

Abstract
Objective
The skeleton has recently emerged as an additional player in the control of whole-body glucose metabolism; however, the mechanism behind this is not clear.
Methods
Here we employ mice lacking neuropeptide Y, Y1 receptors solely in cells of the early osteoblastic lineage (Y1f3.6Cre), to examine the role of osteoblastic Y1 signalling in glycaemic control.
Results
Y1f3.6Cre mice not only have a high bone mass phenotype, but importantly also display altered glucose homeostasis; significantly decreased pancreas weight, islet number and pancreatic insulin content leading to elevated glucose levels and reduced glucose tolerance, but with no effect on insulin induced glucose clearance. The reduced glucose tolerance and elevated bone mass was corrected in Y1f3.6Cre mice by bone marrow transplant from wildtype animals, reinforcing the osteoblastic nature of this pathway. Importantly, when fed a high fat diet, Y1f3.6Cre mice, while equally gaining body weight and fat mass compared to controls, showed significantly improved glucose and insulin tolerance. Conditioned media from Y1f3.6Cre osteoblastic cultures was unable to stimulate insulin expression in MIN6 cells compared to conditioned media from wildtype osteoblast, indicating a direct signalling pathway. Importantly, osteocalcin a secreted osteoblastic factor previously identified as a modulator of insulin secretion was not altered in the Y1f3.6Cre model.
Conclusion
This study identifies the existence of other osteoblast-derived regulators of pancreas function and insulin secretion and illustrates a mechanism by which NPY signalling in bone tissue is capable of regulating pancreatic function and glucose homeostasis.
Keywords: Neuropeptide, Osteoblast, Glucose homeostasis, Y1 receptor, Insulin
1. Introduction
Maintenance of glucose homeostasis requires the coordination of glucose supply to a wide variety of tissues of differing energetic demands, whilst balancing endogenous gluconeogenesis with the periodic supply of glucose from the diet. Appropriate glucose supply is fundamental to effective and consistent cellular function, and can only be ensured through complex communication and feedback pathways regulating insulin-dependent and independent glucose uptake in target tissues. It has become increasingly apparent that whole body insulin action can be disrupted by perturbations within target tissues, such as muscle [1] and white adipose tissue [2], highlighting the importance of tissue feedback communication to control whole body glucose balance. In recent years, skeletal tissue has emerged as an additional regulator of whole body glucose homeostasis. Bone is a known insulin-regulated tissue [3], but importantly, existence of feedback has emerged, with osteocalcin, an osteoblast-derived osteokine, reported to increase insulin release from beta cells and indirectly increasing insulin action through enhanced release of adiponectin from adipose tissue [4]. However, the regulation of glucose metabolism by bone has not been fully elucidated, with evidence emerging of osteocalcin-independent activity. For example, while osteocalcin replacement in osteocalcin null mice can reverse the glucose intolerance and corrected blood glucose levels, it could not restore insulin sensitivity, indicating that other osteoblast-derived factor(s) may also mediate insulin action [5].
A major factor controlling both glucose homeostasis and osteoblast function is neuropeptide Y (NPY). NPY acts both through hypothalamic neuronal circuits [6,7], as well as by directly signalling via Y1 receptors in ®-cells [8] and osteoblasts [9] to control bone and glucose homeostasis. Centrally, NPY has been shown to stimulate insulin secretion from the pancreas [10,11]. The other family members, peptide YY (PYY) and pancreatic polypeptide (PP), are endocrine hormones which act as satiety signals [12] and are involved in the regulation of glucose homeostasis, showing an inhibitory role in glucose-stimulated insulin secretion [13]. NPY also controls bone homeostasis via central Y2 and peripheral Y1 signalling pathways. Particularly, Y1 receptor activation on bone forming osteoblasts suppresses bone formation [9,14] and inhibits the proliferation of mesenchymal progenitor cells [15]. NPY has previously been shown to co-ordinate bone and energy metabolism [16] with increased NPY levels indicating negative energy balance and therefore a reduced need for high bone mass. Consistent with that, lack of NPY in mice, mimicking positive energy balance leads to a dramatic increase in bone mass. However, the role of NPY in co-ordinating bone and glucose homeostasis has not been examined.
Given the dual roles of the NPY system in regulating glucose and bone homeostasis, and the fact that the skeleton has emerged as a modulator of whole-body glucose metabolism, we sought to determine to what extent osteoblastic NPY signalling, via Y1 receptors, contributes to the skeletal control of glucose homeostasis. To investigate this, we utilised conditional Y1 knockout mice where the Y1 receptor is deleted at an early stage of osteoblast differentiation and investigated the consequence on islet function and glucose homeostasis using a range of in vivo and in vitro analyses.
2. Materials and methods
2.1. Mice
All animal experiments were approved by the Garvan Institute/St Vincent's Hospital Animal Experimentation Ethics Committee and conducted in accordance with relevant guidelines and regulations. All data presented are on male mice.
Mice with osteoblast-specific deletion of the Y1 receptor (Y1f3.6Cre) were generated by mating mice expressing Cre under the control of a 3.6 kb fragment of the rat α1(I)-collagen promoter with Y1lox/lox mice. Cre-mediated recombination resulted in osteoblast-specific deletion of the entire coding region of the Y1 gene. Male Y1f3.6Cre mice were bred with female Y1lox/lox mice to generate both Y1f3.6Cre mice and Y1lox/lox littermates to be used as controls. Genotypes were determined by PCR as previously described [15].
2.2. Bone marrow chimeric mice
Mice at 9 weeks of age were lethally irradiated with a split-level dose of 425 rad per dose, with a 5-hour interval between the two doses. 24 h following the initial dose, they were given an intravenous injection of 1.5 × 107 bone marrow cells per mouse isolated from wildtype littermates. Insulin tolerance tests were performed on these mice 7 weeks after irradiation and reconstitution with donor bone marrow, followed by a glucose tolerance test 3 weeks later. The mice were culled at 22 weeks of age following standard protocol outlined below.
2.3. Glucose metabolism
Insulin tolerance tests were performed on male mice at 15 weeks of age (unless otherwise stated) at 14:00–16:00 h. Briefly, the mice were fasted for 6 h and then intraperitoneally injected with insulin (0.5 IU/kg) (Novo Nordisk Pharmaceuticals, Baulkham Hills, NSW, Australia). Tail tip blood samples were then collected at 0, 15, 30, 45, 60, 75 and 90 min after insulin injection, and blood glucose levels were measured using a glucometer (AccuCheck II; Roche, New South Wales, Castle Hill, Australia). 7 days later (unless otherwise stated), mice underwent a glucose tolerance test, where they were fasted for 16–24 h before intraperitoneal injection between 13:00 and 15:00 h of a 10% d-glucose solution (1.0 g/kg) (Astra Zeneca, North Ryde, NSW, Australia). Blood samples were obtained from the tail tip at 0, 20 and 60 min after glucose injection and blood glucose levels were measured using a glucometer. Serum was also separated, immediately frozen and stored at −20 °C for later measurement of serum insulin using an ELISA kit from Linco Research (St Charles, Missouri, USA) or a RIA kit from Millipore (Millipore, Billerica, MA, USA).
2.4. Tissue collection
Male mice were injected with the fluorescent compound calcein (15 mg/kg; Sigma–Aldrich, St Louis, MO, USA) 3 and 10 days prior to tissue collection to enable subsequent calculation of bone formation rate. Whole body lean mass, fat mass, bone mineral content (BMC) and bone mineral density (BMD) were measured in mice anesthetised with isoflurane using a dedicated mouse dual X-ray absorptiometry (DXA) (Lunar Piximus II, GE Medical Systems, Madison WI) 3 days prior to tissue collection. At 16 weeks of age, mice were culled between 13.00 and 16.00 h by cervical dislocation and decapitation for collection of trunk blood. Serum was separated, immediately frozen and stored at −20 °C. The white adipose tissue (WAT) depots (right side inguinal, retroperitoneal, epididymal (gonadal) and mesenteric), and skeletal muscle were removed, weighed, frozen on dry ice and stored at −80 °C. Femurs, tibias and caudal vertebrae were excised, fixed overnight in 4% paraformaldehyde (PFA) in phosphate buffered saline (PBS) at 4 °C and then stored in 70% ethanol at 4 °C before undergoing processing.
2.5. Pancreas histology
Pancreatic tissues were fixed in 4% PFA, embedded in paraffin and sectioned at 5 μm. Immunohistochemistry was then performed using rabbit anti-insulin (Genesearch, 1:200) and biotinylated goat anti-rabbit (Vector Laboratories, California, USA; 1:300) antibodies and a Vectastain ABC kit (Vector Laboratories, California, USA). Insulin-positive islet cell number and size were quantified from at least five sections per pancreas (each 60 μm apart) using a camera attached to a Leica microscope (Leica Microsystems) and ImageJ software (National Institutes of Health, USA). Total pancreatic surface was also measured and used to determine islet cell number per mm2 and ®-cell area (area positive for insulin immunostaining divided by the total pancreatic surface).
2.6. Plasma assays
Unless otherwise stated, serum glucose was measured using a glucose oxidase assay kit (Trace Scientific, Clayton, Victoria, Australia), serum insulin was measured using an ELISA kit from Linco Research (St Charles, Missouri, USA) and serum osteocalcin was measured using an ELISA kit from BTI (Stoughton, Massachusetts, USA).
2.7. Bone histomorphometry
Bone histomorphometry was carried out on 5 μm sagittal sections of the distal half of the right femur as previously described [17]. Briefly, sections were stained for mineralised bone and cancellous bone volume, trabecular thickness and trabecular number were calculated. In addition, fluorescence microscopy was used to calculate mineral apposition rate, mineralising surface and bone formation rate whilst osteoclast surface and osteoclast number were estimated using tartrate-resistant acid phosphatase-stained sections. Cortical mineral apposition rate was measured in an endosteal and a periosteal region both extending 1000 μm distal from the mid-point, as previously described [17].
2.8. Bone micro-computed tomography (micro-CT)
Following fixation, left femora were cleaned of muscle and analyses of the bone were carried out using micro computed tomography (micro-CT) with a Skyscan 1174 scanner and associated analysis software (Skyscan, Aartselaar, Belgium) as previously described [18]. Briefly, analyses of the cortical bone were carried out in 150 slices (0.93 mm) selected 750 slices (4.65 mm) proximally from the distal growth plate resulting in calculations of the following parameters: total tissue area, bone area, marrow area, endosteal perimeter, periosteal perimeter, cortical thickness, and polar moment of inertia (an index of strength). Analyses of the trabecular bone were carried out in 150 slices (0.93 mm) selected 40 slices (0.25 mm) proximally from the distal growth plate resulting in calculations of the following parameters: total tissue volume, bone volume, trabecular number, trabecular thickness and trabecular separation.
2.9. BMSC/MIN6 cell culture
BMSCs were isolated from 7 to 9 week old male mice, cultured and differentiated in osteogenic media as previously described [15]. Supernatant was collected from BMSCs after 14 days in either control or osteogenic media. 24 h before supernatant was collected, cells were switched to media containing 1% FBS. MIN6 cells were maintained in DMEM media containing 25 mM glucose, 10% FBS, 10 mM HEPES, 100 U/mL penicillin and 100 μg/mL streptomycin. For treatment experiments, cells were seeded at 4 × 105 cells/mL, left to recover for 24 h and then media was changed to DMEM low glucose media (5.5 mM) and 1% FBS for 24 h prior to treatment. MIN6 cells were treated for 4 h with 50% BMSC supernatant after which an insulin secretion assay was performed or RNA was extracted from the cells. For the insulin secretion assay, cells were washed and then pre-incubated for 30 min in KRB buffer with 2.8 mM glucose. They were then incubated for 1 h with KRB buffer containing either low (2.8 mM) or high (25 mM) glucose after which the supernatant was collected and stored at −20 °C until assayed for insulin levels using an ELISA kit as detailed above.
2.10. RNA extractions and cDNA synthesis
Unless otherwise stated, all RNA extractions were carried out using TRIzol® reagent (Sigma) according to the manufacturer's instructions. RNA extractions from the mid-shaft of femurs and tibias isolated from male mice were homogenised in 2 mL TRIzol® reagent using a Polytron homogeniser. RNA samples were subsequently checked for consistent quality and quantified using the Agilent 2100 Bioanalyser (Agilent Technologies) according to the manufacturer's instructions. 1 μg of total RNA was taken for cDNA synthesis with oligo(dT)20 and random hexamers using the SuperScript III First-Strand Synthesis System for reverse transcription-PCR (Invitrogen).
2.11. Reverse-transcription PCR
PCR reactions were performed for the number of cycles indicated with denaturing at 94 °C and extension at 72 °C. Mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene to control for variations between samples. The specific primers and annealing temperatures used along with the resultant product sizes obtained are as detailed previously [9,19].
2.12. Quantitative real time-PCR
Quantitative real-time PCR was carried out using either the TaqMan Universal PCR master mix, ABI Prism 7900 HT Sequence Detection System and inventoried kits containing primers and probes (all from Applied Biosystems) or the Lightcycler 480 Probes Mastermix and Universal Probe Library assays on the Lightcycler 480 System (Roche). To control for variability in amplification due to differences in starting mRNA concentrations, either ®-actin or RPL was used as an internal standard. The relative expression of target mRNA was computed from the target Ct values and the housekeeper Ct value using the standard curve method (Applied Biosystems).
2.13. Statistical analyses
All data are expressed as means ± SEM. Differences between groups were assessed by ANOVA. Statistical analyses were performed with SPSS for Mac OS X, version 21.0 (SPSS Inc., Chicago, IL, USA). For all statistical analyses, p < 0.05 was accepted as being statistically significant, with p < 0.1 accepted as showing a trend of change.
3. Results
3.1. Y1 receptor deletion in early osteoblast differentiation enhances bone mass
Whilst the selective deletion of Y1 receptors in mature osteoblasts mimics the bone phenotype seen in germline Y1 deficient mice, these mice did not show any of the other characteristics of global Y1 deletion such as high insulin levels and increased fat mass [9]. Since osteoblasts and adipocytes both mature in a complex way from mesenchymal progenitors and we have previously shown an involvement of Y1 receptor signalling in this process [15], we hypothesised that the ablation of Y1 signalling in an early stage of osteoblast development could give new insights to these processes. For this purpose, we crossed our Y1 floxed mice with mice expressing Cre under the control of a 3.6 kb fragment of the α1(I)-collagen promoter, where Cre expression has been shown to occur early during osteoblast differentiation [20]. As shown in Supplementary Figure 1, Cre expression in Y1f3.6Cre mice was restricted to bone tissue where it resulted in a complete lack of Y1 receptor expression. Importantly, Y1 receptor expression was unaffected in other tissues examined (brain, muscle and white adipose tissue), confirming the generation of mice with the Y1 receptor deleted in the osteoblast lineage (Supplementary Figure 1).
As a consequence of the specific deletion of osteoblastic Y1 receptors, Y1f3.6Cre mice display a marked increase in bone mass similar to that seen in germline Y1−/− mice [21]. The significant increase in cancellous bone volume (Figure 1A) is associated with significant increases in trabecular thickness (Figure 1B) and trabecular number (Figure 1C) at both 18 weeks and 28 weeks of age. Moreover, this high bone mass phenotype is due to significantly increased osteoblast activity as shown by significantly increased mineral apposition rate (MAR) (Figure 1D) and bone formation rate (Figure 1E), with a trend towards increased mineralising surface at 28 weeks of age (Figure 1F). However, in contrast to the increased bone resorption shown by germline Y1−/− mice [21], Y1f3.6Cre mice at 18 weeks of age display significantly decreased osteoclast surface (Figure 1G), suggesting that bone resorption is decreased in these mice. This effect becomes even more pronounced at 28 weeks of age when the lack of osteoblastic Y1 receptors appears to provide protection against an age-induced increase in osteoclast surface (Figure 1G) as well as osteoclast number (Figure 1H). A representative image of the effect of osteoblastic specific Y1 deletion on cancellous bone is shown in Figure 1I.
Figure 1.
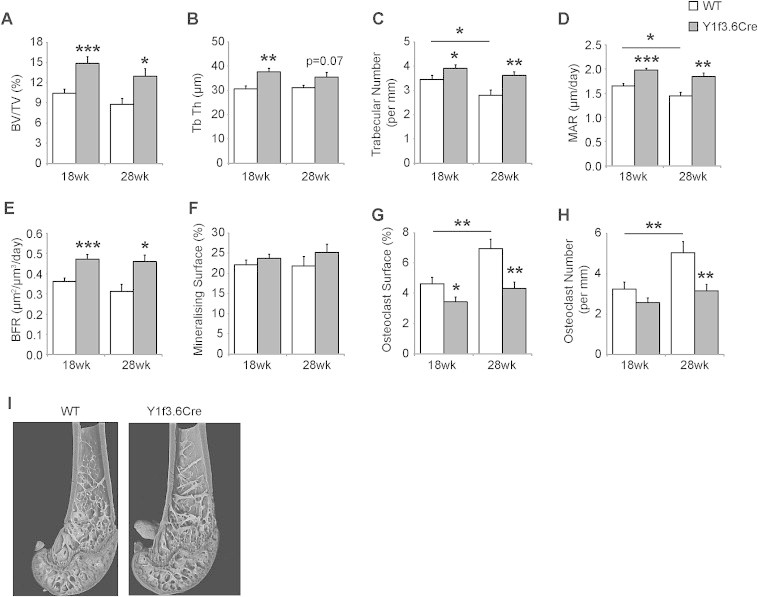
Y1 receptor deletion in early osteoblast differentiation enhances bone mass. Osteoblastic specific Y1 deleted, Y1f3.6Cre mice display a significant increase in cancellous bone volume (BV/TV) (A) associated with significant increases in trabecular thickness (Tb Th) (B) and trabecular number (C) at both 18 weeks and 28 weeks of age. This high bone mass phenotype is due to significantly increased osteoblast activity as shown by significantly increased mineral apposition rate (MAR) (D) and bone formation rate (BFR) (E), with no difference in mineralising surface (F). Y1f3.6Cre mice at 18 weeks of age display significantly decreased osteoclast surface (G), which comes even more pronounced at 28 weeks of age when the lack of osteoblastic Y1 receptors appears to provide significant protection against an age-induced increase in osteoclast surface (G) as well as osteoclast number (H). A representative picture of the effect of osteoblastic specific Y1 deletion on femur tissue is shown in (I). Data are means ± SEM of at least 5 mice per group. * = p < 0.05, ** = p < 0.01, *** = p < 0.001 versus control or as indicated.
3.2. Y1 receptor deletion in early osteoblast differentiation alters insulin production
To further characterise the consequences of Y1 deficiency in early osteoblast differentiation we also analysed parameters of energy homeostasis. Interestingly, we observed a significant decrease in the weight of the pancreas in Y1f3.6Cre mice compared to their wildtype littermates (Figure 2A). This occurred despite no change in body weight or the weights of other organs examined (Supplementary Figure 2). In addition to decreased pancreas weight, histological analysis revealed that Y1f3.6Cre mice also exhibited significant reductions in the number of islets and the total ®-cell area (Figure 2B). Furthermore, Y1f3.6Cre mice showed a significant increase in circulating glucose levels (Figure 2C) associated with a tendency to decreased circulating insulin levels (Figure 2D).
Figure 2.
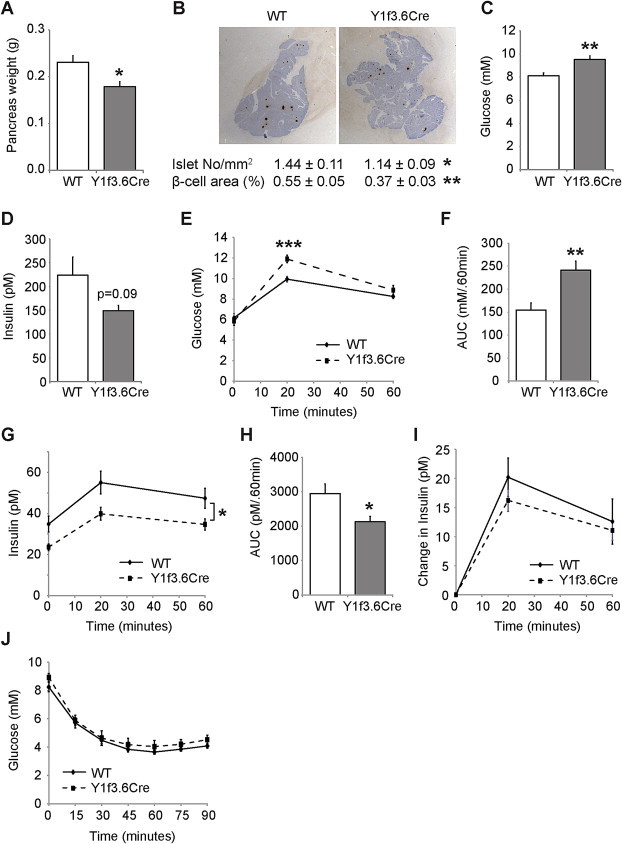
Y1 receptor deletion in early osteoblast differentiation alters insulin release. Y1f3.6Cre mice have a significant smaller pancreas compared to wildtype littermates (A) as well as a significant reduction in pancreas insulin content as demonstrated by significant reductions in the number of islets and the total beta-cell area (B). Furthermore, Y1f3.6Cre mice have a significant increase in circulating glucose levels (C) associated with a trend towards decreased circulating insulin levels (D). Y1f3.6Cre mice also exhibit impaired glucose tolerance with significantly higher serum glucose levels (E) and glucose area under the curve during the glucose tolerance test (F). The impairment in glucose clearance in Y1f3.6Cre mice was associated with significantly reduced serum insulin levels both initially and throughout the glucose tolerance test (G,H) although insulin levels did increase to a similar degree during the test as those of their wildtype littermates (I). Insulin action was unchanged in Y1f3.6Cre mice as their response to insulin during an insulin tolerance test was similar to that of their wildtype littermates (J). Data are means ± SEM of 5–12 mice per group.* = p < 0.05, ** = p < 0.01, *** = p < 0.001 versus control or as indicated.
In order to determine whether the above changes in the pancreas and circulating insulin levels of Y13.6Cre mice affect their ability to regulate glucose homeostasis, we performed glucose tolerance tests. As shown in Figure 2E–I, Y1f3.6Cre mice fasted overnight exhibited impaired glucose tolerance with higher serum glucose levels (Figure 2E) and glucose area under the curve during the glucose tolerance test (Figure 2F). The impairment in glucose clearance in Y1f3.6Cre mice was associated with markedly reduced serum insulin levels both initially and throughout the glucose tolerance test (Figure 2G,H) although insulin levels did increase to a similar degree during the test as those of their wildtype littermates (Figure 2I). To test whether insulin action is also affected by osteoblastic deletion of Y1 receptors, we performed insulin tolerance tests on Y1f3.6Cre mice. However, as shown in Figure 2J, insulin action was unchanged in Y1f3.6Cre mice as their response to insulin was similar to that of their wildtype littermates. Therefore, these data demonstrate that Y1 receptor signalling in early osteoblasts has a significant effect on the regulation of glucose homeostasis through the modulation of islet size, insulin production and secretion.
3.3. Osteoblast specific Y1 deletion is protective against high fat diet induced glucose intolerance and insulin resistance
The decrease in serum insulin levels and glucose tolerance observed in Y1f3.6Cre mice suggested that they could be more susceptible to the detrimental effects of high fat diet induced obesity and glucose intolerance. Therefore we fed Y1f3.6Cre mice and their wildtype littermates a high fat diet for 8 weeks from 9 weeks of age. No differences were observed between genotypes in terms of body weight increase in response to high fat diet (Figure 3A) or fat mass as measured by either DXA (Figure 3B) or the weight of fat depots at cull (Supplementary Figure 2D). In addition, both circulating glucose (Figure 3C) and insulin (Figure 3D) levels were increased by high fat diet feeding to a similar extent in Y1f3.6Cre mice as in their wildtype littermates. However, interestingly and contrary to expectations, while still worsened compared to mice on chow diet, Y1f3.6Cre mice displayed significantly improved glucose tolerance compared to wildtype on a high fat diet despite having impaired glucose tolerance under normal chow conditions (Figure 3E,F). The glucose intolerance in Y1f3.6Cre mice on chow was due to significantly reduced insulin levels. A similar trend towards reduced insulin levels on a high fat diet was seen throughout the glucose tolerance test (Figure 3G,H) however this was not significant. Therefore, Y1f3.6Cre mice on a high fat diet appear to be more responsive to insulin, resulting in improved glucose tolerance as compared to wildtype mice. To further test the insulin sensitivity of these mice on a high fat diet, we performed insulin tolerance tests. As expected, wildtype mice on a high fat diet showed impaired insulin action with elevated glucose levels throughout the insulin tolerance test as compared to chow mice (Figure 3I,J). Remarkably however, despite elevated basal glucose levels, Y1f3.6Cre mice were completely protected against high fat diet induced insulin resistance (Figure 3I,J).
Figure 3.
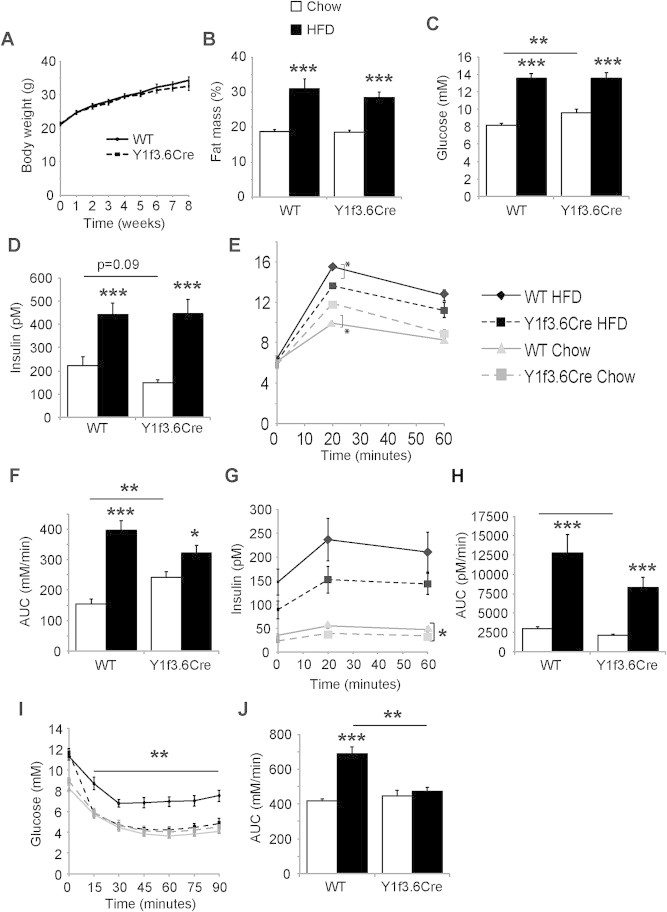
Osteoblast specific Y1 deletion is protective against high fat diet induced glucose intolerance and insulin resistance. Y1F3.6Cre mice fed a high fat diet are not different in terms of bodyweight compared to controls (A). The two groups also similar increases in fat mass (B), Glucose (C) and insulin levels (D). While overall glucose tolerance has been reduced under high fat conditions in both groups, the effect is much less severe in the Y1F3.6Cre mice, which show significant improved glucose tolerance compared to controls (E) and AUC (F). The rise in insulin levels during GTT were also lower in Y1F3.6Cre mice (G) also reflected in AUC (H). Importantly, insulin action was strongly improved in the Y1F3.6Cre mice fed a high fat diet as shown by the significantly reduced glucose levels during insulin tolerance test (I) and AUC (J). Data are means ± SEM of 5–12 mice per group.* = p < 0.05, ** = p < 0.01, *** = p < 0.001 versus control or as indicated.
Furthermore, DXA analysis was used to measure bone mineral density (BMD) and bone mineral content (BMC) at the start and the end of the high fat diet feeding period. Interestingly, despite similar body weights (Figure 3A), mice lacking osteoblastic Y1 receptors had a significantly greater ability to increase whole body BMD and BMC over the high fat fed period (Figure 4A). This led to Y1f3.6Cre mice on high fat diet having significantly increased whole body BMD (Figure 4B) and BMC (Figure 4C) compared to mice on a chow diet whereas wildtype mice had only a trend towards increased BMC with no change in BMD. These changes were also evident when DXA was used to measure BMD (Figure 4D) and BMC (Figure 4E) in isolated femora. Histomorphometric analysis revealed that Y1f3.6Cre mice on a high fat diet retain their high bone mass phenotype with elevated trabecular bone volume (Figure 4F), trabecular thickness (Figure 4G), trabecular number (Figure 4H) and mineral apposition rate (Figure 4I) compared to wildtype mice on a high fat diet. In addition, they displayed a significant increase in trabecular thickness compared to chow Y1f3.6Cre mice (Figure 4G) with no drop in trabecular number as occurred in wildtype mice (Figure 4H). In addition, both Y1f3.6Cre and wild type mice displayed an elevated osteoclast surface on a high fat diet, however, the pattern of reduced osteoclast surface and osteoclast number in Y1f3.6Cre mice remained unchanged by high fat diet (Figure 4I,J). These data suggest that specifically targeting bone Y1 receptors may be a useful therapeutic means of increasing bone formation as well as insulin resistance in overweight patients including those with type 2 diabetes.
Figure 4.
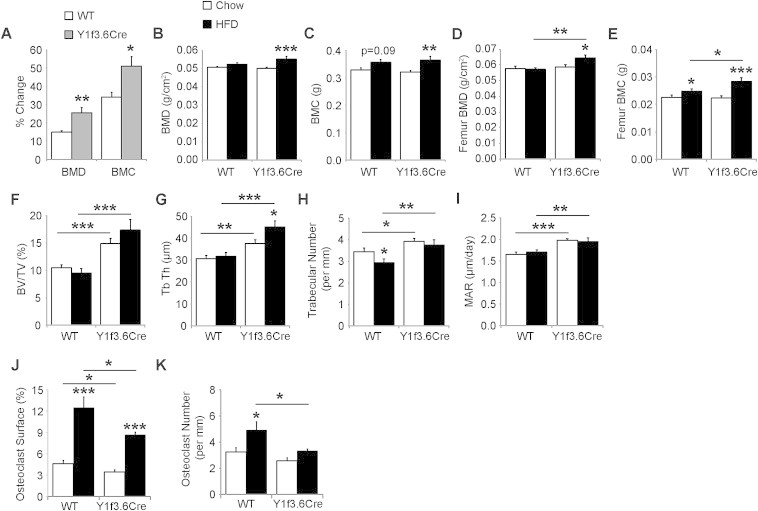
Osteoblast specific Y1 deletion promotes high fat diet induced increases in bone mass. When fed a high fat diet Y1F3.6Cre mice show a significantly increased BMD and BMC over the 8 week treatment period (A). Both, whole body and isolated femur BMD (B,D) as well as BMC (C,E) were also significantly increased in the Y1F3.6Cre mice over controls on the same diet. Y1f3.6Cre mice on a high fat diet also display a significant increase in cancellous bone volume (BV/TV) (F) associated with significant increases in trabecular thickness (Tb Th) (G) but not trabecular number (H). This high bone mass phenotype is due to significantly increased osteoblast activity as shown by significantly increased mineral apposition rate (MAR) (I). Y1f3.6Cre mice fed a high fat diet display significantly decreased osteoclast surface (J), and osteoclast number (K). Data are means ± SEM of 6–8 mice per group.* = p < 0.05, ** = p < 0.01, *** = p < 0.001 versus control or as indicated.
3.4. Reinstating early osteoblastic Y1 receptor signalling normalises glucose tolerance and bone mass
To further confirm that the alterations in insulin production and glucose tolerance seen in Y1f3.6Cre mice are a direct result of the lack of Y1 signalling in osteoblasts, we reinstated osteoblastic Y1 receptors in Y1f3.6Cre mice by generating bone marrow chimeric mice. Y1f3.6Cre mice were lethally irradiated and then reconstituted with bone marrow taken from wildtype donors. As a control, wildtype littermates were also irradiated and reconstituted with wildtype bone marrow. Mice were left to recover for 7 weeks and were then tested for insulin and glucose tolerance. Osteoblastic Y1 receptor deletion had no effect on the response to insulin tolerance tests under chow conditions (Figure 2J) and as expected, reinstating osteoblastic Y1 receptor signalling also had no effect on insulin action (Figure 5A). Importantly however, reconstituting Y1f3.6Cre mice with wildtype bone marrow normalised their glucose and insulin levels throughout the glucose tolerance test (Figure 5B,D) and also when expressed as area under the curve (Figure 5C,E). Mice were culled 3 months following reconstitution to enable sufficient time to observe changes in bone mass. At this time, levels of circulating glucose and insulin were also measured and revealed that reinstating osteoblastic Y1 receptor signalling normalised glucose and insulin levels (Figure 5F,G). Moreover the previously observed difference in pancreas weight and islet size was no longer obvious (data not shown) further confirming that Y1 signalling in osteoblasts critically influences these parameters. Furthermore, the high bone mass phenotype of normal Y1f3.6Cre mice was no longer evident and in fact micro-CT revealed that wildtype-reconstituted Y1f3.6Cre mice had similar trabecular bone mass (Figure 5H) and trabecular number (Figure 5I) with a significant reduction in trabecular thickness (Figure 5J) compared to control mice. A representative image of the effect of reconstituting Y1f3.6Cre mice with wildtype bone marrow is shown in Figure 5K.
Figure 5.
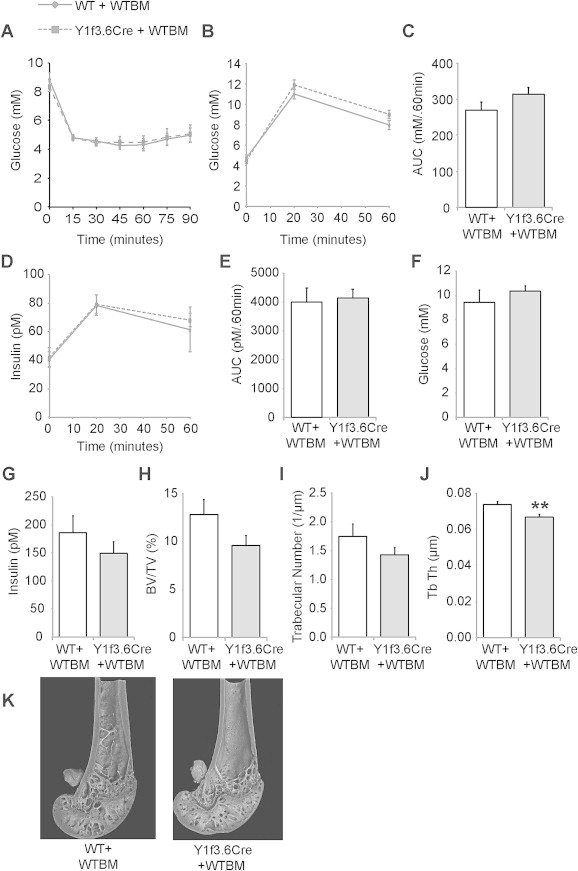
Reinstating early osteoblastic Y1 receptor signalling normalises glucose tolerance and bone mass. Y1F3.6Cre mice which had osteoblastic Y1 receptors reinstated by reconstitution with wildtype bone marrow following lethal irradiation (Y1F3.6Cre + WT) had similar glucose levels throughout an insulin tolerance test (A) to wildtype mice lethally irradiated and reconstituted with wildtype bone marrow (Y1F3.6WT + WT). The 2 groups also displayed similar glucose tolerance with no difference in serum glucose levels (B), glucose area under the curve (C), serum insulin levels (D), or insulin area under the curve (E) during a glucose tolerance test or basal circulating glucose (F) or insulin (G) levels. They also displayed similar femoral trabecular bone volume (H) and trabecular number (I) whilst Y1F3.6Cre + WT mice had a significant reduction in trabecular thickness compared to controls (J) as determined by microCT analysis. A representative picture of a femur with reinstated osteoblastic Y1 receptors is shown in (K). Data are means ± SEM of 7–9 mice per group. ** = p < 0.01 versus control or as indicated.
3.5. Osteoblastic Y1 receptor signalling is mediating insulin production independently of osteocalcin
Insulin is known to act on osteoblasts and amongst other functions has been shown to stimulate osteocalcin production and increase the release of osteocalcin from these cells [3,22]. Conversely, lack of osteocalcin in mice has been reported to affect insulin production and release from the pancreas indicating a feedback loop between these two organs [23]. It is therefore possible that the reduction in pancreatic insulin production observed in Y1f3.6Cre mice is due to decreased circulating osteocalcin as a result of decreased bone resorption.
Indeed, as shown in Figure 6A, qPCR analysis revealed a small but significant increase in Esp expression in the bones of Y1f3.6Cre mice, a gene which has been shown to negatively regulate the decarboxylation of osteocalcin by inhibiting the dephosphorylation of the insulin receptor resulting in increased levels of Foxo1, OPG and subsequently, inhibiting bone resorption [22]. However, mRNA levels of Foxo1 and OPG as well as osteocalcin itself were not different from wildtype levels and, as shown in Figure 6B, serum levels of osteocalcin were not significantly altered in Y1f3.6Cre mice. Therefore, these data suggest that the lack of Y1 receptor signalling in osteoblasts may be regulating pancreas function and related metabolic parameters in a novel and osteocalcin-independent manner. Indeed, osteocalcin deficiency has been previously described to exert additional effects on adiposity and insulin sensitivity through modulating adiponectin levels [4,23], effects which are not evident in these mice, consistent with a non-osteocalcin mediated response. Moreover, the deletion of Y1 receptors from late osteoblast, those which are actively producing osteocalcin, using the 2.3 Kb fragment of the α1(I)-collagen gene to drive Cre expression, did not alter glucose homeostasis, reinforcing the presence of a novel, non-osteocalcin mediated pathway, as suggested previously [5].
Figure 6.
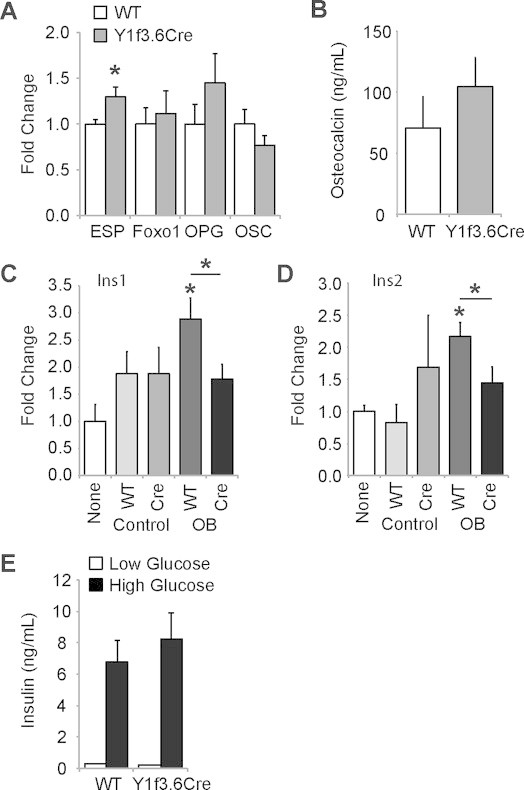
Osteoblastic specific Y1 receptor deletion mediates insulin production independently of osteocalcin. Quantitative real-time PCR expression of ESP, Foxo1, OPG and osteocalcin (OSC) genes in femoral RNA from Y1F3.6Cre and control mice (A). Serum osteocalcin levels in Y1F3.6Cre and control mice (B). Quantitative real-time PCR expression of Ins1 (C) and Ins2 (D) genes in MIN6 cells cultured with the addition of supernatant from wildtype or Y1F3.6Cre bone marrow stromal cells cultured under control or osteogenic (OB) conditions. Glucose-stimulated insulin secretion is unaltered in islets isolated from Y1F3.6Cre mice compared to wildtype (E). Data are means ± SEM of 5–6 mice per group (A), 5-9 mice per group (B), 3 mice per group (C–D), 7 mice per group across 2 experiments (E). * = p < 0.05 versus control or as indicated.
In order to more clearly determine whether the altered insulin production observed in Y1f3.6Cre mice is due directly to altered osteoblast signalling or is a result of reduced bone resorption, in vitro analyses involving cultured bone marrow stromal cells and MIN6 cells, a beta cell line producing insulin, were performed. Bone marrow stromal cells from Y1f3.6Cre mice and their wildtype littermates were isolated and induced to differentiate in vitro into osteoblasts at which point the supernatant was transferred onto MIN6 cells. After 4 h of treatment, RNA was harvested from the MIN6 cells and analysed for the expression of insulin. It has previously been shown that treatment of MIN6 cells for 4 h with osteocalcin results in a significant increase in the expression of the Ins1 and Ins2 mRNA [4]. Interestingly, whilst treatment with supernatant obtained from wildtype osteoblastic cultures resulted in a significant increase in the expression of both Ins1 (Figure 6C) and Ins2 (Figure 6D) in MIN6 cells, no such response in either gene product was observed when the MIN6 cells were treated with supernatant obtained from Y1f3.6Cre osteoblastic cultures (Figure 6C,D). In addition, no effect was observed when MIN6 cells were treated with supernatant from either wildtype or Y1f3.6Cre bone marrow stromal cells cultured under control conditions (Figure 6C,D). Importantly, islet cells isolated from pancreatic tissue of Y1f3.6Cre mice and their wildtype littermates were assessed for their ability to produce insulin in vitro and changes in their mRNA levels of principal ®-cell transcription factors, insulin secretion and glucose metabolism genes and stress genes. As can be seen in Figure 6E, islet cells from Y1f3.6Cre mice had a similar ability to produce insulin in vitro under both low glucose and high glucose conditions, 24 h after isolation. Moreover, the expression of (-cell enriched genes was preserved (Supplementary Figure 3A–L) and stress and pro-apoptotic genes were unchanged (Supplementary Figure 3M–P). Therefore, taken together, these data suggest that the reduction in islet size and reduced insulin production observed in Y1f3.6Cre mice is due to altered secretion of a factor, or several factors, regulated by Y1 receptor signalling in osteoblasts and not due to an intrinsic alteration of beta cell function.
4. Discussion
The findings presented here demonstrate that NPY signalling via osteoblastic Y1 receptors is not only important for the control of bone mass but also contributes significantly to the control of insulin secretion and glucose homeostasis in mice through factor(s) which are different from the previously implicated osteocalcin.
Utilising conditional Y1 knockout mice in which the Y1 receptor was deleted at an early stage of osteoblast differentiation revealed that these mice display high bone mass but also impaired glucose tolerance due to reduced insulin secretion, with a significant reduction in pancreatic insulin content as measured by islet number and β-cell area. Importantly, reinstating osteoblastic Y1 receptor signalling into Y1f3.6Cre mice by bone marrow transplantation was sufficient to restore glucose tolerance and normalise bone mass confirming the osteoblastic nature of this interaction. Importantly, our observation that the effect of osteoblastic Y1 receptor deletion on insulin production and glucose homeostasis is not evident when the Y1 receptor is deleted solely in mature osteoblasts [9] indicates that differential expression of factors during proliferation and commitment of cells at an early stage of osteoblastic differentiation are critical for this interaction. Importantly, the only factor previously implicated with a possible link between osteoblasts and islet function, osteocalcin, was not significantly altered in our model. Furthermore, analysis of the expression of factors previously linked to regulation of glucose homeostasis by osteocalcin, with the exception of Esp [23], were not significantly changed in the osteoblasts of the Y1f3.6Cre mice. The lack of involvement of any of these factors in our model of osteoblastic Y1 deficiency, indicate that insulin and glucose homeostasis in Y1f3.6Cre mice are regulated independent of osteocalcin signalling, and this is rather due to a direct effect of the secretion of another factor(s). While the identity and nature of this factor(s) is still unknown, this study highlights that a more complex interaction between bone and pancreas exist and that this may be linked to developmental stages of osteoblast differentiation.
The potential negative effect of obesity upon bone mass and strength is an area of increasing concern, heightened by the acceleration of rates for obesity. Whilst weight has a positive effect upon bone mass, obesity has been shown to be associated with significant bone loss amongst adults [24] and adolescents [25]. While a number of mechanisms have been developed, the detailed processes are not well understood [26]. However, it is emerging that adipose tissue itself attenuates the positive effects of weight on bone mass [27]. Interestingly, when fed a high fat diet, Y1f3.6Cre mice accrued adipose tissue slightly less compared to wildtype, however, they retained their high bone mass phenotype and furthermore were able to increase their bone mass in line with their higher body weight, whilst this weight-induced bone accrual was markedly attenuated in wildtype mice. Interestingly, despite impaired glucose tolerance on a chow diet, Y1f3.6Cre mice on a high fat diet actually displayed improved glucose tolerance and similar insulin sensitivity to that of chow fed mice. This increase in insulin levels is reflected in elevation in osteoclast surface in the high fat fed groups [28], a likely aid to greater bone accrual during to development of obesity under high fat feeding. These could suggest that under conditions of high fat diet the expression and/or release of potential novel factors from osteoblasts are altered which subsequently could control insulin action in a favourable way.
The research described here explores one of the most novel and interesting recent developments in bone biology, the identification of a regulatory axis from the osteoblast to the pancreatic β-cell. The findings provide critical new mechanistic details that to date have been lacking on how NPY signalling through osteoblastic Y1 receptors is controlling bone formation as well as controlling the release of factor(s) secreted by osteoblasts that communicates with pancreatic β-cells as well as co-ordinately acts with insulin to regulate glucose homeostasis. The identification of a signalling pathway originating from the early osteoblast, which powerfully modulates the glucose economy provides novel insight into the complexity of the interrelationship between bone and glucose homeostasis.
Conflict of interest
None of the authors has a conflict of interest.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
Supplementary Figure 1.
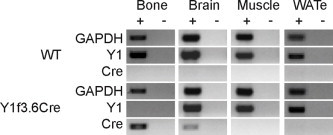
Y1 receptor deletion in early osteoblast differentiation. Cre expression in bone tissue isolated from Y1F3.6Cre mice but not wildtype littermates results in a down regulation of Y1 receptor expression. No Cre expression or alteration in Y1 receptor expression is found in brain, muscle or epididymal white adipose tissue (WATe) of either genotype. Data show results for PCR performed with (+) and without (−) reverse transcription. PCR was performed for 22–26 cycles for GAPDH, 32–26 cycles for Y1 receptor, and 36–40 cycles for Cre varying between tissue types but consistent across genotypes.
Supplementary Figure 2.
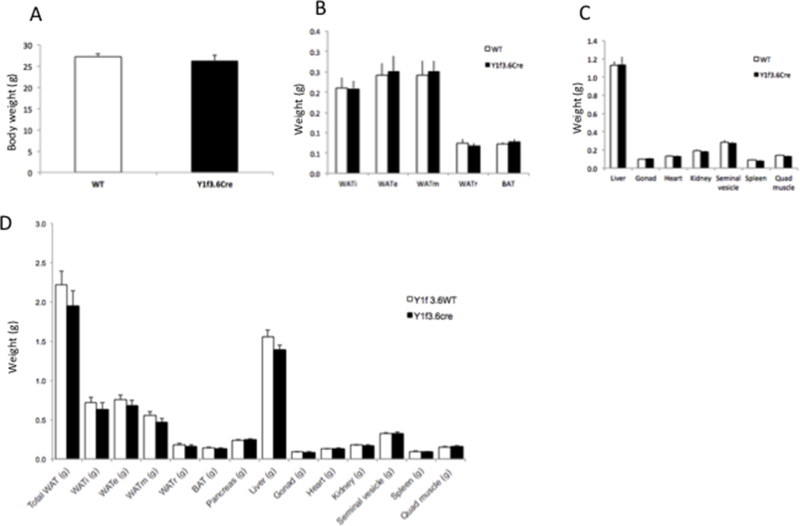
Isolated tissue weights from control and Y1f3.6Cre mice fed a chow diet high fat. Body weight of control and Y1f3.6Cre mice fed a chow diet at 18 weeks of age (A). Weights of isolated different adipose tissues including individual epididymal (WATe), ingunal (WATi), retroperitoneal (WATr) and mesenchymal (WATm), the sum of all with adipose tissues and brown adipose tissue (BAT) (B). Weights of individual organs including liver, gonads, heart, kidney, seminal vesicle, spleen and quadriceps muscle (C). Weights of individual adipose tissue depots and organs of mice fed a high fat diet (D).
Supplementary Figure 3.
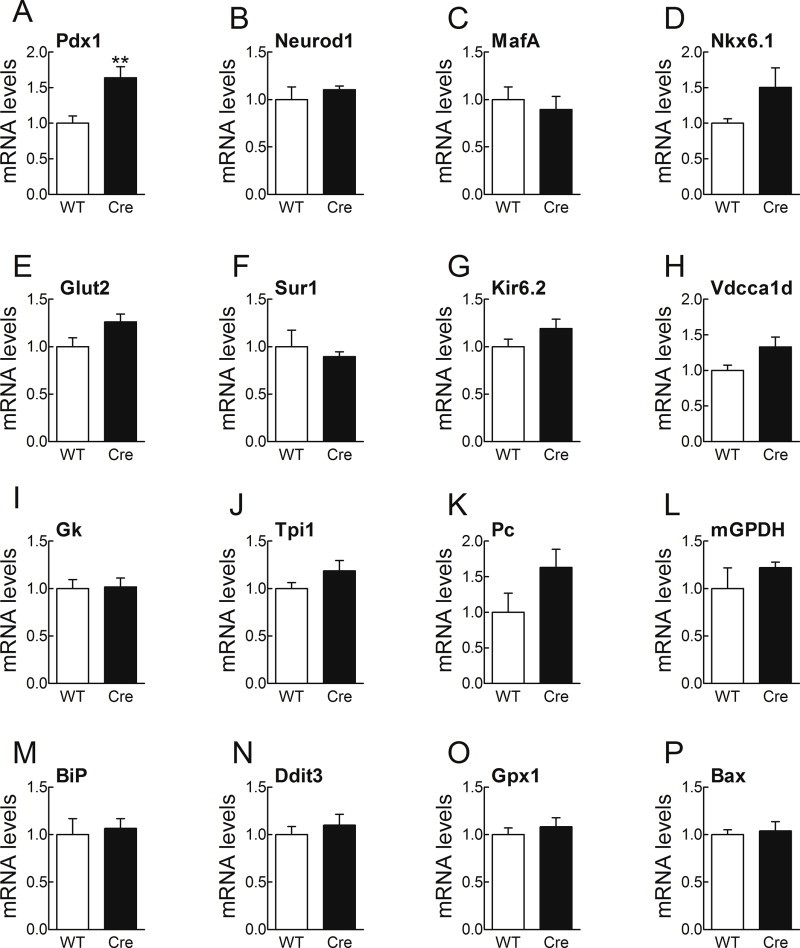
Islets of Y1f3.6Cre mice display a normal differentiated phenotype. The mRNA levels of principal ®-cell transcription factors (A–D) and key insulin secretion and glucose metabolism genes (E–L) were preserved in the islets of Y1f3.6Cre mice in comparison to wild type islets. Furthermore, stress response and pro-apoptotic genes were unchanged (M–P). mRNA levels were assessed by real-time RT-PCR. The value obtained for each specific gene product was normalised to the control gene cyclophilin A and expressed as a fold change of the value in control condition. Primer sequences are listed in Supplementary Table 1. Data are means ± SEM of 5 mice per group. ** = p < 0.01 versus control.
References
- 1.Turner N., Kowalski G.M., Leslie S.J., Risis S., Yang C., Lee-Young C.R.S. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia. 2013;56:1638–1648. doi: 10.1007/s00125-013-2913-1. [DOI] [PubMed] [Google Scholar]
- 2.Wang F., Mullican S.E., DiSpirito J.R., Peed L.C., Lazar M.A. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARgamma. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18656–18661. doi: 10.1073/pnas.1314863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fulzele K., Riddle R.C., DiGirolamo D.J., Cao X., Wan C., Chen D. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. 2010;142:309–319. doi: 10.1016/j.cell.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferron M., Hinoi E., Karsenty G., Ducy P. Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:5266–5270. doi: 10.1073/pnas.0711119105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshikawa Y., Kode A., Xu L., Mosialou I., Silva B.C., Ferron M. Genetic evidence points to an osteocalcin-independent influence of osteoblasts on energy metabolism. Journal of Bone and Mineral Research. 2011;26:2012–2025. doi: 10.1002/jbmr.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin E.J., Sainsbury A., Lee N.J., Boey D., Couzens M., Enriquez R. Combined deletion of Y1, Y2, and Y4 receptors prevents hypothalamic neuropeptide Y overexpression-induced hyperinsulinemia despite persistence of hyperphagia and obesity. Endocrinology. 2006;147:5094–5101. doi: 10.1210/en.2006-0097. [DOI] [PubMed] [Google Scholar]
- 7.Baldock P.A., Sainsbury A., Couzens M., Enriquez R.F., Thomas G.P., Gardiner E.M. Hypothalamic Y2 receptors regulate bone formation. Journal of Clinical Investigation. 2002;109:915–921. doi: 10.1172/JCI14588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan D.G., Kulkarni R.N., Hurley J.D., Wang Z.L., Wang R.M., Ghatei M.A. Inhibition of glucose stimulated insulin secretion by neuropeptide Y is mediated via the Y1 receptor and inhibition of adenylyl cyclase in RIN 5AH rat insulinoma cells. Diabetologia. 1998;41:1482–1491. doi: 10.1007/s001250051095. [DOI] [PubMed] [Google Scholar]
- 9.Lee N.J., Nguyen A.D., Enriquez R.F., Doyle K.L., Sainsbury A., Baldock P.A. Osteoblast specific Y1 receptor deletion enhances bone mass. Bone. 2011;48:461–467. doi: 10.1016/j.bone.2010.10.174. [DOI] [PubMed] [Google Scholar]
- 10.Adeghate E., Ponery A.S., Pallot D.J., Singh J. Distribution of vasoactive intestinal polypeptide, neuropeptide-Y and substance P and their effects on insulin secretion from the in vitro pancreas of normal and diabetic rats. Peptides. 2001;22:99–107. doi: 10.1016/s0196-9781(00)00361-2. [DOI] [PubMed] [Google Scholar]
- 11.Rohner-Jeanrenaud F. Neuroendocrine regulation of nutrient partitioning. Annals of the New York Academy of Sciences. 1999;892:261–271. doi: 10.1111/j.1749-6632.1999.tb07800.x. [DOI] [PubMed] [Google Scholar]
- 12.Katsuura G., Asakawa A., Inui A. Roles of pancreatic polypeptide in regulation of food intake. Peptides. 2002;23:323–329. doi: 10.1016/s0196-9781(01)00604-0. [DOI] [PubMed] [Google Scholar]
- 13.Bottcher G., Ahren B., Lundquist I., Sundler F. Peptide YY: intrapancreatic localization and effects on insulin and glucagon secretion in the mouse. Pancreas. 1989;4:282–288. [PubMed] [Google Scholar]
- 14.Igwe J.C., Jiang X., Paic F., Ma L., Adams D.J., Baldock P.A. Neuropeptide Y is expressed by osteocytes and can inhibit osteoblastic activity. Journal of Cellular Biochemistry. 2009;108:621–630. doi: 10.1002/jcb.22294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee N.J., Doyle K.L., Sainsbury A., Enriquez R.F., Hort Y.J., Riepler S.J. Critical role for Y1 receptors in mesenchymal progenitor cell differentiation and osteoblast activity. Journal of Bone and Mineral Research. 2010;25:1736–1747. doi: 10.1002/jbmr.61. [DOI] [PubMed] [Google Scholar]
- 16.Baldock P.A., Lee N.J., Driessler F., Lin S., Allison S., Stehrer B. Neuropeptide Y knockout mice reveal a central role of NPY in the coordination of bone mass to body weight. PLoS One. 2009;4:e8415. doi: 10.1371/journal.pone.0008415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baldock P.A., Sainsbury A., Allison S., Lin E.J., Couzens M., Boey D. Hypothalamic control of bone formation: distinct actions of leptin and y2 receptor pathways. Journal of Bone and Mineral Research. 2005;20:1851–1857. doi: 10.1359/JBMR.050523. [DOI] [PubMed] [Google Scholar]
- 18.Zhang L., Lee N.J., Nguyen A.D., Enriquez R.F., Riepler S.J., Stehrer B. Additive actions of the cannabinoid and neuropeptide Y systems on adiposity and lipid oxidation. Diabetes, Obesity and Metabolism. 2010;12:591–603. doi: 10.1111/j.1463-1326.2009.01193.x. [DOI] [PubMed] [Google Scholar]
- 19.Lundberg P., Allison A.J., Lee N.J., Baldock P.A., Brouard N., Rost S. Greater bone formation of Y2 knockout mice is associated with increased osteoprogenitor numbers and altered Y1 receptor expression. Journal of Biological Chemistry. 2007;282:19082–19091. doi: 10.1074/jbc.M609629200. [DOI] [PubMed] [Google Scholar]
- 20.Liu F., Woitge H.W., Braut A., Kronenberg M.S., Lichtler A.C., Mina M. Expression and activity of osteoblast-targeted Cre recombinase transgenes in murine skeletal tissues. International Journal of Developmental Biology. 2004;48:645–653. doi: 10.1387/ijdb.041816fl. [DOI] [PubMed] [Google Scholar]
- 21.Baldock P.A., Allison S.J., Lundberg P., Lee N.J., Slack K., Lin E.J. Novel role of Y1 receptors in the coordinated regulation of bone and energy homeostasis. Journal of Biological Chemistry. 2007;282:19092–19102. doi: 10.1074/jbc.M700644200. [DOI] [PubMed] [Google Scholar]
- 22.Ferron M., Wei J., Yoshizawa T., Del Fattore A., DePinho R.A., Teti A., Ducy P. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 2010;142:296–308. doi: 10.1016/j.cell.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee N.K., Sowa H., Hinoi E., Ferron M., Ahn J.D., Confavreux C. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–469. doi: 10.1016/j.cell.2007.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nielson C.M., Srikanth P., Orwoll E.S. Obesity and fracture in men and women: an epidemiologic perspective. Journal of Bone and Mineral Research. 2012;27:1–10. doi: 10.1002/jbmr.1486. [DOI] [PubMed] [Google Scholar]
- 25.Dimitri P., Wales J.K., Bishop N. Fat and bone in children: differential effects of obesity on bone size and mass according to fracture history. Journal of Bone and Mineral Research. 2010;25:527–536. doi: 10.1359/jbmr.090823. [DOI] [PubMed] [Google Scholar]
- 26.Cao J.J. Effects of obesity on bone metabolism. Journal of Orthopaedic Surgery and Research. 2011;6:30. doi: 10.1186/1749-799X-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho-Pham L.T., Nguyen U.D., Nguyen T.V. Association between lean mass, fat mass, and bone mineral density: a meta-analysis. Journal of Clinical Endocrinology & Metabolism. 2014;99:30–38. doi: 10.1210/jc.2014-v99i12-30A. [DOI] [PubMed] [Google Scholar]
- 28.Wei J., Ferron M., Clarke C.J., Hannun Y.A., Jiang H., Blaner W.S. Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. Journal of Clinical Investigation. 2014;124:1–13. doi: 10.1172/JCI72323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.