

Abstract
Androgen receptor (AR) is an attractive target in breast cancer because of its frequent expression in all the molecular subtypes, especially in estrogen receptor (ER)-positive luminal breast cancers. We have previously shown a role for AR overexpression in tamoxifen resistance. We engineered ER-positive MCF-7 cells to overexpress aromatase and AR (MCF-7 AR Arom cells) to explore the role of AR in aromatase inhibitor (AI) resistance. Androstendione (AD) was used as a substrate for aromatization to estrogen. The nonsteroidal AI anastrazole (Ana) inhibited AD-stimulated growth and ER transcriptional activity in MCF-7 Arom cells, but not in MCF-7 AR Arom cells. Enhanced activation of pIGF-1R and pAKT was found in AR-overexpressing cells, and their inhibitors restored sensitivity to Ana, suggesting that these pathways represent escape survival mechanisms. Sensitivity to Ana was restored with AR antagonists, or the antiestrogen fulvestrant. These results suggest that both AR and ERα must be blocked to restore sensitivity to hormonal therapies in AR-overexpressing ERα-positive breast cancers. AR contributed to ERα transcriptional activity in MCF-7 AR Arom cells, and AR and ERα co-localized in AD + Ana-treated cells, suggesting cooperation between the two receptors. AR-mediated resistance was associated with a failure to block ER transcriptional activity and enhanced up-regulation of AR and ER-responsive gene expression. Clinically, it may be necessary to block both AR and ERα in patients whose tumors express elevated levels of AR. In addition, inhibitors to the AKT/IGF-1R signaling pathways may provide alternative approaches to block escape pathways and restore hormone sensitivity in resistant breast tumors.
Keywords: Androgen receptor, Estrogen receptor, Breast cancer, Hormone resistance, Aromatase inhibitor
Introduction
ER-positive breast cancer is treated with the antiestrogen tamoxifen (Tam) and/or aromatase inhibitors (AIs). Currently, a sequence of an AI followed by Tam significantly reduces the risk recurrence and improves overall patient survival [14]. However, in about 40 % of cases, therapy resistance eventually develops and patients recur with metastatic disease. Multiple mechanisms responsible of resistance exist, including deregulation of the estrogen receptor (ER) pathway itself, alterations in cell cycle and cell survival molecules, and activation of escape pathways that provide tumors with alternative proliferative stimuli [30]. One recognized mechanism of Tam resistance is overexpression (OE) of epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) which can be reversed using specific inhibitors [24].
Using microarray analysis comparing Tam-resistant (TamR) metastatic versus Tam-sensitive (TamS) primary breast tumors, we demonstrated that AR levels were relatively higher expressed in the TamR tumors [8]. Tam was an agonist in AR-overexpressing cells, but the AR antagonist bicalutamide restored sensitivity to Tam [8]. AR is a known clinical target in prostate cancer; however, little is known about its role in breast cancer. The majority of ER-positive breast tumors also express AR, and AR may be a new clinical target in ER-negative tumors [21, 25, 28]. A study comparing transcriptomic differences in early stage hormone receptor-positive breast cancer patients treated with adjuvant Tam who experienced early versus late recurrences, showed increased relative expression of AR in the late recurrence group [22].
We hypothesize that AR OE confers resistance, and engineered ERα-positive cells to overexpress both AR and aromatase to study the role of AR in response to AIs. We discovered that AR cooperates with ERα in promoting cells to escape the inhibitory effects of the AI anastrazole (Ana), and define potential mechanisms associated with escape from hormone therapy.
Results
AR overexpression confers resistance to the AI anastrazole (Ana)
Previously, we showed that AR OE drives ERα-positive cells as an escape pathway leading to TamR [8]. To investigate whether AR OE plays a similar role in escape from AIs, we generated ERα-positive MCF-7 breast cancer cells expressing both AR and aromatase proteins. We made stable transfectants of human aromatase (MCF-7 Arom3 cells, Fig. 1a). The Arom3 clone was then stably transfected with a full-length AR cDNA (AR Arom5 and Arom10 subclones). Equivalent levels of ERα were detected, indicating that OE of aromatase with or without AR did not significantly affect ERα levels.
Fig. 1.
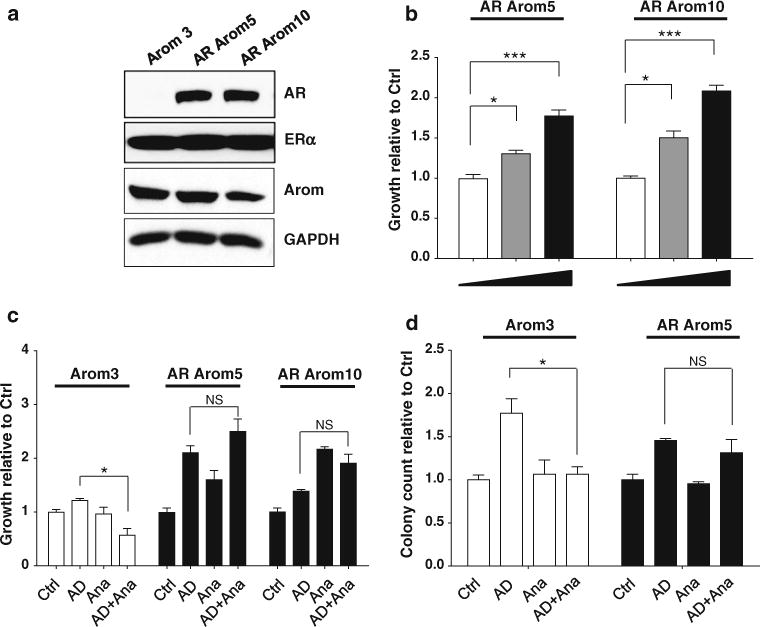
AR overexpression leads to AI resistance. a Immunoblot analysis showing AR, ERα, aromatase, and GAPDH (loading control) in aromatase (Arom)-transfected cells (Arom3) and AR-Aromatase-transfected cells (AR arom5 and AR arom10), b MTT growth assay in cells treated with vehicle (Ctrl, ethanol), and dihydrotestosterone (DHT, 10−9 M and 5 × 10−9 M). Bars SD; (**P < 0.01, ***P < 0.001), c MTT Growth assay in cells treated with Ctrl (ethanol) and androstendione (AD, 10−8 M) ± anastrazole (Ana, 10−6 M). Cell proliferation is expressed as fold change relative to vehicle-treated cells. Data are representative of at least three independent experiments performed in triplicate or quadruplicate. Columns mean, bars SD (*P < 0.05, NS nonsignificant) d Soft agar growth assay in cells treated with Ctrl (ethanol), and AD (10−8 -M ± Ana (10−6 M). Data are representative of three independent experiments performed in triplicate or quadruplicate. Columns mean, bars SD (*P < 0.05, NS nonsignificant)
We performed growth assays-treating cells with either vehicle (Ctrl) or the AR agonist dihydrotestosterone (DHT). Growth of cells overexpressing AR increased in a dose-dependent manner with DHT (Fig. 1b). We then explored growth in the presence of the AI Ana using anchorage-dependent (MTT) and independent (soft agar) growth assays (Fig. 1c, d, respectively). As expected with the expression of aromatase in these cells, treatment with the precursor for estrogen (androstendione, AD)-stimulated growth of all cell lines as compared to control (Ctrl) cells. While AD-stimulated growth in Arom3 cells was significantly blocked by Ana, growth of AR-overexpressing cells was not inhibited. Growth stimulation with Ana treatment was seen in some AR-overexpressing cells (Fig. 1c, Arom10). This demonstrates that AD can act as an androgen-stimulating growth, as has been reported [33]. Similar resistance to Ana treatment was obtained using soft agar assays (Fig. 1d).
Constitutive activation of IGF-1R and downstream phosphorylation of Akt pathway as mechanisms of resistance coincident with AR OE
The HER2 family and IGF-1R growth factor receptors are involved in resistance to endocrine therapy in breast cancer [34, 37, 45]. Activation of these growth factor receptor pathways induces rapid non-genomic effects leading to the phosphorylation of the MAPK and PI3K/Akt pathways [6, 44]. To explore effectors of resistance, we treated cells with AD, either alone or in combination with Ana (Fig. 2a). No differences in the levels of phosphorylated EGFR or HER2 were seen (Supplemental Fig. 1). In contrast, high constitutive IGF-1R phosphorylation was detected with AR OE, which was enhanced with AD or Ana treatments. To determine if pIGF-1R activation was involved in resistance to Ana, an IFG-1R inhibitor (Tyrphostin, AG1024) was used (Fig. 2b). Tyrphostin has been shown to inhibit proliferation in MCF-7 cells [23]. Tyrphostin treatment blocks Akt and Bcl-2 phosphorylation, and up-regulates Bax, p53, and p21 levels [9, 40]. Treatment with this IGF-1R inhibitor alone had little effect on cells, but further decreased the growth of Arom3 cells in combination with AD + Ana (Fig. 2b). The IGF-1R inhibitor in combination with AD + Ana restored sensitivity to Ana in AR-overexpressing cells. Western blot analysis confirmed that AG1024 treatment blocked phosphorylation of IGF-1R and Akt in AR-overexpressing cells (Fig. 2c).
Fig. 2.
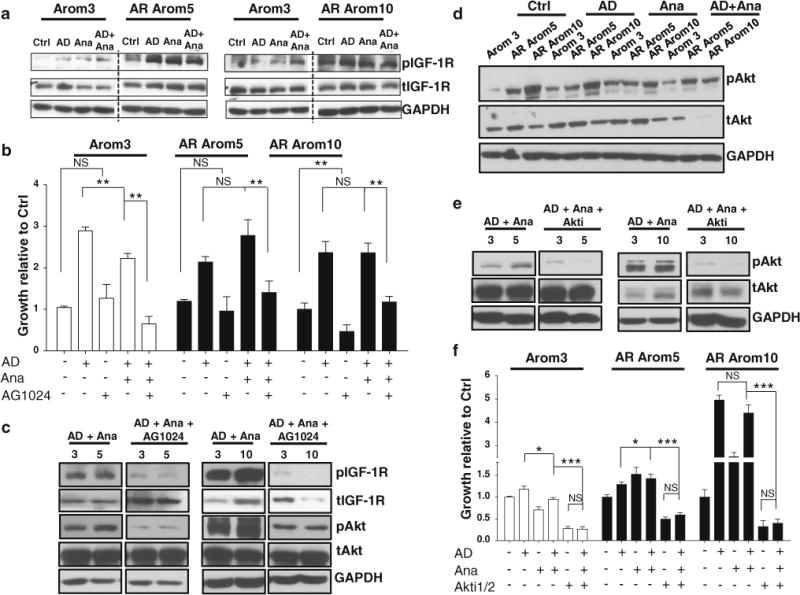
Resistance to AI is associated with constitutive activation of IGF-1R and downstream activation of Akt pathway. a Immunoblot analysis showing constitutive activation of pIGF-1R in AR-overexpressing cell lines (AR Arom5 and AR Arom10) in the presence and in the absence of treatments (5 min) in comparison to Arom3 control cell line. Levels of total IGF-1R (tIGF-1R) are also shown. GAPDH was used as a loading control. Treatments: Ctrl (ethanol), AD (10−8 M), Ana (10−6 M). b MTT growth assay showing AI resistance of AR-overexpressing cells and restoration of sensitivity using an IGF-1R antagonist. Cells were treated for 10 days. Treatments included: Ctrl (ethanol/DMSO), AD (10−8 M), Ana (10−6 M), IGF-1R antagonist (AG1024, 20−6 M). Columns mean, bars SD (**P < 0.01, NS nonsignificant). c Immunoblot analysis showing blockade of pIGF-1R activation and downstream pAkt activation using an IGF-1R antagonist in AR-overexpressing cell lines in comparison to Arom3 control cell line. Cells were treated for 5 min. Levels of total IGF-1R (tIGF-1R) and total Akt (tAkt) are shown. GAPDH was used as a loading control. Treatments included: AD (10−8 M), Ana (10−6 M), AG1024 (20−6 M). d Immunoblot analysis showing constitutive activation of pAkt in AR-overexpressing cell lines (AR Arom5 and AR Arom10) after 5 min of treatment in comparison to Arom3 control cell line. Levels of total Akt (tAkt) are also shown. GAPDH was used as a loading control. Treatments included: Ctrl (ethanol), AD (10−8 M), and Ana (10−6 M). e Immunoblot analysis showing blockade of pAkt activation using an Akt inhibitor in AR-overexpressing cell lines (AR Arom5 and AR Arom10) in comparison to Arom3 control cell line. Cells were treated for 5 min. Levels of total Akt (tAkt) are also shown. GAPDH was used as a loading control. Treatments included: AD (10−8M), Ana (10−6 M), Akt inhibitor 1/2 (Akti, 10−6 M). f MTT growth assay showing AI resistance of AR Arom5 and AR Arom10 cells and restoration of sensitivity using an Akt inhibitor. Cells were treated for 10 days. Treatments included: Ctrl (ethanol/DMSO), AD (10−8 M), Ana (10−6 M), Akti (10−6 M). Columns mean, bars SD (*P < 0.05, ***P < 0.001, NS nonsignificant)
Constitutively higher pAkt levels were seen with AR OE (Fig. 2d). AD-stimulated Akt phosphorylation in Arom3 cells, and Ana blocked these effects (Fig. 2d). Ana treatment of AR-overexpressing cells reduced pAkt levels; however, these levels remained higher with combination treatments. This suggests that AIs were not effective at blocking IGF-1R or downstream Akt activation in AR-overexpressing cells. Next, we looked at the possibility of restoring sensitivity to Ana using an Akt inhibitor (Fig. 2e, f). The dual kinase inhibitor Akti 1/2 can block Akt phosphorylation and signaling [5, 17]. Akti 1/2 combined with AD + Ana drastically reduced Akt phosphorylation levels in AR-overexpressing cells (Fig. 2e). Treatment with Akti 1/2 alone blocked growth, and combined with Ana restored AI sensitivity in AR-overexpressing cells (Fig. 2f). Thus, combination therapy with either an IGF-1R or Akt inhibitor may be required for effective AI therapy of AR-overexpressing cells.
AR and ERα functionally collaborate to induce AIR in AR-overexpressing cells
AR is often co-expressed along with ERα [20, 26], and AR and ERα can physically interact and modulate each other’s activity [31]. AR levels are important for distribution of AR binding sites and sensitivity to AR agonists [38, 39]. To examine the contribution of AR OE to AIR, we next employed a number of AR antagonists, including bicalutamide (Casodex, Cx) and enzalutamide (MDV3100, MDV). MDV is reported to be more effective than Cx in blocking AR nuclear activity and is effective in castration-resistant prostate cancer [16]. We also tested Zytiga® (abiraterone acetate) which works by a different mechanism inhibiting cytochrome P450 (CYP17), the enzyme responsible for the biosynthesis of estrogens and androgens. All three agents are being tested in breast cancer clinical trials. Cells were treated for 9 days with the AR and ER modulators (Fig. 3a). Tam and the pure steroidal antiestrogen fulvestrant (ICI) significantly reduced growth of all cells. Although AR OE alone conferred resistance to Tam in MCF-7 cells [8], aromatase OE in these cells rendered them sensitive to Tam inhibition, possibly through enhanced intracellular synthesis of estrogen. As expected, DHT treatment stimulated growth of AR-overexpressing cells, and Cx was inhibitory when used alone. MDV treatment of AR Arom10 cells slightly stimulated growth. Both Cx and MDV treatments blocked DHT-stimulated growth in AR-overexpressing cells, demonstrating that AR was indeed stimulatory in these cells. Of the AR antagonists, Abi treatment demonstrated the most efficient inhibition of basal growth in this model system. In combination with AD + Ana, Abi and ICI treatments effectively blocked growth. This suggests that complete ER blockade, either using an AI plus the ER degrader fulvestrant, or an AI combined with Abi, would be the most effective approach to restore hormone sensitivity in AR-overexpressing cells. Taken together, these data indicate that both AR and ERα signaling collaborate in acquired AIR.
Fig. 3.
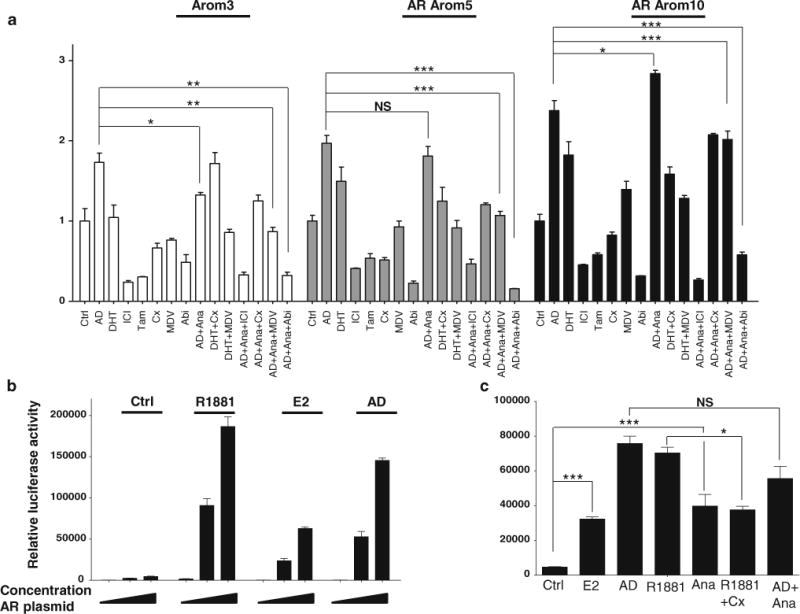
AR and ERα collaborate to induce AI resistance. a MTT growth assay in cells treated with Ctrl (ethanol/DMSO), AD (10−8 M), DHT (10−9 M), fulvestrant (ICI, 10−7 M), Tamoxifen (Tam, 10−7 M), bicalutamide (Cx, 10−5 M), MDV3100 (MDV, 10−5 M), abiraterone (Abi, 10−5 M), AD + Ana, DHT + Cx, DHT + MDV, AD + Ana + ICI, AD + Ana + Cx, AD + Ana + MDV and AD + Ana + Abi. Data are representative of three independent experiments performed in triplicate. Columns mean, bars SD (*P < 0.05, **P < 0.01, ***P < 0.001, NS nonsignificant). b Luciferase reporter assay showing increased AR-transcriptional activity after transfection of Arom3 cells with increased amounts of AR plasmid and treatment with Ctrl (ethanol), E2 (10−8 M), AD (10−8 M), and R1881 (10−8 M). Data are representative of three independent experiments performed in triplicate. c Luciferase reporter assay showing AR-transcriptional activity after transfection of Arom3 cells with AR plasmid and treatment with Ctrl (ethanol/DMSO), E2 (10−8 M), AD (10−8 M), and R1881 (10−8 M), Ana (10−6 M), Cx (10−5 M), R1881 + Cx and AD + Ana. Data are representative of three independent experiments performed in triplicate. Columns mean, bars SD (*P < 0.05, ***P < 0.001, NS nonsignificant)
To explore AR-transcriptional activity, we performed ARE-luciferase reporter assays in Arom3 cells transiently transfected with increasing amounts of AR plasmid. The AR agonist R1881 stimulated activity (Fig. 3b). Both E2 and AD-stimulated AR activity in a dose-dependent manner, suggesting a functional interaction between the receptors in inducing AR activity (Fig. 3b). Ana also stimulated AR activity almost to the same level as E2 (Fig. 3c). As a Ctrl, Cx treatments blocked R1881 activity. Ana failed to completely block AD-stimulated AR activity suggesting that AD could function as an androgen and an estrogen. These results suggest that AR activity could be enhanced by estrogens acting through ERα.
ERα activity is insufficiently blocked by Ana in AIR cells
AD treatment, but not Ana, stimulated ERα activity, as measured by ERE-luciferase reporter assays (Fig. 4a). Ana blocked AD-stimulated ER activity in Arom3, but not in either AR-overexpressing cells (Fig. 4a). This suggests that Ana failed to completely block residual ERα activity which may play a role in AIR. To determine whether AR contributed to AD-stimulated ERα activity, we examined the effects of AR modulators (Fig. 4b). DHT-stimulated activity in a manner similar to AD alone, and Cx blocked both AD and DHT-stimulated activities. Cx treatment had no effect on ERα activity. These data suggest that both AR and ERα contribute to ER activity in AR-overexpressing cells.
Fig. 4.

AR acts as a coactivator for ERα. Luciferase reporter assay showing in a ERα-transcriptional activity in AR OE cells compared to Arom3 cells after treatment with Ctrl (ethanol/DMSO), AD (10−8 M) ± Ana (10−6 M), in b ERα-transcriptional activity in AR Arom5 and AR Arom10 after treatment with AD (10−8 M) ± Cx (10−5 M), DHT (10−8 M) ± Cx (10−5 M). Data are representative of three independent experiments performed in triplicate. Columns mean, bars SD (**P < 0.01). c Proximity ligation assay showing AR-ERα complexes in cells after treatment with AD + Ana (10−8 and 10−6 M respectively). Red box shows a magnification of one representative area, yellow arrows point to AR-ERα complexes. d Quantitative chromatin immunoprecipitation (qChIP) assay showing AR and ERα enrichment on PSA enhancer region and pS2 promoter after treatment with AD (10−8 M) and DHT (10−8 M). AR and ERα enrichment on chromatin was quantified using qPCR and normalized to Ctrl. Data are representative of at least three independent experiments
To determine whether the collaboration between AR and ERα in AIR cells involved nuclear interactions, we performed proximity ligation assays (PLA) (Fig. 4c, and Supplementary Fig. 4). AR-ERα clusters (red-speckled staining, arrows) were seen in the cytoplasm, but were most prominent in the nucleus of AR Arom5 cells. No staining suggesting limited interactions were observed in Arom3 cells. This demonstrates that AR and ERα are in close proximity and were mainly localized in the nucleus of AIR cells.
Next we used ChIP assays to detect AR and ERα associations at the chromatin level on the androgen-regulated PSA enhancer region, and on the classical estrogen-regulated pS2 promoter region in AR Arom5 cells (Fig. 4d). Enrichment of AR on the PSA enhancer region was seen with both AD and DHT treatments. DHT, but not AD, also recruited ERα to the PSA promoter. Since DHT is unable to bind ERα, these results suggest that agonist-bound AR may recruit ERα to the PSA enhancer region. Similarly, both AD and DHT recruited AR to the pS2 promoter region, whereas only AD but not DHT recruited ERα. This suggests that agonist-occupied AR may recruit ERα to androgen-responsive promoters, and that agonist-occupied AR can be recruited to estrogen-responsive promoter regions.
To explore the subcellular localization of AR and ERα in AIR cells, we utilized confocal microscopy (Fig. 5a, b, respectively). AR was mainly localized in the cytoplasm in untreated cells with enhanced staining in AR-overexpressing cells. ERα was localized in the nucleus in both cell lines. AR colocalized with ERα in the nucleus after AD treatment and Ana-stimulated AR nuclear localization. Treatment with AD + Ana enhanced AR-ERα colocalization in the nucleus with some co-staining seen in the cytoplasm (Fig. 5b merge). These combined results demonstrate that AR and ERα can form complexes in the cytoplasm but predominantly in the nucleus, potentially engaging non-genomic and genomic pathways.
Fig. 5.
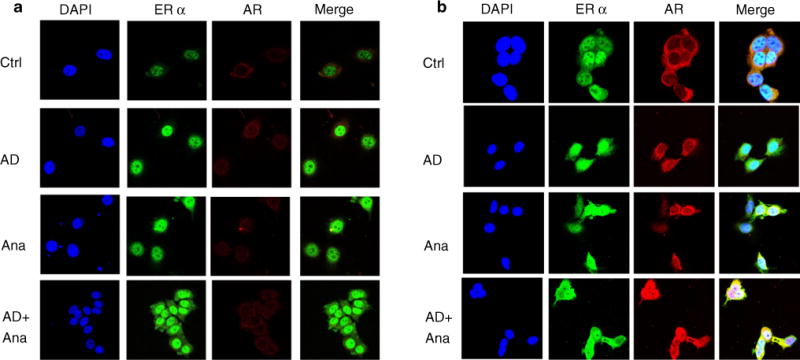
AR and ERα colocalize in AR OE cells. Immunocytochemistry assay showing localization of ERα and AR in cells (Arom3 in panel A and AR OE cells in panel B) after treatment with ethanol (Ctrl), AD (10−8 M) ± Ana (10−6 M) for 10 min
Aromatase is involved in the conversion of androgens into estrogens, and AIs block this activity. In order to identify the most differentially expressed genes in AR Arom-overexpressing cells, we compared gene expression of cells treated with AD and AD + Ana (Table 1, Table S1); 15.4 % (4.58 + 10.96 %) of significant differentially expressed genes were known androgen or estrogen-responsive genes. This suggests that either aromatase blockade was incomplete or AR activity contributed to the differentially expressed signature. Aromatase enzyme was shown to be functional in all cells using activity assays (Fig. S2). These data suggest that both estrogens and androgens contributed to differential gene expression associated with AIR in AR Arom-overexpressing cells.
Table 1.
Percentages of androgen- or estrogen-responsive genes differentially expressed in AR-aromatase-overexpressing cells
| Total number of differentially expressed genes | AR/Androgen-responsive genes (Bolton et al. 2007) | ER/Estrogen-responsive genes (Caroll et al. 2006) | |
|---|---|---|---|
| Up | 182 (36.25 %) | 11 (6.04 %) | 23 (12.64 %) |
| Down | 320 (63.75 %) | 12 (3.75 %) | 32 (10.00 %) |
| Total | 502 (100 %) | 23 (4.58 %) | 55 (10.96 %) |
| AR&ERα | 15.54 % | ||
Genes differentially expressed in AR Arom10 compared to Arom3. Cells were treated with AD + Ana and analyzed using gene expression microarray
P value ≤ 0.01
TamR ER-positive cells endogenously overexpress AR and are resistant to Ana
We previously reported the establishment of acquired TamR sublines that were generated by long-term culture of MCF-7 cells under two experimental conditions simulating pre and post-menopausal estrogen levels (TR1 and TR2, respectively) [2, 13, 35]. Figure 6a shows endogenously high levels of AR in the two cell lines compared to MCF-7 parental cells; ERα levels remain comparable between cells. Figure 6b shows that the two cell lines were indeed resistant to Tam treatment in growth assays in which TR1 and TR2 cells were seeded in the presence of vehicle treatment (ethanol), E2 or Tam. We next questioned whether the acquired TamR lines expressed aromatase Cyp19, and found that TR2 cells expressed higher levels than TR1 and Arom3 cells (Fig. 6c). Therefore, we next examined the hormonal responsiveness of TR1 and TR2 cells (Figs. 6d, e). Treatment with AD-stimulated growth of both cell lines. Ana-stimulated growth, and combined Ana plus AD treatments did not effectively block AD-stimulated growth, like that seen in AR Arom-expressing cells. However, treatment with Abi alone and in combination blocked cell growth of TR1 and TR2 cells. Thus, data from acquired TamR cells confirmed our findings that AR OE in ERα-positive cell lines conferred resistance to an AI, and that targeting ERα and AR restored sensitivity to the inhibitor.
Fig. 6.
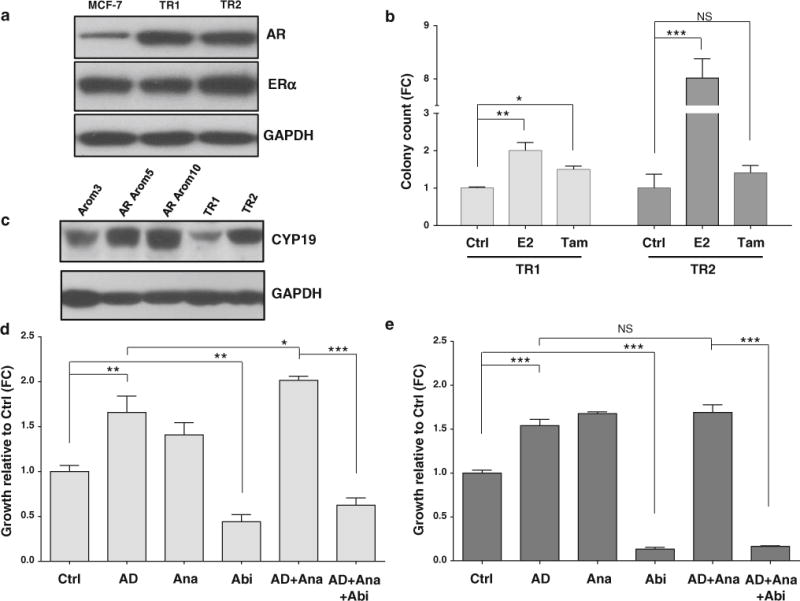
Tamoxifen-resistant (TR) ER-positive cells overexpress endogenous AR and are resistant to an AI. a Immunoblot analysis showing endogenous levels of AR in TamR cells as compared to MCF-7 parental cells. GAPDH was used as a loading control. b Soft agar growth assay showing resistance of TR1 and TR2 cells to tamoxifen. Cells were treated with Ctrl (ethanol), E2 (10−8 M) and Tam (10−7 M). Data are representative of three independent experiments performed in quadruplicate. Columns, mean; bars, SD (*P < 0.05, **P < 0.01, ***P < 0.001, NS nonsignificant). c Immunoblot analysis showing endogenous levels of CYP19 in TamR cells as compared to transfected Arom3, AR Arom5 and AR Arom10 cells. GAPDH was used as a loading control. MTT growth assay showing AI resistance of TR1 (d) and TR2 (e) cells when treated with AD + Ana and sensitivity restoration when combined to Abi. Treatments: Ctrl (ethanol/DMSO), AD (10−8 M), Ana (10−6 M), Abi (10−5 M), AD + Ana, and AD + Ana + Abi for 10 days. Data are representative of three independent experiments performed in triplicate. Columns mean, bars SD (*P < 0.05, **P < 0.01, ***P < 0.001, NS nonsignificant)
Discussion
The optimum care for ERα-positive breast cancers is sequential treatment with Tam and AIs, however, both acquired and de novo resistance to hormone therapy is a major clinical problem in breast cancer. We previously published a role for AR OE in conferring resistance to Tam in breast cancer patients [8]. Herein we report a role for AR in AIR via collaboration with ERα, involving constitutive activation of IGF-1R and AKT signaling pathways.
In prostate cancer, AR has been extensively studied and its role as a critical effector of tumor development and progression is well documented, therefore, AR antagonists are effective for the treatment of advanced prostate cancer [1]. In breast cancer, the role of AR is complex because androgens can stimulate or inhibit cell growth [4, 7, 8, 18, 19, 22, 25, 28, 32]. Because of the dual actions of androgens in breast cancer, it is very important to identify biomarkers of AR activity to manage patients in AR-targeted studies, like the use of PSA monitoring in prostate cancer. An AR-targeted trial using bicalutamide has been recently reported in AR-positive, ER-negative breast cancer patients demonstrating proof-of-principle clinical benefit in metastatic breast cancer [15], and a Phase II clinical trial with enzalutamide (MDV3100) has also recently opened in ER-positive, HER2 normal breast cancer patients [43]. A study exploring whether abiraterone acetate can extend clinical benefit with an AI is planned to be completed in July 2014 (NCT01381874). Our results suggest that AR OE may be a novel determinant of resistance to AIs in breast cancer. Here we show that overexpressing AR in ERα-positive MCF-7BK cells rendered them refractory to Ana. In this model, Ana alone stimulated anchorage-independent growth assays. This effect could be due to the known increase in androgens with Ana treatment [11].
Resistance to hormonal therapy is often due to the emergence of escape survival pathways [27, 30, 41]. In ERα-positive AR-overexpressing cells, IGF-1R was constitutively activated along with downstream activation of pAkt. Mechanisms of escape in our AR-overexpressing model are unique in that we did not observe activation of the HER growth factor receptor family, which is the dominant escape pathway in other models of hormone resistance. Furthermore, IGF-1R or Akt inhibitors were very effective in restoring Ana sensitivity. Clinically, this suggests that pIGF-1R and/or pAkt may be biomarkers of resistance associated with AR OE. Of course this will need to be tested in prospective clinical studies. We do not yet understand mechanistically how AR OE leads to constitutive activation of pIGF-1R, but potentially this could include activation of this pathway at several levels as we observed in another model of AI resistance due to a specific somatic mutation in ESR1 (K303R ERα) [3, 10]. Blocking AR with any of the AR modulators or synthesis inhibitors tested (bicalutamide, enzalutamide, or abiraterone) effectively restored hormone sensitivity in AR-overexpressing cells. The involvement of both AR and ERα in resistance was evident when AR activity was blocked with ERα antagonists, and similarly ER transcriptional activity was affected by AR antagonists, suggesting cooperation between the two steroid receptors. Ana treatment failed to completely block residual ERα activity in AR-overexpressing cells, suggesting that the failure to block estrogen-stimulated activity could define AIR. Thus, we conclude that both AR and ERα functionally cooperate and contribute to enhanced residual ERα activity in resistant cells.
Immunocytochemistry and PLA assays demonstrated collaboration between AR and ERα at the nuclear level. PLA data suggested that AR and ERα may be in close proximity in the nucleus. ChIP assays also confirmed these findings demonstrating that AR and ERα cooperate on known androgen and estrogen-responsive gene promoters. Collectively this suggests that agonist-occupied AR may recruit, either directly or indirectly, ERα to androgen-responsive promoters, and that AR can also be recruited to estrogen-responsive promoter regions. We also demonstrated that ER-positive breast cancer cells up-regulate endogenous AR with long-term Tam treatment (TR1 and TR2), and growth of these cells was effectively blocked with Abi treatment. This is consistent with a role for both AR and ERα in the resistant phenotype, and provides confirmation of the role of AR in resistance using cells with endogenous increases in AR.
An important clinically relevant finding is that resistance was associated with an increase in androgen and estrogen-responsive gene expression, and that AIR may be associated with a failure to completely block AR- and/or ER-regulated gene expression. Our preclinical data suggest that simultaneous and complete blockade of AR and ER-regulated escape survival pathways may be required to provide clinical benefit. Thus, we predict that AR OE is an important marker of resistance to both Tam and AIs in patients with ER-positive breast cancer, depending on the expression of aromatase in the microenvironment and/or intratumoral aromatase levels. Targeting AR signaling, which can impact on ERα function, may provide a unique strategy for more effective treatment of ER-positive breast cancer, or a longer extension of clinical benefit from ER-targeted therapy in metastatic patients. With the rapid expansion of new AR-targeted agents in the cancer armamentarium, this is an exciting possibility in targeted breast cancer therapy.
Materials and methods
Reagents, hormones, and antibodies
Methyltrienolone (R1881), 17β-estradiol (E2), and androstenedione (AD) were obtained from Sigma (St. Louis, MO). Anastrazole (Arimidex, Ana) was obtained from AstraZeneca (London, UK), and bicalutamide (Casodex®, Cx) was purchased from LKT Laboratories Inc. (St. Paul, MN). Abiraterone acetate was a kind gift from Janssen Pharmaceuticals (Beerse, Belgium). Antibodies used for immunoblotting were anti-GAPDH from Santa Cruz Biotechnology (Santa Cruz, CA), anti-ERα 6F11 from Vector Laboratories Inc. (Burlingame, CA), anti-PR from DAKO (Carpinteria, CA), anti- total and phosphorylated IRS-1, HER2, IGF-1R, AKT, and MAPK from Cell Signaling Technology (Beverly, MA), anti-AR clone 441 from Santa Cruz Biotechnology, and anti-aromatase (CYP19) from Serotec (Oxford, UK). MTT (Thiazoyl blue tetrazolium bromide) was obtained from Sigma.
Cells and stable transfection
MCF-7BK cells have been maintained in our laboratory for over 20 years. Cells were grown in MEM (Invitrogen, Carlsbad, CA) supplemented with 10 % fetal bovine serum (FBS; Summit Biotechnology, Fort Collins, CO), 200 units/ml penicillin, 200 μg/ml streptomycin, and 10 μg/ml insulin. Cells were incubated at 37 °C in 5 % CO2. To generate MCF-7 cells stably overexpressing the CYP19 aromatase enzyme (designated Arom cells), pCR3.1-CYP19 cDNA, or empty pCR3.1 vector (Invitrogen) were transfected as described in [4], and positive clones were identified using immunoblot analysis with the anti-CYP19 antibody. To generate MCF-7BK cells that stably overexpress aromatase and AR, we transfected Arom cells as described above, and clones were identified using immunoblot analysis with the anti-AR antibody clone 441.
Cell extraction and immunoblot analysis
Cells were starved in phenol red- and serum-free MEM for 48 h, and then treated for 24–48 h with different compounds as indicated. For rapid signaling studies, cells were starved as above followed by 20 min treatments with vehicle (C, ethanol or DMSO), estrogen (E2, 10 nM), AD (10 nM), Ana (1 μM), R1881 (10 nM), or bicalutamide (Cx, 10 μM). After these varying treatments, cells were rinsed twice with ice cold phosphate-buffered saline (PBS) and were then lysed immediately with 200μL of cell lysis buffer (20 mM Tris–HCl, pH 7.4, 15 mM NaCl, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate, and 10 % glycerol) plus 1:100 proteinase inhibitor cocktail III (Calbiochem, La Jolla, CA) per 100 mm tissue culture plate. Cell lysates were cleared by centrifugation at 16,0009g for 10 min at 4 °C. Protein concentrations were determined using the BCA Protein Assay kit (Pierce, Rockford, IL) according to the manufacturer’s directions. Equal amounts of cell extracts were resolved under denaturing conditions by electrophoresis in 8–10 % polyacrylamide gels containing SDS (SDS-PAGE) and transferred to nitrocellulose membranes by electroblotting (Protran® Nitrocellulose Membranes, GE Healthcare, Piscataway, NJ). The blots were first stained with Ponceau S to confirm uniform transfer of all samples. After blocking of the transferred nitrocellulose membranes, they were incubated with primary antibodies overnight at 4 °C, incubated with secondary antibodies for 1 h at room temperature, and developed with enhanced chemiluminescence reagents (ProteinSimple, San Jose, CA).
Anchorage-independent growth assays
Cells were starved for 2 days in phenol red and serum-free MEM supplemented with 5 % charcoal-stripped FBS, and 200 μg/ml streptomycin. Soft agar assays were performed in six-well plates using 5 × 103 cells per well. After 14 days, the colonies with greater than 50 cells from triplicate assays were counted. Data shown are the mean colony numbers of three wells, and are representative of two independent experiments.
Aromatase activity assays
Aromatase activity in medium from subconfluent cell cultures was measured by a tritiated water release assay using 0.5 μM [1β-3H]androst-4-ene-3, 17-dione (PerkinElmer Life Sciences) as substrate. Incubations were performed at 37 °C for 2 h under a 95:5 % air/CO2 atmosphere. Results obtained were expressed as picomoles/h and normalized to milligrams of protein (picomoles/h/mg of protein).
Transient transfection assay and ERE-luciferase reporter gene assays
MCF-7BK, Arom, and AR Arom cells were serum-starved using phenol red and serum-free MEM containing 5 % charcoal-stripped serum for 48 h, then plated in 24-well plates (7 × 104 cells per well). After incubation overnight at 37 °C, cells were co-transfected with a plasmid containing an estrogen-responsive element (ERE) linked to luciferase (ERE-Luc, 0.25 μg per well), and a plasmid expressing β-galactosidase (β -gal, 0.1 μg/well) to normalize transfection efficiency through the whole assay. Lipofectamin™ LTX with Plus™ reagent (Invitrogen) was used following the manufacturer’s instructions. Eight hours after transfection, cells were treated with the indicated ligands for 16–24 h, then lysed and the luciferase assay was performed using a luciferase reporter assay kit (Promega). β-galactosidase activity was used to normalize the transfection efficiencies. Transfections were performed in triplicate.
MTT assays
MCF-7BK, Arom, and AR Arom cells were plated in 96-well plates, then serum-starved as described above for 48 h. Cells were treated with the indicated ligands for 7 days then MTT (Thiazoyl blue tetrazolium bromide) solution (5 g/L) was added to each well, and plates were re-incubated for 4 h. The violet precipitate (formazan) that appears during incubation, proportional to the number of living cells, was solubilized using DMSO and the optical density (OD) of the violet solution was measured using a colorimeter. The OD values (570–650 nm) were normalized using the ODs obtained on at the day of treatment. Each condition of treatment was performed at least in triplicate.
Immunofluorescence
For immunocytochemical staining, cells were grown on coverslips in full media then serum-starved using charcoal-stripped media for 48 h. Cells were fixed in 4 % paraformaldehyde in PBS buffer for 10 min at room temperature. After fixation, cells were washed 3 times with PBS then permeabilized using a detergent solution (0.2 %Triton X-100, 0.2 % BSA prepared in PBS) for 10 min at room temperature, washed 3× with PBS, and incubated in blocking solution (1 % BSA) for 30 min at room temperature. Incubation of primary antibodies diluted in blocking buffer was carried out overnight at 4 °C. We used anti-ERα (1/20) and anti-AR (1/100) antibodies. Coverslips were washed three times in PBS. Cells were then incubated with an Alexa 488-conjugated goat-anti-mouse (1:200) (Molecular Probes, Eugene, OR) and an Alexa 546-conjugated goat-anti-rabbit (1:500) (Molecular Probes, Eugene, OR). Nuclei were stained for 5 min with DAPI (10 μg/ml) (Vectashield®, Vector laboratories). Immunofluorescence microscopy was performed using a Leica confocal microscope TCS SP5 (Leica Microsystems, Buffalo Grove, IL) and LAS software to collect images.
Proximity ligation assays (PLA)
PLA was performed using the Duolink kit according to manufacturer’s instructions (Olink Bioscience, Uppsala, Sweden). Briefly, cells were plated and treated with indicated hormones in chamber slides. After fixation with 4 % paraformaldehyde, cells were permeabilized and non-specific binding sites blocked with 1 % bovine serum albumin solution (BSA). Samples were then incubated with specific primary antibodies AR rabbit antibody D6F11 (Cell Signaling), and ERα clone 6F11 (Vector Laboratories). Samples were incubated with secondary antibodies linked to PLA probes. Next, a ligase was added to the mixture allowing formation of complete circularized oligonucleotides from the juxtaposed probes. Finally, Detection Reagent Red was added to the mixture to recognize the amplified DNA sequences and allow visualization under microscope of AR-ERα complexes. Sequential washings with PBS were performed after each step.
Immunoprecipitation (IP)
Dynabeads® Protein A and a Protein G kit (Invitrogen, Carlsbad, CA) were used following manufacturer’s instructions. Briefly, 10 μg of antibody was incubated with 50 lL of a 1:1 Protein A:Protein G Dynabeads® mixture. 200 μg of protein was then added to the complex beads plus antibody and rotated for 15 min at room temperature. After washing, mixture was resuspended in SDS sample buffer and boiled for 7 min to dissociate the immuno-complex from the beads. The supernatant was then collected and subjected to Western blot analysis.
Quantitative Chromatin IP (ChIP)
Rabbit polyclonal AR N-20 and ERα HC-20 (Santa Cruz Biotechnology) antibodies were used for ChIP. Dynabeads® Protein A and Protein G kit (Invitrogen) were used. Cells plated at 80 % confluence were subjected to different treatments (Ctrl, AD or DHT) for 90 min. Protein-Chromatin crosslinking was performed using 37 % formaldehyde solution for 10 min then quenched with Glycine (0.375 M). Cells were then lysed and subjected to 4 sonication steps to shear chromatin to an average length of about 500–1000 bp. Cell debris was removed after centrifugation and the supernatant diluted 109 with ChIP dilution buffer. The input fraction was set aside at this step (150 μL), the remaining lysate was split into 2 equal aliquots (one for each antibody) and incubated overnight at 4 °C in the presence of 10 μg of antibody. The next day, samples were incubated with 50 μL of precleared Dynabeads mixture for 2 h at 4 °C with rotation. Samples were then washed sequentially with low salt immune buffer– high salt immune buffer–TE buffer. To collect DNA–protein complexes, beads were extracted 3 times with 50 μL of elution buffer. To reverse crosslinking, a solution of NaHCO3 was added to all samples, and incubated for 6 h at 65 °C. Immunoprecipitated DNA was purified using Qiaex II gel extraction Kit (Qiagen, Valencia, CA) following manufacturer’s instructions. Fold enrichment of nuclear receptors at promoter regions in Ctrl samples versus treated samples was calculated by ΔΔCT using quantitative PCR (Applied Biosystems 7500 Fast Real-Time PCR instrument). Specific promoter regions assayed were pS2 (Cell Signaling) and the PSA enhancer region (Applied Biosystems, Foster City, CA).
Microarray analyses
For microarray analyses, vector Ctrl and AR-overexpressing cells were cultured in 5 % CSM for 48 h, and then cells were treated overnight with Ana with or without AD. After 3× PBS washes, cells were pelleted and RNA was extracted using RNeasy micro kit (Qiagen) following manufacturer’s instructions. Labeled cRNA was hybridized onto Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays (Affymetrix Inc. Santa Clara, CA). Three chips per group were used. Chips were normalized with probeset-level estimates of expression previously generated using tools in Bioconductor (www.bioconductor.org) and/or BRB Array tools (http://linus.nci.nih.gov/BRB-ArrayTools.html) [12]. Analyses employed a ‘moderated’ linear model [36], and contrasts to detect changes with treatment (Ana, Ana + AD) that differ between vector Ctrl and AR OE lines. Analyses focused on the overlap of gene expression associated with treatments between sensitive and resistant cell line models using meta-analytic methods [29, 42].
Statistics
Experimental data were analyzed for statistical significance using a two-tailed Student’s t test or one-way ANOVA test, and p-value was corrected for multiple comparisons.
Supplementary Material
Acknowledgments
We thank Amanda Beyer and Sasha Pejerrey for excellent editorial assistance. This work was supported by NIH/NCI R01-CA72038, CPRIT RP120732 to SAWF and Komen for the Cure – Promise KG081694 to PB. We are grateful to Janssen Pharmaceuticals Inc. for providing abiraterone acetate and a research grant for the studies with this agent.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10549-014-3082-8) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare that they have no conflict of interest.
Contributor Information
Yassine Rechoum, Lester and Sue Smith Breast Center, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA.
Daniela Rovito, Department of Pharmacy, Health, and Nutritional Sciences, University of Calabria, Rende, Italy.
Domenico Iacopetta, Department of Pharmacy, Health, and Nutritional Sciences, University of Calabria, Rende, Italy.
Ines Barone, Department of Cellular Biology, University of Calabria, Rende, Italy; Centro Sanitario, University of Calabria, Rende, Italy.
Sebastiano Andò, Department of Cellular Biology, University of Calabria, Rende, Italy.
Nancy L. Weigel, Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX, USA
Bert W. O’Malley, Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX, USA
Powel H. Brown, Department of Clinical Cancer Prevention, UT M. D. Anderson Cancer Center, Houston, TX, USA Department of Breast Medical Oncology, UT M. D. Anderson Cancer Center, Houston, TX, USA.
Suzanne A. W. Fuqua, Email: sfuqua@bcm.edu, Lester and Sue Smith Breast Center, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA.
References
- 1.Balk SP, Knudsen KE. AR, the cell cycle, and prostate cancer. Nucl Recept Signal. 2008;6:e001. doi: 10.1621/nrs.06001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barone I, Brusco L, Gu G, Selever J, Beyer A, Covington KR, Tsimelzon A, Wang T, Hilsenbeck SG, Chamness GC, Ando S, Fuqua SA. Loss of Rho GDIalpha and resistance to tamoxifen via effects on estrogen receptor alpha. J Natl Cancer Inst. 2011;103:538–552. doi: 10.1093/jnci/djr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barone I, Cui Y, Herynk MH, Corona-Rodriguez A, Giordano C, Selever J, Beyer A, Ando S, Fuqua SA. Expression of the K303R estrogen receptor-alpha breast cancer mutation induces resistance to an aromatase inhibitor via addiction to the PI3K/Akt kinase pathway. Cancer Res. 2009;69:4724–4732. doi: 10.1158/0008-5472.CAN-08-4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buchanan G, Birrell SN, Peters AA, Bianco-Miotto T, Ramsay K, Cops EJ, Yang M, Harris JM, Simila HA, Moore NL, Bentel JM, Ricciardelli C, Horsfall DJ, Butler LM, Tilley WD. Decreased androgen receptor levels and receptor function in breast cancer contribute to the failure of response to medroxyprogesterone acetate. Cancer Res. 2005;65:8487–8496. doi: 10.1158/0008-5472.CAN-04-3077. [DOI] [PubMed] [Google Scholar]
- 5.Cherrin C, Haskell K, Howell B, Jones R, Leander K, Robinson R, Watkins A, Bilodeau M, Hoffman J, Sanderson P, Hartman G, Mahan E, Prueksaritanont T, Jiang G, She QB, Rosen N, Sepp-Lorenzino L, Defeo-Jones D, Huber HE. An allosteric Akt inhibitor effectively blocks Akt signaling and tumor growth with only transient effects on glucose and insulin levels in vivo. Cancer Biol Ther. 2010;9:493–503. doi: 10.4161/cbt.9.7.11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheskis BJ. Regulation of cell signalling cascades by steroid hormones. J Cell Biochem. 2004;93:20–27. doi: 10.1002/jcb.20180. [DOI] [PubMed] [Google Scholar]
- 7.Cochrane DR, Bernales S, Jacobsen BM, Cittelly DM, Howe EN, D’Amato NC, Spoelstra NS, Edgerton SM, Jean A, Guerrero J, Gomez F, Medicherla S, Alfaro IE, McCullagh E, Jedlicka P, Torkko KC, Thor AD, Elias AD, Protter AA, Richer JK. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014;16:R7. doi: 10.1186/bcr3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Amicis F, Thirugnansampanthan J, Cui Y, Selever J, Beyer A, Parra I, Weigel NL, Herynk MH, Tsimelzon A, Lewis MT, Chamness GC, Hilsenbeck SG, Ando S, Fuqua SA. Androgen receptor overexpression induces tamoxifen resistance in human breast cancer cells. Breast Cancer Res Treat. 2010;121:1–11. doi: 10.1007/s10549-009-0436-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deutsch E, Maggiorella L, Wen B, Bonnet ML, Khanfir K, Frascogna V, Turhan AG, Bourhis J. Tyrosine kinase inhibitor AG1024 exerts antileukaemic effects on STI571-resistant Bcr-Abl expressing cells and decreases AKT phosphorylation. Br J Cancer. 2004;91:1735–1741. doi: 10.1038/sj.bjc.6602190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuqua SA, Gu G, Rechoum Y. Estrogen receptor (ER) alpha mutations in breast cancer: hidden in plain sight. Breast Cancer Res Treat. 2014 doi: 10.1007/s10549-014-2847-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallicchio L, Macdonald R, Wood B, Rushovich E, Helzlsouer KJ. Androgens and musculoskeletal symptoms among breast cancer patients on aromatase inhibitor therapy. Breast Cancer Res Treat. 2011;130:569–577. doi: 10.1007/s10549-011-1611-2. [DOI] [PubMed] [Google Scholar]
- 12.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giordano C, Catalano S, Panza S, Vizza D, Barone I, Bonofiglio D, Gelsomino L, Rizza P, Fuqua SA, Ando S. Farnesoid X receptor inhibits tamoxifen-resistant MCF-7 breast cancer cell growth through downregulation of HER2 expression. Oncogene. 2011;30:4129–4140. doi: 10.1038/onc.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goss PE, Ingle JN, Martino S, Robert NJ, Muss HB, Piccart MJ, Castiglione M, Tu D, Shepherd LE, Pritchard KI, Livingston RB, Davidson NE, Norton L, Perez EA, Abrams JS, Cameron DA, Palmer MJ, Pater JL. Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: updated findings from NCIC CTG MA.17. J Natl Cancer Inst. 2005;97:1262–1271. doi: 10.1093/jnci/dji250. [DOI] [PubMed] [Google Scholar]
- 15.Gucalp A, Tolaney S, Isakoff SJ, Ingle JN, Liu MC, Carey LA, Blackwell K, Rugo H, Nabell L, Forero A, Stearns V, Doane AS, Danso M, Moynahan ME, Momen LF, Gonzalez JM, Akhtar A, Giri DD, Patil S, Feigin KN, Hudis CA, Traina TA, Translational Breast Cancer Research C Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin Cancer Res. 2013;19:5505–5512. doi: 10.1158/1078-0432.CCR-12-3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guerrero J, Alfaro IE, Gomez F, Protter AA, Bernales S. Enzalutamide, an androgen receptor signaling inhibitor, induces tumor regression in a mouse model of castration-resistant prostate cancer. Prostate. 2013;73:1291–1305. doi: 10.1002/pros.22674. [DOI] [PubMed] [Google Scholar]
- 17.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discovery. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 18.Hickey TE, Robinson JL, Carroll JS, Tilley WD. Minireview: the androgen receptor in breast tissues: growth inhibitor, tumor suppressor, oncogene? Mol Endocrinol. 2012;26:1252–1267. doi: 10.1210/me.2012-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iacopetta D, Rechoum Y, Fuqua SAW. The role of androgen receptor in breast cancer. Drug Discovery Today: Dis Mech. 2012;9:8. doi: 10.1016/j.ddmec.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lea OA, Kvinnsland S, Thorsen T. Improved measurement of androgen receptors in human breast cancer. Cancer Res. 1989;49:7162–7167. [PubMed] [Google Scholar]
- 21.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Investig. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim E, Metzger-Filho O, Winer EP. The natural history of hormone receptor-positive breast cancer. Oncology. 2012;26(688–694):696. [PubMed] [Google Scholar]
- 23.Mahajan K, Coppola D, Rawal B, Chen YA, Lawrence HR, Engelman RW, Lawrence NJ, Mahajan NP. Ack1-mediated androgen receptor phosphorylation modulates radiation resistance in castration-resistant prostate cancer. J Biol Chem. 2012;287:22112–22122. doi: 10.1074/jbc.M112.357384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68:826–833. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 25.Micello D, Marando A, Sahnane N, Riva C, Capella C, Sessa F. Androgen receptor is frequently expressed in HER2-positive, ER/PR-negative breast cancers. Virchows Arch. 2010;457:467–476. doi: 10.1007/s00428-010-0964-y. [DOI] [PubMed] [Google Scholar]
- 26.Moinfar F, Okcu M, Tsybrovskyy O, Regitnig P, Lax SF, Weybora W, Ratschek M, Tavassoli FA, Denk H. Androgen receptors frequently are expressed in breast carcinomas: potential relevance to new therapeutic strategies. Cancer. 2003;98:703–711. doi: 10.1002/cncr.11532. [DOI] [PubMed] [Google Scholar]
- 27.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 28.Ni M, Chen Y, Lim E, Wimberly H, Bailey ST, Imai Y, Rimm DL, Liu XS, Brown M. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell. 2011;20:119–131. doi: 10.1016/j.ccr.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ochsner SA, Steffen DL, Hilsenbeck SG, Chen ES, Watkins C, McKenna NJ. GEMS (Gene Expression MetaSignatures), a web resource for querying meta-analysis of expression microarray datasets: 17beta-estradiol in MCF-7 cells. Cancer Res. 2009;69:23–26. doi: 10.1158/0008-5472.CAN-08-3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–247. doi: 10.1146/annurev-med-070909-182917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panet-Raymond V, Gottlieb B, Beitel LK, Pinsky L, Trifiro MA. Interactions between androgen and estrogen receptors and the effects on their transactivational properties. Mol Cell Endocrinol. 2000;167:139–150. doi: 10.1016/s0303-7207(00)00279-3. [DOI] [PubMed] [Google Scholar]
- 32.Peters AA, Buchanan G, Ricciardelli C, Bianco-Miotto T, Centenera MM, Harris JM, Jindal S, Segara D, Jia L, Moore NL, Henshall SM, Birrell SN, Coetzee GA, Sutherland RL, Butler LM, Tilley WD. Androgen receptor inhibits estrogen receptor-alpha activity and is prognostic in breast cancer. Cancer Res. 2009;69:6131–6140. doi: 10.1158/0008-5472.CAN-09-0452. [DOI] [PubMed] [Google Scholar]
- 33.Raman JD, Schlegel PN. Aromatase inhibitors for male infertility. J Urol. 2002;167:624–629. doi: 10.1016/S0022-5347(01)69099-2. [DOI] [PubMed] [Google Scholar]
- 34.Santen RJ, Fan P, Zhang Z, Bao Y, Song RX, Yue W. Estrogen signals via an extra-nuclear pathway involving IGF-1R and EGFR in tamoxifen-sensitive and -resistant breast cancer cells. Steroids. 2009;74:586–594. doi: 10.1016/j.steroids.2008.11.020. [DOI] [PubMed] [Google Scholar]
- 35.Selever J, Gu G, Lewis MT, Beyer A, Herynk MH, Covington KR, Tsimelzon A, Dontu G, Provost P, Di Pietro A, Boumendjel A, Albain K, Miele L, Weiss H, Barone I, Ando S, Fuqua SA. Dicer-mediated upregulation of BCRP confers tamoxifen resistance in human breast cancer cells. Clin Cancer Res. 2011;17:6510–6521. doi: 10.1158/1078-0432.CCR-11-1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 37.Song RX, Chen Y, Zhang Z, Bao Y, Yue W, Wang JP, Fan P, Santen RJ. Estrogen utilization of IGF-1-R and EGF-R to signal in breast cancer cells. J Steroid Biochem Mol Biol. 2010;118:219–230. doi: 10.1016/j.jsbmb.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urbanucci A, Marttila S, Janne OA, Visakorpi T. Androgen receptor overexpression alters binding dynamics of the receptor to chromatin and chromatin structure. Prostate. 2012;72:1223–1232. doi: 10.1002/pros.22473. [DOI] [PubMed] [Google Scholar]
- 39.Urbanucci A, Sahu B, Seppala J, Larjo A, Latonen LM, Waltering KK, Tammela TL, Vessella RL, Lahdesmaki H, Janne OA, Visakorpi T. Overexpression of androgen receptor enhances the binding of the receptor to the chromatin in prostate cancer. Oncogene. 2012;31:2153–2163. doi: 10.1038/onc.2011.401. [DOI] [PubMed] [Google Scholar]
- 40.Van Kolen K, Gilany K, Moens L, Esmans EL, Slegers H. P2Y12 receptor signalling towards PKB proceeds through IGF-I receptor cross-talk and requires activation of Src, Pyk2 and Rap1. Cell Signal. 2006;18:1169–1181. doi: 10.1016/j.cellsig.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 41.Vilquin P, Villedieu M, Grisard E, Larbi SB, Ghayad SE, Heudel PE, Bachelot T, Corbo L, Treilleux I, Vendrell JA, Cohen PA. Molecular characterization of anastrozole resistance in breast cancer: pivotal role of the Akt/mTOR pathway in the emergence of de novo or acquired resistance and importance of combining the allosteric Akt inhibitor MK-2206 with an aromatase inhibitor. Int J Cancer/Journal international du cancer. 2013;133:1589–1602. doi: 10.1002/ijc.28182. [DOI] [PubMed] [Google Scholar]
- 42.Whitlock MC. Combining probability from independent tests: the weighted Z-method is superior to Fisher’s approach. J Evol Biol. 2005;18:1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x. [DOI] [PubMed] [Google Scholar]
- 43.Yardley DA, Awada A, Cortes J, Burris HA, Peterson A, Tudor IC, Stopatschinskaja S, Gianni L, Miller K, Winer E. A Phase 2 randomized, double-blind, placebo-controlled multicenter trial evaluating the efficacy and safety of enzalutamide in combination with exemestane in estrogen or progesterone receptor-positive and HER2-normal advanced breast cancer. Cancer Res. 2013;73(24 Suppl) [Google Scholar]
- 44.Yee D, Lee AV. Crosstalk between the insulin-like growth factors and estrogens in breast cancer. J Mammary Gland Biol Neoplasia. 2000;5:107–115. doi: 10.1023/a:1009575518338. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Moerkens M, Ramaiahgari S, de Bont H, Price L, Meerman J, van de Water B. Elevated insulin-like growth factor 1 receptor signaling induces antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling routes. Breast Cancer Res. 2011;13:R52. doi: 10.1186/bcr2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.