. D, There is a nonsignificant increase in the number of paranodes disrupted at 3 d hypoperfusion (n = 5) compared with shams (n = 5), which is significant at 1 month after hypoperfusion (n = 4) compared with shams (n = 6). Analysis was conducted in the corpus callosum. *p < 0.05 (unpaired t test, two-tailed). E, The overlap coefficient after Manders, which is insensitive to differences in signal intensities between the two channels, shows significant loss in colocalization after 3 d and after 1 month of hypoperfusion (3 d sham: n = 13, hypoperfusion: n = 12; 1 month sham: n = 9, hypoperfusion: n = 9). Analysis was conducted in the corpus callosum. *p < 0.05, **p < 0.005 (unpaired t test, two-tailed). F, A significant increase in paranodes without septate-like junctions was observed after 1 month of hypoperfusion. *p < 0.05 (unpaired t test, two-tailed).
To further determine whether the loss of Caspr/Nfasc double-immunolabeling indicates paranodal disruption, we performed ultrastructural analysis by EM on longitudinal optic nerve sections (Fig. 1C). EM images of paranodes showed a significant loss of septate-like junctions of paranodal loops in hypoperfused samples compared with sham controls (Fig. 1F).
Increased voltage-gated sodium channel cluster length with hypoperfusion
Breakdown of paranodal septate-like junctions could lead to changes in the structural organization of the nodes of Ranvier, which depend on a complex interplay between axons and glia. To determine this, we delineated nodal regions by double-labeling sections with antibodies to voltage-gated sodium channel (Nav1.6) and Caspr, an integral component of paranodal junctions. Voltage-gated sodium channel (Nav1.6) clusters are normally bounded by the paranodal protein Caspr and this was observed in the sham group (Fig. 2A). However, in response to hypoperfusion, after 3 d and 1 month, the distribution of the Nav1.6 channels was markedly altered compared with shams. In sham-controls, Nav1.6 staining is confined to the nodes of Ranvier and does not exceed beyond the localization of the paranodal barrier, as outlined by the protein Caspr. In contrast, in response to hypoperfusion, overlap between the Nav1.6 and Caspr staining was observed (Fig. 2A). To quantify the extent and progression of these changes, we measured the length of the Nav1.6 clusters in three white matter tracts (corpus callosum, internal capsule, and optic tract) after 3 d and 1 month of hypoperfusion (Fig. 2B). We found that there was a significant increase in the length of the Nav1.6 domain after 3 d hypoperfusion in the corpus callosum and internal capsule, which was exacerbated with a longer duration of hypoperfusion at 1 month. At this later time, the length of the Nav1.6 channels was also significantly increased in the optic tract after hypoperfusion (Fig. 2B).
Figure 2.

Voltage-gated sodium channel distribution is rapidly disrupted by hypoperfusion. A, Nav1.6 sodium channels, which outline the node of Ranvier, are normally bounded by the paranodal protein Caspr, as shown in the sham. In response to hypoperfusion, after 3 d and 1 month, the distribution of the Nav1.6 sodium channels is markedly altered and laterally spreads along the axon (corpus callosum). Scale bar, 1 μm. B, There is a significant increase in the length of the Nav1.6 clusters after 3 d of hypoperfusion in the corpus callosum and, after 1 month, in the internal capsule and optic tract. Forty nodes per animal were analyzed and these 240–400 nodes are plotted as cumulative frequency distribution, tabulated as relative frequencies as percentage (two-sample Kolmogorov–Smirnov test). Number of nodes of Ranvier remains unchanged after 3 d or 1 month of hypoperfusion. C, Confocal stack of nodes of Ranvier in the corpus callosum, double-labeled with Nav1.6 and Caspr. Left, Sham; right, 1 month of hypoperfusion. Scale bar, 1 μm. D, Numbers of nodes of Ranvier are not altered in response to hypoperfusion. After 3 d and after 1 month, the number of Nav1.6 immunopositive nodes was not changed between the sham and hypoperfused groups. Analysis was conducted by confocal laser microscopy in the corpus callosum, internal capsule, and optic tract in a 47 × 47 × 5 μm3 confocal stack (Mann–Whitney, two-tailed).
Number of nodes does not change after hypoperfusion
We next determined whether the number of nodes of Ranvier is changed in response to hypoperfusion. Analysis of a 47 × 47 × 5 μm3 confocal stack of the corpus callosum (Fig. 2C), internal capsule, and optic tract by stereologically counting the number of nodes of Ranvier for both time points revealed that their number was not altered in response to hypoperfusion (Fig. 2D). This indicates that chronic cerebral hypoperfusion does not lead to a loss in the number of nodes of Ranvier but a disruption of the distribution of Nav1.6 normally contained within the nodal region.
Nodal anchoring proteins remain unaltered after hypoperfusion
At the node of Ranvier, the scaffolding protein AnkyrinG interacts with Nfasc186 and Nav1.6 channels. This interaction is proposed to be involved in maintenance of the nodal Nav1.6 complex (Susuki and Rasband, 2008). Nfasc186 connects the complex to the extracellular matrix and AnkyrinG via βIV-spectrin to the underlying cytoskeleton. Therefore, we assessed changes in the nodal distribution of these proteins. Surprisingly, in response to hypoperfusion, at either 3 d or 1 month, AnkyrinG and Nfasc186 show no change in localization (Fig. 3A,B). Furthermore, the length of the AnkyrinG and Nfasc186 domains after 3 d or 1 month of hypoperfusion were not significantly different to shams, i.e., there was no lateral spreading of these protein domains along the axon (Fig. 3C).
Figure 3.
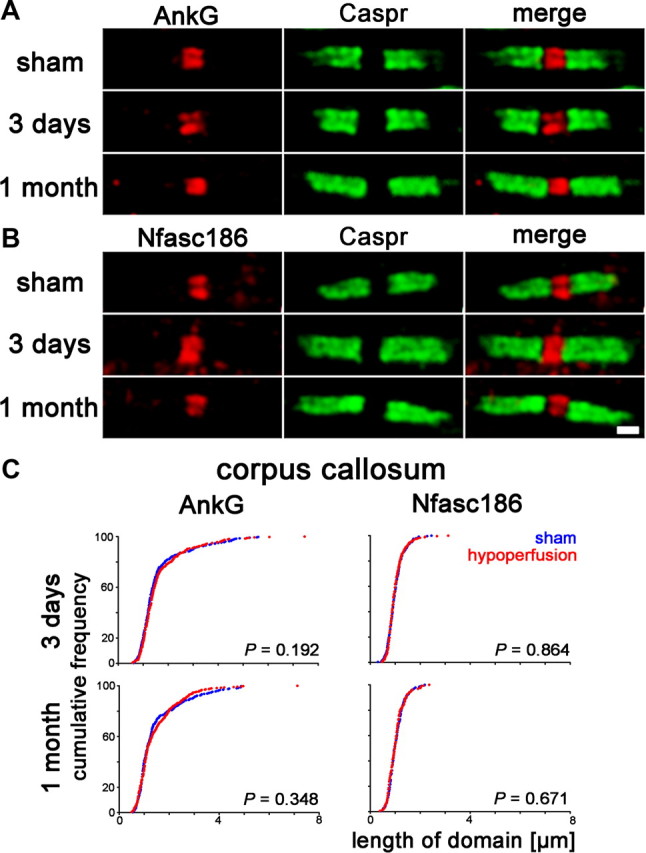
AnkyrinG and Neurofascin186 distribution remains unchanged with hypoperfusion. A, Axonal AnkyrinG remains within the node of Ranvier in response to hypoperfusion. B, The spatial distribution of Neurofascin186, which connects nodal proteins to the extracellular matrix, does not change. Scale bar, 1 μm. C, There is no significant increase in the length of the AnkyrinG or Neurofascin186 domains after 3 d or 1 month of hypoperfusion in the corpus callosum. Forty nodes per animal were analyzed and these 160–400 nodes are plotted as cumulative frequency (two-sample Kolmogorov–Smirnov test).
Internodal axon–glial alterations after hypoperfusion
The axon–glial connection at the internode between nodes of Ranvier consists of the glial cell membrane glycoprotein MAG and its axonal binding partners. As the paranodal axon–glial integrity appeared to be compromised due to hypoperfusion, we investigated whether the internodal axon–glial connection is similarly affected. We analyzed the spatial distribution of the glial MAG protein in response to hypoperfusion and found it to be markedly changed compared with the sham group. A discontinuous granular accumulation of the MAG labeling in the hypoperfused group was observed (Fig. 4A). To quantify these changes, we determined the fluorescent intensity of single optical confocal sections in the same white matter regions that displayed a breakdown in paranodal integrity, and found a significant reduction in the intensity of MAG staining at both 3 d and at 1 month of hypoperfusion compared with shams (Fig. 4B). These results build on and extend our previous findings in which MAG was shown to be disrupted in response to 1 month of hypoperfusion (Coltman et al., 2011). Together, these findings suggest that the internodal axon–glial connection is compromised due to chronic cerebral hypoperfusion.
Figure 4.
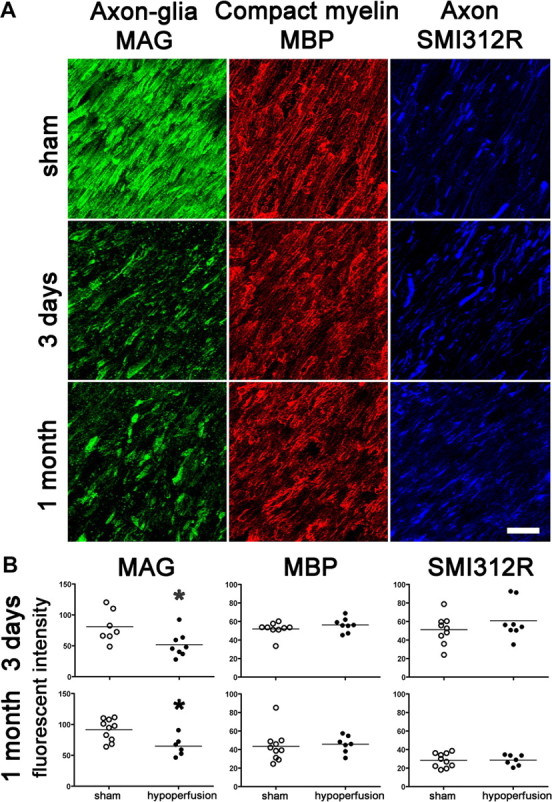
Axon–glial integrity is disrupted, whereas myelin and axonal integrity remains intact. A, Disruption of axon–glial integrity was defined as reduced and discontinuous granular accumulation of the MAG staining in response to hypoperfusion compared with shams. In contrast, the integrity of the myelin sheath (assessed by MBP) and integrity of axons (assessed by SMI312R) remains intact at 3 d and at 1 month. Scale bar, 10 μm. B, Measurement of the fluorescent intensity of MAG, MBP, and SMI312R staining was conducted by confocal laser microscopy in the corpus callosum, internal capsule, and optic tract in sham and hypoperfused groups at 3 d (sham, n = 9; hypoperfused, n = 8) and 1 month (sham, n = 10; hypoperfused, n = 7). There was a significant reduction in the intensity of MAG staining at both 3 d and at 1 month hypoperfusion compared with shams. There were no alterations in MBP and SMI312R immunostaining in response to hypoperfusion at any time studied. *p < 0.05 (Mann–Whitney, two-tailed).
Similarly, we assessed the spatial distribution and measured the intensity of the MBP, which is located in the compact myelin sheath, in single optical confocal sections. However, we found that neither the spatial distribution nor the fluorescent intensity of MBP was changed in response to hypoperfusion after 3 d or 1 month compared with shams (Fig. 4A,B).
In addition, we assessed whether the underlying axons, which would contain the binding partners for the MAG protein, show obvious morphological changes with hypoperfusion. The axons were labeled using the panaxonal neurofilament marker SMI312R and measured in the same way as the myelin on single optical confocal sections. There was no evidence that there were either structural changes or differences in the fluorescent intensity in response to hypoperfusion compared with shams (Fig. 4).
No evidence of increased apoptosis with hypoperfusion
To analyze the extent of apoptosis in our model, we counted activated caspase-3-positive cells in white matter regions after 3 d and 1 month of hypoperfusion (Fig. 5). No significant increase in numbers of apoptotic cells were determined in any of the regions studied or at any time point.
Figure 5.

Increased apoptosis does not occur after 3 d or 1 month of chronic cerebral hypoperfusion. The number of active caspase-3-positive cells in the corpus callosum, internal capsule, and optic tract is not increased in response to hypoperfusion. Colocalization of active caspase-3 with DAPI is shown. *p < 0.05 (Mann–Whitney, two-tailed).
Multiple molecular events in white matter associated with hypoperfusion
To investigate potential molecular events in response to hypoperfusion that may contribute to the axon–glial damage, we undertook a microarray study in white matter-enriched tissue at 3 d posthypoperfusion, a time when we detected early changes. Of the 16,711 fully annotated genes, 129 were found to be regulated differently within the white matter-enriched samples in response to hypoperfusion at p < 0.001. Hypoperfusion is associated with alterations in multiple biological processes. Enrichment analysis of the 129 regulated genes presented in Table 1 demonstrates the multiple processes regulated in response to hypoperfusion. In particular, genes associated with inflammatory responses (Table 1), such as those represented by the annotations Jak–STAT signaling pathway (seven genes) and cytokine–cytokine receptor interaction (eight genes), which included Stat3, Il2rg, Osmr, and Tnfrsf1a, were found to be regulated. Cardiovascular development, represented by the functional annotations angiogenesis (nine genes) and blood vessel development (10 genes) and including the genes Angpt2, Vegfa, Serpine1, and Dll4, were also altered. Cell proliferation processes were altered as represented by regulation of cell proliferation (15 genes) and included Cdh5, Adm, Igfb3, and Cdkn1a. Other highlighted processes were anatomical structure formation/morphogenesis and cell adhesion.
Table 1.
Microarray analysis of enriched white matter from the corpus callosum indicates multiple pathways involved in the response to hypoperfusion
| Database | Biological process | Observed | Expected | p value |
|---|---|---|---|---|
| GO | Regulation of cell proliferation | 15 | 3.66 | 0.001 |
| GO | Angiogenesis | 9 | 1.23 | 0.001 |
| KEGG | Jak–STAT signaling pathway | 7 | 1.06 | 0.0027 |
| KEGG | Complement and coagulation cascades | 5 | 0.54 | 0.0029 |
| GO | Organ development | 28 | 12.89 | 0.0037 |
| GO | Blood vessel morphogenesis | 9 | 1.7 | 0.0037 |
| GO | Blood vessel development | 10 | 2.07 | 0.0037 |
| GO | Vasculature development | 10 | 2.11 | 0.0037 |
| GO | Anatomical structure formation/morphogenesis | 12 | 2.88 | 0.0037 |
| KEGG | Cytokine–cytokine receptor interaction | 8 | 1.79 | 0.0039 |
| GO | Cell adhesion | 14 | 4.34 | 0.0048 |
| GO | Cell proliferation | 15 | 4.99 | 0.0048 |
| GO | Anatomical structure development | 32 | 16.39 | 0.0048 |
| KEGG | Acute myeloid leukemia | 4 | 0.42 | 0.0058 |
Genes (n = 129) altered (p < 0.001) were uploaded to WebGestalt enrichment analysis based on Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. Biological processes [adjusted p < 0.01 (Benjamini and Hochberg, 1995)] from both Gene Ontology and Kyoto Encyclopedia of Genes and Genomes databases, along with the observed and expected number of molecules represented for each category, are listed.
Hypoperfusion is a complex and multifactorial process, as demonstrated by the number of biological processes listed in Table 1. These affect genes involved in cell death and plasticity across multiple cell types even within white matter-enriched samples. The exact mechanism leading to the nodal changes observed remains to be established, but the complexity of action of the molecules involved would indicate that no single molecular event is responsible.
Discussion
Our results demonstrate that within days of cerebral hypoperfusion, there is a rapid and selective disruption of key proteins within the paranodal axon–glial junctions, which are critical to the stability and function of myelinated axons. Furthermore, in the early response to hypoperfusion, there are several biological pathways altered (notably inflammatory, vascular development, and cell adhesion) that could contribute to the axon–glial damage.
These results are relevant to age-related changes in white matter to which cerebral hypoperfusion is suggested to contribute. Although there is some evidence that the protein architecture may be altered at nodal regions in the aging brain (Lasiene et al., 2009), specific protein changes have yet to be identified. The present study identifies key changes in paranodal and nodal proteins in response to cerebral hypoperfusion. Normally, Nfasc155 is located at the paranodal loops and is known to form the paranodal septate-like junctions with the axonal protein Caspr and contactin. The stability of these proteins is critical to maintain nodal structure. We determined a disruption of the paranodes occurs in response to hypoperfusion, with a loss of the glial protein Nfasc155 at the paranode concomitant with a loss of septate-like junctions. This selective damage to paranodal integrity was also associated with a compromised nodal structure, as demonstrated by an increase in Nav1.6 channel cluster length. This increase in nodal length was dependent on the white matter tract examined. The corpus callosum and internal capsule appeared particularly susceptible early in response to hypoperfusion, with a significant increase in nodal length, detectable within 3 d, while the optic tract remained unaffected. After 1 month of hypoperfusion, the effect appeared to be progressive and alterations within the optic tract were subsequently determined. One explanation for this observation is regional differences of blood supply due to the cerebrovascular architecture and increased energy demand of myelinated fibers during even modest hypoxia (Dorr et al., 2007; Trapp and Stys, 2009). In addition to vascular alterations, dynamic distribution of blood and turnover of oligodendrocytes might result in the differences of progression between the analyzed white matter tracts.
Consistent with changes at the paranodes, there was a marked and rapid disruption of the axon–glial connection at the internodal region of myelinated axons in response to hypoperfusion. MAG plays an important role in signaling and maintenance of the axon–glial connection and the myelin sheath (for review, see Quarles, 2007; Schnaar and Lopez, 2009). The cellular distribution of glial MAG protein was disrupted in response to hypoperfusion and determined to accumulate in discontinuous granular staining. Interestingly, the protein level of MAG was not altered after 1 month of chronic cerebral hypoperfusion. This suggests that the observed changes could be a result of protein misdistribution rather than a loss of protein. A preferential loss of MAG in hypoxia-like white matter damage after stroke, multiple sclerosis, and other inflammatory brain diseases has been previously described (Aboul-Enein et al., 2003). A link between myelin-associated glycoprotein disruption and paranodal integrity has been shown in mice deficient in both MAG and myelin galactolipids, which display impaired maintenance of nodes of Ranvier (Marcus et al., 2002).
At the node of Ranvier, the scaffolding protein AnkyrinG interacts with Nfasc186 and Nav1.6 channels, and this interaction is proposed to be involved in maintenance of the nodal Nav1.6 complex (Susuki and Rasband, 2008). Nfasc186 connects the complex to the extracellular matrix and AnkyrinG via βIV-spectrin to the underlying cytoskeleton. Thus, it was surprising to determine in the present study that alterations in Nav1.6 were not accompanied by alterations in Nfasc186 and the anchoring protein AnkyrinG. The lack of changes in AnkyrinG and Nfasc186 despite alterations in Nav1.6 channels might indicate a dissociation of Nav1.6 channels from the AnkyrinG/Nfasc186 complex. There are a number of mechanisms by which voltage-gated sodium channels could dissociate from its AnkyrinG scaffolding. Previously, it has been demonstrated that phosphorylation of the Ankyrin-binding motif of Nav1.2 channels via protein kinase CK2 regulates the sodium channel interaction with Ankyrins (Bréchet et al., 2008). Alternatively, calpain-mediated proteolysis of the voltage-gated sodium channel α-subunit has been described for Nav1.2 (von Reyn et al., 2009). Similar events to Nav1.6 at the nodes of Ranvier could explain the dissociation of sodium channels from nodal AnkyrinG.
Additionally, we did not detect axonal damage nor demyelination in response to hypoperfusion, which is in agreement with our previous studies using cellular pathology and MRI approaches (Holland et al., 2011).The observation that AnkyrinG and Neurofascin186 are unaltered after hypoperfusion supports the absence of detectable alteration of the underlying axonal cytoskeleton. This is in contrast to more severe models of cerebral ischemia where myelin/axonal pathology is evident, [such as focal cerebral ischemia (Yam et al., 2000) and global cerebral ischemia (Jiwa et al., 2010)] in which blood flow is severely reduced (70%), and axonal and neuronal perikaryal damage occur within hours to days of the initial blood flow reduction. In the present study, hypoperfusion induced by carotid stenosis is associated with modest reductions in blood flow (<30%) within the first 24 h, and recovers gradually over weeks and is reduced by 10–20% compared with baseline (Shibata et al., 2004). This is more akin to changes that occur in the normal aging brain, where blood flow reductions are modest (∼20%) and sustained over years. The myelin sheath and underlying axon do not appear to be changed in our model of hypoperfusion but instead there is rapid axon–glial disruption. Western blot analysis revealed that there was no change in the levels of the proteins studied (MBP, SMI312, MAG, Nfasc155, and Nav1.6; our unpublished observations), providing further support that the hypoperfusion causes disruption of the cellular distribution and coupling of axon–glial proteins. Several mechanisms of damage, including excitotoxicity and caspase activation, have been suggested (Craner et al., 2004; Fu et al., 2009) to lead to axonal degeneration in models of severe ischemia. Since we found no evidence of axonal injury or caspase-3-labeled oligodendrocytes at the times studied (Fig. 5) in our model, alternative mechanisms may contribute to the axon–glial disruption.
Vascular brain injury has been shown to cause white matter deterioration and response of the oligodendrocyte lineage in a human cohort (Back et al., 2011). This highlights the link between vascular disturbances and changes in white matter integrity. Furthermore, dynamic communication between endothelial cells and oligodendrocytes is suggested to maintain their homeostasis and the integrity of white matter (Arai and Lo, 2009). This current view of an oligovascular niche suggests a highly coordinated exchange mediated by a diversity of signaling molecules between cerebral blood vessels and oligodendrocytes (Arai and Lo, 2009). Thus, it is likely that not one molecular event can explain the axon–glial damage in response to hypoperfusion. Indeed, within our microarray analysis of white matter-enriched tissue from the corpus callosum, we determined significant alterations in a number of genes and biological processes in response to hypoperfusion, including inflammatory, vascular, and cell-adhesion pathways. Each of these may contribute to the degenerative processes. The extent of their participation in axon–glial disruption requires intensive future studies.
It is well recognized that axon–glial integrity is critical to maintenance of cognitive function (Nave, 2010). Previously, we demonstrated that selective disruption of white matter integrity in response to hypoperfusion results in impairment of spatial working memory (Coltman et al., 2011). Although these distinct changes to the protein architecture at a subcellular level might appear subtle, they might be sufficient to explain the previously described cognitive dysfunction. In development, conduction velocities are significantly increased with clustering of Nav1.6 sodium channels at the node of Ranvier (Rasband et al., 1999) and disrupted paranodes lead to changes in electrophysiological measurements (Susuki et al., 2007). Disruption of nodal structures would be expected to result in changes in action potential propagation and thus could lead to functionally impaired connections between different brain regions.
We propose that the axon–glial connection at the paranodes and internodes in white matter tracts are rapidly disrupted in response to hypoperfusion relevant to the aging brain. The onset of this disruption is fast, which may indicate a narrow window of opportunity to prevent damage from occurring. However, in future studies, it will be important to determine whether this damage is reversible. In our model, the underlying axon remains intact, as does the compact myelin in the myelin sheaths. Although it is not clear whether long-term axonal degeneration and myelin sheath loss occurs with progressive hypoperfusion, there is emerging evidence that it leads to neuronal cell loss (Nishio et al., 2010). The complexity of the molecular mechanisms elicited by hypoperfusion suggests that there is no single molecular target. However, stabilizing the axon–glial connection at the early onset of disruption to myelinated axons could be crucial to prevent further degenerative processes and cognitive impairment.
Notes
Supplemental material for this article is available at http://www.cnr.ed.ac.uk/Research/horsburgh.html. Supplemental Table 1: Genes regulated by hypoperfusion in white matter-enriched microsamples. This material has not been peer reviewed.
Footnotes
This work was supported by the Disconnected Mind program (supported by Age, UK and Alzheimer's Research UK). We gratefully acknowledge the University of Edinburgh Centre for Cognitive Ageing and Cognitive Epidemiology (part of the cross council Lifelong Health and Wellbeing Initiative).
References
- Aboul-Enein F, Rauschka H, Kornek B, Stadelmann C, Stefferl A, Brück W, Lucchinetti C, Schmidbauer M, Jellinger K, Lassmann H. Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol. 2003;62:25–33. doi: 10.1093/jnen/62.1.25. [DOI] [PubMed] [Google Scholar]
- Arai K, Lo EH. An oligovascular niche: cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci. 2009;29:4351–4355. doi: 10.1523/JNEUROSCI.0035-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Kroenke CD, Sherman LS, Lawrence G, Gong X, Taber EN, Sonnen JA, Larson EB, Montine TJ. White matter lesions defined by diffusion tensor imaging in older adults. Ann Neurol. 2011;70:465–476. doi: 10.1002/ana.22484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartzokis G, Sultzer D, Lu PH, Nuechterlein KH, Mintz J, Cummings JL. Heterogeneous age-related breakdown of white matter structural integrity: implications for cortical “disconnection” in aging and Alzheimer's disease. Neurobiol Aging. 2004;25:843–851. doi: 10.1016/j.neurobiolaging.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Bastin ME, Clayden JD, Pattie A, Gerrish IF, Wardlaw JM, Deary IJ. Diffusion tensor and magnetization transfer MRI measurements of periventricular white matter hyperintensities in old age. Neurobiol Aging. 2009;30:125–136. doi: 10.1016/j.neurobiolaging.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Bréchet A, Fache MP, Brachet A, Ferracci G, Baude A, Irondelle M, Pereira S, Leterrier C, Dargent B. Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J Cell Biol. 2008;183:1101–1114. doi: 10.1083/jcb.200805169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coltman R, Spain A, Tsenkina Y, Fowler JH, Smith J, Scullion G, Allerhand M, Scott F, Kalaria RN, Ihara M, Daumas S, Deary IJ, Wood E, McCulloch J, Horsburgh K. Selective white matter pathology induces a specific impairment in spatial working memory. Neurobiol Aging. 2011;32:2324.e7–2324.e12. doi: 10.1016/j.neurobiolaging.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Craner MJ, Hains BC, Lo AC, Black JA, Waxman SG. Co-localization of sodium channel Nav1.6 and the sodium-calcium exchanger at sites of axonal injury in the spinal cord in EAE. Brain. 2004;127:294–303. doi: 10.1093/brain/awh032. [DOI] [PubMed] [Google Scholar]
- Dorr A, Sled JG, Kabani N. Three-dimensional cerebral vasculature of the CBA mouse brain: a magnetic resonance imaging and micro computed tomography study. Neuroimage. 2007;35:1409–1423. doi: 10.1016/j.neuroimage.2006.12.040. [DOI] [PubMed] [Google Scholar]
- Fernando MS, Simpson JE, Matthews F, Brayne C, Lewis CE, Barber R, Kalaria RN, Forster G, Esteves F, Wharton SB, Shaw PJ, O'Brien JT, Ince PG. White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke. 2006;37:1391–1398. doi: 10.1161/01.STR.0000221308.94473.14. [DOI] [PubMed] [Google Scholar]
- Franklin K, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic Press; 1997. [Google Scholar]
- Fu Y, Sun W, Shi Y, Shi R, Cheng JX. Glutamate excitotoxicity inflicts paranodal myelin splitting and retraction. PLoS One. 2009;4:e6705. doi: 10.1371/journal.pone.0006705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland PR, Bastin ME, Jansen MA, Merrifield GD, Coltman RB, Scott F, Nowers H, Khallout K, Marshall I, Wardlaw JM, Deary IJ, McCulloch J, Horsburgh K. MRI is a sensitive marker of subtle white matter pathology in hypoperfused mice. Neurobiol Aging. 2011;32:2325.e1–2325.e6. doi: 10.1016/j.neurobiolaging.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Howell OW, Palser A, Polito A, Melrose S, Zonta B, Scheiermann C, Vora AJ, Brophy PJ, Reynolds R. Disruption of neurofascin localization reveals early changes preceding demyelination and remyelination in multiple sclerosis. Brain. 2006;129:3173–3185. doi: 10.1093/brain/awl290. [DOI] [PubMed] [Google Scholar]
- Jiwa NS, Garrard P, Hainsworth AH. Experimental models of vascular dementia and vascular cognitive impairment: a systematic review. J Neurochem. 2010;115:814–828. doi: 10.1111/j.1471-4159.2010.06958.x. [DOI] [PubMed] [Google Scholar]
- Lasiene J, Matsui A, Sawa Y, Wong F, Horner PJ. Age-related myelin dynamics revealed by increased oligodendrogenesis and short internodes. Aging Cell. 2009;8:201–213. doi: 10.1111/j.1474-9726.2009.00462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus J, Dupree JL, Popko B. Myelin-associated glycoprotein and myelin galactolipids stabilize developing axo-glial interactions. J Cell Biol. 2002;156:567–577. doi: 10.1083/jcb.200111047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA. Myelination and support of axonal integrity by glia. Nature. 2010;468:244–252. doi: 10.1038/nature09614. [DOI] [PubMed] [Google Scholar]
- Nishio K, Ihara M, Yamasaki N, Kalaria RN, Maki T, Fujita Y, Ito H, Oishi N, Fukuyama H, Miyakawa T, Takahashi R, Tomimoto H. A mouse model characterizing features of vascular dementia with hippocampal atrophy. Stroke. 2010;41:1278–1284. doi: 10.1161/STROKEAHA.110.581686. [DOI] [PubMed] [Google Scholar]
- Quarles RH. Myelin-associated glycoprotein (MAG): past, present and beyond. J Neurochem. 2007;100:1431–1448. doi: 10.1111/j.1471-4159.2006.04319.x. [DOI] [PubMed] [Google Scholar]
- Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE, Shrager P. Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci. 1999;19:7516–7528. doi: 10.1523/JNEUROSCI.19-17-07516.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman C, Medine CN, Bergmann A, Duncan RR. Functionally and spatially distinct modes of munc18-syntaxin 1 interaction. J Biol Chem. 2007;282:12097–12103. doi: 10.1074/jbc.M700227200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios JC, Rubin M, St Martin M, Downey RT, Einheber S, Rosenbluth J, Levinson SR, Bhat M, Salzer JL. Paranodal interactions regulate expression of sodium channel subtypes and provide a diffusion barrier for the node of Ranvier. J Neurosci. 2003;23:7001–7011. doi: 10.1523/JNEUROSCI.23-18-07001.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaar RL, Lopez PH. Myelin-associated glycoprotein and its axonal receptors. J Neurosci Res. 2009;87:3267–3276. doi: 10.1002/jnr.21992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Ohtani R, Ihara M, Tomimoto H. White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke. 2004;35:2598–2603. doi: 10.1161/01.STR.0000143725.19053.60. [DOI] [PubMed] [Google Scholar]
- Susuki K, Rasband MN. Molecular mechanisms of node of Ranvier formation. Curr Opin Cell Biol. 2008;20:616–623. doi: 10.1016/j.ceb.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susuki K, Baba H, Tohyama K, Kanai K, Kuwabara S, Hirata K, Furukawa K, Furukawa K, Rasband MN, Yuki N. Gangliosides contribute to stability of paranodal junctions and ion channel clusters in myelinated nerve fibers. Glia. 2007;55:746–757. doi: 10.1002/glia.20503. [DOI] [PubMed] [Google Scholar]
- Tait S, Gunn-Moore F, Collinson JM, Huang J, Lubetzki C, Pedraza L, Sherman DL, Colman DR, Brophy PJ. An oligodendrocyte cell adhesion molecule at the site of assembly of the paranodal axo-glial junction. J Cell Biol. 2000;150:657–666. doi: 10.1083/jcb.150.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009;8:280–291. doi: 10.1016/S1474-4422(09)70043-2. [DOI] [PubMed] [Google Scholar]
- von Reyn CR, Spaethling JM, Mesfin MN, Ma M, Neumar RW, Smith DH, Siman R, Meaney DF. Calpain mediates proteolysis of the voltage-gated sodium channel alpha-subunit. J Neurosci. 2009;29:10350–10356. doi: 10.1523/JNEUROSCI.2339-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci. 2006;7:932–941. doi: 10.1038/nrn2023. [DOI] [PubMed] [Google Scholar]
- Yam PS, Dunn LT, Graham DI, Dewar D, McCulloch J. NMDA receptor blockade fails to alter axonal injury in focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:772–779. doi: 10.1097/00004647-200005000-00003. [DOI] [PubMed] [Google Scholar]