

Abstract
Objective
During pregnancy, women normally increase their food intake and body fat mass, and exhibit insulin resistance. However, an increasing number of women are developing metabolic imbalances during pregnancy, including excessive gestational weight gain and gestational diabetes mellitus. Despite the negative health impacts of pregnancy-induced metabolic imbalances, their molecular causes remain unclear. Therefore, the present study investigated the molecular mechanisms responsible for orchestrating the metabolic changes observed during pregnancy.
Methods
Initially, we investigated the hypothalamic expression of key genes that could influence the energy balance and glucose homeostasis during pregnancy. Based on these results, we generated a conditional knockout mouse that lacks the suppressor of cytokine signaling-3 (SOCS3) only in leptin receptor-expressing cells and studied these animals during pregnancy.
Results
Among several genes involved in leptin resistance, only SOCS3 was increased in the hypothalamus of pregnant mice. Remarkably, SOCS3 deletion from leptin receptor-expressing cells prevented pregnancy-induced hyperphagia, body fat accumulation as well as leptin and insulin resistance without affecting the ability of the females to carry their gestation to term. Additionally, we found that SOCS3 conditional deletion protected females against long-term postpartum fat retention and streptozotocin-induced gestational diabetes.
Conclusions
Our study identified the increased hypothalamic expression of SOCS3 as a key mechanism responsible for triggering pregnancy-induced leptin resistance and metabolic adaptations. These findings not only help to explain a common phenomenon of the mammalian physiology, but it may also aid in the development of approaches to prevent and treat gestational metabolic imbalances.
Keywords: Leptin, Suppressor of cytokine signaling, Gestational diabetes, Obesity, Leptin resistance, Hypothalamus
Abbreviations: ARH, arcuate nucleus of the hypothalamus; DIO, diet-induced obesity; DMH, dorsomedial nucleus of the hypothalamus; EGWG, excessive gestational weight gain; GDM, gestational diabetes mellitus; GTT, glucose tolerance test; IR, insulin receptor; ITT, insulin tolerance test; LepR, leptin receptor; PKC, protein kinase C; GH-V, placental growth hormone; pSTAT3, phosphorylation of the signal transducer and activator of transcription 3; pSTAT3-ir, pSTAT3-immunoreactive; RP, retroperitoneal; SOCS3, suppressor of cytokine signaling-3; STZ, streptozotocin; VMH, ventromedial nucleus of the hypothalamus
Graphical abstract
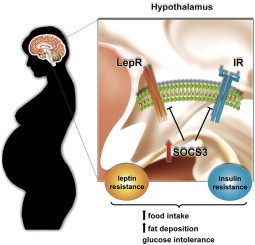
1. Introduction
The developing embryo and subsequent fetus impose energy demands on pregnant women. Consequently, increased food intake is expected during gestation [1,2]. Additionally, pregnancy leads to benign and transitory insulin resistance [3]. These metabolic adaptations are thought to have evolved to ensure better conditions for offspring development [2]. However, an increasing number of women are developing metabolic imbalances during pregnancy, including excessive gestational weight gain (EGWG) and gestational diabetes mellitus (GDM) [3,4]. These conditions represent a serious health threat to women and their offspring and are becoming a major obstetric complication worldwide. For example, EGWG increases the risk of maternal mortality, GDM, pre-eclampsia, thromboembolism, postpartum hemorrhage and other gestational complications [3]. Furthermore, long-term postpartum weight retention predisposes women to obesity, and GDM increases the risk of diabetes mellitus later in life [4]. Therefore, metabolic imbalances during pregnancy may also aggravate the obesity and diabetes epidemics.
Despite the negative health impacts of pregnancy-induced metabolic imbalances, the precise mechanisms responsible for orchestrating these changes remain largely unknown. Previous studies have reported lower responsiveness to leptin in pregnant animals [5–7]. Nonetheless, no compelling evidence has been provided about the real contribution of leptin resistance for the onset of pregnancy-induced metabolic changes. Therefore, the objective of the present study was to identify key molecular mechanisms that trigger the metabolic changes observed during pregnancy.
2. Material and methods
2.1. Generation of conditional knockout mice
All mouse strains were backcrossed at least 4 times to the C57BL/6 background before the initiation of breeding. To induce the Socs3 gene deletion exclusively in leptin-responsive cells, we bred the LepR-IRES-Cre strain (B6.129-Leprtm2(cre)Rck/J, Jackson Laboratories) with mice carrying loxP-flanked Socs3 alleles (B6.129S4-Socs3tm1Ayos/J, Jackson Laboratories). The SOCS3 KO group was composed of animals homozygous for the loxP-flanked Socs3 allele and homozygous for the LepR-IRES-Cre allele. The control group was composed of animals homozygous for the LepR-IRES-Cre allele. We used only littermates as controls. The mice were weaned at 4 weeks of age, and the genomic DNA was extracted from tail tips for PCR genotyping (Sigma). The loxP-flanked Socs3 allele and the wild-type allele were identified by the presence of a 420 bp or 272 bp PCR fragment, respectively. The LepR-IRES-Cre allele and the wild-type allele were identified by the presence of a 213 bp or 800 bp PCR fragment, respectively. After weaning, the mice received a regular low-fat rodent chow diet (2.99 kcal/g; 9.4% calories from fat; Quimtia, Brazil). All animal procedures were approved by the Ethics Committee on the Use of Animals of the Institute of Biomedical Sciences at the University of São Paulo, São Paulo, Brazil.
2.2. Breeding strategy and tissue collection
Two-month-old females were bred with sexually experienced males, and we assessed the presence of copulatory plugs on a daily basis. Females that did not mate within 3 weeks were discarded. A breeding strategy was designed to assure that all pups in both groups would have the same genotype (homozygous for LepR-IRES-Cre and heterozygous for LoxP-flanked Socs3 allele). The day that the copulatory plug was detected was considered to be the first day of pregnancy (P1), and the females were single-housed. All females were euthanized after a 4-h fasting period during the middle of the light phase. Non-pregnant mice (nulliparous and primiparous) were euthanized on the second day of diestrus. To determine adiposity levels, we measured the masses of the uterine, ovarian and retroperitoneal (RP) fat pads. The uterine pad was collected from the midpoint of the cervix and trimmed away along the horn length to the infundibulum region. Ovarian pad was well defined by a circular fat deposit around the ovary. The retroperitoneal pad was removed as a triangular section extending from a vertex in the inguinal region up the midline and across at the lower pole of the kidney, extending laterally as far as fat was visible. The deposits were collected bilaterally and the values presented represent their average.
2.3. Relative gene expression
The tissues were quickly dissected for relative gene expression analysis as previously described in Ref. [8]. Specific primers were designed for each target gene according to the sequences obtained from GenBank or acquired from Applied Biosystems (Supplemental Table 1). The data were reported as fold changes compared with the values obtained from the control group (set at 1.0).
2.4. Leptin sensitivity tests
For all experiments, we used mouse recombinant leptin purchased from Dr. A.F. Parlow (National Hormone and Peptide Program, National Institute of Diabetes and Digestive and Kidney Diseases). On P5, mice were anaesthetized (isoflurane) and we performed a surgical procedure, in which osmotic micro-pumps (model 1002; Alzet) were implanted s.c. to deliver 0.5 μg leptin/h. Body weight and food intake were measured daily before (P1–P5) and after (P6–P16) surgery. To determine the region-specific responses to leptin, pregnant control and SOCS3 KO mice received a leptin injection after a 4 h fasting period (2.5 μg leptin/g; s.c.). Three hours later, the mice were perfused with 10% buffered formalin, and their brains were processed to detect leptin-induced phosphorylation of the Signal Transducer and Activator of Transcription 3-immunoreactive (pSTAT3-ir) as previously described in Ref. [7]. We counted the number of pSTAT3-ir cells on one side of a representative rostrocaudal level.
2.5. Western blot
The tissues were homogenized in RIPA buffer (Sigma) containing a cocktail of proteases and phosphatase inhibitors (1:100, Sigma). Protein (40 or 50 μg) was resolved on a 10% SDS-PAGE gel and transferred to a nitrocellulose membrane (Bio-Rad). The membranes were blocked with 5% BSA and incubated overnight at 4 °C with primary antibodies (1:1,000; pSTAT3Tyr705, Cell Signaling; STAT3, Santa Cruz; SOCS3, Cell Signaling; pIRTyr1162/1163, Santa Cruz; pAKTSer473, Cell Signaling; GAPDH, Santa Cruz). Next, the membranes were incubated for 45 min with secondary antibody (1:10,000; IRDye 800CW, Li-COR). The proteins were detected and analyzed using the Li-COR Odyssey system.
2.6. Hormone levels
ELISA kits were used to determine the serum concentrations of leptin (Crystal Chem), insulin (Crystal Chem) and glucagon (Sigma).
2.7. Glucose homeostasis
A glucose tolerance test (GTT; 2 g glucose/kg; s.c.) and an insulin tolerance test (ITT; 1 IU insulin/kg; s.c.) were performed on P14 and P16, respectively. Insulin sensitivity was evaluated in different tissues by infusing 5 IU insulin/kg s.c. into pregnant mice and euthanizing them 15 min after the injection. GDM was induced by a single intraperitoneal injection of freshly prepared streptozotocin (STZ; 200 mg/kg; Amresco) dissolved in 50 mM sodium citrate (pH 4.5) on P5 [9]. Glycemia was assessed before (on P1 and P4) and after (on P7 and P13) the STZ injection, followed by a GTT (0.5 g glucose/kg; s.c.) on P14.
2.8. Statistics
The results are expressed as mean ± SEM. The differences between the groups were compared using an unpaired two-tailed Student's t-test. The data obtained from the leptin and insulin sensitivity tests were analyzed by two-way ANOVA and the Bonferroni post hoc test. Glycemic changes after the STZ treatment were assessed using repeated measures ANOVA. Statistical analyses were performed using GraphPad Prism software. We considered p values of less than 0.05 to be statistically significant.
3. Results
3.1. Identification of proteins potentially related with leptin resistance during pregnancy
We investigated the hypothalamic expression of several genes potentially related to leptin resistance including components of the suppressor of cytokine signaling (SOCS) family, adapter proteins such as SH2B1, protein-tyrosine phosphatases and LepR isoforms. Among them, only SOCS3 and Ob-Re expression were increased in the hypothalamus of pregnant mice (Figure 1A). Interestingly, SOCS3 is the main member of the SOCS family capable of inhibiting leptin receptor (LepR) signaling [10,11]. Its expression is increased in the hypothalamus of obese animals, and the brain-specific ablation of SOCS3 increases leptin sensitivity in diet-induced obesity (DIO) [12–15]. Therefore, SOCS3 is a strong candidate to induce both leptin resistance and the metabolic changes during pregnancy.
Figure 1.
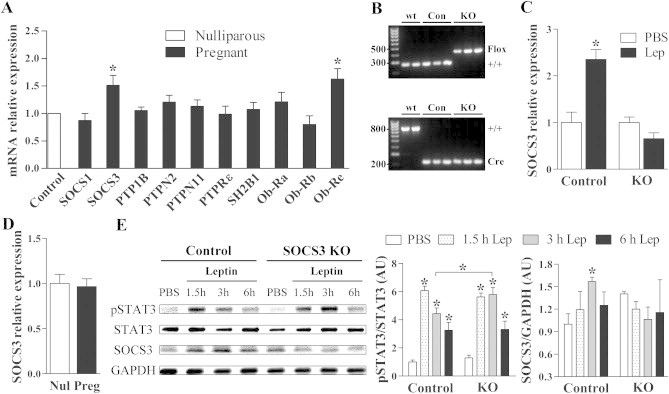
Validation of mice lacking SOCS3 in LepR-expressing cells. (A) Gene expression analysis in the hypothalamus of nulliparous (n = 8) and pregnant (n = 7) wild-type mice to determine candidate genes involved in leptin resistance during pregnancy. (B) PCR genotyping from tail biopsies to identify the loxP-flanked Socs3 allele (upper picture) or the LepR-IRES-Cre allele (lower picture) in wild-type, control and SOCS3 KO mice. (C) Deletion of SOCS3 in LepR-expressing cells prevented the leptin-induced increase in hypothalamic SOCS3 expression after 1 h (n = 6/group). (D) SOCS3 KO mice did not show the typical increase in hypothalamic SOCS3 expression during pregnancy (n = 7–8). (E) SOCS3 KO mice exhibited longer, sustained leptin-induced phosphorylation of STAT3 (pSTAT3) and did not exhibit changes in SOCS3 protein levels in the hypothalamus compared to control mice (n = 3/group). *p < 0.05.
3.2. Generation of mice lacking SOCS3 only in LepR-expressing cells
Mice carrying LoxP-flanked Socs3 alleles were bred with animals expressing Cre recombinase under the Lepr promoter control (Figure 1B). Thus, SOCS3 was inactivated specifically in LepR-expressing cells (SOCS3 KO mice). Littermate mice carrying the wild-type Socs3 gene were used as controls. Both groups were composed of animals homozygous for the Cre allele (Figure 1B). To functionally confirm the genetic deletion, we assessed the capacity of an acute leptin injection to induce hypothalamic SOCS3 mRNA expression [10,11]. We found that leptin increased SOCS3 expression only in control animals (Figure 1C). Then, we compared hypothalamic SOCS3 expression in nulliparous and pregnant SOCS3 KO mice. In contrast to the elevated SOCS3 mRNA expression seen during pregnancy in wild-type mice (Figure 1A), pregnant SOCS3 KO mice did not show increased SOCS3 hypothalamic expression (Figure 1D). These results indicate that pregnancy-induced changes in hypothalamic SOCS3 expression are restricted to LepR-expressing cells. Gene expression analysis in the hypothalamus of pregnant mice confirmed that SOCS3 KO females did not exhibit compensatory alterations in the expression of other components potentially related to leptin resistance, although pregnant SOCS3 KO mice had a higher hypothalamic expression of Ob-Ra compared to control animals (Supplemental Figure 1).
Because SOCS3 inhibits LepR signaling [10,11], SOCS3 KO mice are expected to be more responsive to leptin. To determine leptin sensitivity in the hypothalamus, non-pregnant control and SOCS3 KO mice received a leptin injection to induce pSTAT3. Leptin increased pSTAT3 expression 1.5 h after the injection. However, SOCS3 KO mice showed a longer and sustained increase in the pSTAT3 expression compared to control animals, indicating a prolonged signaling through the LepR (Figure 1E). Additionally, hypothalamic SOCS3 was increased in control mice 3 h after leptin injection, whereas no such changes were detected in SOCS3 KO mice (Figure 1E). These results confirm the efficiency of SOCS3 inactivation in LepR-responsive cells.
3.3. Metabolic changes during pregnancy are attenuated in SOCS3 KO mice
Despite the increased sensitivity to leptin (Figure 1E), non-pregnant SOCS3 KO females have a similar food intake (Control: 4.5 ± 0.3 g/day; SOCS3 KO: 4.6 ± 0.3 g/day; p = 0.73) and body weight (Figure 2B) compared with control animals, and only a slight decrease in fat mass (Figure 2C). To investigate whether SOCS3 plays a role in the leptin resistance and metabolic changes observed during pregnancy, females were bred and monitored daily to detect the copulatory plug, indicating the first day of pregnancy (P1). Then, they were single-housed, and their body weight and food intake were recorded daily during the gestation, lactation and postweaning periods. SOCS3 KO females exhibited normal fertility rates (data not shown) and did not experience any noticeable pregnancy complications. As expected, control animals showed a progressive increase in food intake during gestation (Figure 2A). Remarkably, SOCS3 KO mice maintained their food intake significantly lower than control animals from P5 to P17 (Figure 2A). During pregnancy, body weight increased progressively in control mice, whereas in SOCS3 KO females this increase was attenuated (Figure 2B). Because adiposity increased during gestation in control animals (Figure 2C), we hypothesized that the lower weight gain of pregnant SOCS3 KO females was due to the prevention of fat mass accumulation. Accordingly, deletion of SOCS3 from LepR-expressing cells reduced the accumulation of fat mass by approximately 80% during pregnancy (Figure 2C). Consequently, pregnant SOCS3 KO females exhibited the same degree of adiposity as nulliparous controls.
Figure 2.
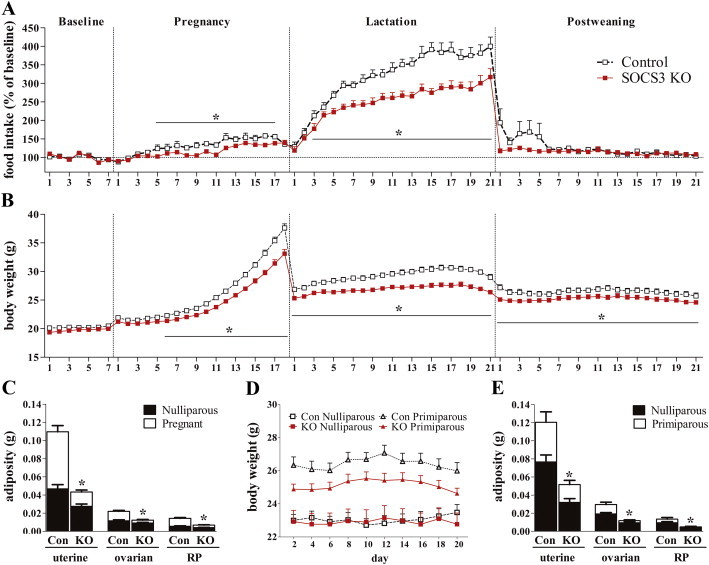
Energy balance changes during pregnancy are attenuated in SOCS3 KO mice. (A and B) Relative food intake and body weight at baseline and during pregnancy, lactation and postweaning periods in control and SOCS3 KO mice (n = 20–30/group). (C) Bar graphs comparing fat pad masses of nulliparous and pregnant mice at P16 from the control (n = 12) and SOCS3 KO (n = 12) groups. (D and E) Graphs comparing the body weights and adiposities of age-matched nulliparous and primiparous mice from the control (n = 12) and SOCS3 KO (n = 12) groups. *p < 0.05.
Pups had similar body weight at embryo stage (E16) or at birth in control and SOCS3 KO groups (Supplemental Figure 2). To ensure comparable metabolic demands during lactation, we standardized to 6 pups per litter. Lactating control animals exhibited profound increases in food intake (Figure 2A). However, hyperphagia during lactation was markedly reduced by SOCS3 deletion suggesting that ongoing leptin resistance contributes to the hyperphagia of lactation. Food intake returned to baseline levels after weaning in both groups (Figure 2A). In addition, control animals remained significantly heavier than SOCS3 KO females during lactation (Figure 2B). Pups from SOCS3 KO dams showed a lower weight gain rate resulting in a significantly lower body weight from day 14 on compared to control litters (Supplemental Figure 2).
Pregnancy is considered an important risk factor for obesity in women [4]. To investigate whether SOCS3 deletion prevented postpartum weight retention, we compared the body weight of age-matched nulliparous and primiparous mice. In nulliparous mice, we found no differences in body weight between the groups (Figure 2D). However, primiparous control animals showed sustained increases in body weight and adiposity than nulliparous controls (Figure 2D,E). Notably, SOCS3 deletion mitigated pregnancy-induced increases in body weight and fat accumulation (Figure 2D,E). Therefore, long-term postpartum fat retention was prevented by SOCS3 conditional deletion.
3.4. SOCS3 deletion in LepR-expressing cells precludes the development of leptin resistance during pregnancy
It remains unknown whether the development of leptin resistance leads to the metabolic changes observed during pregnancy. Control mice exhibited a marked increase in serum leptin concentration during late pregnancy (Figure 3A). Since hyperleptinemia is a common feature of leptin resistance [12,14,15], these results corroborate previous findings indicating that late pregnant animals are leptin resistant [5–7]. Notably, no changes in leptin levels were observed in pregnant SOCS3 KO mice suggesting improved leptin sensitivity (Figure 3A). To directly evaluate leptin sensitivity, osmotic pumps delivering leptin were implanted subcutaneously at day 5 of pregnancy in control and SOCS3 KO mice. Blood analysis at day 16 of pregnancy indicated that leptin-treated control and SOCS3 KO mice had similarly high leptin levels (Supplemental Figure 3). Control mice showed reduced weight gain at the beginning of leptin treatment. However, by late gestation, their weight gain was similar to non-treated animals (Figure 3B). These results are in accordance with the fact that leptin resistance arises at midpregnancy towards the end of gestation, coinciding with the period of highest weight gain rate and hyperphagia (Supplemental Figure 4) [1,2,16,17]. In contrast, leptin-treated SOCS3 KO mice had a lower rate of weight gain by late pregnancy (Figure 3C). By the end of pregnancy, leptin-treated controls showed similar rate of weight gain as the non-treated controls (Figure 3D and Supplemental Figure 4). Conversely, leptin-treated SOCS3 KO mice gained significantly less weight during pregnancy than non-treated animals (Figure 3D and Supplemental Figure 4). Although leptin treatment was able to reduce food intake of control animals, the anorexigenic effects of leptin were significantly stronger in SOCS3 KO mice, completely preventing hyperphagia during late pregnancy (Figure 3E). Moreover, leptin drastically reduced the adiposity of pregnant SOCS3 KO mice by nearly eliminating their fat deposits (Figure 3F).
Figure 3.
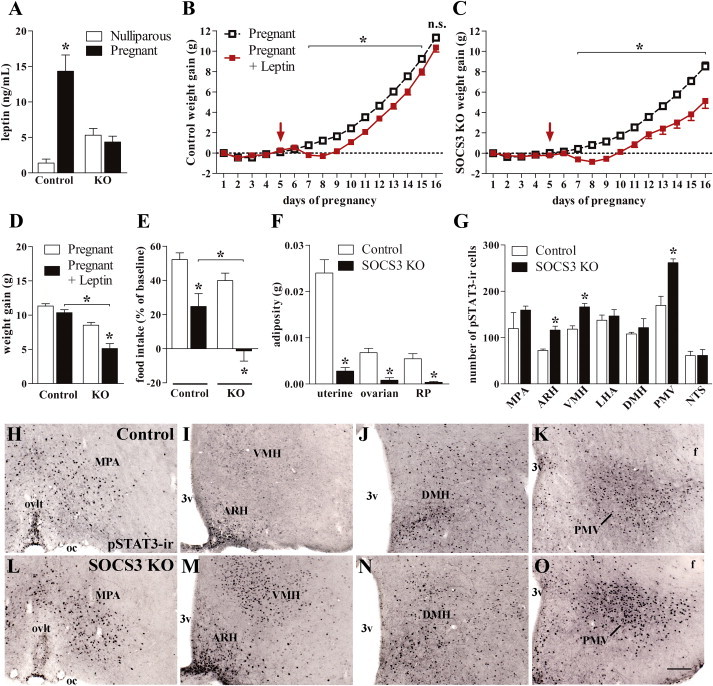
Deletion of SOCS3 in LepR-expressing cells precludes the development of leptin resistance during pregnancy. (A) Serum leptin levels of nulliparous and pregnant mice at P16 (n = 8/group). (B and C) Weight change differences between non-treated and leptin-treated mice during pregnancy (n = 8–22/group). The arrow indicates the day (P5) that the osmotic pumps containing leptin were implanted into the animals. (D and E) Changes in body weight and food intake between leptin-treated and non-treated mice at baseline (P1–P5) and during late pregnancy (P14–P16; n = 30–33/group). (F) Fat pad masses of leptin-treated control and SOCS3 KO mice (n = 8–11/group). (G) The numbers of pSTAT3-ir cells in different brain nuclei of pregnant control (n = 6) and SOCS3 KO (n = 5) mice at 3 h after leptin injection. (H–O) Brain photomicrographs of representative animals showing the distribution of leptin-induced pSTAT3-ir cells. Abbreviations: 3v, third ventricle; ARH, arcuate nucleus of the hypothalamus; DMH, dorsomedial nucleus of the hypothalamus; f, fornix; LHA, lateral hypothalamic area; MPA, medial preoptic area; NTS, nucleus of the solitary tract; oc, optic chiasm; ovlt; organum vasculosum of the lamina terminalis; PMV, ventral premammillary nucleus; VMH, ventromedial nucleus of the hypothalamus. Scale bar = 100 μm. *p < 0.05.
To assess region-specific changes in leptin sensitivity, another group of pregnant control and SOCS3 KO mice received an acute leptin injection to determine the activation of the STAT3 signaling pathway in the brain (Figure 3G). No differences between control and SOCS3 KO mice were observed in the number of leptin-induced pSTAT3-ir cells in the medial preoptic area (Figure 3H,L), lateral hypothalamic area, dorsomedial nucleus of the hypothalamus (DMH; Figure 3J,N) and nucleus of the solitary tract. However, SOCS3 KO mice exhibited increased pSTAT3-ir cells in the arcuate nucleus of the hypothalamus (ARH; Figure 3I,M), ventromedial nucleus of the hypothalamus (VMH; Figure 3I,M) and ventral premammillary nucleus (Figure 3K,O). Therefore, mice carrying SOCS3 deletion in LepR-expressing cells exhibited improved leptin sensitivity during pregnancy, which was likely responsible for the prevention of metabolic changes during pregnancy. Similarly to that observed in DIO [18,19], some brain areas are not affected by leptin resistance during gestation (i.e., DMH). However, pregnant SOCS3 KO mice showed improved responsiveness to leptin in hypothalamic nuclei related to energy balance regulation including the ARH and VMH [20]. Interestingly, these same areas are prone to developing leptin resistance induced by high-fat diets [18,19] or during pregnancy [5,6].
3.5. Improved glucose homeostasis and protection against GDM in SOCS3 KO mice
Pregnancy leads to the development of insulin resistance [3], although the mechanisms that cause this condition are unclear. No significant changes in serum insulin or glucagon levels were observed in pregnant control or SOCS3 KO mice (Supplemental Figure 5). However, SOCS3 KO mice showed improved glucose tolerance during pregnancy (Figure 4A). Remarkably, pregnant SOCS3 KO mice required less insulin secretion during the GTT to control their glucose levels (Figure 4B). Additionally, SOCS3 KO mice subjected to an ITT exhibited increased insulin sensitivity during late pregnancy (Figure 4C), which is the period associated with the highest risk of developing GDM [1]. Although these results indicate significant improvements in glucose homeostasis in pregnant SOCS3 KO mice, it remains unclear whether these changes can protect them from GDM. Thus, we studied GDM using a previously described protocol [9]. Mice received a single injection of STZ on P5 to induce lesions in their pancreatic β-cells. Although no changes were observed between the groups in serum insulin levels after the STZ treatment (p = 0.98; data not shown), control animals were hyperglycemic by P13 (>200 mg/dL), whereas SOCS3 KO mice showed no significant changes in glycemia (Figure 4D). Accordingly, the results of a GTT performed at day 14 indicated considerably better glucose tolerance in STZ-injected SOCS3 KO mice compared with controls (Figure 4E).
Figure 4.
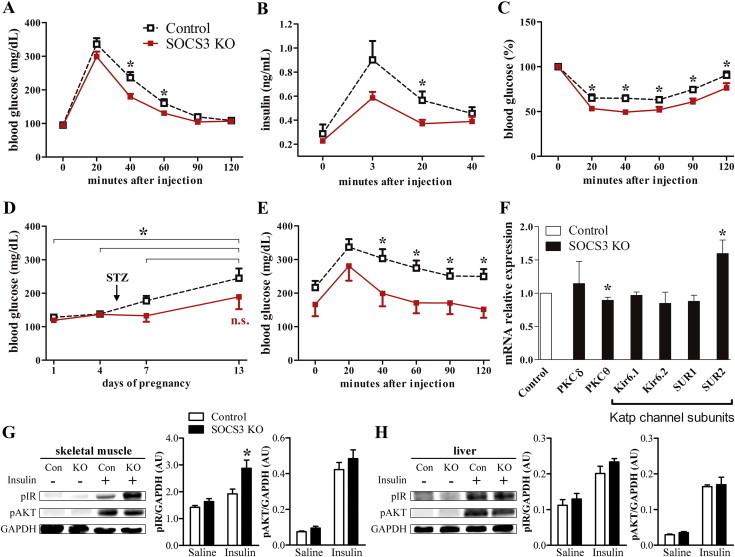
Improved glucose homeostasis and protection against gestational diabetes in the SOCS3 KO mice. (A and B) glycemic changes (n = 14–18/group) and serum insulin levels (n = 6/group) in pregnant mice during a GTT. (C) ITT in pregnant mice (n = 14–16/group). (D and E) Glycemic changes and GTT in the streptozotocin (STZ)-injected mice (n = 8–20/group). The arrow indicates the day STZ was injected in the animals. (F) Hypothalamic expression of genes involved in the central regulation of glucose homeostasis in pregnant mice (n = 8/group). (G and H) Activation of the insulin intracellular pathway in the gastrocnemius/soleus muscle and liver of saline- or insulin-treated pregnant mice (n = 4/group). *p < 0.05.
Central leptin signaling modulates insulin sensitivity in peripheral tissues [14,20–22]. The regulation of glucose homeostasis by hypothalamic neurons requires the activity of Katp channels [23,24] and is affected by specific isoforms of protein kinase C (PKC) [25,26]. We observed an upregulation of the SUR2 Katp channel subunit as well as a reduced expression of PKCθ in the hypothalamus of pregnant SOCS3 KO mice (Figure 4F). To investigate whether these changes affected insulin sensitivity in peripheral tissues, pregnant mice received an acute insulin stimulus to assess the activation of the insulin receptor (IR) signaling pathway. Insulin induced the phosphorylation of the IR and AKT in the skeletal muscle (Figure 4G) and liver (Figure 4H). However, SOCS3 KO mice showed increased IR phosphorylation in the skeletal muscle, suggesting higher insulin responsiveness (Figure 3G). No differences between control and SOCS3 KO groups were observed in the insulin sensitivity (Figure 4H) or the expression of genes involved in glucose and lipid metabolism in the liver (Supplemental Figure 6). These results indicate that alterations in hypothalamic SOCS3 expression are responsible for inducing insulin resistance during pregnancy. These effects involve changes in the hypothalamic expression of Katp channels and PKCθ, leading to better insulin responsiveness in skeletal muscles. Remarkably, SOCS3 deletion in LepR-expressing cells is sufficient to prevent the development of GDM in the STZ-induced model.
4. Discussion
In the present study, we identified a key molecular mechanism responsible for inducing metabolic changes during pregnancy. By preventing SOCS3 expression only in LepR-expressing cells, we turned off most of the pregnancy-induced metabolic changes without affecting the ability of the females to carry their gestation to term. Thus, SOCS3 may represent an integrative protein recruited by multiple signals to induce gestational metabolic changes. In the present obesogenic environment, this response may have become maladaptive leading many women to present metabolic imbalances during pregnancy such as EGWG and GDM. These conditions are becoming a common complication for obstetric practice as well as a risk factor of intergenerational programming of metabolic disorders [3,4,9]. In this case, consumption of high-caloric diets or prior obesity may increase hypothalamic SOCS3 expression leading to an aggravation of gestational metabolic imbalances. On the other hand, in physiological conditions, SOCS3 expression is induced by hormones secreted during gestation including estradiol [27], leptin [10], prolactin/placental lactogens [27] and growth hormone/placental growth hormone (GH-V) [28]. Therefore, increased hypothalamic SOCS3 expression and the consequent leptin resistance may represent a downstream mechanism common for several hormones to induce physiological adaptations of pregnancy. This fact is important because, from an evolutionary perspective, pregnancy-induced metabolic changes help female mammals to cope with the energy demands of gestation and lactation [2]. Our results corroborate this idea because pups from SOCS3 KO dams showed a lower weight gain rate, despite presenting the same genotype and body weight at birth compared with control pups. Fat accretion during pregnancy and food intake during lactation were decreased in SOCS3 KO dams which probably compromised their ability to nurture the offspring.
Among the hormones possibly involved in the physiological adaptations of pregnancy, prolactin has received special attention [2,16,29]. For example, central prolactin infusion causes leptin resistance in pseudopregnant rats [30]. Since LepR-expressing cells are directly responsive to prolactin in some brain areas [7], a crosstalk between leptin and prolactin signaling may affect the regulation of energy balance during pregnancy [2,7,16,29,30]. An important aspect to be considered is that some hormonal changes during pregnancy are different between rodents and primates. For instance, primates, including humans, present a tonic secretion of GH-V which is absent in rodents [31]. GH-V has a diabetogenic effect and at least part of the changes in glucose homeostasis in pregnant women may be caused by this hormone [31]. Therefore, the extrapolation of our findings to primates must be carefully evaluated. Overall, future studies are still necessary to determine the precise role of each hormone in the onset of metabolic changes observed during pregnancy.
Another important aspect is that our study cannot distinguish whether the prevention of gestational metabolic changes is caused by the deletion of SOCS3 in LepR-expressing cells or by the resulting improvement in leptin sensitivity. Therefore, the rescue of leptin sensitivity during pregnancy may represent a key aspect responsible for mitigating the development of gestational metabolic changes. Although previous studies have demonstrated that hypothalamic SOCS3 expression is increased during pregnancy [32,33], our study is the first to analyze several other genes that could modulate leptin sensitivity and to show that only SOCS3 expression is significantly affected by gestation. In addition, SOCS3 KO females did not exhibit the pregnancy-induced changes in hypothalamic SOCS3 expression indicating that this increase is restricted to LepR-expressing cells. Therefore, our findings indicate that SOCS3 likely plays a major role in inducing the leptin resistance during pregnancy and consequently controlling the onset of pregnancy-induced metabolic changes. An interesting finding is that pregnant SOCS3 KO mice have increased hypothalamic expression of Ob-Ra. This isoform is believed to be involved in the transport of leptin from the systemic circulation to the central nervous system and its expression is reduced in the choroid plexus of pregnant mice [5]. Thus, an increased transport of leptin to the central nervous system may have contributed to the better leptin sensitivity observed in pregnant SOCS3 KO mice. However, a recent study showed that a knockout mice specific for the Ob-Ra have only a modest phenotype indicating that this isoform plays a minor role in the regulation of metabolism [34].
Earlier studies have described the role of SOCS3 in modulating leptin sensitivity in DIO models [12–15]. Haploinsufficiency or neuronal SOCS3 deficiency partially protects mice against DIO and insulin resistance [12–15]. Surprisingly, deletion of SOCS3 from LepR-expressing cells did not prevent DIO, although improved diet-induced insulin resistance [14]. In accordance, overexpression of SOCS3 in LepR-expressing cells did not lead to obesity [35]. These results contrast with the present findings indicating that SOCS3 deficiency in LepR-expressing cells leads to drastic consequences in the regulation of energy balance of pregnant mice. Therefore, although SOCS3 modulates leptin sensitivity in distinct conditions, the consequences of manipulating SOCS3 in LepR-expressing cells depend on the situation. Regarding this, SOCS3 seems to influence glucose homeostasis in both DIO and pregnancy. However, SOCS3 expression in LepR cells modulates the energy balance more significantly during pregnancy.
Based on our findings, therapies that target SOCS3 could become a promising approach to prevent and treat gestational metabolic diseases. Of note, currently there is no specific treatment for pregnancy-induced metabolic imbalances. Nonetheless, SOCS3 is involved in different biological functions, and systemic manipulations of SOCS3 may cause unpredicted effects. For example, SOCS3 ablation may influence placental functions [36]. Therefore, more studies are still necessary to assess the risk-benefits of manipulating SOCS3 during pregnancy. It seems, however, that central inhibition of SOCS3 may produce more benefits than risks not only for the treatment of gestational metabolic imbalances, but for obesity and diabetes mellitus as well. Because LepR expression is also found in different organs [37], peripheral tissues may have been affected in SOCS3 KO females. However, this fact did not cause any apparent complications during pregnancy. Regarding the phenotype of pregnant SOCS3 KO females, we cannot rule out that part of it was caused by peripheral SOCS3 deletion. For example, liver- or skeletal muscle-specific SOCS3 deletion increases insulin sensitivity in those particular tissues [38,39]. However, this fact is unlikely because liver-specific SOCS3 ablation paradoxically leads to obesity [38] which is incompatible with the phenotype of SOCS3 KO females observed in our study. Additionally, since the recovery of leptin sensitivity during pregnancy may represent a major aspect in the prevention of metabolic changes in pregnant SOCS3 KO females, and it is well-known that the key metabolic effects of leptin are mediated by the brain [40,41], the metabolic phenotype of pregnant SOCS3 KO mice is likely caused by central mechanisms.
In conclusion, our study identified the increased hypothalamic expression of SOCS3 as a key mechanism responsible for inducing pregnancy-induced leptin resistance and metabolic adaptations. These findings not only help to explain a common phenomenon of the mammalian physiology, but it may also aid in the development of approaches to prevent and treat gestational metabolic imbalances.
Disclosure statement
The authors have nothing to disclose.
Acknowledgments
We would like to thank Ana Campos, Deborah P. Romeu and Vanessa Nagaishi for their technical assistance.
Footnotes
Grants: This study was supported by the São Paulo Research Foundation (FAPESP-Brazil, 10/18086-0, 12/15517-6, 13/21722-4, 13/25032-2 and 14/11752-6) and the International Brain Research Organization (IBRO; Return Home Fellowship).
Conflict of interest
No conflicts of interest, financial or otherwise, are declared by the authors.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Sarwer D.B., Allison K.C., Gibbons L.M., Markowitz J.T., Nelson D.B. Pregnancy and obesity: a review and agenda for future research. Journal of Women's Health. 2006;15(6):720–733. doi: 10.1089/jwh.2006.15.720. [DOI] [PubMed] [Google Scholar]
- 2.Ladyman S.R., Augustine R.A., Grattan D.R. Hormone interactions regulating energy balance during pregnancy. Journal of Neuroendocrinology. 2010;22(7):805–817. doi: 10.1111/j.1365-2826.2010.02017.x. [DOI] [PubMed] [Google Scholar]
- 3.Norman J.E., Reynolds R.M. The consequences of obesity and excess weight gain in pregnancy. Proceedings of the Nutrition Society. 2011;70(4):450–456. doi: 10.1017/S0029665111003077. [DOI] [PubMed] [Google Scholar]
- 4.Nehring I., Schmoll S., Beyerlein A., Hauner H., von Kries R. Gestational weight gain and long-term postpartum weight retention: a meta-analysis. American Journal of Clinical Nutrition. 2011;94(5):1225–1231. doi: 10.3945/ajcn.111.015289. [DOI] [PubMed] [Google Scholar]
- 5.Ladyman S.R., Grattan D.R. Suppression of leptin receptor messenger ribonucleic acid and leptin responsiveness in the ventromedial nucleus of the hypothalamus during pregnancy in the rat. Endocrinology. 2005;146(9):3868–3874. doi: 10.1210/en.2005-0194. [DOI] [PubMed] [Google Scholar]
- 6.Ladyman S.R., Fieldwick D.M., Grattan D.R. Suppression of leptin-induced hypothalamic JAK/STAT signalling and feeding response during pregnancy in the mouse. Reproduction. 2012;144(1):83–90. doi: 10.1530/REP-12-0112. [DOI] [PubMed] [Google Scholar]
- 7.Nagaishi V.S., Cardinali L.I., Zampieri T.T., Furigo I.C., Metzger M., Donato J., Jr. Possible crosstalk between leptin and prolactin during pregnancy. Neuroscience. 2014;259(0):71–83. doi: 10.1016/j.neuroscience.2013.11.050. [DOI] [PubMed] [Google Scholar]
- 8.Zampieri T.T., Pedroso J.A., Furigo I.C., Tirapegui J., Donato J., Jr. Oral leucine supplementation is sensed by the brain but neither reduces food intake nor induces an anorectic pattern of gene expression in the hypothalamus. PLoS One. 2013;8(12):e84094. doi: 10.1371/journal.pone.0084094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steculorum S.M., Bouret S.G. Maternal diabetes compromises the organization of hypothalamic feeding circuits and impairs leptin sensitivity in offspring. Endocrinology. 2011;152(11):4171–4179. doi: 10.1210/en.2011-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bjorbaek C., El-Haschimi K., Frantz J.D., Flier J.S. The role of SOCS-3 in leptin signaling and leptin resistance. Journal of Biological Chemistry. 1999;274(42):30059–30065. doi: 10.1074/jbc.274.42.30059. [DOI] [PubMed] [Google Scholar]
- 11.Bjorbaek C., Elmquist J.K., Frantz J.D., Shoelson S.E., Flier J.S. Identification of SOCS-3 as a potential mediator of central leptin resistance. Molecular Cell. 1998;1(4):619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- 12.Mori H., Hanada R., Hanada T., Aki D., Mashima R., Nishinakamura H. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nature Medicine. 2004;10(7):739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- 13.Briancon N., McNay D.E., Maratos-Flier E., Flier J.S. Combined neural inactivation of suppressor of cytokine signaling-3 and protein-tyrosine phosphatase-1B reveals additive, synergistic, and factor-specific roles in the regulation of body energy balance. Diabetes. 2010;59(12):3074–3084. doi: 10.2337/db10-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pedroso J.A., Buonfiglio D.C., Cardinali L.I., Furigo I.C., Ramos-Lobo A.M., Tirapegui J. Inactivation of SOCS3 in leptin receptor-expressing cells protects mice from diet-induced insulin resistance but does not prevent obesity. Molecular Metabolism. 2014;3(6):608–618. doi: 10.1016/j.molmet.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howard J.K., Cave B.J., Oksanen L.J., Tzameli I., Bjorbaek C., Flier J.S. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nature Medicine. 2004;10(7):734–738. doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- 16.Augustine R.A., Ladyman S.R., Grattan D.R. From feeding one to feeding many: hormone-induced changes in bodyweight homeostasis during pregnancy. Journal of Physiology. 2008;586(2):387–397. doi: 10.1113/jphysiol.2007.146316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mounzih K., Qiu J., Ewart-Toland A., Chehab F.F. Leptin is not necessary for gestation and parturition but regulates maternal nutrition via a leptin resistance state. Endocrinology. 1998;139(12):5259–5262. doi: 10.1210/endo.139.12.6523. [DOI] [PubMed] [Google Scholar]
- 18.Münzberg H., Flier J.S., Bjørbæk C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145(11):4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- 19.Enriori P.J., Sinnayah P., Simonds S.E., Garcia Rudaz C., Cowley M.A. Leptin action in the dorsomedial hypothalamus increases sympathetic tone to brown adipose tissue in spite of systemic leptin resistance. Journal of Neuroscience. 2011;31(34):12189–12197. doi: 10.1523/JNEUROSCI.2336-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams K.W., Elmquist J.K. From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nature Neuroscience. 2012;15(10):1350–1355. doi: 10.1038/nn.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berglund E.D., Vianna C.R., Donato J., Jr., Kim M.H., Chuang J.C., Lee C.E. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. Journal of Clinical Investigation. 2012;122(3):1000–1009. doi: 10.1172/JCI59816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koch C., Augustine R.A., Steger J., Ganjam G.K., Benzler J., Pracht C. Leptin rapidly improves glucose homeostasis in obese mice by increasing hypothalamic insulin sensitivity. Journal of Neuroscience. 2010;30(48):16180–16187. doi: 10.1523/JNEUROSCI.3202-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miki T., Liss B., Minami K., Shiuchi T., Saraya A., Kashima Y. ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nature Neuroscience. 2001;4(5):507–512. doi: 10.1038/87455. [DOI] [PubMed] [Google Scholar]
- 24.Pocai A., Lam T.K.T., Gutierrez-Juarez R., Obici S., Schwartz G.J., Bryan J. Hypothalamic KATP channels control hepatic glucose production. Nature. 2005;434(7036):1026–1031. doi: 10.1038/nature03439. [DOI] [PubMed] [Google Scholar]
- 25.Benoit S.C., Kemp C.J., Elias C.F., Abplanalp W., Herman J.P., Migrenne S. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-θ subcellular localization in rodents. Journal of Clinical Investigation. 2009;119(9):2577–2589. doi: 10.1172/JCI36714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cabou C., Vachoux C., Campistron G., Drucker D.J., Burcelin R. Brain GLP-1 signaling regulates femoral artery blood flow and insulin sensitivity through hypothalamic PKC-δ. Diabetes. 2011;60(9):2245–2256. doi: 10.2337/db11-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steyn F.J., Anderson G.M., Grattan D.R. Hormonal regulation of suppressors of cytokine signaling (SOCS) messenger ribonucleic acid in the arcuate nucleus during late pregnancy. Endocrinology. 2008;149(6):3206–3214. doi: 10.1210/en.2007-1623. [DOI] [PubMed] [Google Scholar]
- 28.Krebs D.L., Hilton D.J. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19(5):378–387. doi: 10.1634/stemcells.19-5-378. [DOI] [PubMed] [Google Scholar]
- 29.Woodside B., Budin R., Wellman M.K., Abizaid A. Many mouths to feed: the control of food intake during lactation. Frontiers in Neuroendocrinology. 2012;33(3):301–314. doi: 10.1016/j.yfrne.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 30.Augustine R.A., Grattan D.R. Induction of central leptin resistance in hyperphagic pseudopregnant rats by chronic prolactin infusion. Endocrinology. 2008;149(3):1049–1055. doi: 10.1210/en.2007-1018. [DOI] [PubMed] [Google Scholar]
- 31.Newbern D., Freemark M. Placental hormones and the control of maternal metabolism and fetal growth. Current Opinion in Endocrinology, Diabetes and Obesity. 2011;18(6):409–416. doi: 10.1097/MED.0b013e32834c800d. [DOI] [PubMed] [Google Scholar]
- 32.Anderson G.M., Beijer P., Bang A.S., Fenwick M.A., Bunn S.J., Grattan D.R. Suppression of prolactin-induced signal transducer and activator of transcription 5b signaling and induction of suppressors of cytokine signaling messenger ribonucleic acid in the hypothalamic arcuate nucleus of the rat during late pregnancy and lactation. Endocrinology. 2006;147(10):4996–5005. doi: 10.1210/en.2005-0755. [DOI] [PubMed] [Google Scholar]
- 33.Trujillo M.L., Spuch C., Carro E., Senaris R. Hyperphagia and Central mechanisms for leptin resistance during pregnancy. Endocrinology. 2011;152(4):1355–1365. doi: 10.1210/en.2010-0975. [DOI] [PubMed] [Google Scholar]
- 34.Li Z., Ceccarini G., Eisenstein M., Tan K., Friedman J.M. Phenotypic effects of an induced mutation of the ObRa isoform of the leptin receptor. Molecular Metabolism. 2013;2(4):364–375. doi: 10.1016/j.molmet.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reed A.S., Unger E.K., Olofsson L.E., Piper M.L., Myers M.G., Jr., Xu A.W. Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes. 2010;59(4):894–906. doi: 10.2337/db09-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts A.W., Robb L., Rakar S., Hartley L., Cluse L., Nicola N.A. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(16):9324–9329. doi: 10.1073/pnas.161271798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fei H., Okano H.J., Li C., Lee G.H., Zhao C., Darnell R. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(13):7001–7005. doi: 10.1073/pnas.94.13.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sachithanandan N., Fam B.C., Fynch S., Dzamko N., Watt M.J., Wormald S. Liver-specific suppressor of cytokine signaling-3 deletion in mice enhances hepatic insulin sensitivity and lipogenesis resulting in fatty liver and obesity. Hepatology. 2010;52(5):1632–1642. doi: 10.1002/hep.23861. [DOI] [PubMed] [Google Scholar]
- 39.Jorgensen S.B., O'Neill H.M., Sylow L., Honeyman J., Hewitt K.A., Palanivel R. Deletion of skeletal muscle SOCS3 prevents insulin resistance in obesity. Diabetes. 2013;62(1):56–64. doi: 10.2337/db12-0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen P., Zhao C., Cai X., Montez J.M., Rohani S.C., Feinstein P. Selective deletion of leptin receptor in neurons leads to obesity. Journal of Clinical Investigation. 2001;108(8):1113–1121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Luca C., Kowalski T.J., Zhang Y., Elmquist J.K., Lee C., Kilimann M.W. Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. Journal of Clinical Investigation. 2005;115(12):3484–3493. doi: 10.1172/JCI24059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.