

Abstract
Objective
Dysregulated muscle metabolism is a cardinal feature of human insulin resistance (IR) and associated diseases, including type 2 diabetes (T2D). However, specific reactions contributing to abnormal energetics and metabolic inflexibility in IR are unknown.
Methods
We utilize flux balance computational modeling to develop the first systems-level analysis of IR metabolism in fasted and fed states, and varying nutrient conditions. We systematically perturb the metabolic network to identify reactions that reproduce key features of IR-linked metabolism.
Results
While reduced glucose uptake is a major hallmark of IR, model-based reductions in either extracellular glucose availability or uptake do not alter metabolic flexibility, and thus are not sufficient to fully recapitulate IR-linked metabolism. Moreover, experimentally-reduced flux through single reactions does not reproduce key features of IR-linked metabolism. However, dual knockdowns of pyruvate dehydrogenase (PDH), in combination with reduced lipid uptake or lipid/amino acid oxidation (ETFDH), does reduce ATP synthesis, TCA cycle flux, and metabolic flexibility. Experimental validation demonstrates robust impact of dual knockdowns in PDH/ETFDH on cellular energetics and TCA cycle flux in cultured myocytes. Parallel analysis of transcriptomic and metabolomics data in humans with IR and T2D demonstrates downregulation of PDH subunits and upregulation of its inhibitory kinase PDK4, both of which would be predicted to decrease PDH flux, concordant with the model.
Conclusions
Our results indicate that complex interactions between multiple biochemical reactions contribute to metabolic perturbations observed in human IR, and that the PDH complex plays a key role in these metabolic phenotypes.
Keywords: Muscle insulin resistance, Muscle metabolism, Flux balance analysis, Computational modeling
Graphical abstract
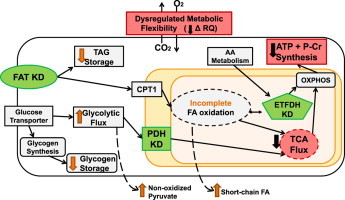
Schematic of targeted knockdowns that model phenotypes of insulin resistance. Knockdowns (KD; green) of fatty acid transporter (FAT), pyruvate dehydrogenase (PDH), and electron transfer flavoprotein (ETFDH) recapitulated the metabolic phenotypes of insulin resistance (red), defined as reduced ATP + P-Cr synthesis, TCA flux, and metabolic flexibility (RQ). Additional metabolic alterations identified are depicted by orange arrows.
1. Introduction
Insulin resistance (IR), or reduced responsiveness to insulin, is a key feature of aging, cardiovascular disease, obesity, type 2 diabetes (T2D), lipid disorders, some cancers, and other components of the metabolic syndrome. IR contributes to the pathophysiology of each of these increasingly prevalent disorders, being present years before disease onset and predicting disease development decades later [1,2]. Although IR occurs in many tissues, skeletal muscle is a key tissue, being responsible for over 80% of insulin-stimulated glucose uptake [3].
Both whole-body and muscle IR are typically defined by its effects on tissue glucose uptake. However, muscle IR is also intimately linked to altered energy metabolism, including (i) reduced ATP and creatine phosphate (Cr-P) synthesis at rest [4] and in response to insulin [5] and exercise [6]; (ii) reduced tricarboxylic acid (TCA) cycle activity [7,8]; and (iii) reduced ability to toggle between fatty acid and carbohydrate metabolism (termed “metabolic inflexibility”) in the fasting-to-fed transition [9]. Transcriptome analysis demonstrates only modest dysregulation of mitochondrial regulatory genes in IR and T2D [10–14], suggesting that regulation is likely achieved at a post-transcriptional level. However, the specific metabolic reactions responsible for driving these metabolic defects related to IR remain unknown. Moreover, it is not known which reactions could be targeted to improve insulin sensitivity and metabolic wellness. Given the complexity and redundancy of metabolism, and the interactions between genetic and environmental risk factors which contribute to metabolic defects in IR, it is difficult to address these key questions experimentally.
To approach this important challenge, we have utilized an integrative approach, using flux balance analysis (FBA) computational modeling [15,16], together with experimental validation. FBA is a constraint-based approach which has been used successfully to study both bacterial and human metabolism by modeling relationships between input and output fluxes through biochemical reactions in metabolic networks under different conditions and objectives [16–30]. We now apply FBA to the study of muscle metabolism, the fasted to fed transition, and the impact of increased substrate availability (as in IR conditions). Furthermore, we systematically perturb the metabolic network by performing unbiased in silico knockdowns of each of the model's 388 reactions in order to identify those perturbations which yield IR-associated metabolic phenotypes.
2. Material and methods
2.1. Model development
We utilized our muscle flux balance model [29], a model which built upon the prior work of Ramakrishna et al. [31] with the addition of critical muscle-specific components of metabolism, including amino acid metabolism, protein synthesis, fatty acid oxidation, and pentose phosphate metabolism (Figure 1). To more fully study metabolism associated with insulin resistance, we updated the model and expanded the fatty acid synthesis reactions, as detailed in Supplemental Experimental Procedures. The model was optimized in MATLAB (www.mathworks.com) using the open-source GLPK solver with default settings. The objective function was set to maximize flux through ATP, Cr-P, TAG, and glycogen storage with the sum of ATP + Cr-P weighted twice that of the other molecules, as detailed in Supplemental Experimental Procedures. The model is provided in SBML format (Supplemental File Muscle Model.zip).
Figure 1.
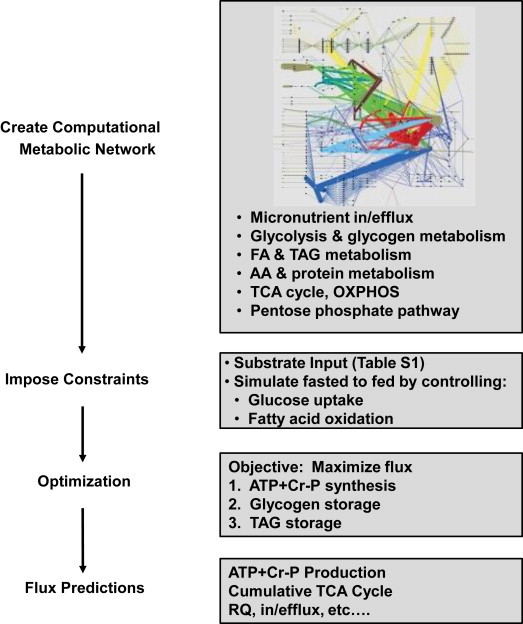
Schematic of steps in model development.
To simulate metabolic transitions between fasting and fed conditions, lipid oxidation (CPT1) and glucose uptake were reciprocally modulated. CPT1 activity was limited stepwise from a maximum of 100% (fasted) to minimum of 0% (fed), while glucose uptake was modeled in two distinct components: basal (GUbas; constant 5%) and modifiable (GUmod; limited stepwise from 0% to 100% of plasma concentration). Three metabolic states were defined in particular: fasted (90% CPT1, 10% GUmod), mixed (50% CPT1, 50% GUmod), and fed (10% CPT1, 90% GUmod). Since the obesity/diabetes metabolic milieu is characterized by increased plasma levels of nutrient substrates, which may themselves alter flux, we also tested the impact of 2-fold increases in glucose (Gluc) and fatty acid (FA) availability (designated as 2XGluc and 2XFA), both independently and in combination.
2.2. Modeling IR phenotypes and the impact of In silico knockdowns
We defined IR metabolism using three distinct phenotypes derived from studies of IR humans: (1) reduced ATP + Cr-P production [4–6], (2) reduced TCA cycle flux [7,8], and (3) reduced metabolic flexibility [9]. These IR phenotypes were defined mathematically, as described in Supplemental Experimental Procedures.
To identify metabolic reactions which could recapitulate these predefined metabolic features of IR, we performed systematic in silico knockdowns (KDs) of each reaction in the model by constraining the flux for each reaction to 50% of its normal, optimal value for each of the three defined states (fasted, mixed, and fed). With this additional constraint, the model was re-optimized to obtain another set of fluxes (Figure 3A), and fluxes and IR phenotype scores were compared to the control (no knockdown).
Figure 3.
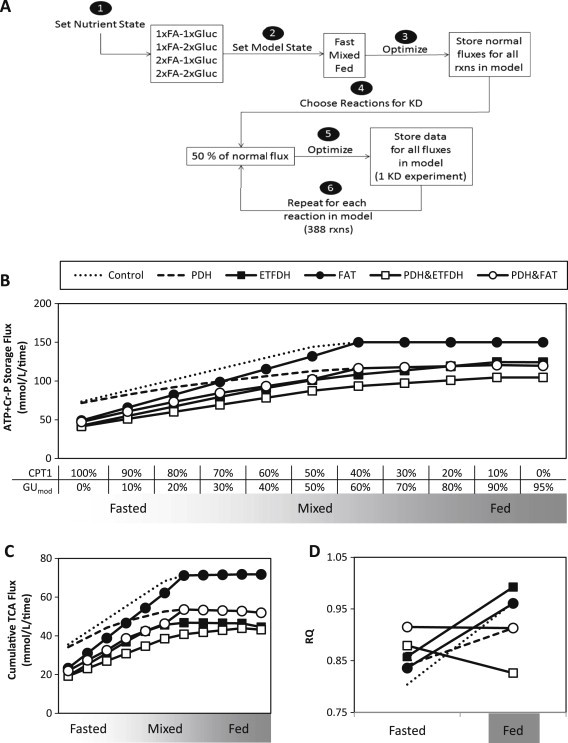
Combined knockdowns can induce metabolic phenotypes associated with insulin resistance. (A) Workflow for in silico knockdown experiments. (B) ATP + Cr-P storage, (C) TCA cycle flux, and (D) respiratory quotient (RQ) under control conditions and in response to single or combined knockdowns.
2.3. Pathway analysis
Each of the 388 model reactions were assigned to KEGG pathway(s) (using EC number if available) or manually curated pathways (Supplemental Experimental Procedures). The 47 pathways are delineated in Supplemental File All KDs-fluxes, scores, and p-values.xls, pathways tab. For each pathway and phenotype, we counted the number of knockdowns of reactions within the pathway that caused a phenotype change, and assessed enrichment using Fisher's exact test to determine whether the ratio (number of KD affecting phenotype per pathway divided by the total number of KD affecting phenotype) differed statistically from the expected ratio (number of reactions in pathway/total number of reactions). Benjamini-Hochberg method was used to calculate false discovery rate (FDR).
2.4. Primary cell preparation, transfection, and validation studies
Experimental protocols are described in Supplemental Experimental Procedures. In brief, myocytes were isolated from 4-week-old wild type or PDK2/4-KO mice [32] by digesting leg muscles with 2.4 U/mL dispase and 1% collagenase. Myoblasts were fed daily with F10 with 20% FBS and 2.5 ng/mL basic FGF. Scrambled or ETFDH-specific siRNA were transfected using HiPerFect (Qiagen, Valencia, CA). Knockdown efficacy was analyzed at 72 h by qRT-PCR and western blotting. ATP content was measured using CellTiter-Glo luminescent assay (Promega, Fitchburg, WI, USA), normalized to protein. CS activity was measured using CS0720 kit (Sigma, St. Louis, MO, USA), normalized to protein. Metabolic flexibility was assessed using a Seahorse XF24 Flux Analyzer. Cells were treated overnight with 250 uM palmitate/BSA, prior to incubation for 1 h in KRB buffer with 5.5 mM glucose and 250 uM palmitate/BSA. OCR and ECAR were measured prior to and after addition of 5.5 mM glucose; data were normalized to DNA content. Significance was assessed using 2-tailed Student's t-test, with p < 0.05 as threshold.
Gene expression was assessed in human muscle biopsy samples from a cohort of 52 metabolically characterized individuals obtained in the fasting state using Affymetrix HG-U133 2.0 Plus microarrays as previously described [33]. Data were normalized using the MAS5.0 algorithm and are available at the Gene Expression Omnibus (GEO, GSE25462). Significance of expression differences between groups was assessed in log-transformed data using a moderated t-test with the limma package in R software [34], and association with insulin sensitivity (log SI) was tested using Pearson correlation. Both tests were one-tailed (predicted downregulation for PDH and ETF complex genes, upregulation for PDKs). Untargeted metabolomic analysis was performed in the same biopsy samples using multi-platform mass spectroscopy, as described (Metabolon, Durham, NC) [35]. The association of pyruvate with log SI was tested using Spearman correlation.
3. Results
3.1. The model captures fasted to fed transitions
We simulated dynamic shifts across the fasting-fed transition by modulating fatty acid and glucose uptake (Figure 2A). Specifically, we constrained CPT1 (the rate-limiting step of mitochondrial fatty acid uptake), assigning maximal activity in fasting, with progressive stepwise reductions toward the fed spectrum. For glucose uptake, we assigned both a constant basal component (5% of plasma glucose concentration, Table S1) and a modifiable component that increased progressively from fasted to fed states (Figure 2A). In each state, the model allows free uptake of fatty acids and amino acids up to concentrations in fasting human plasma (Table S1) [36,37].
Figure 2.
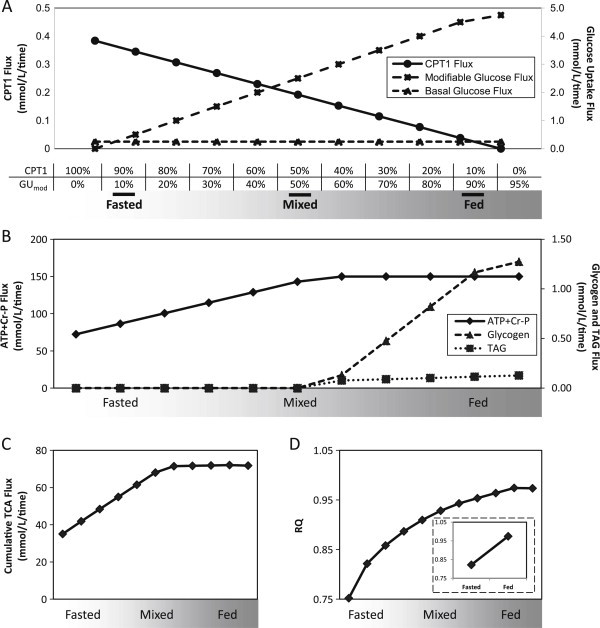
Modeling muscle metabolism under normal nutrient conditions: (A) Simulation of fasting-to-fed transition by modulation of flux through carnitine palmitoyl-transferase 1 (CPT1) and a modifiable component of glucose uptake (GUmod) at discrete percentages of maximal flux through CPT1 and GUmod. (B) Fluxes for ATP + Cr-P, glycogen storage, and TAG storage. (C) Cumulative TCA flux increases across the fasted to fed spectrum. (D) Respiratory quotient (RQ) across the fasted to fed spectrum.
We next examined flux through key reactions across the fasting-fed spectrum to confirm that our model mirrored expected metabolic profiles. ATP + Cr-P production increases to maximum in the mixed/fed states, with excess energy stored as glycogen and triglycerides (Figure 2B). Similarly, TCA cycle cumulative flux increases from fasting to fed states (Figure 2C). Other changes in flux across the fasted to fed transition are visualized in Figure S1. As expected, glycolytic flux increases in the fed state, concurrent with increased glucose availability, while amino acid uptake and oxidation remain constant (Figure S2C/D). As expected, respiratory quotient (RQ) increases across the fasted to fed transition, reflecting increased reliance on glucose-derived substrate oxidation (Figure 2D).
3.2. Impact of increased nutrient availability on metabolic fluxes
The obesity/diabetes metabolic milieu is characterized by increased plasma levels of nutrient substrates, which may themselves alter flux. We tested the impact of 2-fold increases in glucose and fatty acid availability (2XGluc, 2XFA), both independently and in combination (Table 1A, Figure S1). As expected, nutrient excess conditions increased ATP + Cr-P production and TCA flux during the fasted to fed transition (Figure S1A/B), paralleling oxygen consumption and OXPHOS flux (Figure S1D/E). Increased glucose availability (2xGluc) results in increased flux through glycolysis, glycogen storage, and TAG storage, and shifts the RQ curve leftward (Figure S1C), indicating increased glucose oxidation even in the fasting spectrum. Similarly, increased fatty acid availability (2XFA) increased flux through TCA, OXPHOS, glycogen storage, and TAG storage (Figure S1B/E/G/I) and decreased RQ (Figure S1C), reflecting increased lipid oxidation. With increased availability of both glucose and fatty acids (2XGluc/2XFA), effects of increased glucose predominate (Table 1A), with increased flux through glycolysis, glycogen storage, and TAG storage, even in fasting conditions.
Table 1.
A. Major qualitative changes induced by increased glucose and fatty acid substrate availability (1xFA = 0.3838 mmol/L; 1xGluc = 5.0 mmol/L). B. Top KDs causing reduced metabolic flexibility in basal nutrient condition (1xFA-1xGluc). For metabolic flexibility, data indicate cumulative difference in RQ (Eq. 8 in Supplemental Experimental Procedures). For each KD, data indicate ratio of KD compared to control (Eq. 6 and 7 in Supplemental Experimental Procedures).
| Condition | Major qualitative changes compared to 1xFA-1xGluc |
|---|---|
| A. | |
| 2× glucose |
|
| 2× fatty acid |
|
| 2× fatty acid-2× glucose |
|
| KD Reaction | EC Number | Met. Flex |
ATP + Cr-P Production |
Cumulative TCA Flux |
||||
|---|---|---|---|---|---|---|---|---|
| Δ RQ | Fasted | Mixed | Fed | Fasted | Mixed | Fed | ||
| B. | ||||||||
| PDH | 1.2.4.1 | 0.09 | 0.94 | 0.78 | 0.80 | 0.93 | 0.77 | 0.73 |
| FAT-CD36 | n/a | 0.03 | 0.75 | 0.92 | 1.00 | 0.74 | 0.91 | 1.00 |
| PalTK | 6.2.1.3 | 0.03 | 0.75 | 0.92 | 1.00 | 0.74 | 0.91 | 1.00 |
| ASTM | 2.6.1.1 | 0.01 | 0.96 | 0.73 | 0.68 | 0.97 | 0.91 | 0.64 |
| ASTC | n/a | 0.01 | 0.95 | 0.73 | 0.68 | 0.96 | 0.72 | 0.64 |
| Glu–Asp Ex | n/a | 0.01 | 0.96 | 0.73 | 0.68 | 0.97 | 0.72 | 0.64 |
3.3. Reduction in glucose uptake does not alter metabolic flexibility
Since reduced glucose uptake is a cardinal feature of IR, we asked whether experimental reduction in cellular glucose uptake could contribute to metabolic phenotypes associated with IR. While a 50% reduction in the modifiable glucose uptake reduced ATP + Cr-P, cumulative TCA flux, and other glucose-dependent pathways as expected (Figure S1), it did not modify RQ substantially at any point along the fasting-fed spectrum, and thus did not alter metabolic flexibility (Figure S1C).
3.4. Identification of network perturbations resulting in IR metabolism
Given that neither nutrient excess nor reduced glucose uptake fully mimicked IR metabolism, we aimed to identify reactions and pathways which could confer these metabolic phenotypes by systematically perturbing flux in each of the network reactions. We performed in silico knockdowns (KD), defined as a 50% reduction in the normal (control) flux for each nutrient condition and state (Figure 3A), and examined the impact of each knockdown on three predefined metabolic endpoints: ATP + Cr-P production, TCA flux, and the change in RQ from fasting to fed conditions.
In the basal nutrient condition (1xFA-1xGluc), KDs in 34 reactions in fasted state, 30 in mixed state, and 26 in fed state significantly reduced ATP + Cr-P production (KD/control <1, p ≤ 0.05, Table S2A). Top-ranking reactions included all OXPHOS complexes, cytosol-mitochondrial NADH shuttles, and TCA cycle. KDs in lipid and branched chain amino acid oxidation reduced ATP + Cr-P production in fasting and mixed, but not fed states, consistent with the more important role for these substrates during fasting. KDs that reduced cumulative TCA cycle flux (KD/control <1, p ≤ 0.05) almost completely overlapped those influencing ATP + Cr-P (Table S2B).
We assessed metabolic flexibility by assessing RQ patterns in the fasted and fed states; values indicating reduced RQ change in the fasting-fed transition were considered to reflect the metabolic inflexibility characteristic of IR, with significance determined by comparison to the distribution of RQ changes (details, section 2). KD of only one reaction, pyruvate dehydrogenase (PDH) significantly reduced metabolic flexibility (Table S2C). KD of fatty acid transport (e.g., Ex_pal; FAT-CD36), long-chain acyl-CoA synthase (ACSL), glutamate-aspartate mitochondrial exchange (GLAST), and both cytosolic and mitochondrial aspartate transaminase (AST) also reduced metabolic flexibility scores (Table S2C).
Given that reduced insulin-stimulated glucose uptake is a hallmark of IR, we also asked whether knockdowns of any reaction could reduce glucose uptake. Interestingly, knockdowns in hexokinase, glycerol-3-phosphate dehydrogenase, and phosphoglycerokinase each reduced glucose uptake, in a pattern similar to reducing modifiable glucose uptake (described above in Section 3.3).
Under conditions of increased glucose and fatty acid availability (2xFA-2xGluc), we observed that objective functions of the model can be achieved more readily (even in the fasting state), and fewer KDs had significant effects on energy production. Significant reductions in ATP + Cr-P production were observed with 31 KDs in fasting, 18 in mixed, and 17 in fed state (Table S2D), with similar patterns for TCA flux (Table S2E). Once again, only KD in PDH yielded significant alterations in metabolic flexibility, with similar trends for FAT and ACSL (Tables S2F/G). Results for individual nutrient excess conditions are provided in Supplemental File All KDs-fluxes, scores, and p-values.xls.
3.5. Pathway analysis
To determine whether knockdowns within specific metabolic pathways were more likely to yield metabolic phenotypes linked to IR, we performed a statistical pathway-based analysis (Supplemental File All KDs-fluxes, scores, and p-values.xls, pathways tab), and ranked pathways using Fisher's exact test (with FDR correction). Under control nutrient conditions, three pathways were significantly enriched for number of reactions for which KD caused a reduction in ATP + Cr-P production (Q < 0.25) in the fasting and mixed states: BCAA, butanoate, and lysine metabolism (Table S3A/D). Similarly, 7 pathways were significantly enriched for reducing TCA flux: BCAA, butanoate, lysine, propanoate, tryptophan, FAD metabolism, and TCA cycle itself (Table S3B/D). Only three pathways were enriched for reductions in metabolic flexibility: glycerophospholipid metabolism, lysine metabolism, and NADH shuttle (Table S3C/D).
With increased glucose and fatty acid availability (2xFA-2xGluc), the dominant energy-producing pathways were significantly enriched for affecting ATP + Cr-P production (e.g., OXPHOS, TCA, glycolysis, fatty acid metabolism, NADH shuttles), while the BCAA pathway was no longer enriched (Table S3E/H). Increased nutrient availability also reduced the number of pathways significantly enriched for reduced TCA flux (Table S3F/H): glycerophospholipid, glycolysis, lysine, OXPHOS, and NADH shuttle. The only pathway enriched (nominal p < 0.05) for reduced metabolic flexibility with increased glucose and fatty acid availability was glycerophospholipid metabolism; this did not remain significant after adjustment (Q = 0.73) (Table S3G/H).
3.6. Multiple simultaneous knockdowns are required to recapitulate IR metabolic phenotypes
Our results indicate that reduced flux in any single reaction cannot produce all three metabolic phenotypes associated with IR in all conditions, a likely result of the extensive redundancy in cellular metabolism. Thus, we asked whether simultaneous KDs in more than one reaction could more readily induce IR-related phenotypes. We performed KD of each of the top three reactions causing decreased metabolic flexibility (PDH, FAT, AST), and then systematically performed a simultaneous knockdown of all other reactions. Only two knockdown pairs, either PDH and electron-transferring-flavoprotein dehydrogenase (ETFDH), or PDH and FAT, reduced ATP + Cr-P production, cumulative TCA flux, and metabolic flexibility (Figure 3B–D, open shapes). While the PDH&ETFDH and PDH&FAT double KDs both led to the IR-related phenotype in the basal nutrient state, the PDH&ETFDH double KD was the only combination which yielded all three IR-related phenotypes in all four nutrient conditions (Table S4).
Interestingly, reduced flux through PDH&ETFDH or PDH&FAT also modulated glucose, lipid, and amino acid metabolism, yielding phenotypes commonly observed in IR humans. These included decreased PDH activity and increased non-oxidized pyruvate (lactate) production (Figure 4A and B), most striking with the PDH&ETFDH combination. Although these combined KD did not alter modifiable glucose uptake (Figure S2A), glycogen storage was reduced [38] (Figure 4C). Strikingly, double KDs in PDH&ETFDH or PDH&FAT reduced complete fatty acid oxidation (Figure 4D) and increased release of short-chain fatty acids (Figure 4E), suggesting incomplete fatty acid oxidation [39]. Combined KD also reduced FA uptake (Figure S2B) and TAG storage (Figure 4F and S2E-H). ETFDH knockdown reduced both amino acid uptake and BCAA and lysine oxidation (Figure S2C/D). Knockdown of PDH alone, or together with FAT, increased amino acid uptake in the fed spectrum, potentially allowing maintenance of cellular energetics when lipid supplies are limited (Figure S2C). Thus, perturbations in flux identified by the model yielded many robust metabolic phenotypes associated with IR (Figure 7).
Figure 4.
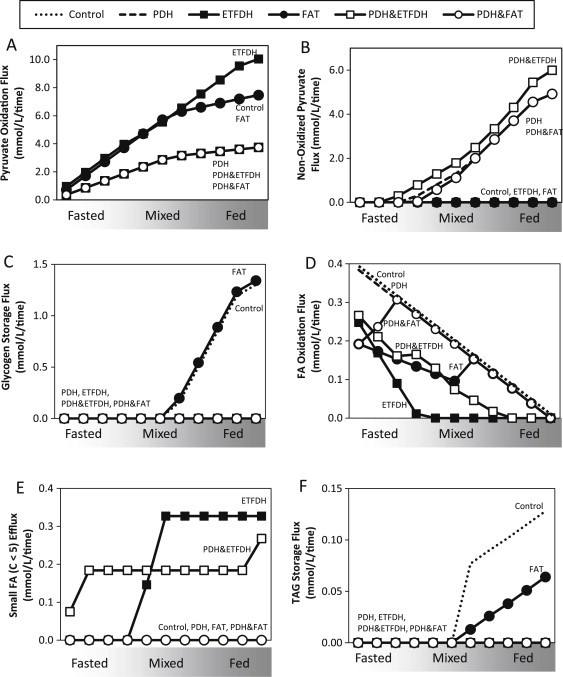
Double knockdowns (PDH/ETFDH or PDH/FAT) also alter additional parameters of carbohydrate and fatty acid metabolism, including (A) PDH activity, (B) reduced non-oxidized pyruvate, (C) reduced glycogen storage, (D) reduced complete fatty acid oxidation, (E) increased release of short-chain acylcarnitines, and (F) TAG storage.
Figure 7.
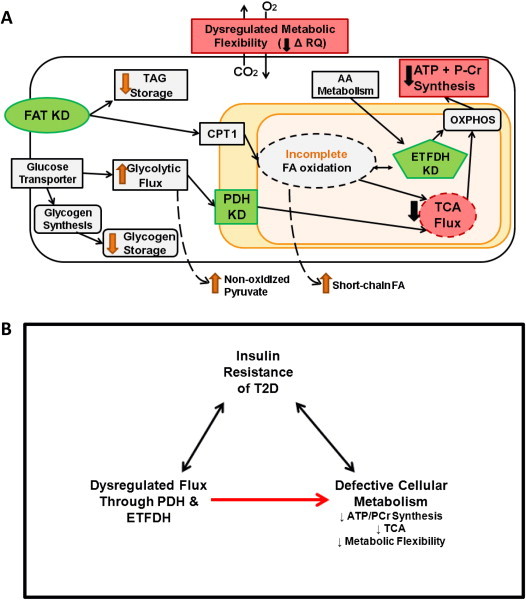
Schematic depiction of targeted knockdowns that model phenotypes of IR. (A) Knockdowns (KD; green) of fatty acid transporter (FAT), pyruvate dehydrogenase (PDH), and electron transfer flavoprotein (ETFDH) recapitulated the metabolic phenotypes of IR (red), defined as reduced ATP + P-Cr synthesis, TCA flux, and metabolic flexibility (RQ). Additional metabolic alterations identified are depicted by orange arrows. (B) Model depicting the role of PDH and ETFDH in metabolic phenotypes associated with IR.
3.7. Biological validation of in silico results in cultured mouse myocytes
To determine if alterations in flux through the enzymes identified in our in silico analysis would also recapitulate IR metabolic phenotypes in vivo, we analyzed the impact of modulating candidate enzymes in mouse muscle cells. We isolated primary myoblasts derived from mice in which PDH flux was modulated by dual genetic ablation of the PDH inhibitors pyruvate dehydrogenase kinase 2 and 4 (PDK2/4). PDKs are important regulators of PDH, deactivating PDH via phosphorylation [40]. Without PDK's inhibitory effect, PDH phosphorylation and flux are increased, as compared to WT (Figure 5A); thus, comparison of PDK2/4-KO and WT cells permits analysis of the metabolic impact of relative reductions in PDH activity. We then superimposed siRNA-mediated KD of ETFDH in PDK2/4-KO and WT cells to examine the impact of decreased activity of both candidate enzymes, independently and in combination. We confirmed decreased mRNA expression of PDK2/4 and ETFDH by qRT-PCR (Figure 5B). Western blots demonstrated reduced PDH phosphorylation in PDK2/4-KO cells and reduced ETFDH protein in ETFDH siRNA-treated cells (Figure 5C).
Figure 5.
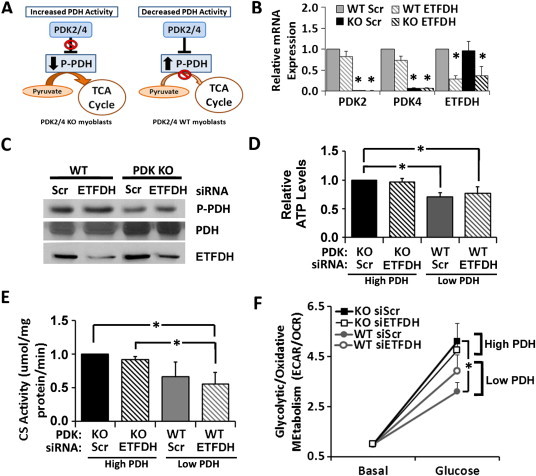
Double knockdowns in myoblasts recapitulate the IR phenotypes predicted by in silico experiments. (A) Schematic of PDK2/4 regulation of PDH. (B) mRNA expression by qRT-PCR analysis. (C) Western blot analysis of phosphorylated and total PDH and ETFDH protein expression. (D) Changes in ATP levels (normalized to protein). (E) Changes in citrate synthase (CS) activity (normalized to protein). (F) Changes in ECAR/OCR values (normalized to DNA) under basal conditions (250 μM palmitate) and after addition of 5.5 mM glucose, shown relative to the basal level. *p < 0.05.
Consistent with model predictions, cells with lower PDH activity (WT) had significantly lower ATP content (Figure 5D). Myoblasts with decreased PDH and ETFDH activity had decreased activity of citrate synthase, a key TCA cycle enzyme (Figure 5E). To determine the impact of experimental manipulation of PDH and ETFDH on metabolic flexibility, we assessed cellular substrate utilization and responses to nutrient modulation. Specifically, we measured extracellular acidification rate (ECAR), a proxy for glycolytic glucose metabolism, and oxygen consumption rate (OCR), a measure of aerobic respiration; we determined the ECAR/OCR ratio as a surrogate for relative glycolytic/oxidative substrate utilization. In fasted conditions, lipid substrate utilization dominates, with more oxygen consumption and less glycolysis, yielding a lower ECAR/OCR ratio. By contrast, in the fed state, higher ECAR/OCR ratio is consistent with predominant glucose utilization. Cells were incubated with palmitate (promoting lipid oxidation) and then acutely treated with glucose to provoke a switch from lipid to carbohydrate metabolism. As expected, glucose increased ECAR/OCR ratio, consistent with shift to glycolytic metabolism (Figure 5F). Consistent with model predictions, cells with lower PDH flux displayed a reduced shift to glycolytic metabolism (decreased slope of ECAR/OCR change) upon addition of glucose (Figure 5F).
3.8. Human data also demonstrate perturbation at the PDH complex
To determine whether perturbation in subunits of the PDH and ETFDH complexes could also be observed in humans with IR metabolism, we assessed gene expression in muscle biopsies from patients with T2D as compared with healthy controls. One of 2 probesets for ETFB was reduced by 69% (p = 0.04). Strikingly, 5 of 7 probesets for PDH were significantly downregulated in T2D by 21–30% (Table S5); data for one probeset for PDHB is shown in Figure 6A (p = 0.009). Expression of PDHB also correlated with insulin sensitivity (Figure 6B, p = 0.026).
Figure 6.
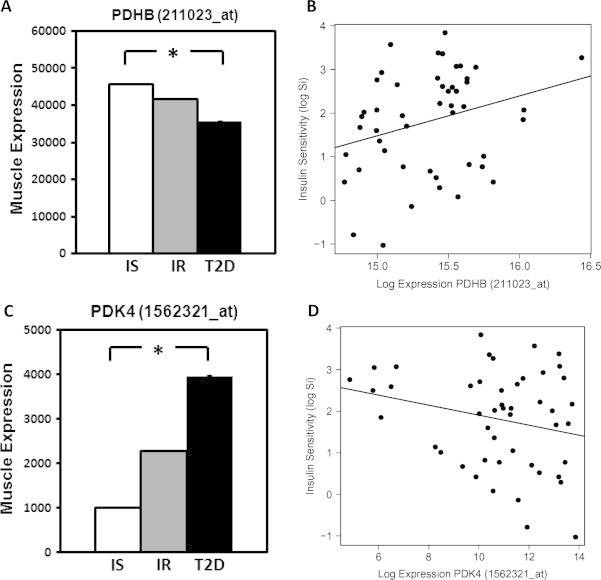
Expression of PDHB and PDK4 are perturbed in human insulin resistance and T2D. (A) Expression of PDHB and (B) correlation with insulin sensitivity (log Si) in muscle from humans with normal insulin sensitivity (IS), insulin resistance (IR), and T2D. (C) Expression of PDK4 and (D) correlation with insulin sensitivity in IS, IR, and T2D humans (representative probesets). *p < 0.05.
Recent studies have underscored the importance of inhibitory regulation of PDH by PDH kinases (PDK), particularly the muscle-dominant PDK4 [41,42]. PDK4 is upregulated in diet-induced IR, and PDK4 deficiency improves glucose tolerance in mice [41]. Thus, we examined expression of PDKs in human muscle (Table S5). PDK4 was significantly upregulated in T2D, with 3 of 3 probesets revealing increases of 2.7–4-fold (Figure 6C). Importantly, PDK4 expression was also increased by 1.5–2.3 fold in individuals with isolated IR (p < 0.05 for 2 of 3 probesets, Figure 6C, middle bars) and was correlated inversely with insulin sensitivity in 3 of 3 probesets (Figure 6D, p ≤ 0.05 for all). Metabolomic analysis of muscle samples also demonstrated an inverse correlation between pyruvate levels and insulin sensitivity (Spearman correlation coefficient −0.32, not shown). Together, these data demonstrate that pyruvate dehydrogenase is a key regulatory node disrupted in human IR and T2D, and may contribute to defective energy metabolism and impaired metabolic flexibility characteristic of human IR.
4. Discussion
Muscle insulin resistance is associated with dysregulation in multiple metabolic pathways. However, it remains unknown which specific metabolic reactions are the most important contributors to these phenotypes. We have approached this important question by developing a contextually novel in silico flux-based analysis model of IR muscle metabolism. As with any modeling approach, FBA requires several key assumptions [16]. Most importantly, FBA assumes that flux across each metabolic reaction is modulated to achieve the defined objectives of the entire system. We defined the objective function (goal) of muscle as both energy production (ATP + Cr-P) and storage (glycogen, triglycerides) for subsequent contraction. In addition, FBA modeling also assumes that a steady state can be achieved. While this is typically achieved in vivo during prolonged fasting, we examined steady-state fluxes at discrete points along the fasting-to-fed transition. To model the fasting state, characterized by increased fatty acid utilization, we allow maximal flux through CPT1. Similarly, we relax the constraint on modifiable glucose uptake to mimic increased glucose uptake in the fed state. Thus, our model mirrors alterations in flux in response to prevailing nutrient and metabolic conditions in vivo.
We first used our model to determine whether isolated increases in availability of extracellular glucose and fatty acids, as seen in patients with IR and T2D, would induce metabolic phenotypes associated with IR. A two-fold increase in glucose availability increased flux through glycolysis, glycogen storage, and TAG storage, while a two-fold increase in fatty acid availability increased TCA cycle, OXPHOS, and glycogen and TAG storage. With increases in both glucose and fatty acids, glucose effects dominate, with increases in glycolytic fluxes even in fasting states. The predominant effect of glucose is likely to result from its higher extracellular concentration (2xGluc = 10 mM) as compared with fatty acids (2xFA = 0.77 mM). Indeed, further increases in FA availability (7.8x FA, or 3 mM, predicted to provide ATP equivalent to 2xGluc) increase energy availability and shift toward lipid metabolism (Figure S3). Thus, isolated increases in nutrient availability do not reproduce the metabolic phenotypes associated with IR. Whether IR could ultimately develop as a protective response to limit nutrient overload in this setting remains an intriguing possibility [43].
Given that insulin signaling results in increased glucose uptake into muscle tissue, both whole-body and muscle IR are often defined and quantified by reduced glucose uptake, an important endpoint for insulin signaling. Such reductions in intracellular glucose availability have been proposed to contribute to reduced glucose metabolism in the insulin-stimulated (fed) state, a cardinal feature of metabolic inflexibility. Surprisingly, we find that reduced glucose availability or uptake does reduce ATP production (as expected), but does not alter relative fuel metabolism (as indicated by RQ). Thus, these data suggest that reduced glucose uptake is not sufficient to induce metabolic inflexibility, and that some key metabolic features of IR can be dissociated from glucose uptake.
We next performed in silico knockdown (KD) of each reaction within the model, both singly and in combination, in order to identify reactions and pathways which modulate IR-linked metabolic phenotypes. Not surprisingly, ATP + Cr-P production was sensitive to KDs within key energy-transducing pathways (e.g., TCA, OXPHOS, and NADH shuttles). By contrast, reduced flux in lipid and branched chain amino acid oxidation pathways only reduced ATP + Cr-P production in the fasting state—consistent with the more important role for these nutrients as energy sources when glucose is limited. KDs that reduced cumulative TCA cycle flux were very similar to those reducing ATP production, a finding likely related to the role for TCA and OXPHOS as the “final common pathway” for energy production.
Conversely, relatively few KDs altered metabolic flexibility. Notably, reductions in OXPHOS flux did not alter metabolic flexibility; thus, isolated defects in OXPHOS are unlikely to be solely responsible for all IR-linked metabolic phenotypes [44]. Interestingly, metabolic inflexibility was induced by regulators of glucose oxidative metabolism (PDH), lipid uptake and metabolism (CD36 and ACSL), aspartate metabolism (AST), and glutamate-aspartate shuttles that transport electrons into the mitochondrion for oxidative phosphorylation. Of these, only knockdown of PDH also yielded significant reductions in ATP production and TCA cycle flux in the basal state.
In general, cellular energy metabolism was less sensitive to experimental knockdowns when glucose or fatty acid availability was increased; by contrast, increased nutrient availability increased sensitivity to metabolic inflexibility. However, no single knockdown (even PDH) yielded all 3 IR metabolic phenotypes under all conditions sampled. This is intuitive given that metabolic flux differs in each nutrient condition, and redundancy could attenuate effects of a single knockdown.
We thus hypothesized that altered flux at multiple key reactions would be required to fully recapitulate the three metabolic phenotypes. Consistent with this hypothesis, KD in PDH, when combined with either electron-transferring-flavoprotein dehydrogenase (ETFDH) or FAT, did reduce ATP + Cr-P production, TCA flux, and metabolic flexibility (Figure 7A). Combined knockdowns also yielded additional features of IR metabolism, including reduced glycogen storage and fatty acid oxidation. Combined knockdowns also increased short-chain fatty acid release, suggesting incomplete fatty acid oxidation [39]. Knockdown of ETFDH also had unique effects on amino acid metabolism, reducing both uptake and metabolism of branched chain amino acids and lysine. This is particularly notable since plasma levels of branched chain amino acids are increased in IR [45,46], perhaps related to reduced tissue BCAA metabolism [47].
We validated our in silico predictions in vivo using cultured myocytes by modulating flux through PDH and ETFDH, singly and in combination, using a combination of genetic ablation (of PDK2/4) and siRNA targeting ETFDH. Lower PDH activity and ETFDH KD reduced ATP production, reduced citrate synthase activity, and reduced metabolic flexibility (approximated by change in ECAR/OCR in response to glucose). Thus, altered flux through PDH and ETFDH can recapitulate elements of IR-linked metabolism in vivo.
Our data highlight the central regulatory role for pyruvate dehydrogenase (PDH), a multi-enzyme complex that converts pyruvate to acetyl-CoA. Activity of PDH is regulated by its coenzymes NAD+ and CoA, post-translational modifications (e.g., lysine acetylation [48], O-glycosylation [49], glutathionylation [50], and PDH kinases/phosphatases). Randle first proposed a key regulatory role for PDH in muscle; increased ATP and NADH availability, as with excess lipid substrates, would inhibit PDH, reducing glucose uptake and metabolism [51]. More recently, cardiac mitochondrial network analysis predicted reduced PDH flux under conditions of increased fatty acid uptake [19]. PDH activity is reduced in IR humans [52] and in lipid-induced IR [53], and has been linked with metabolic inflexibility in rodent cardiac muscle [54]. By contrast, activation of PDH by PDK knockout [41], pharmacologic inhibition of PDH kinase [55], or exercise [56] improves glycemia. Interestingly, constitutively increased PDH activity in chow-fed mice with combined PDK2/4 knockout increases glucose oxidation, yet also induces insulin resistance [57]. These data suggest that the PDH complex may also contribute to adaptive responses to sustained increases in glucose oxidation, including induction of insulin resistance. We now demonstrate that expression of multiple PDH subunits is also reduced in parallel with IR in humans with T2D, in parallel with increased expression of the inhibitory kinase PDK4 in both T2D and in isolated IR. Thus, multi-level dysregulation of PDH activation may further contribute to metabolic defects associated with IR and T2D.
Electron transfer flavoprotein (ETF) accepts electrons from dehydrogenases in fatty acid, choline, and amino acid metabolism, shuttling them via electron-transferring-flavoprotein dehydrogenase (ETFDH) for ubiquinone reduction. Mutations in either of these enzymes cause multiple acyl-CoA dehydrogenase deficiency [58], an inherited disorder causing impaired oxidation of amino acids and lipids and increased plasma acylcarnitines [59]. Recent genome-wide association studies also link the ETFDH locus to plasma acylcarnitines in healthy individuals [60]. Expression of ETFDH is downregulated in IR mice [61] and humans [62], and upregulated by fatty acids and the PPARγ agonist rosiglitazone [63,64]. Thus, ETFDH plays a central role in lipid and amino acid oxidation; our model now identifies this enzyme complex as a candidate regulatory node that can interact with PDH to potentiate IR-associated metabolic phenotypes.
Our model predicts that reductions in fatty acid transport flux also augment effects of PDH. Reduced intracellular lipid substrate availability in this scenario would likely limit adaptive responses to maintain normal energetics. This concept is supported by experimental data indicating that reduced muscle fatty acid uptake (via CD36 or LPL) reduces mitochondrial function [65]. Moreover, fatty acid transporters (e.g., FATP1) can modulate PDH activity in myotubes [66]. In our model, reductions in fatty acid transport also reduce triglyceride storage. This can be seen either with direct FAT knockdown or with knockdowns that reduce fatty acid utilization. While such responses are logical in the context of the model, where lipid supply can be appropriately modulated, in vivo IR is accompanied by increased triglyceride storage, potentially due to excessive and sustained fatty acid supply with unrestrained adipose lipolysis in obesity.
Our model also demonstrates the utility of novel systems biology approaches for studying metabolic disease. While incremental improvements are possible to improve our analysis, significant advances in both methodological and experimental methods (e.g. integration of insulin signaling pathways) are needed to take full advantage of metabolic models for the study of diabetes and related disorders. Nevertheless, our first-in-kind application of metabolic networks to model dysregulated metabolism linked to insulin resistance appears to be a promising approach to identify key reactions which may contribute to disease risk. The next major step in this high-risk but high-reward discipline will include development within a genome-scale modeling framework with incorporation of multi–omics data, hormonal responses, and signaling into the analysis [20,67,68]. Future metabolic network modeling may be used to predict the impact of differentially expressed or regulated enzymes in diabetes, thus helping to prioritize variants for deeper exploration, and to identify novel pharmacologic targets [69–71]. Moreover, the database of affected fluxes that induce IR-related metabolic phenotypes can serve as a resource to screen genomic variants for both causal and associated variants.
In summary, we have developed an in silico model of muscle metabolism that incorporates reciprocal modulation of lipid oxidation and glucose uptake in order to model the fasted to fed transition. Using this approach, we demonstrate several novel findings. First, while reduced glucose uptake is a major hallmark of IR, model-based reductions in either extracellular glucose availability or uptake do not alter metabolic flexibility, and thus are not sufficient to fully recapitulate IR-linked metabolism. Secondly, experimentally-reduced flux through single reactions does not reproduce key features of IR-linked metabolism. Finally, key metabolic phenotypes characteristic of human IR can be recapitulated by experimental reductions in activity of flux through pyruvate dehydrogenase, in combination with reductions in cellular lipid availability (fatty acid transport) and/or lipid or amino acid metabolism (ETFDH) (Figure 7A). The combined knockdowns also yield additional metabolic phenotypes linked to IR, including reduced fatty acid oxidation, increased incomplete fatty acid oxidation, and decreased glycogen storage. Importantly, we also provide experimental support for model predictions; reduced flux through PDH, ETFDH, and the combination (PDH&ETFDH) reduce cellular ATP levels, citrate synthase activity, and metabolic flexibility in cultured myocytes, mirroring in silico predictions. PDH is also modulated at a transcriptional level in humans with T2D, with decreased expression paralleling IR, and by robust increases in the PDH inhibitory kinase PDK4 in both isolated IR and T2D (Figure 6C).
Together, our findings suggest that metabolic phenotypes linked to IR are not likely to arise from a single gene defect, but rather from dysregulation of metabolic adaptations to energy/nutrient stress at several key nodes. Disruption at these key nodes may contribute mechanistically to the defective energy metabolism observed in IR humans (Figure 7B). More broadly, our analysis illustrates how computational approaches can be used to model subtle changes in metabolism and also provides a valuable resource that can be queried and expanded for further studies to identify novel candidate enzymes and pathways for IR/T2D pathogenesis and treatment.
Acknowledgments
We gratefully acknowledge grant support from the Graetz Foundation (to MEP), the American Diabetes Association (to MEP, Grant no. 7-12-MN-66), NIH T32DK007260-34 (AB, Joslin Diabetes Center), Harold Whitworth Pierce Charitable Trust Postdoctoral Fellowship (to AB), NIH P30DK 036836 (Diabetes Research Center) and NIH U54-LM008748 (to SK). We also thank Daniel Segre, Tomer Shlomi, Mary-Ellen Harper, Allison Goldfine, and Ron Kahn for helpful discussions. We thank Dr. Robert Harris (Indiana University) for providing the PDK2/4 knockout mice.
Contributor Information
Christopher Nogiec, Email: Nogiec@bu.edu.
Alison Burkart, Email: Alison.burkart@joslin.harvard.edu.
Carles Lerin, Email: clerin@fsjd.edu.
Simon Kasif, Email: Kasif@bu.edu.
Mary-Elizabeth Patti, Email: mary.elizabeth.patti@joslin.harvard.edu.
Conflict of interest
The authors declare no conflicts of interest.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Martin B.C., Warram J.H., Krolewski A.S., Bergman R.N., Soeldner J.S., Kahn C.R. Role of glucose and insulin resistance in development of type II diabetes mellitus: results of a 25-year follow-up study. Lancet. 1992;340:925–929. doi: 10.1016/0140-6736(92)92814-v. [DOI] [PubMed] [Google Scholar]
- 2.Tabak A.G., Jokela M., Akbaraly T.N., Brunner E.J., Kivimaki M., Witte D.R. Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. Lancet. 2009;373(9682):2215–2221. doi: 10.1016/S0140-6736(09)60619-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeFronzo R.A., Gunnarson R., Biorkman O., Olsson M., Warren J. Effects of insulin on peripheral and splanchnic glucose metabolism in non-insulin dependent (Type II) diabetes mellitus. The Journal of Clinical Investigation. 1985;76:149–155. doi: 10.1172/JCI111938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petersen K.F., Dufour S., Befroy D., Garcia R., Shulman G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. The New England Journal of Medicine. 2004;350(7):664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petersen K.F., Dufour S., Shulman G.I. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Medicine. 2005;2(9):e233. doi: 10.1371/journal.pmed.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kacerovsky-Bielesz G., Chmelik M., Ling C., Pokan R., Szendroedi J., Farukuoye M. Short-term exercise training does not stimulate skeletal muscle ATP synthesis in relatives of humans with type 2 diabetes. Diabetes. 2009;58(6):1333–1341. doi: 10.2337/db08-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Befroy D.E., Petersen K.F., Dufour S., Mason G.F., de Graaf R.A., Rothman D.L. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56(5):1376–1381. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simoneau J.A., Kelley D.E. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. Journal of Applied Physiology. 1997;83(1):166–171. doi: 10.1152/jappl.1997.83.1.166. [DOI] [PubMed] [Google Scholar]
- 9.Kelley D.E., Mandarino L.J. Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes. 2000;49(5):677–683. doi: 10.2337/diabetes.49.5.677. [DOI] [PubMed] [Google Scholar]
- 10.Patti M.E., Butte A.J., Crunkhorn S., Cusi K., Berria R., Kashyap S. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genetics. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 12.Boyle K.E., Zheng D., Anderson E.J., Neufer P.D., Houmard J.A. Mitochondrial lipid oxidation is impaired in cultured myotubes from obese humans. International Journal of Obesity (London) 2012;36(8):1025–1031. doi: 10.1038/ijo.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pihlajamaki J., Lerin C., Itkonen P., Boes T., Floss T., Schroeder J. Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metabolism. 2011;14(2):208–218. doi: 10.1016/j.cmet.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pihlajamaki J., Boes T., Kim E.Y., Dearie F., Kim B.W., Schroeder J. Thyroid hormone-related regulation of gene expression in human fatty liver. The Journal of Clinical Endocrinology and Metabolism. 2009;94(9):3521–3529. doi: 10.1210/jc.2009-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varma A., Palsson B.O. Metabolic flux balancing: basic concepts, scientific and practical use. Nature Biotechnology. 1994;12:994–998. [Google Scholar]
- 16.Oberhardt M.A., Palsson B.O., Papin J.A. Applications of genome-scale metabolic reconstructions. Molecular Systems Biology. 2009;5:320. doi: 10.1038/msb.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duarte N.C., Becker S.A., Jamshidi N., Thiele I., Mo M.L., Vo T.D. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(6):1777–1782. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jerby L., Shlomi T., Ruppin E. Computational reconstruction of tissue-specific metabolic models: application to human liver metabolism. Molecular Systems Biology. 2010;6:401. doi: 10.1038/msb.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thiele I., Price N.D., Vo T.D., Palsson B.O. Candidate metabolic network states in human mitochondria. Impact of diabetes, ischemia, and diet. Journal of Biological Chemistry. 2005;280(12):11683–11695. doi: 10.1074/jbc.M409072200. [DOI] [PubMed] [Google Scholar]
- 20.Thiele I., Swainston N., Fleming R.M., Hoppe A., Sahoo S., Aurich M.K. A community-driven global reconstruction of human metabolism. Nature Biotechnology. 2013;31(5):419–425. doi: 10.1038/nbt.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shlomi T., Cabili M.N., Herrgard M.J., Palsson B.O., Ruppin E. Network-based prediction of human tissue-specific metabolism. Nature Biotechnology. 2008;26(9):1003–1010. doi: 10.1038/nbt.1487. [DOI] [PubMed] [Google Scholar]
- 22.Shlomi T., Cabili M.N., Ruppin E. Predicting metabolic biomarkers of human inborn errors of metabolism. Molecular Systems Biology. 2009;5:263. doi: 10.1038/msb.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shlomi T., Benyamini T., Gottlieb E., Sharan R., Ruppin E. Genome-scale metabolic modeling elucidates the role of proliferative adaptation in causing the Warburg effect. PLoS Computational Biology. 2011;7(3):e1002018. doi: 10.1371/journal.pcbi.1002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gille C., Bolling C., Hoppe A., Bulik S., Hoffmann S., Hubner K. HepatoNet1: a comprehensive metabolic reconstruction of the human hepatocyte for the analysis of liver physiology. Molecular Systems Biology. 2010;6:411. doi: 10.1038/msb.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewis N.E., Schramm G., Bordbar A., Schellenberger J., Andersen M.P., Cheng J.K. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nature Biotechnology. 2010;28(12):1279–1285. doi: 10.1038/nbt.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bordbar A., Feist A.M., Usaite-Black R., Woodcock J., Palsson B.O., Famili I. A multi-tissue type genome-scale metabolic network for analysis of whole-body systems physiology. BMC Systems Biology. 2011;5:180. doi: 10.1186/1752-0509-5-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agren R., Bordel S., Mardinoglu A., Pornputtapong N., Nookaew I., Nielsen J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Computational Biology. 2012;8(5):e1002518. doi: 10.1371/journal.pcbi.1002518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mardinoglu A., Agren R., Kampf C., Asplund A., Nookaew I., Jacobson P. Integration of clinical data with a genome-scale metabolic model of the human adipocyte. Molecular Systems Biology. 2013;9:649. doi: 10.1038/msb.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nogiec C.D., Kasif S. To supplement or not to supplement: a metabolic network framework for human nutritional supplements. PLoS ONE. 2013;8(8):e68751. doi: 10.1371/journal.pone.0068751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bordbar A., Monk J.M., King Z.A., Palsson B.O. Constraint-based models predict metabolic and associated cellular functions. Nature Reviews Genetics. 2014;15(2):107–120. doi: 10.1038/nrg3643. [DOI] [PubMed] [Google Scholar]
- 31.Ramakrishna R., Edwards J.S., McCulloch A., Palsson B.O. Flux-balance analysis of mitochondrial energy metabolism: consequences of systemic stoichiometric constraints. The American Journal of Physiology – Regulatory, Integrative and Comparative Physiology. 2001;280(3):R695–R704. doi: 10.1152/ajpregu.2001.280.3.R695. [DOI] [PubMed] [Google Scholar]
- 32.Jeoung N.H., Rahimi Y., Wu P., Lee W.N., Harris R.A. Fasting induces ketoacidosis and hypothermia in PDHK2/PDHK4-double-knockout mice. Biochemical Journal. 2012;443(3):829–839. doi: 10.1042/BJ20112197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin W., Goldfine A.B., Boes T., Henry R.R., Ciaraldi T.P., Kim E.Y. Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. The Journal of Clinical Investigation. 2011;121(3):918–929. doi: 10.1172/JCI41940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology. 2004;3 doi: 10.2202/1544-6115.1027. Article3- [DOI] [PubMed] [Google Scholar]
- 35.Gall W.E., Beebe K., Lawton K.A., Adam K.P., Mitchell M.W., Nakhle P.J. alpha-hydroxybutyrate is an early biomarker of insulin resistance and glucose intolerance in a nondiabetic population. PLoS ONE. 2010;5(5):e10883. doi: 10.1371/journal.pone.0010883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cynober L.A. Plasma amino acid levels with a note on membrane transport: characteristics, regulation, and metabolic significance. Nutrition. 2002;18(9):761–766. doi: 10.1016/s0899-9007(02)00780-3. [DOI] [PubMed] [Google Scholar]
- 37.MacLaren D.P., Nevill A.M., Thake C.D., Campbell I.T., Cheetham E., Keegan M.A. Human erythrocyte and plasma amino acid concentrations during exercise. Medicine & Science in Sports & Exercise. 2000;32(7):1244–1249. doi: 10.1097/00005768-200007000-00010. [DOI] [PubMed] [Google Scholar]
- 38.Eriksson J., Franssila-Kallunki A., Ekstrand A., Saloranta C., Widen E., Schalin C. Early metabolic defects in persons at increased risk for non-insulin-dependent diabetes mellitus. The New England Journal of Medicine. 1989;321(6):337–343. doi: 10.1056/NEJM198908103210601. [DOI] [PubMed] [Google Scholar]
- 39.Koves T.R., Ussher J.R., Noland R.C., Slentz D., Mosedale M., Ilkayeva O. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metabolism. 2008;7(1):45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 40.Bowker-Kinley M.M., Davis W.I., Wu P., Harris R.A., Popov K.M. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochemical Journal. 1998;329(Pt 1):191–196. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeoung N.H., Harris R.A. Pyruvate dehydrogenase kinase-4 deficiency lowers blood glucose and improves glucose tolerance in diet-induced obese mice. The American Journal of Physiology – Endocrinology and Metabolism. 2008;295(1):E46–E54. doi: 10.1152/ajpendo.00536.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang S., Hulver M.W., McMillan R.P., Cline M.A., Gilbert E.R. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutrition & Metabolism (London) 2014;11(1):10. doi: 10.1186/1743-7075-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim K.S., Lee Y.M., Lee I.K., Kim D.J., Jacobs D.R., Jr., Lee D.H. Paradoxical associations of insulin resistance with total and cardiovascular Mortality in humans. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2014 Oct 17 doi: 10.1093/gerona/glu194. [pii glu194, epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 44.Patti M.E., Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocrine Reviews. 2010;31(3):364–396. doi: 10.1210/er.2009-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Newgard C.B., An J., Bain J.R., Muehlbauer M.J., Stevens R.D., Lien L.F. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metabolism. 2009;9(4):311–326. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang T.J., Larson M.G., Vasan R.S., Cheng S., Rhee E.P., McCabe E. Metabolite profiles and the risk of developing diabetes. Nature Medicine. 2011;17(4):448–453. doi: 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herman M.A., She P., Peroni O.D., Lynch C.J., Kahn B.B. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. The Journal of Biological Chemistry. 2010;285(15):11348–11356. doi: 10.1074/jbc.M109.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327(5968):1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kohda Y., Umeki M., Kono T., Terasaki F., Matsumura H., Tanaka T. Thiamine ameliorates diabetes-induced inhibition of pyruvate dehydrogenase (PDH) in rat heart mitochondria: investigating the discrepancy between PDH activity and PDH E1alpha phosphorylation in cardiac fibroblasts exposed to high glucose. Journal of Pharmacological Sciences. 2010;113(4):343–352. doi: 10.1254/jphs.09359fp. [DOI] [PubMed] [Google Scholar]
- 50.Odin J.A., Huebert R.C., Casciola-Rosen L., LaRusso N.F., Rosen A. Bcl-2-dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. The Journal of Clinical Investigation. 2001;108(2):223–232. doi: 10.1172/JCI10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Randle P.J., Garland P.B., Hales C.N., Newsholme F.A. The glucose fatty-acid cycle: its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 52.Mostert M., Rabbone I., Piccinini M., Curto M., Vai S., Musso A. Derangements of pyruvate dehydrogenase in circulating lymphocytes of NIDDM patients and their healthy offspring. Journal of Endocrinological Investigation. 1999;22(7):519–526. doi: 10.1007/BF03343603. [DOI] [PubMed] [Google Scholar]
- 53.Pehmoller C., Brandt N., Birk J.B., Hoeg L.D., Sjoberg K.A., Goodyear L.J. Exercise alleviates lipid-induced insulin resistance in human skeletal muscle-signaling interaction at the level of TBC1 domain family member 4. Diabetes. 2012;61(11):2743–2752. doi: 10.2337/db11-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vadvalkar S.S., Baily C.N., Matsuzaki S., West M., Tesiram Y.A., Humphries K.M. Metabolic inflexibility and protein lysine acetylation in heart mitochondria of a chronic model of type 1 diabetes. Biochemical Journal. 2013;449(1):253–261. doi: 10.1042/BJ20121038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mayers R.M., Leighton B., Kilgour E. PDH kinase inhibitors: a novel therapy for type II diabetes? Biochemical Society Transactions. 2005;33(Pt 2):367–370. doi: 10.1042/BST0330367. [DOI] [PubMed] [Google Scholar]
- 56.Kiilerich K., Birk J.B., Damsgaard R., Wojtaszewski J.F., Pilegaard H. Regulation of PDH in human arm and leg muscles at rest and during intense exercise. The American Journal of Physiology – Endocrinology and Metabolism. 2008;294(1):E36–E42. doi: 10.1152/ajpendo.00352.2007. [DOI] [PubMed] [Google Scholar]
- 57.Rahimi Y., Camporez J.P., Petersen M.C., Pesta D., Perry R.J., Jurczak M.J. Genetic activation of pyruvate dehydrogenase alters oxidative substrate selection to induce skeletal muscle insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(46):16508–16513. doi: 10.1073/pnas.1419104111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olsen R.K., Olpin S.E., Andresen B.S., Miedzybrodzka Z.H., Pourfarzam M., Merinero B. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. 2007;130(Pt 8):2045–2054. doi: 10.1093/brain/awm135. [DOI] [PubMed] [Google Scholar]
- 59.Rinaldo P., Matern D., Bennett M.J. Fatty acid oxidation disorders. The Annual Review of Physiology. 2002;64:477–502. doi: 10.1146/annurev.physiol.64.082201.154705. [DOI] [PubMed] [Google Scholar]
- 60.Illig T., Gieger C., Zhai G., Romisch-Margl W., Wang-Sattler R., Prehn C. A genome-wide perspective of genetic variation in human metabolism. Nature Genetics. 2010;42(2):137–141. doi: 10.1038/ng.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sena S., Hu P., Zhang D., Wang X., Wayment B., Olsen C. Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. The Journal of Molecular and Cellular Cardiology. 2009;46(6):910–918. doi: 10.1016/j.yjmcc.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Viguerie N., Montastier E., Maoret J.J., Roussel B., Combes M., Valle C. Determinants of human adipose tissue gene expression: impact of diet, sex, metabolic status, and cis genetic regulation. PLOS Genetics. 2012;8(9):e1002959. doi: 10.1371/journal.pgen.1002959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lockridge J.B., Sailors M.L., Durgan D.J., Egbejimi O., Jeong W.J., Bray M.S. Bioinformatic profiling of the transcriptional response of adult rat cardiomyocytes to distinct fatty acids. The Journal of Lipid Research. 2008;49(7):1395–1408. doi: 10.1194/jlr.M700517-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilson-Fritch L., Nicoloro S., Chouinard M., Lazar M.A., Chui P.C., Leszyk J. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. The Journal of Clinical Investigation. 2004;114(9):1281–1289. doi: 10.1172/JCI21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morino K., Petersen K.F., Sono S., Choi C.S., Samuel V.T., Lin A. Regulation of mitochondrial biogenesis by lipoprotein lipase in muscle of insulin-resistant offspring of parents with type 2 diabetes. Diabetes. 2012;61(4):877–887. doi: 10.2337/db11-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khajah M., Millen B., Cara D.C., Waterhouse C., McCafferty D.M. Granulocyte-macrophage colony-stimulating factor (GM-CSF): a chemoattractive agent for murine leukocytes in vivo. Journal of Leukocyte Biology. 2011;89(6):945–953. doi: 10.1189/jlb.0809546. [DOI] [PubMed] [Google Scholar]
- 67.Becker S.A., Palsson B.O. Context-specific metabolic networks are consistent with experiments. PLoS Computational Biology. 2008;4(5):e1000082. doi: 10.1371/journal.pcbi.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yizhak K., Benyamini T., Liebermeister W., Ruppin E., Shlomi T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics. 2010;26(12):i255–i260. doi: 10.1093/bioinformatics/btq183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frezza C., Zheng L., Folger O., Rajagopalan K.N., MacKenzie E.D., Jerby L. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477(7363):225–228. doi: 10.1038/nature10363. [DOI] [PubMed] [Google Scholar]
- 70.Folger O., Jerby L., Frezza C., Gottlieb E., Ruppin E., Shlomi T. Predicting selective drug targets in cancer through metabolic networks. Molecular Systems Biology. 2011;7:501. doi: 10.1038/msb.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yizhak K., Le Devedec S.E., Rogkoti V.M., Baenke F., de Boer V.C., Frezza C. A computational study of the Warburg effect identifies metabolic targets inhibiting cancer migration. Molecular Systems Biology. 2014;10:744. doi: 10.15252/msb.20134993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.