

Abstract
Objective
Elevated very low-density lipoprotein (VLDL)-triglyceride (TG) secretion from the liver contributes to an atherogenic dyslipidemia that is associated with obesity, diabetes and the metabolic syndrome. Numerous models of obesity and diabetes are characterized by increased central nervous system (CNS) neuropeptide Y (NPY); in fact, a single intracerebroventricular (icv) administration of NPY in lean fasted rats elevates hepatic VLDL-TG secretion and does so, in large part, via signaling through the CNS NPY Y1 receptor. Thus, our overarching hypothesis is that elevated CNS NPY action contributes to dyslipidemia by activating central circuits that modulate liver lipid metabolism.
Methods
Chow-fed Zucker fatty (ZF) rats were pair-fed by matching their caloric intake to that of lean controls and effects on body weight, plasma TG, and liver content of TG and phospholipid (PL) were compared to ad-libitum (ad-lib) fed ZF rats. Additionally, lean 4-h fasted rats with intact or disrupted hepatic sympathetic innervation were treated with icv NPY or NPY Y1 receptor agonist to identify novel hepatic mechanisms by which NPY promotes VLDL particle maturation and secretion.
Results
Manipulation of plasma TG levels in obese ZF rats, through pair-feeding had no effect on liver TG content; however, hepatic PL content was substantially reduced and was tightly correlated with plasma TG levels. Treatment with icv NPY or a selective NPY Y1 receptor agonist in lean fasted rats robustly activated key hepatic regulatory proteins, stearoyl-CoA desaturase-1 (SCD-1), ADP-ribosylation factor-1 (ARF-1), and lipin-1, known to be involved in remodeling liver PL into TG for VLDL maturation and secretion. Lastly, we show that the effects of CNS NPY on key liporegulatory proteins are attenuated by hepatic sympathetic denervation.
Conclusions
These data support a model in which CNS NPY modulates mediators of hepatic PL remodeling and VLDL maturation to stimulate VLDL-TG secretion that is dependent on the Y1 receptor and sympathetic signaling to the liver.
Keywords: NPY Y1 receptor, Triglyceride, Phospholipid, Lipin-1, Sympathetic denervation, VLDL
Abbreviations: VLDL, very low-density lipoprotein; TG, triglyceride; CNS, central nervous system; NPY, neuropeptide Y; POMC, proopiomelanocortin; icv, intracerebroventricular; SNS, sympathetic nervous system; FFA(s), free fatty acid(s); ApoB, apolipoprotein B; ER, endoplasmic reticulum; PL, phospholipid; ARF-1, ADP-ribosylation factor-1; PLD, phospholipase D; PA, phosphatidic acid; AGPAT, 1-acyl-glycerol-3-phosphate acyltransferase; PAP-1, phosphatidic acid phosphatase-1; DAG, diacylglycerol; DGAT, diacylglycerol acyltransferase; SCD-1, stearoyl-CoA desaturase-1; ZF, Zucker fatty; ad-lib, ad-libitum; PF, pair-fed; Veh, vehicle; Sham, sham-denervation; Sx, sympathetic denervation; NE, norepinephrine; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HDAC-1, histone deacetylase-1; RT-PCR, real-time PCR; RPL13A, ribosomal protein L13a; Nuc, nuclear; Cyto, cytoplasmic
1. Introduction
Overproduction of very low-density lipoprotein (VLDL)-triglyceride (TG) from the liver contributes to an atherogenic dyslipidemia that is associated with obesity, diabetes, and the metabolic syndrome [1], [2], [3], and is an increasingly recognized component of cardiovascular risk [2]. While peripheral factors, such as visceral adiposity and insulin resistance [4], [5], [6], clearly contribute to this disorder, mounting evidence points to central mechanisms contributing to these endemic diseases. We hypothesize, therefore, that regulation of hepatic lipid homeostasis is normally subject to a degree of central nervous system (CNS) input, and dyslipidemia may arise from defective CNS control.
Neuropeptide Y (NPY) expressing neurons are highly concentrated in the mediobasal hypothalamus and have important effects on feeding and energy homeostasis [7], [8], [9], [10], [11]. NPY neurons are increasingly recognized to have an additional role in the regulation of hepatic lipid [12], [13], [14] and glucose metabolism [12], [15], [16], [17], [18]. Elevated hypothalamic NPY tone (with a concomitant reduction of proopiomelanocortin (POMC) tone) is associated with obesity and diabetes in rodent models [19], [20] and humans [21], [22], [23] due to defects in inhibitory feedback signaling to the CNS [24], [25]. This elevated NPY tone within these neural circuits may contribute to dyslipidemia independently of positive energy balance and increased visceral adiposity. In fact, a single intracerebroventricular (icv) injection of NPY directly to the third ventricle of the brain rapidly doubles hepatic VLDL-TG secretion in lean fasted rats, and does so, in large part, via the CNS NPY Y1 receptor [13], [14]. Bruinstroop and colleagues [12] recently demonstrated that increases in hypothalamic NPY tone during fasting increases sympathetic nervous system (SNS) outflow to the liver to promote hepatic VLDL-TG secretion. Coupled with this observation, we [13], [14] and others [12] have shown that CNS NPY increases hepatic VLDL-TG secretion independent of changes in levels of plasma free fatty acids (FFAs) and glucoregulatory hormones that are known to modulate VLDL production (reviewed in Ref. [26]). Thus, our overarching hypothesis is that elevated CNS NPY action contributes to dyslipidemia by activating central circuits that modulate liver lipid metabolism. Therefore, we sought to identify novel hepatic mechanisms by which NPY promotes VLDL particle maturation and secretion.
The assembly and secretion of TG-rich VLDL is a tightly regulated and complex process (reviewed in Ref. [27], [28]). First, a small quantity of TG is assembled onto the structural protein apolipoprotein B (apoB) in the rough endoplasmic reticulum (ER) through the action of microsomal triglyceride transfer protein [27]. This process is accompanied by the acquisition of a phospholipid (PL) monolayer to encase the TG core, which results in the formation of a smaller-sized, immature, pre-VLDL lipoprotein. Next, via a less well characterized step, TG is added to the apoB-containing pre-VLDL to form a mature VLDL particle [27], [29]. Increasing evidence suggests that TG utilized for VLDL assembly and secretion can originate from sources other than hepatic de novo fatty acid synthesis and extrahepatic FFAs [28], [30]. Indeed, it has been reported that a substantial proportion of TG that ends up in VLDL particles is derived from a pool of intracellular PL [31]. The implication is that the formation of the lipid core of VLDL, which is required for its maturation and secretion, involves the remodeling of PL in the microsomal membranes to TG [32], [33].
The process of pre-VLDL maturation and transit to the Golgi apparatus for secretion is dependent on the activity of ADP-ribosylation factor-1 (ARF-1), a member of the Ras superfamily of GTP binding proteins, that is also known to activate phospholipase D (PLD) [32], [34]. PLD, in turn, catalyzes the formation of phosphatidic acid (PA) from phosphatidylcholine [32], [34]. Indeed, agents that inhibit ARF-1 activity (e.g., Brefeldin A) or block the formation of PA generated by PLD suppress VLDL maturation and secretion [32], [35]. The PA generated by PLD action or formed by 1-acyl-glycerol-3-phosphate acyltransferase (AGPAT) via the glycerol phosphate pathway (reviewed in Ref. [36]) is then dephosphorylated by the rate-controlling enzyme phosphatidic acid phosphatase (PAP-1), also known as lipin-1, producing the TG precursor diacylglycerol (DAG) [37], [38]. The addition of a third acyl chain to DAG is catalyzed by diacylglycerol acyltransferase (DGAT), which has been shown to interact with stearoyl-CoA desaturase-1 (SCD-1) in the ER membrane to channel monounsaturated fatty acids (MUFA) to DGAT for the final step in TG formation [39]. Additionally, a major product of SCD-1, oleic acid, controls the activity of lipin-1 by promoting its translocation from the cytosol to the ER membrane, to engage its PAP-1 activity [40], [41]. Thus, lipin-1 generated DAG serves as substrate for the synthesis and formation of TG and of PLs (phosphatidylcholine and phosphatidylethanolamine) that are required for lipidation of the nascent VLDL particle resulting in the maturation and secretion of a TG-rich VLDL particle [37], [38].
Altogether, the aforementioned findings suggest the possibility that CNS NPY signaling may modulate VLDL secretion via the control of key regulatory steps involved in hepatic PL remodeling and VLDL maturation. Therefore, we sought to test the hypothesis that elevated CNS NPY signaling increases sympathetic tone to the liver, to modulate key regulatory proteins for a coordinated and multifaceted response to facilitate hepatic PL remodeling and VLDL maturation, thereby increasing VLDL secretion. As a first step to determine whether liver PL stores are related to plasma VLDL-TG concentration, we manipulated plasma TG levels through pair-feeding obese Zucker fatty (ZF) rats (characterized by elevated NPY tone and dyslipidemia [42], [43], [44], [45]). Next, we determined whether the rapid increase in plasma TG levels in response to an acute modulation of CNS NPY signaling in lean fasted rats corresponds to changes in liver PL stores and key regulatory proteins involved in hepatic PL remodeling and VLDL maturation. Then we determined whether this NPY effect could be recapitulated by acute activation of the NPY Y1 receptor with a selective Y1 receptor agonist ([F7, P34]-NPY). Finally, we determined whether CNS NPY modulation of regulatory proteins involved in liver lipid metabolism requires hepatic sympathetic innervation.
2. Material and methods
2.1. Experimental animals
Male rats, Long-Evans (HsdBlu:LE) weighing 250–274 g (Harlan; Indianapolis, IN), Obese, Zucker fatty (ZF; fa/fa) and their lean Zucker controls (fa/+) weighing 300–340 g (Crl:ZUC-Leprfa; Charles River laboratories; Wilmington, MA), and Wistar weighing 280–310 g (Crl:WI; Charles River laboratories) were maintained with free access to water and a standard rodent chow diet under temperature- and humidity-controlled conditions with a 12-h light–dark cycle. Study protocols were approved by the Institutional Animal Care and Use Committee of the Tennessee Valley Veterans Affairs (VA) Healthcare System (Vanderbilt University, Nashville, TN) and the Royal Netherlands Academy of Arts and Sciences (Amsterdam, the Netherlands).
2.2. ZF rat pair-feeding study
To control for hyperphagia and positive energy balance in ad-libitum (ad-lib) chow-fed obese, ZF rats (fa/fa), a group of ZF rats was pair-fed (PF) to the caloric intake of the control lean Zucker rats (fa/+). Body weights were measured daily. At study termination, 30-h fasted rats were euthanized followed by collection of trunk blood and liver samples.
2.3. Intracerebroventricular cannulation
The icv cannulations of Long-Evans rats were performed under inhalational isoflurane anesthesia. Buprenex (0.05 mg/kg body weight) was administered postoperatively to mitigate pain and distress. Each animal received a 22 gauge guide cannula (Plastics One, Roanoke, VA) that was implanted into the third cerebral ventricle at stereotaxic coordinates: midline, 2.2 mm posterior to Bregma and 7.5 mm below the skull surface based on the rat brain atlas of Paxinos and Watson [46].
2.4. Intracerebroventricular infusions
Studies on Long-Evans rats were performed 5–7 days after surgery when food intake and body weight curves returned to a pre-surgery trajectory. NPY was purchased from GenScript (human; Piscataway, NJ). A selective agonist for the NPY Y1 receptor, [F7, P34]-NPY, was synthesized as described previously in Ref. [47]. Freshly prepared NPY, the NPY Y1 receptor agonist, or vehicle (Veh) consisting of 0.9% normal saline were administered using a (28 gauge) needle that extends 1 mm beyond the tip of the icv guide cannula over a period of 60 s in a final volume of 2 μl. We matched the icv dose of NPY (1 nmol) and the Y1 receptor agonist (1 nmol) for potency to stimulate feeding behavior in ad-lib chow-fed rats to that of the food intake of Veh-treated rats after a 12-h fast, which confirmed that we used physiologically relevant doses in our lipid studies [13].
2.5. NPY and sympathetic denervation experiments
Wistar rats were implanted with a stainless steel guide cannula (22 gauge; Plastics One, Roanoke, VA) into the lateral cerebral ventricle (Bregma: 0.8 mm posterior, 2.0 mm lateral, and 3.2 mm ventral) as previously described in Ref. [15]. Subsequently, the animals received either a control sham-denervation (Sham) or sympathetic denervation (Sx) of the liver according to the methodology described in Ref. [12]. The effectiveness of the hepatic Sx was assessed by measuring norepinephrine (NE) content [12]. A Sx rat was included in the analysis only if the NE content was below 10% of the mean NE concentration in Sham-operated controls. After recovery to pre-surgery body weight and a day before the study, rats were connected and allowed to adapt to the metal collar guiding the icv infusion tubing. The next morning at 8 AM, food was removed. At 12 PM, icv NPY (1 μg/μl, human, rat; Bachem) or Veh (purified water; Milli-Q) was infused for 2-h (bolus 5 μl/5 min, followed by 5 μl/h). At study termination, animals were euthanized and liver samples were collected.
2.6. Lipid assays
Lipids were assayed from plasma with enzymatic colorimetric assays using the following reagent kits: TG and total cholesterol from Raichem (San Diego, CA), non-esterified FFA from Wako Diagnostics (Richmond, VA) and glycerol from Sigma–Aldrich (St. Louis, MO).
2.7. Plasma hormones and metabolites
Plasma levels of insulin and glucagon were quantified by the Vanderbilt Diabetes Center Hormone Assay & Analytical Services Core (Nashville, TN) using radioimmunoassay kits from Millipore (Billerica, MA). Blood glucose concentration was measured using a glucometer from Roche Diagnostics (Accu-Chek Advantage, Indianapolis, IN) on trunk blood at study termination.
2.8. Plasma and hepatic lipid content
Lipids were quantified from plasma and liver tissues as previously described in Ref. [48], extracted using the method of Folch [49], followed by separation by thin layer chromatography and methylated using BF3/methanol as described by Morrison and Smith [50]. The methylated fatty acids were extracted and analyzed by gas chromatography. The lipid profiles were performed in the lipid subcore of the Vanderbilt Diabetes Center Hormone Assay & Analytical Services Core.
2.9. Western blot analysis
Western blots from liver homogenates were probed with the following antibodies using the methodology described in Ref. [13]: from Cell Signaling Technology (Danvers, MA), β-tubulin; from Enzo Life Sciences (Farmingdale, NY), constitutive cytosolic Hsc70 (HSP73); and from Santa Cruz Biotechnology (Dallas, TX), ARF-1, calnexin, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), histone deacetylase-1 (HDAC-1), and lipin-1. Nuclear–cytoplasmic fractionation was conducted using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer's protocol. Calnexin, HDAC-1, and β-tubulin were used as marker proteins of ER membrane, nuclear, and cytoplasmic fractions, respectively. Western blots were analyzed by densitometry utilizing Image J Software (National Institutes of Health).
2.10. RNA isolation and quantitative real-time PCR (RT-PCR)
RT-PCR was performed with IQ SYBR Green Supermix, as previously described in Ref. [13]. Quantification results of RNA of interest were normalized to the housekeeping gene ribosomal protein L13a (RPL13A); for comparative analysis, RNA ratios of the treatment group were normalized to the Veh control group. PCR primer sequences are available upon request.
2.11. Statistical analysis
Data are presented as mean ± SEM. Two-group comparisons were performed using Student's t-test (unpaired, two-tailed) and three-group (or more) comparisons by one-way ANOVA with Bonferroni's multiple comparisons post-test analysis. All analyses were performed using GraphPad Prism, version 6 (GraphPad Software, San Diego, CA). Differences with p < 0.05 were considered statistically significant.
3. Results
3.1. Liver PL levels are tightly associated with plasma TG in ZF rats
As a first step to investigating the hypothesis that liver PL stores may provide the lipid source for hepatic VLDL-TG secretion, we pair-fed one cohort of ZF rats (ZF PF) by carefully matching their caloric intake to that of the lean (fa/+) rats to control for hyperphagia and positive energy balance. Ad-lib fed ZF rats (ZF ad-lib) rapidly developed obesity (Figure 1A; final body weights, 426 ± 8.5 g for ZF ad-lib vs. 390 ± 5.7 g for Control; p < 0.05), whereas pair-feeding (i.e. caloric restriction) successfully matched the body weight of the ZF PF rats to that of lean control animals (Figure 1A; final body weights, ZF PF, 396 ± 2.5 g vs. Control, p = ns). At the end of the study (day 38), ad-lib fed ZF rats developed severe hypertriglyceridemia (456 ± 55 mg/dl), whereas pair-fed ZF rats had plasma TG levels reduced by 84% (Figure 1B). Despite being calorically matched to controls and having the same body weight, TG levels in ZF PF rats remained ∼2.6-fold higher compared with controls (72.2 ± 9.3 mg/dl for ZF PF vs. 28.2 ± 3.8 mg/dl for Control; p < 0.01), suggesting this modest elevation in TG levels in ZF PF rats (relative to lean controls) is independent of positive energy balance and visceral adiposity. These effects were specific to plasma TG since plasma cholesterol levels were not reduced by pair-feeding (Control, 77.8 ± 5.4 mg/dl; ZF ad-lib, 198 ± 8.5 mg/dl; ZF PF, 245 ± 8.9 mg/dl). Although pair-feeding substantially reduced plasma TG concentrations in ZF rats (Figure 1B), it had no effect on liver TG content (Figure 1C), while liver PL content fell to below control levels (Figure 1D; 15.3 ± 0.6 μg/mg liver in ZF PF vs. 26.5 ± 0.8 μg/mg liver in Control, p < 0.001). Thus, hepatic PL (r = 0.95; p = 0.0125; Figure 1F) and not TG (r = −0.088; p = ns; Figure 1E) content tightly correlated with plasma TG levels in ZF PF rats. This observation of a tight correlation between hepatic PL content and plasma TG concentration led us to the hypothesis that liver PL may be an important determinant in VLDL secretion. We next sought to determine whether the hypertriglyceridemia induced by an acute modulation of CNS NPY signaling in lean fasted rats may also correspond to changes in liver PL stores and/or key regulatory proteins involved in hepatic PL remodeling.
Figure 1.
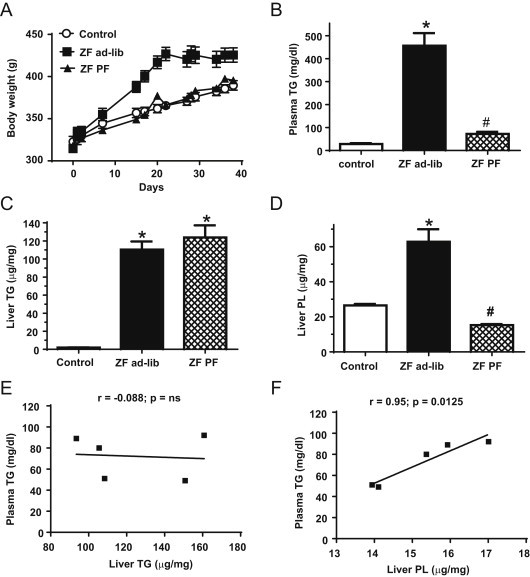
PL and not TG levels in liver are tightly associated with plasma TGs in ZF rats. (A) Chow-fed ZF rats were pair-fed (ZF PF, black triangles) by matching their caloric intake to that of lean controls (Control, white circles) and an effect on daily body weight (n = 5/group) is illustrated in comparison to ad-lib fed ZF rats (ZF ad-lib, black squares). (B–D) Plasma and liver samples were collected from 30-h fasted ad-lib fed ZF (black bars), PF ZF (hatched bars), and lean control (white bars) rats to measure plasma TG (B), liver TG (C) and liver PL (D). Data are presented as mean ± SEM and were analyzed by one way ANOVA; *indicates a significant difference (p < 0.05) between ZF ad-lib or ZF PF vs. Control; #indicates a significant difference (p < 0.05) between ZF ad-lib and ZF PF. Correlational analyses are illustrated for ZF PF rats of plasma TG vs. liver TG levels (E) and of plasma TG vs. liver PL levels (F).
3.2. Acute elevation of CNS NPY via Y1 receptor signaling increases both plasma TG and PL levels without a significant change in total liver TG, FFA or PL content
Previously, we demonstrated that icv administration of NPY or a selective NPY Y1 receptor agonist into the third ventricle of lean rats rapidly increases hepatic VLDL-TG secretion independently of feeding and energy balance [13], [14]. To test the hypothesis that liver PL stores may be tightly associated with plasma TG levels in response to an acute modulation of CNS NPY signaling via the NPY Y1 receptor, lean 4-h fasted rats were given an icv injection of either NPY (1 nmol), a selective Y1 receptor agonist ([F7, P34]-NPY; 1 nmol), or Veh (saline), and at 60 min post-injection, trunk blood and liver samples were collected. Of note, NPY and the Y1 receptor agonist were tested in a separate cohort of animals, and each cohort was compared to its corresponding Veh control group. Both NPY and the Y1 receptor agonist treatment increased plasma TG levels by 2.0- and 1.6-fold, respectively (Figure 2A,B). When FFA composition of plasma TG was analyzed for changes in individual FFA concentration after icv treatment with NPY (Figure 2C) or its Y1 receptor agonist (Figure 2D), most individual FFA concentrations increased, further confirming an increase in plasma TG. In addition, plasma PL increased with NPY and the Y1 receptor agonist treatment (Figure 2A,B), while there was no change in plasma FFA (Figure 2A,B). While we did not detect increases in plasma FFA levels, we did not specifically assess FFA flux. Moreover, consistent with our previous findings [13], this increase in plasma TG in response to icv NPY treatment did not alter blood glucose, plasma insulin or glucagon concentrations (Table 1). Although icv Y1 receptor agonist treatment did not alter plasma glucagon and blood glucose levels, plasma insulin levels were moderately increased (Table 1), suggesting that even in the face of a modest increase in insulin levels, increased central Y1 receptor tone can increase hepatic VLDL-TG secretion.
Figure 2.
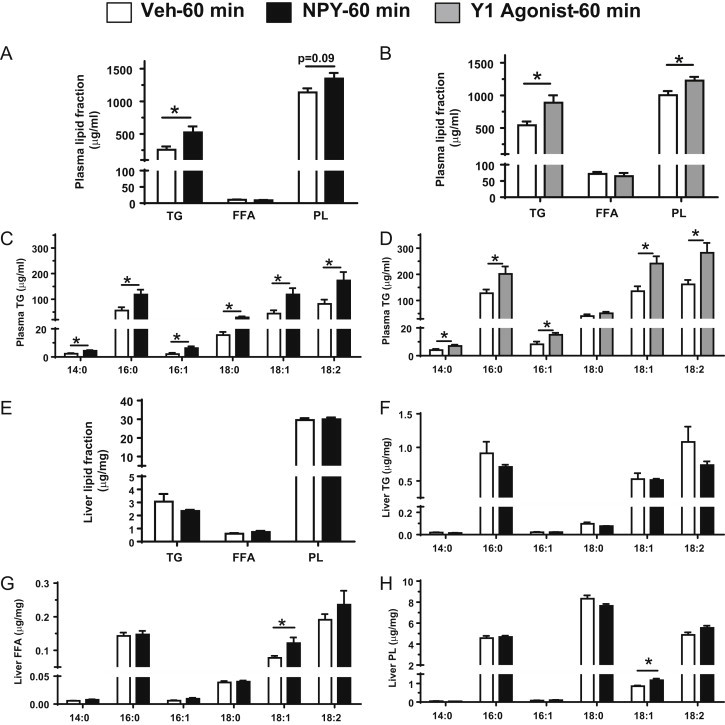
Changes in plasma and liver lipid content in response to CNS NPY and Y1 receptor agonist signaling. Plasma and liver samples from lean 4-h fasted rats (n = 5–6/group) were collected 60 min post-icv injection of either NPY (1 nmol; black bars) or the Y1 receptor agonist [F7, P34]-NPY (1 nmol; gray bars) or Veh (white bars) and were used to measure the following: plasma TG, FFA, and PL content in icv NPY- (A) or Y1 receptor agonist- (B) or Veh-treated rats. FFA composition of plasma TG was analyzed for changes in individual FFA levels of myristic acid (14:0), palmitic acid (16:0), palmitoleic acid (16:1), stearic acid (18:0), oleic acid (18:1), and linoleic acid (18:2) after icv treatment with NPY (C) or the Y1 receptor agonist (D). Levels of total TG, FFA, and PL fractions in liver (E) were measured from icv NPY- or Veh-treated rats. FFA composition of liver TG (F), FFA (G), or PL (H) were analyzed for changes in individual FFA levels of 14:0, 16:0, 16:1, 18:0, 18:1, and 18:2 after icv treatment with NPY. Data are presented as mean ± SEM and were analyzed by Student's t-test (unpaired, two-tailed); *indicates a significant difference (p < 0.05) between icv treatment vs. Veh.
Table 1.
Effects of NPY or the Y1 receptor agonist on glucoregulatory hormones and blood glucose at 60 min post-icv injection.
| NPY |
Y1 agonist |
|||
|---|---|---|---|---|
| Veh | NPY | Veh | Y1 | |
| Insulin (ng/ml) | 0.80 ± 0.2 | 1.18 ± 0.2 | 2.99 ± 0.5 | 5.45 ± 0.4* |
| Glucagon (pg/ml) | 101 ± 13 | 120 ± 13 | 100 ± 10 | 117 ± 10 |
| Blood glucose (mg/dl) | 127 ± 6.0 | 130 ± 9.2 | 153 ± 2.6 | 152 ± 4.0 |
Data are presented as mean ± SEM (n = 6–7/group) and were analyzed by Student's t-test (unpaired, two-tailed); *p < 0.01 for icv treatment vs. Veh comparison.
We then determined the liver content of TG, FFA and PL and found no detectable changes in intrahepatic content of these lipid fractions in icv NPY treated rats relative to Veh controls (Figure 2E). We also measured the abundance of several individual FFAs derived from these hepatic lipid fractions (Figure 2F–H). We found that at 60 min after icv NPY treatment in lean 4-h fasted rats, there was an increase in oleic acid (18:1) within the liver FFA and PL pools (Figure 2G,H) but no detectable increase in the TG pool (Figure 2F). Oleic acid has been shown to promote the final steps in the assembly of VLDL particles in hepatocytes [51].
3.3. An acute increase in CNS NPY signaling via the Y1 receptor modulates SCD-1 expression and activity
SCD-1 desaturates palmitic (16:0) and stearic (18:0) acids to palmitoleic (16:1) and oleic (18:1) acids, respectively, which are the major MUFAs of PLs, TGs, and cholesteryl esters that serve as constituents for the synthesis of lipoproteins [52], [53]. We have previously shown that CNS NPY treatment robustly induces liver SCD-1 mRNA levels at 60 min post-icv injection, an effect that is recapitulated by NPY Y1 receptor activation [13]. A corresponding increase in SCD-1 activity should accompany the observed increase in expression. That being said, a good estimate of in vivo hepatic SCD-1 activity is the desaturation index, which calculates the ratio of SCD-1 products to its precursors [51], [53]. Indeed, icv NPY treatment significantly increased the desaturation index of liver TG, FFA, and PL fractions within 60 min post-injection (Figure 3A) as well as increasing the desaturation index in both plasma TG and PL pools (Figure 3B). Similarly, icv Y1 receptor agonist treatment increased the desaturation index in plasma TGs but no increase was detected in plasma PLs (Figure 3C). As expected, neither icv NPY treatment nor icv Y1 receptor agonist treatment altered the plasma FFA desaturation index (Figure 3B,C). Altogether, these data suggest that an acute increase in CNS NPY signaling increases the enrichment of the liver lipid pools with MUFA through the modulation of SCD-1 expression and activity that would facilitate an increase in VLDL-TG secretion.
Figure 3.
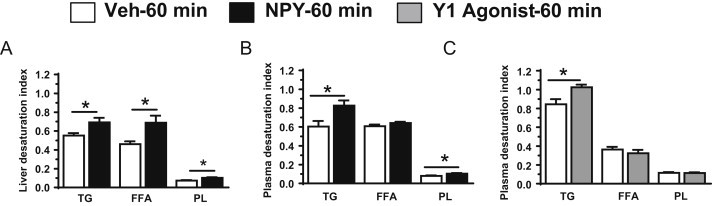
Changes in desaturation index associated FFA, TG, and PL fractions in liver and plasma in response to CNS NPY and Y1 receptor signaling. Plasma and/or liver samples from lean 4-h fasted rats (n = 5–6/group) were collected 60 min post icv injection of NPY (1 nmol; black bars) or the Y1 receptor agonist [F7, P34]-NPY (1 nmol; gray bars) or Veh (white bars). Desaturation index [(16:1 + 18:1)/(16:0 + 18:0)] of TG, FFA, and PL from liver (A) as well as plasma (B) in lean 4-h fasted NPY- (black bars) or Veh-treated (white bars) rats. Desaturation index of plasma TG, FFA, and PL from plasma (C) in lean 4-h fasted Y1 receptor agonist- (gray bars) or Veh-treated (white bars) rats. Data are presented as mean ± SEM and were analyzed by Student's t-test (unpaired, two-tailed); *indicates a significant difference (p < 0.05) between icv treatment vs. Veh.
3.4. CNS NPY signaling via the Y1 receptor modulates ARF-1 and lipin-1 expression
We next sought to investigate changes in expression of two key regulatory proteins, ARF-1 [32], [34] and lipin-1 [38], involved in liver PL remodeling as well as VLDL maturation and secretion. We measured mRNA levels at 60 min and protein levels at 120 min post-injection. This later time point (120 min) was included to facilitate the detection by Western blot analysis of any change in ARF-1 or lipin-1 protein levels, since 120 min post-icv injection of NPY and Y1 receptor agonist also elevated plasma TG levels 2-fold above control values [13]. When compared to Veh, icv NPY or Y1 receptor agonist treatment resulted in an approximate 1.7-fold increase in ARF-1 mRNA levels at 60 min post-treatment (Figure 4A). By 120 min, compared to Veh, NPY treatment resulted in a 3.1-fold increase in ARF-1 protein levels (Figure 4B,C). Lipin-1 mRNA levels were induced 3.8-fold, relative to Veh levels, by icv NPY treatment at 60 min post-injection (Figure 4D), and a 2.3-fold increase in protein levels was detected at 120 min (Figure 4E,F). Lipin-1 protein levels were induced 2.2-fold relative to Veh by 120 min post-icv Y1 receptor agonist treatment (Figure 4E,F). Collectively, these data raise the possibility that increased CNS NPY signaling via the Y1 receptor may promote liver PL remodeling and VLDL maturation to enhance VLDL-TG secretion through the modulation of ARF-1 and lipin-1 expression.
Figure 4.
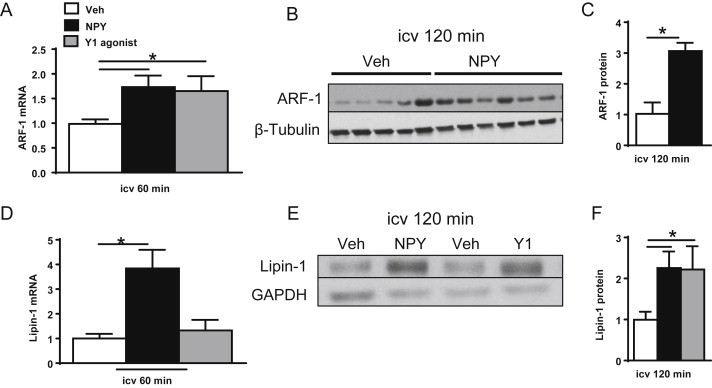
CNS NPY and Y1 receptor signaling promote expression of ARF-1 and lipin-1. The levels of mRNA and protein of ARF-1 and lipin-1, two key regulatory proteins involved in PL remodeling, were assessed from livers of 4-h fasted lean rats (n = 5–7/group) isolated 60 or 120 min after icv treatment with NPY (black bars), Y1 receptor agonist (gray bars) or Veh (white bars). Relative mRNA levels for liver ARF-1 (A) and lipin-1 (D) were obtained by quantitative RT-PCR. Individual mRNA levels for the protein of interest were first normalized to a non-regulated reference RNA, RPL13A, and then normalized to the Veh group for comparative analysis. Protein extracts prepared from livers of 4-h fasted lean rats isolated 120 min after icv injection of NPY or Y1 receptor agonist or Veh were immunoblotted to detect levels of ARF-1 (B and C) and lipin-1 (E and F). Images of Western blots [representative blots shown for ARF-1 (B) and Lipin-1 (E)] were analyzed by densitometry and data shown relative to Veh levels after normalization to loading controls, β-Tubulin or GAPDH. Data are presented as mean ± SEM and were analyzed by Student's t-test (unpaired, two-tailed); *indicates a significant difference (p < 0.05) between icv treatment and Veh.
3.5. CNS NPY signaling alters hepatic lipin-1 subcellular localization
The association of lipin-1 with the ER membrane governs the availability of substrate (PA) for its PAP-1 activity [38], generating DAG for the synthesis of the lipid substrates (TG and PLs) required for VLDL maturation and secretion; therefore, we determined lipin-1 protein content in subcellular fractions of livers from rats treated with either icv NPY (or Veh) at 60 or 120 min post-injection. Since we found that the ER-membrane protein, calnexin, was enriched only in the nuclear (Nuc) and not the cytoplasmic (Cyto) fraction, we denoted the ER fraction as Nuc/ER fraction (Figure 5A). Icv NPY treatment increased lipin-1 protein content by 1.7- and 1.9-fold in the Nuc/ER fraction at 60 and 120 min post-injection, respectively, with a detectable drop in cytoplasmic levels observed at 60 min (Figure 5A,B). The fall in cytoplasmic levels with a corresponding rise in Nuc/ER levels at 60 min suggests the possibility that CNS NPY signaling promotes the translocation and association of lipin-1 with the ER membranes, and, thus, allows for lipin-1 directed remodeling of liver PL for VLDL maturation and secretion.
Figure 5.
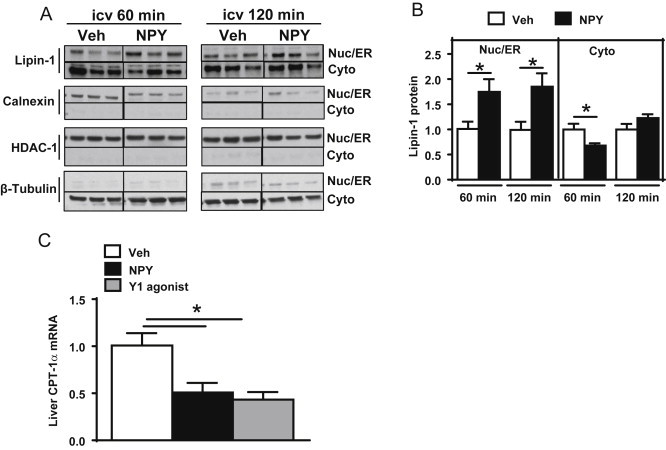
CNS NPY signaling alters lipin-1 subcellular localization. Western blot analysis to detect levels of lipin-1 in nuclear (Nuc)/endoplasmic reticulum (ER) and cytoplasmic (Cyto) fractions (A and B) from livers of 4-h fasted lean rats (n = 5–6/group) isolated 60 or 120 min after icv injection of NPY (black bars) or Veh (white bars). Representative Western blots for lipin-1 are shown (A), as well as for calnexin, HDAC-1, and β-Tubulin, which were used as marker proteins of ER membrane, nuclear, and cytoplasmic fractions, respectively. Western blots were analyzed by densitometry and data shown for lipin-1 are relative to levels of Veh controls after normalization to β-Tubulin or HDAC1 (B). RNA was isolated from livers of lean 4-hour fasted rats (n = 5–6/group) 60 min after icv treatment with NPY (black bars), Y1 receptor agonist (gray bars), or Veh (white bars). Relative mRNA levels for liver CPT-1α (C) were obtained by quantitative RT-PCR and normalized to RPL13A. For comparative analysis, CPT-1α mRNA ratios were normalized to that of the Veh group. Data are presented as mean ± SEM and were analyzed by Student's t-test (unpaired, two-tailed); *indicates a significant difference (p < 0.05) between icv treatment and Veh.
Intriguingly, our finding that lipin-1 protein levels are elevated in the Nuc/ER membrane fraction, raises the possibility that nuclear localized lipin-1 may also be present, which would allow it to act as a transcriptional co-activator with peroxisome proliferator activated receptor-ɣ-coactivator-1α (PGC-1α) and peroxisome proliferator activated receptor-alpha (PPARα) to upregulate genes involved in β-oxidation [54], [55]; this would oppose its function as a glycerolipid biosynthetic enzyme. However, we found that after a 60 min treatment with either icv NPY or Y1 receptor agonist (Figure 5C), mRNA levels of carnitine palmitoyltransferase-1 alpha (CPT-1α), a regulatory protein of FFA oxidation [56], were reduced by 50 or 58%, respectively, suggesting a decrease in β-oxidation that is consistent with previous studies [12], [57]. The mRNA levels of other proteins involved in FFA oxidation, such as PPARα and acyl-CoA oxidase-1, were unaltered with icv NPY treatment (data not shown), which lessens the possibility that nuclear localized lipin-1 was present to promote β-oxidation. Finally, we found that icv NPY treatment did not alter mRNA expression of key hepatic enzymes (data not shown) involved in the glycerol phosphate pathway (DGAT-1, DGAT-2, AGPAT, and GPAT [36]) and the hepatic uptake of fatty acids, Cluster of Differentiation 36 (CD36) [58].
3.6. Hepatic sympathetic denervation attenuates the stimulatory effect of CNS NPY on key liporegulatory proteins
To test our hypothesis that CNS NPY rapidly modulates key liporegulatory proteins via sympathetic inputs to liver, we combined a 2-h icv NPY (or Veh) infusion with either a selective sympathetic denervation (Sx) or Sham denervation of liver in lean 4-h fasted rats. A marked reduction of NE levels in liver confirmed the effectiveness of Sx (Figure 6A). An icv NPY infusion in Sham rats robustly elevated SCD-1 mRNA (Figure 6B) and lipin-1 protein expression (Figure 6C) by 2.6- and 3.4-fold, respectively. These NPY induced levels of SCD-1 and lipin-1 are substantially reduced in livers of Sx rats (Figure 6B,C). Finally, given that the association of lipin-1 with the ER membrane is crucial to engage in its PAP-1 activity, we measured the ER membrane marker calnexin by Western blot and found that sympathetic denervation did not alter protein levels (data not shown). Altogether, these data suggest that CNS NPY stimulates hepatic VLDL-TG secretion at least in part by increasing sympathetic outflow to the liver to rapidly modulate key hepatic liporegulatory proteins.
Figure 6.
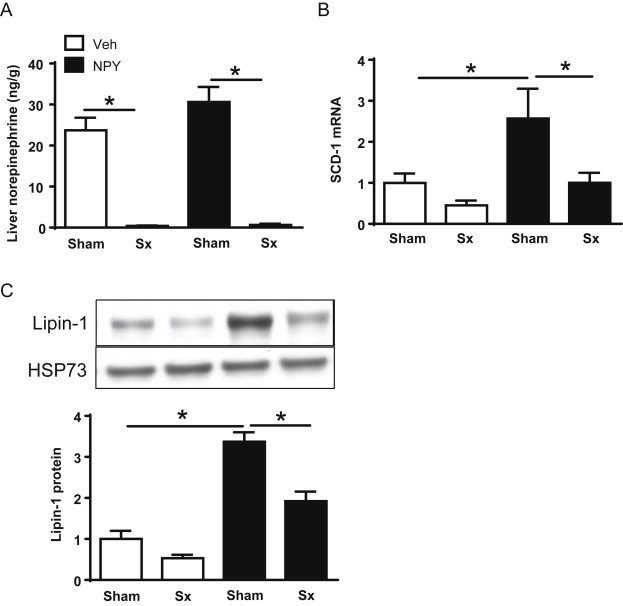
Hepatic sympathetic denervation (Sx) attenuates the stimulatory effect of icv NPY treatment on liver SCD-1 and lipin-1 expression. Lean 4-h fasted Sham or Sx rats received either a 2-h icv infusion of NPY (black bars; n = 5–6/group) or Veh (white bars; n = 6–7/group). Data are presented as mean ± SEM and were analyzed by one-way ANOVA with Bonferroni's post-test analysis; *p < 0.05. (A) Liver norepinephrine levels of all treatment groups are shown. (B) Relative gene expression in liver is shown for SCD-1 mRNA that was assessed by quantitative RT-PCR and normalized to the expression level of a reference gene, RPL13A. For comparative analysis, SCD-1 mRNA ratios were normalized to that of the Sham Veh group. (C) Protein extracts prepared from livers were immunoblotted and analyzed by densitometry to detect expression levels of lipin-1, which were normalized to a loading control, constitutive cytosolic Hsc70 (HSP73). For comparative analysis of lipin-1 expression, levels of each group were normalized to the Sham Veh group.
4. Discussion
Increasing evidence implicates CNS NPY as a regulator of hepatic lipoprotein metabolism [12], [13], [14] beyond its effects on feeding, energy homeostasis [7], [8], [9], [10], [11] and glucose metabolism [12], [15], [16], [17], [18]. Specifically, acute modulation of CNS NPY and NPY Y1 receptor signaling rapidly increases hepatic VLDL-TG secretion in lean, fasted rats, while not altering peripheral factors, glucoregulatory hormones and adipocyte lipolysis [12], [13], [14] that can affect VLDL secretion by liver (reviewed in Ref. [26]). Recent evidence from Bruinstroop and colleagues [12] revealed that central infusion of NPY under postprandial conditions (characterized by low endogenous NPY tone and nearly undetectable chylomicrons in plasma [14]) cannot increase hepatic VLDL-TG secretion in Sx rats. Furthermore, VLDL-TG secretion decreased in overnight fasted Sx rats characterized by a physiologic elevation in hypothalamic NPY tone, whereas parasympathetic denervation had no effect on VLDL-TG secretion [12]. Clearly, this work demonstrates a neural-hepatic circuit mediated by the SNS to control liver lipid metabolism under fasting conditions characterized by elevated CNS NPY tone [12]. We, therefore, sought to identify novel hepatic mechanisms by which CNS NPY promotes VLDL particle maturation and secretion.
VLDL is the chief carrier of TG in the postabsorptive state [30]. The TG utilized for VLDL assembly and secretion can be derived either from fatty acids produced by hepatic de novo lipogenesis (DNL), by the uptake and re-esterification of plasma FFAs, or by the uptake of chylomicron and VLDL lipoprotein remnants by the liver [30]. Using a combination of stable isotope-labeled tracer and indirect calorimetry, Diraison et al. [30] estimated in normal human subjects in the postabsorptive state, that hepatic re-esterification of plasma FFA accounted for 50–55% of TG secretion, whereas DNL was a minor contributor. The remaining lipids were presumed to be provided by stored lipids (TG and PL) or lipoprotein remnants taken up by the liver. Although, the method used by Diraison et al. [30] was semi-quantitative and did not allow for the determination of the overall contribution of these potential lipid sources to TG secretion, it provides suggestive evidence that TGs for VLDL secretion may additionally be derived from PL stores. Furthermore, it has been reported that up to 70% of secreted VLDL-TG by the liver is attributable to the hydrolysis and re-esterification of pre-existing PL and cytosolic TG [31]. This and our current work increase the likelihood that some of the TG which ends up in VLDL is derived from a pool of intracellular PL, a novel concept given the common view that intracellular membrane PL plays mostly a structural role for the assembly of VLDL particles.
Our initial findings that the reduction in plasma TGs that occurred via pair-feeding (i.e. caloric restriction) obese, hyperlipidemic ZF rats correlated with substantially reduced liver PLs (while not altering hepatic TG content), and raised the possibility that liver PLs are a readily mobilized lipid pool for hepatic VLDL-TG secretion. While peripheral factors may be involved, based collectively upon these findings, we hypothesized that elevated CNS NPY tone induces hepatic PL remodeling, generating a local pool of DAG for the generation of TG, as well as PL, both required for the maturation and rapid secretion of VLDL-TG by the liver. We further hypothesized that this process is sustained by hepatic sympathetic innervation.
Although the acute modulation of CNS NPY and NPY Y1 receptor signaling in lean 4-h fasted rats doubled plasma TGs in the absence of changes in intrahepatic lipid (PL, TG, FFA) stores, we found that oleic acid (18:1), a MUFA known to enhance VLDL secretion [38], [51], was not only enriched in secreted TGs but in liver FFA and PL fractions as well. Moreover, the enrichment of MUFA in liver and plasma lipids in icv NPY and Y1 receptor agonist treated rats was attributed, in part, to the rapid induction of liver SCD-1 expression and activity and the concomitant attenuation of CPT-1α expression (the key protein involved in hepatic fat oxidation). Clearly, this NPY effect on liver is contrary to the normal effects of fasting, in which MUFAs are preferentially oxidized [59], [60]. In support of our observation, Zhang et al. [57] found that Y1 receptor null mice had greater utilization of lipid as an oxidative fuel source. This most likely involved increases in liver and muscle CPT-1 protein levels as well as increases in the activity of enzymes involved in β-oxidation, suggesting that Y1-receptor-signaling might control mitochondrial capacity for FFA transport and oxidation. Lastly, icv NPY and the Y1 receptor agonist treatment rapidly increases the expression and/or activity of proteins (ARF-1, lipin-1) involved in PL remodeling as well as VLDL-TG maturation and secretion [32], [34], [38]. All of these findings support a coordinated and multifaceted CNS NPY modulated mechanism to increase VLDL-TG secretion that is dependent on Y1 receptor signaling and would not necessarily require the utilization of hepatic TG stores. Future studies aimed at specifically manipulating PL remodeling pathways will be required to cement this hypothesis.
SCD-1 catalyzes the rate limiting step of the synthesis of MUFAs and is an important mediator of lipoprotein metabolism [51], [52], [61]. Mice with genetic deletion of liver SCD-1 have impaired hepatic TG synthesis and VLDL-TG secretion [52]. Indeed, we observed a robust induction of hepatic SCD-1 expression and activity (as measured by the desaturation index [51], [53]) in response to CNS NPY treatment and this effect was recapitulated by NPY Y1 receptor activation [13]. Building upon these findings, we speculate that increased CNS NPY tone may rapidly induce SCD-1 activity to increase the availability of MUFAs, particularly oleic acid, required for cellular PL synthesis, which, in turn, determines the rate of PL remodeling and generates the lipid substrate for VLDL-TG maturation and secretion. In support of this notion, a study by Tran et al. [33] using rat hepatoma (McA-RH7777) cells that stably express human apoB found that treatment with oleic acid robustly stimulated the turnover of [14C] oleate-labeled PL and the transfer into cytosolic TGs which were subsequently incorporated into secreted VLDL. Moreover, the fact that hepatic SCD-1 induction can be effectively blocked by hepatic sympathetic denervation indicates that NPY increases sympathetic outflow to the liver to modulate hepatic SCD-1 and VLDL-TG secretion. Clearly, the effect we observe would likely be opposed by glucose [51] and glycine [62] metabolism possibly within the same hypothalamic circuits engaged by NPY.
The expression of ARF-1 and lipin-1, key proteins involved in liver PL remodeling were robustly elevated in the livers of icv NPY treated rats, an effect recapitulated by the activation of NPY Y1 receptor. ARF-1 is involved in the process of pre-VLDL maturation and transit to the Golgi apparatus for secretion as well as the activation of PLD, which catalyzes the production of PA from the liver PL pool [32], [34]. Indeed, overexpression of ARF-1 or PLD in cultured rat hepatocytes can increase VLDL secretion, whereas hepatic overexpression of a dominant negative ARF-1 results in a suppressive effect [34]. Lipin-1 performs the critical step of converting PA generated by PLD or formed by AGPAT via the glycerol phosphate pathway into DAG [36] that serves as substrate for the synthesis of TG and PL for VLDL maturation and secretion [37], [38]. This model is in contrast to those suggesting that mobilization and re-esterification of liver TG stores is the major pathway involved in VLDL maturation [27].
The subcellular localization and compartmentalization of lipin-1 determines its molecular function as either a glycerolipid biosynthetic enzyme or a transcriptional co-activator (reviewed in Ref. [37]). Insulin stimulates the phosphorylation of lipin-1 [63], sequestering it within the cytosol and attenuating its intrinsic PAP-1 activity [64]. Dephosphorylation of lipin-1, which has been shown to occur in response to fatty acids (i.e. oleic acid) or epinephrine, leads to its translocation from the cytosol to ER membrane [40], [41], where it engages in its PAP-1 activity, generating the lipid substrates (DAGs derived from PAs) required for VLDL maturation and secretion [38]. The observation that CNS NPY increases liver oleic acid content strengthens the feasibility of our model that NPY enhanced oleic acid production may promote lipin-1 translocation from the cytosol to the ER membrane to augment VLDL assembly and secretion. Consistent with this observation, we found that lipin-1 protein expression was robustly elevated in the isolated cellular fraction that contained the ER membrane in response to NPY treatment. In turn, we speculate that this translocation facilitates the conversion of PA into DAG, which serves as a substrate for the synthesis of TG, via DGAT, that is then assembled onto the nascent VLDL particle leading to its maturation and secretion. Indeed, overexpression of lipin-1 in cultured rat hepatocytes, in the presence of oleic acid, markedly increases TG synthesis and secretion, whereas siRNA mediated knockdown of lipin-1 decreases VLDL assembly and secretion [38]. The definitive demonstration that NPY-induced hypertriglyceridemia is dependent on liver lipin-1, however, will require additional studies.
An additional novel finding of our study is that CNS NPY regulation of hepatic lipin-1 is mediated by sympathetic inputs to the liver. Although our study does not dissect the intracellular signaling mechanisms, it is interesting to speculate that liver NE may modulate lipin-1 activity by promoting lipin-1 dephosphorylation and translocation to the ER [40]. Indeed, NE may activate adrenergic signaling in rat liver via the α1-adrenergic receptor which is robustly coupled to the activation of the protein kinase C/cAMP-response-element binding protein (CREB) pathway [65]. In turn, lipin-1 and (potentially SCD-1) expression are targets of CREB mediated gene transcription [66], [67].
4.1. Conclusions
Collectively, our findings reveal NPY signaling to be important in the central regulation of hepatic lipoprotein metabolism. Although previous work documented that the activation of the SNS is required for the effect of increased hypothalamic NPY tone to modulate hepatic VLDL-TG secretion [12], the novel liver-specific mechanisms leading to the rapid maturation and secretion of TG-rich VLDL particles in response to CNS NPY were not completely identified. Here, we provide the first direct evidence that in lean fasted rats, CNS NPY signaling modulates the expression and/or activity of key hepatic regulatory proteins (SCD-1, ARF-1, and lipin-1) involved in remodeling of PL and VLDL maturation to enhance VLDL-TG secretion. Moreover, we show that these effects of central NPY are mediated by NPY Y1 receptor signaling and sympathetic innervation of the liver. Altogether, this work has overarching implications in further understanding how obesity-related CNS dysfunction contributes to the pathophysiology of atherogenic dyslipidemia associated with obesity, diabetes and the metabolic syndrome.
Acknowledgments
This work was supported by resources of the Tennessee Valley Healthcare System, a Veterans Affairs Merit Review Award and NIH grant, DK085712 (K.N.). J.M.R. received a National Research Service Award, DK089906, from the National Institutes of Health (NIH). This work was partially supported by resources of Hormone Assay & Analytical Services Core of the Vanderbilt Diabetes Research and Training Center (DK020593).
Conflict of interest
None declared.
References
- 1.Ginsberg H.N. Insulin resistance and cardiovascular disease. Journal of Clinical Investigation. 2000;106:453–458. doi: 10.1172/JCI10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Isomaa B., Almgren P., Tuomi T., Forsen B., Lahti K., Nissen M. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 2001;24:683–689. doi: 10.2337/diacare.24.4.683. [DOI] [PubMed] [Google Scholar]
- 3.Ginsberg H.N., Zhang Y.L., Hernandez-Ono A. Metabolic syndrome: focus on dyslipidemia. Obesity (Silver Spring) 2006;14(Suppl. 1):41S–49S. doi: 10.1038/oby.2006.281. [DOI] [PubMed] [Google Scholar]
- 4.Riches F.M., Watts G.F., Naoumova R.P., Kelly J.M., Croft K.D., Thompson G.R. Hepatic secretion of very-low-density lipoprotein apolipoprotein b-100 studied with a stable isotope technique in men with visceral obesity. International Journal of Obesity and Related Metabolic Disorders. 1998;22:414–423. doi: 10.1038/sj.ijo.0800602. [DOI] [PubMed] [Google Scholar]
- 5.Kissebah A.H., Alfarsi S., Adams P.W., Wynn V. Role of insulin resistance in adipose tissue and liver in the pathogenesis of endogenous hypertriglyceridaemia in man. Diabetologia. 1976;12:563–571. doi: 10.1007/BF01220632. [DOI] [PubMed] [Google Scholar]
- 6.Laws A., Hoen H.M., Selby J.V., Saad M.F., Haffner S.M., Howard B.V. Differences in insulin suppression of free fatty acid levels by gender and glucose tolerance status. Relation to plasma triglyceride and apolipoprotein b concentrations. Insulin resistance atherosclerosis study (iras) investigators. Arteriosclerosis Thrombosis and Vascular Biology. 1997;17:64–71. doi: 10.1161/01.atv.17.1.64. [DOI] [PubMed] [Google Scholar]
- 7.Vettor R., Zarjevski N., Cusin I., Rohner-Jeanrenaud F., Jeanrenaud B. Induction and reversibility of an obesity syndrome by intracerebroventricular neuropeptide y administration to normal rats. Diabetologia. 1994;37:1202–1208. doi: 10.1007/BF00399793. [DOI] [PubMed] [Google Scholar]
- 8.Raposinho P.D., Pierroz D.D., Broqua P., White R.B., Pedrazzini T., Aubert M.L. Chronic administration of neuropeptide y into the lateral ventricle of c57bl/6j male mice produces an obesity syndrome including hyperphagia, hyperleptinemia, insulin resistance, and hypogonadism. Molecular and Cellular Endocrinology. 2001;185:195–204. doi: 10.1016/s0303-7207(01)00620-7. [DOI] [PubMed] [Google Scholar]
- 9.Tiesjema B., Adan R.A., Luijendijk M.C., Kalsbeek A., La Fleur S.E. Differential effects of recombinant adeno-associated virus-mediated neuropeptide y overexpression in the hypothalamic paraventricular nucleus and lateral hypothalamus on feeding behavior. Journal of Neuroscience. 2007;27:14139–14146. doi: 10.1523/JNEUROSCI.3280-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Den Heuvel J.K., Eggels L., Van Rozen A.J., Luijendijk M.C., Fliers E., Kalsbeek A. Neuropeptide y and leptin sensitivity is dependent on diet composition. Journal of Neuroendocrinology. 2014;26:377–385. doi: 10.1111/jne.12155. [DOI] [PubMed] [Google Scholar]
- 11.Van Den Heuvel J.K., Furman K., Gumbs M.C., Eggels L., Opland D.M., Land B.B. Neuropeptide y activity in the nucleus accumbens modulates feeding behavior and neuronal activity. Biological Psychiatry. 2014 doi: 10.1016/j.biopsych.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruinstroop E., Pei L., Ackermans M.T., Foppen E., Borgers A.J., Kwakkel J. Hypothalamic neuropeptide y (npy) controls hepatic vldl-triglyceride secretion in rats via the sympathetic nervous system. Diabetes. 2012;61:1043–1050. doi: 10.2337/db11-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rojas J.M., Stafford J.M., Saadat S., Printz R.L., Beck-Sickinger A.G., Niswender K.D. Central nervous system neuropeptide y signaling via the y1 receptor partially dissociates feeding behavior from lipoprotein metabolism in lean rats. American Journal of Physiology. Endocrinology and Metabolism. 2012;303:E1479–E1488. doi: 10.1152/ajpendo.00351.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stafford J.M., Yu F., Printz R., Hasty A.H., Swift L.L., Niswender K.D. Central nervous system neuropeptide y signaling modulates vldl triglyceride secretion. Diabetes. 2008;57:1482–1490. doi: 10.2337/db07-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Den Hoek A.M., Van Heijningen C., Schroder-Van Der Elst J.P., Ouwens D.M., Havekes L.M., Romijn J.A. Intracerebroventricular administration of neuropeptide y induces hepatic insulin resistance via sympathetic innervation. Diabetes. 2008;57:2304–2310. doi: 10.2337/db07-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marks J.L., Waite K. Intracerebroventricular neuropeptide y acutely influences glucose metabolism and insulin sensitivity in the rat. Journal of Neuroendocrinology. 1997;9:99–103. doi: 10.1046/j.1365-2826.1997.00554.x. [DOI] [PubMed] [Google Scholar]
- 17.Van Den Hoek A.M., Voshol P.J., Karnekamp B.N., Buijs R.M., Romijn J.A., Havekes L.M. Intracerebroventricular neuropeptide y infusion precludes inhibition of glucose and vldl production by insulin. Diabetes. 2004;53:2529–2534. doi: 10.2337/diabetes.53.10.2529. [DOI] [PubMed] [Google Scholar]
- 18.Yi C.X., Foppen E., Abplanalp W., Gao Y., Alkemade A., La Fleur S.E. Glucocorticoid signaling in the arcuate nucleus modulates hepatic insulin sensitivity. Diabetes. 2012;61:339–345. doi: 10.2337/db11-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sipols A.J., Baskin D.G., Schwartz M.W. Effect of intracerebroventricular insulin infusion on diabetic hyperphagia and hypothalamic neuropeptide gene expression. Diabetes. 1995;44:147–151. doi: 10.2337/diab.44.2.147. [DOI] [PubMed] [Google Scholar]
- 20.Wilding J.P., Gilbey S.G., Mannan M., Aslam N., Ghatei M.A., Bloom S.R. Increased neuropeptide y content in individual hypothalamic nuclei, but not neuropeptide y mrna, in diet-induced obesity in rats. Journal of Endocrinology. 1992;132:299–304. doi: 10.1677/joe.0.1320299. [DOI] [PubMed] [Google Scholar]
- 21.Baranowska B., Wolinska-Witort E., Martynska L., Chmielowska M., Mazurczak-Pluta T., Boguradzka A. Sibutramine therapy in obese women–effects on plasma neuropeptide Y (NPY), insulin, leptin and beta-endorphin concentrations. Neuro Endocrinology Letters. 2005;26:675–679. [PubMed] [Google Scholar]
- 22.Baranowska B., Wolinska-Witort E., Wasilewska-Dziubinska E., Roguski K., Martynska L., Chmielowska M. The role of neuropeptides in the disturbed control of appetite and hormone secretion in eating disorders. Neuro Endocrinology Letters. 2003;24:431–434. [PubMed] [Google Scholar]
- 23.Saderi N., Salgado-Delgado R., Avendano-Pradel R., Basualdo Mdel C., Ferri G.L., Chavez-Macias L. NPY and VGF immunoreactivity increased in the arcuate nucleus, but decreased in the nucleus of the tractus solitarius, of type-ii diabetic patients. PLoS One. 2012;7:e40070. doi: 10.1371/journal.pone.0040070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Posey K., Clegg D.J., Printz R.L., Byun J., Morton G.J., Vivekanandan-Giri A. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. American Journal of Physiology. Endocrinology and Metabolism. 2009;296:E1003–E1012. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Picardi P.K., Calegari V.C., Prada Pde O., Moraes J.C., Araujo E., Marcondes M.C. Reduction of hypothalamic protein tyrosine phosphatase improves insulin and leptin resistance in diet-induced obese rats. Endocrinology. 2008;149:3870–3880. doi: 10.1210/en.2007-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nielsen S., Karpe F. Determinants of vldl-triglycerides production. Current Opinion in Lipidology. 2012;23:321–326. doi: 10.1097/MOL.0b013e3283544956. [DOI] [PubMed] [Google Scholar]
- 27.Gibbons G.F., Wiggins D., Brown A.M., Hebbachi A.M. Synthesis and function of hepatic very-low-density lipoprotein. Biochemical Society Transactions. 2004;32:59–64. doi: 10.1042/bst0320059. [DOI] [PubMed] [Google Scholar]
- 28.Sundaram M., Yao Z. Recent progress in understanding protein and lipid factors affecting hepatic vldl assembly and secretion. Nutrition & Metabolism (London) 2010;7:35. doi: 10.1186/1743-7075-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olofsson S.O., Asp L., Boren J. The assembly and secretion of apolipoprotein b-containing lipoproteins. Current Opinion in Lipidology. 1999;10:341–346. doi: 10.1097/00041433-199908000-00008. [DOI] [PubMed] [Google Scholar]
- 30.Diraison F., Beylot M. Role of human liver lipogenesis and reesterification in triglycerides secretion and in ffa reesterification. American Journal of Physiology. 1998;274:E321–E327. doi: 10.1152/ajpendo.1998.274.2.E321. [DOI] [PubMed] [Google Scholar]
- 31.Wiggins D., Gibbons G.F. Origin of hepatic very-low-density lipoprotein triacylglycerol: the contribution of cellular phospholipid. Biochemical Journal. 1996;320(Pt 2):673–679. doi: 10.1042/bj3200673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asp L., Claesson C., Boren J., Olofsson S.O. Adp-ribosylation factor 1 and its activation of phospholipase d are important for the assembly of very low density lipoproteins. Journal of Biological Chemistry. 2000;275:26285–26292. doi: 10.1074/jbc.M003520200. [DOI] [PubMed] [Google Scholar]
- 33.Tran K., Sun F., Cui Z., Thorne-Tjomsland G., St Germain C., Lapierre L.R. Attenuated secretion of very low density lipoproteins from mca-rh7777 cells treated with eicosapentaenoic acid is associated with impaired utilization of triacylglycerol synthesized via phospholipid remodeling. Biochimica et Biophysica Acta. 2006;1761:463–473. doi: 10.1016/j.bbalip.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 34.Asp L., Magnusson B., Rutberg M., Li L., Boren J., Olofsson S.O. Role of adp ribosylation factor 1 in the assembly and secretion of apob-100-containing lipoproteins. Arteriosclerosis Thrombosis and Vascular Biology. 2005;25:566–570. doi: 10.1161/01.ATV.0000154135.21689.47. [DOI] [PubMed] [Google Scholar]
- 35.Rustaeus S., Lindberg K., Boren J., Olofsson S.O. Brefeldin a reversibly inhibits the assembly of apob containing lipoproteins in mca-rh7777 cells. Journal of Biological Chemistry. 1995;270:28879–28886. doi: 10.1074/jbc.270.48.28879. [DOI] [PubMed] [Google Scholar]
- 36.Takeuchi K., Reue K. Biochemistry, physiology, and genetics of gpat, agpat, and lipin enzymes in triglyceride synthesis. American Journal of Physiology. Endocrinology and Metabolism. 2009;296:E1195–E1209. doi: 10.1152/ajpendo.90958.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bou Khalil M., Blais A., Figeys D., Yao Z. Lipin – the bridge between hepatic glycerolipid biosynthesis and lipoprotein metabolism. Biochimica et Biophysica Acta. 2010;1801:1249–1259. doi: 10.1016/j.bbalip.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 38.Bou Khalil M., Sundaram M., Zhang H.Y., Links P.H., Raven J.F., Manmontri B. The level and compartmentalization of phosphatidate phosphatase-1 (lipin-1) control the assembly and secretion of hepatic vldl. Journal of Lipid Research. 2009;50:47–58. doi: 10.1194/jlr.M800204-JLR200. [DOI] [PubMed] [Google Scholar]
- 39.Man W.C., Miyazaki M., Chu K., Ntambi J. Colocalization of scd1 and dgat2: implying preference for endogenous monounsaturated fatty acids in triglyceride synthesis. Journal of Lipid Research. 2006;47:1928–1939. doi: 10.1194/jlr.M600172-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.Harris T.E., Huffman T.A., Chi A., Shabanowitz J., Hunt D.F., Kumar A. Insulin controls subcellular localization and multisite phosphorylation of the phosphatidic acid phosphatase, lipin 1. Journal of Biological Chemistry. 2007;282:277–286. doi: 10.1074/jbc.M609537200. [DOI] [PubMed] [Google Scholar]
- 41.Cascales C., Mangiapane E.H., Brindley D.N. Oleic acid promotes the activation and translocation of phosphatidate phosphohydrolase from the cytosol to particulate fractions of isolated rat hepatocytes. Biochemical Journal. 1984;219:911–916. doi: 10.1042/bj2190911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phillips M.S., Liu Q., Hammond H.A., Dugan V., Hey P.J., Caskey C.J. Leptin receptor missense mutation in the fatty zucker rat. Nature Genetics. 1996;13:18–19. doi: 10.1038/ng0596-18. [DOI] [PubMed] [Google Scholar]
- 43.Cleary M.P., Vasselli J.R., Greenwood M.R. Development of obesity in zucker obese (fafa) rat in absence of hyperphagia. American Journal of Physiology. 1980;238:E284–E292. doi: 10.1152/ajpendo.1980.238.3.E284. [DOI] [PubMed] [Google Scholar]
- 44.Beck B., Burlet A., Bazin R., Nicolas J.P., Burlet C. Elevated neuropeptide y in the arcuate nucleus of young obese zucker rats may contribute to the development of their overeating. Journal of Nutrition. 1993;123:1168–1172. doi: 10.1093/jn/123.6.1168. [DOI] [PubMed] [Google Scholar]
- 45.Sanacora G., Kershaw M., Finkelstein J.A., White J.D. Increased hypothalamic content of preproneuropeptide y messenger ribonucleic acid in genetically obese zucker rats and its regulation by food deprivation. Endocrinology. 1990;127:730–737. doi: 10.1210/endo-127-2-730. [DOI] [PubMed] [Google Scholar]
- 46.Paxinos G., Watson C. The rat brain in stereotaxic coordinates. Academic Press; San Diego: 1998. [DOI] [PubMed] [Google Scholar]
- 47.Soll R.M., Dinger M.C., Lundell I., Larhammer D., Beck-Sickinger A.G. Novel analogues of neuropeptide y with a preference for the y1-receptor. European Journal of Biochemistry. 2001;268:2828–2837. doi: 10.1046/j.1432-1327.2001.02161.x. [DOI] [PubMed] [Google Scholar]
- 48.Saraswathi V., Hasty A.H. The role of lipolysis in mediating the proinflammatory effects of very low density lipoproteins in mouse peritoneal macrophages. Journal of Lipid Research. 2006;47:1406–1415. doi: 10.1194/jlr.M600159-JLR200. [DOI] [PubMed] [Google Scholar]
- 49.Folch J., Lees M., Sloane Stanley G.H. A simple method for the isolation and purification of total lipides from animal tissues. Journal of Biological Chemistry. 1957;226:497–509. [PubMed] [Google Scholar]
- 50.Morrison W.R., Smith L.M. Preparation of fatty acid methyl esters and dimethylacetals from lipids with boron fluoride–methanol. Journal of Lipid Research. 1964;5:600–608. [PubMed] [Google Scholar]
- 51.Lam T.K., Gutierrez-Juarez R., Pocai A., Bhanot S., Tso P., Schwartz G.J. Brain glucose metabolism controls the hepatic secretion of triglyceride-rich lipoproteins. Nature Medicine. 2007;13:171–180. doi: 10.1038/nm1540. [DOI] [PubMed] [Google Scholar]
- 52.Miyazaki M., Kim Y.C., Gray-Keller M.P., Attie A.D., Ntambi J.M. The biosynthesis of hepatic cholesterol esters and triglycerides is impaired in mice with a disruption of the gene for stearoyl-coa desaturase 1. Journal of Biological Chemistry. 2000;275:30132–30138. doi: 10.1074/jbc.M005488200. [DOI] [PubMed] [Google Scholar]
- 53.Attie A.D., Krauss R.M., Gray-Keller M.P., Brownlie A., Miyazaki M., Kastelein J.J. Relationship between stearoyl-coa desaturase activity and plasma triglycerides in human and mouse hypertriglyceridemia. Journal of Lipid Research. 2002;43:1899–1907. doi: 10.1194/jlr.m200189-jlr200. [DOI] [PubMed] [Google Scholar]
- 54.Barroso E., Rodriguez-Calvo R., Serrano-Marco L., Astudillo A.M., Balsinde J., Palomer X. The pparbeta/delta activator gw501516 prevents the down-regulation of ampk caused by a high-fat diet in liver and amplifies the pgc-1alpha-lipin 1-pparalpha pathway leading to increased fatty acid oxidation. Endocrinology. 2011;152:1848–1859. doi: 10.1210/en.2010-1468. [DOI] [PubMed] [Google Scholar]
- 55.Finck B.N., Gropler M.C., Chen Z., Leone T.C., Croce M.A., Harris T.E. Lipin 1 is an inducible amplifier of the hepatic pgc-1alpha/pparalpha regulatory pathway. Cell Metabolism. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Bonnefont J.P., Djouadi F., Prip-Buus C., Gobin S., Munnich A., Bastin J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Molecular Aspects of Medicine. 2004;25:495–520. doi: 10.1016/j.mam.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 57.Zhang L., Macia L., Turner N., Enriquez R.F., Riepler S.J., Nguyen A.D. Peripheral neuropeptide y y1 receptors regulate lipid oxidation and fat accretion. International Journal of Obesity (London) 2010;34:357–373. doi: 10.1038/ijo.2009.232. [DOI] [PubMed] [Google Scholar]
- 58.Koonen D.P., Jacobs R.L., Febbraio M., Young M.E., Soltys C.L., Ong H. Increased hepatic cd36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes. 2007;56:2863–2871. doi: 10.2337/db07-0907. [DOI] [PubMed] [Google Scholar]
- 59.Leyton J., Drury P.J., Crawford M.A. Differential oxidation of saturated and unsaturated fatty acids in vivo in the rat. British Journal of Nutrition. 1987;57:383–393. doi: 10.1079/bjn19870046. [DOI] [PubMed] [Google Scholar]
- 60.Lynn W.S., Brown R.H. Oxidation and activation of unsaturated fatty acids. Archives of Biochemistry and Biophysics. 1959;81:353–362. doi: 10.1016/0003-9861(59)90213-9. [DOI] [PubMed] [Google Scholar]
- 61.Cohen P., Miyazaki M., Socci N.D., Hagge-Greenberg A., Liedtke W., Soukas A.A. Role for stearoyl-coa desaturase-1 in leptin-mediated weight loss. Science. 2002;297:240–243. doi: 10.1126/science.1071527. [DOI] [PubMed] [Google Scholar]
- 62.Yue J.T., Mighiu P.I., Naples M., Adeli K., Lam T.K. Glycine normalizes hepatic triglyceride-rich vldl secretion by triggering the cns in high-fat fed rats. Circulation Research. 2012;110:1345–1354. doi: 10.1161/CIRCRESAHA.112.268276. [DOI] [PubMed] [Google Scholar]
- 63.Huffman T.A., Mothe-Satney I., Lawrence J.C., Jr. Insulin-stimulated phosphorylation of lipin mediated by the mammalian target of rapamycin. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:1047–1052. doi: 10.1073/pnas.022634399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eaton J.M., Mullins G.R., Brindley D.N., Harris T.E. Phosphorylation of lipin 1 and charge on the phosphatidic acid head group control its phosphatidic acid phosphatase activity and membrane association. Journal of Biological Chemistry. 2013;288:9933–9945. doi: 10.1074/jbc.M112.441493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thonberg H., Fredriksson J.M., Nedergaard J., Cannon B. A novel pathway for adrenergic stimulation of camp-response-element-binding protein (creb) phosphorylation: mediation via alpha1-adrenoceptors and protein kinase c activation. Biochemical Journal. 2002;364:73–79. doi: 10.1042/bj3640073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chanda D., Kim Y.H., Kim D.K., Lee M.W., Lee S.Y., Park T.S. Activation of cannabinoid receptor type 1 (cb1r) disrupts hepatic insulin receptor signaling via cyclic amp-response element-binding protein h (crebh)-mediated induction of lipin1 gene. Journal of Biological Chemistry. 2012;287:38041–38049. doi: 10.1074/jbc.M112.377978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ryu D., Oh K.J., Jo H.Y., Hedrick S., Kim Y.N., Hwang Y.J. Torc2 regulates hepatic insulin signaling via a mammalian phosphatidic acid phosphatase, lipin1. Cell Metabolism. 2009;9:240–251. doi: 10.1016/j.cmet.2009.01.007. [DOI] [PubMed] [Google Scholar]