

Summary
The bacteria that colonize humans and our built environments have the potential to influence our health. Microbial communities associated with seven families and their homes over six weeks were assessed, including three families that moved home. Microbial communities differed significantly among homes, and the home microbiome was largely sourced from humans. The microbiota in each home were identifiable by family. Network analysis identified humans as the primary bacterial vector, and a Bayesian method significantly matched individuals to their dwellings. Draft genomes of potential human pathogens were observed on a kitchen counter could be matched to the hands of occupants. Following a house move, the microbial community in the new house rapidly converged on the microbial community of the occupants’ former house, suggesting rapid colonization by the family’s microbiota.
Keywords: Indoor Microbiology, Bacteria, Human Microbiome
The global trend towards urbanization has increasingly bound humanity, as a species, to the indoor environment (1,2). We spend much of our time in our homes, but know little about how microbial transmission influences the home and its occupants. Each human maintains a unique microbial ‘fingerprint’ (3–7), which should transfer to a new indoor space with skin shedding, respiratory activity, and skin-surface contact (8), the latter of which can transfer millions of microbial cells per event (9). The microbial diversity of the home likely affects immune defense (10) and disease transmission (11) among its residents, so that tracking how people microbially interact with the indoor environment may provide a ‘road-map’ to defining the health in our homes.
In the Home Microbiome Project (www.homemicrobiome.com), we microbially monitored seven ethnically diverse US families and their homes over six weeks by sampling their skin and home surface bacterial communities. Eighteen participants were trained in the collection of 1,625 microbial samples from body and home sites of interest over a 4–6 week period from 10 houses (Table S1), three dogs, and one cat. For three families, samples were taken immediately before and after moving to a new home. Approximately 15 million high-quality 16S rRNA V4 amplicons represented 136,957 distinct operational taxonomic units (OTUs; 97% nucleotide identity). We subsampled this database at 2500 sequences/sample, omitting OTUs represented by <10 reads, yielding 4 million sequences comprising 21,997 OTUs (97 % identity) from 1,586 samples.
Samples from different sites within the same home differed less than samples from the same site in different homes (ANOSIM R=0.210, p<0.0001 vs R=0.408, p<0.0001). A density plot of weighted UniFrac distances between all home and human samples (Fig. 1A) showed that microbial communities of human hands, noses and bare feet resemble those of home surfaces. However, microbial communities found on home surfaces varied less than those found on humans. In each analyzed home surface the microbial communities of different houses differed significantly (p<0.0001, Fig. 1B), but the extent depended on the surface sampled and was highest for floor environments (ANOSIM R= 0.757 and 0.716 for kitchen and bedroom floors respectively), while door knobs were the most similar (R = 0.379 for front and 0.402 for bedroom doorknob). ANOSIM tests of the differences between the microbial community structure (weighted UniFrac) of the surfaces of each of the three pre- and post-move house combinations (homes 5, 6, 7) were insignificant, suggesting rapid colonization of the new home by the microbial signature of the family. Strikingly, one of the pre-move homes was a hotel room.
Figure 1. Differentiation in microbial community structure between homes and individuals.
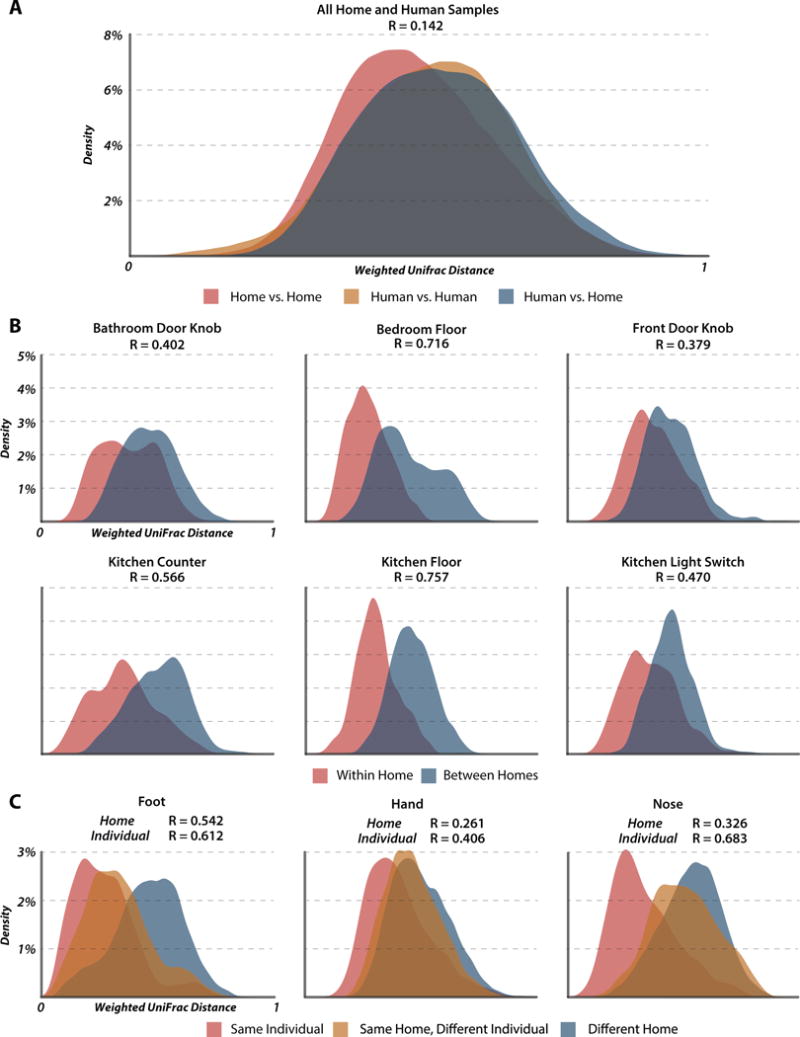
Density plots comparing the distributions of weighted UniFrac distances calculated within and between various criteria with accompanying ANOSIM tests of differentiation (all p values are less than 0.0001 based on 10,000 permutations of the randomized dataset). (A) Distribution of distances calculated between two human samples, between two home samples, and between a human sample and a home sample. An ANOSIM test on the effect of source produced a low Rvalue of 0.142, suggesting that home and human surfaces share a large degree of their microbial communities. (B) Distribution of distances for within home and between home comparisons of all samples taken from individual home surfaces. (C) Distribution of distances between human samples for the three sampled surfaces. Comparisons are segregated by whether a sample was compared to another from the same person, to a sample taken from an occupant of the same house, or to a sample taken from a resident of a different home. ANOSIM results are for tests on the effect of the home the sample was taken from (top) and of the individual the sample was taken from (bottom).
Humans sharing a home were more microbially similar than those not sharing a home, with samples taken from the same individual having the greatest similarity (Fig. 1C). Of the three human environments analyzed in this study, foot samples were differentiated most by home (R=0.542) and hand samples least (R=0.261). Hand samples were also least differentiated by individual (R=0.406) and nose samples differed most between individuals (R=0.683). ANOSIM statistics were robust to sequencing depth, with rarefaction to even 100 reads per sample having little effect on the observed strength of differentiation (Fig. S1A)
A third of all abundant OTUs (564 OTUs with >500 reads) had relative abundances that did not significantly differ between human and inanimate environments (nonparametric t-test with FDR correction >0.05; ρ = 0.88; Fig. 2A). Abundant OTUs were less likely to differ significantly in abundance between human and home surfaces when homes were analyzed individually and rarer OTUs (100 reads/OTU) were included (average=60% undifferentiated OTUs, Fig. S2A). Interestingly, although relative OTU abundances always correlated tightly between human and home environments, they varied in their correlations with pets (Fig. S2B).
Figure 2. Widespread sharing of microbial taxa between human and home surfaces.
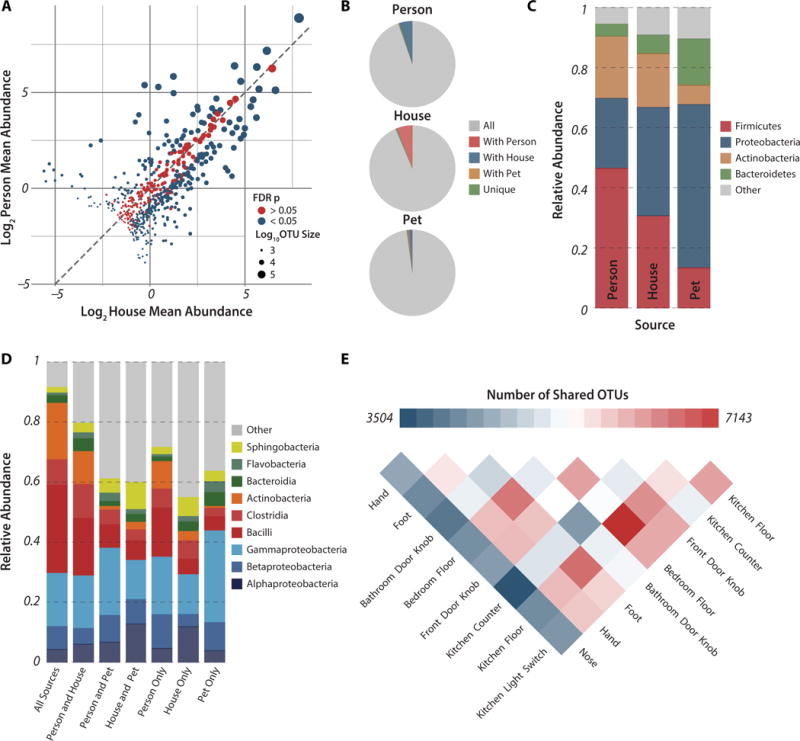
(A) Plot of log2-transformed average relative abundances in the human and home environments for all OTUs in the study with greater than 500 reads. OTUs are colored by whether their average relative abundance is significantly different between the home and person environments based on the FDR corrected p-value from a non-parametric t-test run with 1,000 permutations, and are sized based on their log10-transformed number of reads. The dashed line is y=x, indicating an equal average relative abundance. (B) Fraction of all reads from within a source belonging to OTUs shared with other sources, demonstrating the ubiquitous sharing of OTUs between homes and the humans and pets that occupy them. The percent of reads that cluster within source-specific OTUs is less that 0.6% for all three sources. (C) Taxonomic summary of observed relative abundance of abundant phyla across all samples divided by source. (D) Taxonomic summary of observed relative abundance of taxa at class level for all reads in the study by source-specific OTU overlap. (E) Shared phylotypes heatmap for individual surfaces after consolidation of samples taken from the same surface type across temporal sampling series and homes. Pooled samples were rarified to an even depth of 277,500 reads.
Only about one third of OTUs were detected in all three sources, yet these 7,200 OTUs comprised between 93.6–97.8% of sequences in each source (Fig. 2B). OTUs detected exclusively in a single source, although numerous (4137 OTUs), comprised <0.6% of sequences in each sample.
Relative abundances of dominant bacterial phyla differed among sources (Fig. 2C, split by sample in Fig. S3). Firmicutes and Actinobacteria were enriched in human samples relative to the home, Proteobacteria dominated home and pet samples, while Bacteroidetes were abundant in pets. However, the relative abundances of the nine most abundant bacterial classes had no significant relationship with the number of sources that shared them (ANOSIM p>0.05; Fig. 2D). Pairwise comparison of OTU sharing between surfaces across all homes revealed greatest phylotype overlap between the two floor environments, with the nose sharing the least OTUs with other surfaces (Fig. 2E). Interestingly, the number of OTUs shared by the surfaces with greatest overlap and by the surfaces with the least overlap differed only by a factor of two.
We tested whether microbial community profiles could identify the house or surface a sample originated from by using random forest classifiers (Table S2). Floor samples were highly diagnostic of the family associated with that sample (ratios of random error to model error of 53.62 and 40.17 for kitchen and bedroom floors, respectively), and, even considering all home surface samples together, the family that a sample was taken from was easily predicted (error ratio of 19.91). Models trained to predict the surface type from which a sample was taken were comparatively unsuccessful (error ratio of 3.29), with less predictive accuracy than those trained to predict family origin using broader taxonomic groupings. Families 5, 6 and 7 showed no significant difference between pre- and post-move homes, with error ratios <1.75 for each model. The relative success of predicting family of origin, even when models are trained on broader taxonomic levels, suggests that even error-prone reads from degraded DNA might still be a strong signal of an individual family’s microbiota. Rarefaction to lower sequencing depth resulted in a steep decline in the models’ ability to classify the home a sample was taken from (Fig. S1B), suggesting that greater sequencing depth than employed in this study might significantly strengthen the models’ predictive ability.
We matched human-associated microbial communities to home surfaces using a Bayesian technique known as SourceTracker (12) (Fig. 3). Hand samples were pooled by family, and considered ‘vectors’ to the bathroom doorknob, front doorknob, and kitchen light switch ‘recipient’ communities. Bare-foot samples were pooled by family and treated as vectors for the bedroom and kitchen floor communities. On average, 76.7% of models successfully attributed the recipient community to the correct vector (68.6% of hand samples were identified as vectors to the correct home’s kitchen light switch; 82.9% of pooled family foot samples were identified as vectors to the correct home’s bedroom floor). We also estimated the contribution of individual occupants to their home’s surface communities, which appears to be highly variable between surfaces, between homes and over time. The effect of an individual leaving his or her residence for three sampling days, as occurred in homes 1 and 4, resulted in a decline in that individual’s predicted contribution to a number of the home surfaces, which varied between homes, during their absence (Fig. S4). This suggests the human microbiome signature on home surfaces (e.g., bathroom, front doorknob, and kitchen counter) decays or is replaced rapidly. Because different surfaces respond differently to a human leaving, careful sampling of each surface could provide a metric for assessing the time course of events related to that house and those persons.
Figure 3. Summary of predictive accuracy of Source Tracker models.
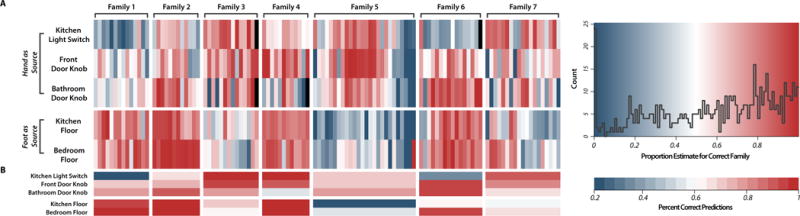
(A) Percent composition estimate for the correct source for each home surface sample in the study. Samples within each block are ordered by collection date and black boxes occur where a sample is missing because it did not pass quality filtering standards. Across all surfaces, the models averaged a 59% prediction for the correct source. (B) Heatmap of model success across individual surface timeseries. The model was considered to be successful when the proportion of the sink community attributed to the correct source was greater than that attributed to any other source.
We tested the direction of microbial transfer among surfaces in the four homes where the subjects did not move houses using dynamic Bayesian networks (Fig. S5; Table S3). Humans were more likely to be sources of OTUs than physical surfaces, with an average of 26 taxonomic edges leaving a human skin surface and arriving at a physical surface, versus 8 edges in the opposite direction (p<0.001). By contrast, human and home surfaces were equally likely to be recipients (Human=20.6 taxonomic edges; Home=19.3, n.s.). OTUs sourced from humans were mainly Actinobacteria and Proteobacteria (Table S3), which are major components of the human skin microbial community (6).
To assess whether personal relationships affect sharing of microbial taxa, we focused on home 4, where none of the residents were genetically related. The two occupants who were in a relationship shared more of their microbiota with each other than with the third occupant, who resided in a separate part of the house (Fig. S6). This differentiation was observed across all surfaces, being greatest in nose samples (R = 0.690) and smallest in hand samples (R = 0.300) (all p<0.0001). In contrast, only weak or insignificant differentiation was observed between married couples and their young children.
Overall, there were significant differences in the volatility of microbial communities associated with each surface type (Kruskal-Wallis chi-squared = 21.6, p = 0.0057) (Fig. S7). However, following a pairwise Wilcoxon test and FDR correction, the only significant differences were between hand and bedroom floor, hand and foot, and hand and nose. We can consequently conclude that the hand is especially variable over time relative to other body habitats and surfaces, presumably reflecting high inputs from the various surfaces with which it comes in contact, and/or more frequent disruption due to washing.
To determine whether taxa transferred between surfaces and human occupants maintained genes associated with pathogenicity, we selected 56 samples from home 4 for longitudinal analysis via shallow shotgun metagenomic sequencing, including 18 home surface samples, 23 human samples, and 15 dog samples (Fig. S8). Genes associated with phage and transposable elements were enriched in human samples. Taxonomic analysis of un-assembled metagenomic reads revealed Corynebacterium on all human samples, Enhydrobacter-, Corynebacterium- and Streptococcus-like bacteria on all bathroom doorknob samples, and Enterobacter-like bacteria on the kitchen counter. Enterobacter-like sequences were also identified on the hands of two occupants on days 2 and 4, further supporting the dynamic Bayesian network analysis above (in addition to genome reconstructions) that indicated a close link between these surfaces (Fig. S4). Following deeper metagenomic sequencing, multiple draft genomes were assembled from hand and kitchen counter samples, including uncultivated Enterobacteraceae and Acinetobacter genomes, and associated bacteriophage. These latter genomes shared 99.7 and 99.9% reconstructed 16S rRNA gene sequence similarity with the respective opportunistic human pathogens Pantoea agglomerans and Acinetobacter baumannii, and maintained genes associated with pathogenicity and antibiotic resistance. Representatives of these genomes sourced from both the kitchen counter and one household occupant’s hand on day 2 shared >2,400 genes with 100% protein sequence identity. When considering the whole bacterial community, and including phage sequence, a total of 84% (7,671) of genes from the hand were shared with the countertop, suggesting a multi-organism transference event between these surfaces. A further 24–29% of the community genes (>3,100) were also identical between the countertop across days (between the 2nd and 4th days), and between the countertop on day 4 and the hand of one of the other occupants sampled on the same day.
There is strikingly little research into relationships between microbial communities associated with home surfaces, and their potential origins. Most studies explore fungal contamination of damp surfaces (13–16), the role of hygiene in removing microbial communities (7,17), and the length of time microbes can survive on surfaces (18,19). Here, we present an intensive longitudinal analysis of the microbial communities associated with the home environment, and present evidence for substantial interaction among human, home, and pet microbiota. Such interactions could have considerable human and animal health implications. Further, we suggest that homes harbor a unique microbial fingerprint which can be predicted by their occupants, and that supersedes inter-surface differentiation within the home. We further show the rapidity and extent to which a human population can influence the microbial diversity of the space they inhabit.
Supplementary Material
Supplementary Figure 1: Effect of rarefaction to lower sequencing depth on ANOSIM R statistics (A) and random forest predictive accuracy (B).
Supplementary Figure 2: (A) Plot of log2-transformed average relative abundances in the human and home samples of individual homes (similar to Figure 2A, except divided by house). For an OTU to be included, it must have been detected at least 100 times within the home. OTUs are colored by whether their average relative abundance is significantly different between the home and person environments based on the FDR corrected p-value from a non-parametric t-test run with 1,000 permutations, and are sized based on their log10-transformed number of reads. The dashed line is y=x, indicating an equal average relative abundance. (B) Ternary plots of average relative abundances of OTUs with greater than 500 reads in the two homes with pets. OTUs are colored by taxonomy (phylum, with Proteobacteria split by class) and scaled to reflect OTU size. Both plots depict strong correlation between the relative abundances of large OTUs in person and home samples, but varying degrees of overlap with pet samples.
Supplementary Figure 3: Summary of observed relative abundance of taxa for every sample in the study, divided by surface. Colors represent the 7 most abundant phyla in the study, and colored bars at base indicate which home a sample was taken from. Home surface samples are ordered by house and then by collection date while human samples are ordered by house, then individual, and then by collection date.
Supplementary Figure 4: Time series of SourceTracker source contribution estimates for the 6 home surfaces in houses 1 (A) and 4 (B). All samples taken from the occupants of each home were consolidated and treated as sources impacting the home surface sinks. Data is visualized as a stacked area chart of the proportion of each sink sample estimated to originate from each source, with samples ordered by sampling date along the x-axis. In both houses, Person 1 was traveling and did not interact with the house for the time indicated by the black bar below each time series.
Supplementary Figure 5: Dynamic Bayesian networks depicting causal interactions between surfaces in homes 1 though 4. Population data was considered at the phylum level and statistically significant Bayesian co-dependent relationships were defined between surfaces (nodes) in each family and home by phyla level taxonomic associations. Nodes are distributed using a spring-embedded layout that clusters highly-linked surfaces together. The direction of transfer is indicated by yellow arrowheads pointing from source to sink. Edges connecting the same source and sink are binned together and appear darker the more edges they encompass.
Supplementary Figure 6: PCoA plots of human samples taken from home 3 based on weighted Unifrac distance. The left plot includes all human samples while the right three are subdivided by surface. R values are ANOSIM tests for significant differentiation between the two residents in a couple (individuals 1 and 2) and a third roommate (person 3). All p-values are less that 0.0001 based on 10,000 permutations of the randomized dataset.
Supplementary Figure 7: Temporal community volatility of individual surfaces, measured as the median weighted UniFrac distance between consecutive samples in a timeseries.
Supplementary Figure 8: Summary of house 4 shotgun meteganomic data, analyzed as “level 2” functional abundance with a minimum e-value cutoff of 10−15. (A) Heatmap of functional abundance data, with samples (columns) ordered by similarity (dendrogram at top) and colored by source. (B) Principal coordinate plot of the 56 samples colored by source and based on Euclidean distances between functional abundance data.
Supplementary Figure 9: Relative abundance of antibiotic resistance genes in the shotgun metagenomes of house 4 samples, divided by sample surface.
Overview of the 1,586 samples included in the study. Samples were collected from six home surfaces and the foot, nose and hands of study participants across a two-week time series. Three of the seven participating families moved houses during the course of sampling; those samples taken before the move are designated “A” and those taken after are designated “B”. Samples were also collected from the foot and nose of three dogs living in home 4 and the fur and paw of a cat living in home 7.
Ten-fold cross validation models were constructed with 1,000 trees using OTUs generated from evenly rarified samples as predictors of sample origin. In the bottom 5 models, OTUs were collapsed to the taxonomic level indicated in the “Taxonomic Level” column.
Summary of all edges in the dynamic Bayesian network, including taxonomy and direction of transfer. Pairwise comparisons in the overview table are denoted “NA” if the particular transfer was not allowed by model parameters (see supplementary methods).
Acknowledgments
Alfred P Sloan Foundation’s Microbiology of the Built Environment Program. NIH Director’s New Innovator Award (DP2-DK-098089) to G.D. S.M.G was supported by an EPA STAR Fellowship. M.K.G. is a NSF graduate research fellow (award number DGE-11143954). This work was supported in part by the U.S. Dept. of Energy under Contract DE-AC02-06CH11357. We acknowledge the University of Chicago Research Computing Center for support of this work. All sequence data was submitted to the EBI’s database under accession number ERP005806
Footnotes
References and Notes
- 1.Höppe P, Martinac I. Indoor climate and air quality. Review of current and future topics in the field of ISB study group 10. Int J Biometeorol. 1998;42:1–7. doi: 10.1007/s004840050075. [DOI] [PubMed] [Google Scholar]
- 2.Custovic A, Taggart SCO, Woodcock A. House duet mite and cat allergen in different indoor environments. Clin Exp Allergy. 1994;24:1164–1168. doi: 10.1111/j.1365-2222.1994.tb03323.x. [DOI] [PubMed] [Google Scholar]
- 3.Dekio I, et al. Detection of potentially novel bacterial components of the human skin microbiota using culture-independent molecular profiling. J Med Microbiol. 2005;54:1231–8. doi: 10.1099/jmm.0.46075-0. [DOI] [PubMed] [Google Scholar]
- 4.Gao Z, Tseng C, Pei Z, Blaser MJ. Molecular analysis of human forearm superficial skin bacterial biota. Proc Natl Acad Sci. 2007;104:2927–32. doi: 10.1073/pnas.0607077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grice EA, et al. A diversity profile of the human skin microbiota. Genome Res. 2008;18:1043–50. doi: 10.1101/gr.075549.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grice EA, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–2. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grice EA, Segre Ja. The skin microbiome. Nat Rev. 2011;9:244–53. doi: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tringe SG, et al. The airborne metagenome in an indoor urban environment. PLoS One. 2008;3:e1862. doi: 10.1371/journal.pone.0001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawson P, Han I, Cox M, Black C, Simmons L. Residence time and food contact time effects on transfer of Salmonella typhimurium from tile, wood and carpet: testing the five-second rule. J Appl Microbiol. 2007;102:945–53. doi: 10.1111/j.1365-2672.2006.03171.x. [DOI] [PubMed] [Google Scholar]
- 10.Fujimura KE, Demoor T, Rauch M, Faruqi AA, Jang S, Johnson CC, Boushey HA, Zoratti E, Ownby D, Lukacs NW, Lynch SV. House dust exposure mediates gut microbiome Lactobacillus enrichment and airway immune defense against allergens and virus infections. PNAS. 2014;111(2):805–810. doi: 10.1073/pnas.1310750111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boone SA, Gerba CP. Significance of Fomites in the Spread of Respiratory Enteric Viral Disease. Applied and Environmental Microbiology. 2007;73(6):1687–1696. doi: 10.1128/AEM.02051-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, Bushman FD, Knight R, Kelley ST. Bayesian community-wide culture-independent microbial source tracking. Nature Methods. 2011;8(9):761–763. doi: 10.1038/nmeth.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaffal AA, et al. Residential Indoor Airborne Microbial Populations in the United Arab Emirates. Environ Int. 1997;23:529–533. [Google Scholar]
- 14.Hyvarinen A, Meklin T, Vepsalainen a, Nevalainen a. Fungi and actinobacteria in moisture-damaged building materials — concentrations and diversity. Int Biodeterior Biodegradation. 2002;49:27–37. [Google Scholar]
- 15.Nevalainen M, Seuri A. Of Microbes and Men. Indoor Air. 2005;15:58–64. doi: 10.1111/j.1600-0668.2005.00344.x. [DOI] [PubMed] [Google Scholar]
- 16.Lignell U, et al. Evaluation of quantitative PCR and culture methods for detection of house dust fungi and streptomycetes in relation to moisture damage of the house. Lett Appl Microbiol. 2008;47:303–308. doi: 10.1111/j.1472-765x.2008.02431.x. [DOI] [PubMed] [Google Scholar]
- 17.Bright KR, Boone Sa, Gerba CP. Occurrence of bacteria and viruses on elementary classroom surfaces and the potential role of classroom hygiene in the spread of infectious diseases. J Sch Nurs. 2010;26:33–41. doi: 10.1177/1059840509354383. [DOI] [PubMed] [Google Scholar]
- 18.Kramer A, Schwebke I, Kampf G. How long do nosocomial pathogens persist on inanimate surfaces? A systematic review. BMC Infect Dis. 2006;6:130. doi: 10.1186/1471-2334-6-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moschandreas DJ, Pagilla KR, Storino LV. Time and Space Uniformity of Indoor Bacteria Concentrations in Chicago Area Residences. Aerosol Science and Technology. 2003;37:11, 899–906. [Google Scholar]
- 20.Caporaso JG, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–4. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caporaso JG, et al. QIIME allows analysis of high- throughput community sequencing data Intensity normalization improves color calling in SOLiD sequencing. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lozupone C, Knight R. UniFrac: a New Phylogenetic Method for Comparing Microbial Communities UniFrac: a New Phylogenetic Method for Comparing Microbial Communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards RA. The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–8. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 25.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasan NA, Young BA, Minard-Smith AT, Saeed K, Li H, Heizer EM, McMillan NJ, Isom R, Abdullah AS, Bornman DM, Faith SA, Choi SY, Dickens ML, Cebula TA, Colwell RR. Microbial Community Profiling of Human Saliva Using Shotgun Metagenomic Sequencing. PLOS ONE. 2014;9(5):e97699. doi: 10.1371/journal.pone.0097699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lohse M, et al. RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic acids research. 2012;40:W622–627. doi: 10.1093/nar/gks540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu B, Pop M. ARDB–Antibiotic Resistance Genes Database. Nucleic acids research. 2009;37:D443–447. doi: 10.1093/nar/gkn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forslund K, et al. Country-specific antibiotic use practices impact the human gut resistome. Genome research. 2013;23:1163–1169. doi: 10.1101/gr.155465.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Effect of rarefaction to lower sequencing depth on ANOSIM R statistics (A) and random forest predictive accuracy (B).
Supplementary Figure 2: (A) Plot of log2-transformed average relative abundances in the human and home samples of individual homes (similar to Figure 2A, except divided by house). For an OTU to be included, it must have been detected at least 100 times within the home. OTUs are colored by whether their average relative abundance is significantly different between the home and person environments based on the FDR corrected p-value from a non-parametric t-test run with 1,000 permutations, and are sized based on their log10-transformed number of reads. The dashed line is y=x, indicating an equal average relative abundance. (B) Ternary plots of average relative abundances of OTUs with greater than 500 reads in the two homes with pets. OTUs are colored by taxonomy (phylum, with Proteobacteria split by class) and scaled to reflect OTU size. Both plots depict strong correlation between the relative abundances of large OTUs in person and home samples, but varying degrees of overlap with pet samples.
Supplementary Figure 3: Summary of observed relative abundance of taxa for every sample in the study, divided by surface. Colors represent the 7 most abundant phyla in the study, and colored bars at base indicate which home a sample was taken from. Home surface samples are ordered by house and then by collection date while human samples are ordered by house, then individual, and then by collection date.
Supplementary Figure 4: Time series of SourceTracker source contribution estimates for the 6 home surfaces in houses 1 (A) and 4 (B). All samples taken from the occupants of each home were consolidated and treated as sources impacting the home surface sinks. Data is visualized as a stacked area chart of the proportion of each sink sample estimated to originate from each source, with samples ordered by sampling date along the x-axis. In both houses, Person 1 was traveling and did not interact with the house for the time indicated by the black bar below each time series.
Supplementary Figure 5: Dynamic Bayesian networks depicting causal interactions between surfaces in homes 1 though 4. Population data was considered at the phylum level and statistically significant Bayesian co-dependent relationships were defined between surfaces (nodes) in each family and home by phyla level taxonomic associations. Nodes are distributed using a spring-embedded layout that clusters highly-linked surfaces together. The direction of transfer is indicated by yellow arrowheads pointing from source to sink. Edges connecting the same source and sink are binned together and appear darker the more edges they encompass.
Supplementary Figure 6: PCoA plots of human samples taken from home 3 based on weighted Unifrac distance. The left plot includes all human samples while the right three are subdivided by surface. R values are ANOSIM tests for significant differentiation between the two residents in a couple (individuals 1 and 2) and a third roommate (person 3). All p-values are less that 0.0001 based on 10,000 permutations of the randomized dataset.
Supplementary Figure 7: Temporal community volatility of individual surfaces, measured as the median weighted UniFrac distance between consecutive samples in a timeseries.
Supplementary Figure 8: Summary of house 4 shotgun meteganomic data, analyzed as “level 2” functional abundance with a minimum e-value cutoff of 10−15. (A) Heatmap of functional abundance data, with samples (columns) ordered by similarity (dendrogram at top) and colored by source. (B) Principal coordinate plot of the 56 samples colored by source and based on Euclidean distances between functional abundance data.
Supplementary Figure 9: Relative abundance of antibiotic resistance genes in the shotgun metagenomes of house 4 samples, divided by sample surface.
Overview of the 1,586 samples included in the study. Samples were collected from six home surfaces and the foot, nose and hands of study participants across a two-week time series. Three of the seven participating families moved houses during the course of sampling; those samples taken before the move are designated “A” and those taken after are designated “B”. Samples were also collected from the foot and nose of three dogs living in home 4 and the fur and paw of a cat living in home 7.
Ten-fold cross validation models were constructed with 1,000 trees using OTUs generated from evenly rarified samples as predictors of sample origin. In the bottom 5 models, OTUs were collapsed to the taxonomic level indicated in the “Taxonomic Level” column.
Summary of all edges in the dynamic Bayesian network, including taxonomy and direction of transfer. Pairwise comparisons in the overview table are denoted “NA” if the particular transfer was not allowed by model parameters (see supplementary methods).