

Abstract
MicroRNAs have been identified and analyzed in various model species, but an investigation of miRNAs in nonmodel species is required for a more complete understanding of miRNA evolution. In this study, we investigated the miRNAs of the nonmodel species Triops cancriformis (tadpole shrimp), a “living fossil,” whose morphological form has not changed in almost 200 million years. Dramatic ontogenetic changes occur during its development. To clarify the evolution of miRNAs, we comparatively analyzed its miRNAs and the components of its RNAi machinery. We used deep sequencing to analyze small RNA libraries from the six different developmental stages of T. cancriformis (egg, first–fourth instars, and adult), and also analyzed its genomic DNA with deep sequencing. We identified 180 miRNAs (87 conserved miRNAs and 93 novel candidate miRNAs), and deduced the components of its RNAi machinery: the DICER1, AGO1–3, PIWI, and AUB proteins. A comparative miRNA analysis of T. cancriformis and Drosophila melanogaster showed inconsistencies in the expression patterns of four conserved miRNAs. This suggests that although the miRNA sequences of the two species are very similar, their roles differ across the species. An miRNA conservation analysis revealed that most of the conserved T. cancriformis miRNAs share sequence similarities with those of arthropods, although T. cancriformis is called a “living fossil.” However, we found that let-7 and DICER1 of T. cancriformis are more similar to those of the vertebrates than to those of the arthropods. These results suggest that miRNA systems of T. cancriformis have evolved in a unique fashion.
Keywords: microRNA, deep sequencing, development, evolution, T. cancriformis
INTRODUCTION
MicroRNAs (miRNAs) are noncoding RNAs (ncRNAs) of ∼22 nucleotides (nt) that regulate the expression of target messenger RNAs (mRNAs), mainly at the post-transcriptional level (Bartel 2004). It has been reported that sequences of ∼7 nt in the 5′ region of miRNAs, the so-called “seed sequences,” are important in the recognition of miRNA targets (Lewis et al. 2003; Bartel 2009; Nahvi et al. 2009). In canonical miRNA biogenesis, primary miRNAs (pri-miRNAs) are processed by DROSHA and subsequently by DICER to produce double-stranded RNAs. The mature miRNAs are then incorporated into the RNA-induced silencing complex (RISC) containing the AGO proteins to perform their functions (Meister 2013). It is well known that miRNAs are related to various cellular processes, including cell differentiation and development. For instance, in Caenorhabditis elegans, two evolutionarily conserved miRNAs (cel-lin-4 and cel-let-7) are expressed in a stage-specific manner and regulate developmental timing (Reinhart et al. 2000). In Drosophila melanogaster, dme-miR-2 is required to suppress apoptosis during embryonic development by repressing grim mRNA, which encodes a proapoptotic factor (Leaman et al. 2005). In addition to miRNAs, another class of ncRNAs, PIWI-interacting RNAs (piRNAs), have been reported to associate with the PIWI family of proteins (PIWI, AUB, and AGO3), which are involved in gonad development (Siomi et al. 2011).
Some miRNA genes are conserved in various species. We have previously shown that five miRNAs (let-7, miR-1, miR-34, miR-124, and miR-125/lin-4) are evolutionarily conserved in bilaterian animals, and that these conserved miRNAs target orthologous mRNAs in corresponding species (Takane et al. 2010). It has also been reported that Nematostella vectensis, which diverged before the appearance of the bilaterian animals, possesses almost no miRNAs with sequences similar to those of bilaterian animals, except for miR-100 (Grimson et al. 2008). Because the number of N. vectensis miRNAs is small (49 precursor miRNAs) as compared with the number in most bilaterian animals, and because bilaterian animals are morphologically more complex than nonbilaterians, miRNAs are considered to be strongly related to the morphological evolution of animals. Based on these observations, miRNAs have been studied not only in typical model species but also in nonmodel species with interesting characteristics. For instance, an evolutionary analysis of miRNAs was performed in the amphioxus Branchiostoma belcheri, which is a key animal in the evolution of chordates, and a phylogenetic analysis of miRNAs demonstrated that B. belcheri is more similar to vertebrates than to tunicates (Chen et al. 2009). In addition to analyses of miRNA sequences, it is also important to focus on the components of the RNAi machinery. For example, Ascaris suum reportedly lacks the piRNAs, Piwi-clade Argonautes, and other proteins associated with the piRNA pathway, indicating that the piRNA pathway was lost in A. suum (Wang et al. 2011). These findings indicate that the investigation of miRNAs and components of RNAi machinery in nonmodel species is important for elucidating their evolutionary pathways.
In this study, our analyses focused on the tadpole shrimp Triops cancriformis. The morphological form of T. cancriformis is considered to have remained unchanged for 200 million years, making it a so-called “living fossil.” In contrast, recent reports have indicated that T. cancriformis appeared <50 million years ago (Korn et al. 2013; Mathers et al. 2013), so the use of the term “living fossil” for T. cancriformis is controversial. The larvae of T. cancriformis change dramatically during development (Fig. 1A), progressing from the first to the fourth instar in only ∼26 h, and at the same time doubling in size. Important morphological changes occur during that time, including the appearance of compound eyes and an increase in the number of body segments (Igarashi 1971). Despite these interesting characteristics of T. cancriformis, almost no genome or transcriptome data are currently available for this organism. To investigate the evolutionary history of miRNAs, we conducted a comparative analysis of miRNAs and the components of RNAi machinery. We identified conserved miRNAs and novel candidate miRNAs and deduced the components of the RNAi machinery in T. cancriformis. Most of the conserved miRNAs of T. cancriformis share sequence similarities with those of the arthropods, although T. cancriformis is called a “living fossil.” However, a comparative analysis revealed that T. cancriformis let-7 and DICER1 are more similar to those of the vertebrates than to those of the arthropods, suggesting that the evolution of miRNA systems in T. cancriformis has been unique, differing from those of other model species.
FIGURE 1.

Summary of the deep-sequencing analysis in each developmental stage of T. cancriformis. (A) Morphological changes during T. cancriformis development. Scale bars represent 0.5 mm for the egg and larvae from the first to fourth instars (black bars), and 1.5 cm for the adult (red bar). (a) Egg; (b) first instar larva; (c) second instar larva; (d) third instar larva; (e) fourth instar larva; (f) adult. (B) Relative numbers of reads based on small RNA lengths. The y-axis represents the ratio of small RNA reads and 100% represents the total small RNA reads that perfectly matched T. cancriformis genome contigs in each stage (see Materials and Methods). The color indicates each stage (pink for egg, red for first instar, yellow for second instar, green for third instar, light blue for fourth instar, and purple for adult). The miRNA fraction (18–24 nt) is shaded in gray. (C) Pie charts summarizing the proportions of small RNA annotations in the miRNA fraction in each stage.
RESULTS AND DISCUSSION
Identification of 87 evolutionarily conserved miRNAs and 93 novel candidate miRNAs in T. cancriformis
To clarify the relationships between miRNA expression and the dramatic morphological changes that occur in T. cancriformis (Fig. 1A), we performed a deep sequencing analysis of small RNA libraries from the six different developmental stages (egg, first, second, third, and fourth instar, and adult) of T. cancriformis. Illumina deep sequencing yielded 151 million small RNA reads, predominantly 18–35 nt in length, from the six developmental stages. Reads with identical sequences were collapsed into a unique read to calculate the variation in the unique reads per stage, but information on the read numbers of the identical sequences was retained. The numbers of unique reads from which conserved miRNAs were extracted are summarized in Supplemental Table S1. First, small RNA reads of poor quality or with <5 counts were discarded. Because the genomic DNA sequence was required to reliably identify both the miRNAs and the components of the RNAi machinery, deep sequencing of the genomic DNA was performed. We obtained 133 million genomic DNA reads (100 nt) in total, and discarded the low-quality reads. Reads with identical sequences were collapsed into a unique read to reduce the repeated sequences, and the number of unique reads was 86,279,282. These genomic DNA sequences were assembled with Velvet v. 1.2.10 (Zerbino and Birney 2008), generating 60,629 contigs with N50 of 12,784 bp, a largest contig of 133,027 bp, and ∼109 Mb of assembled sequence. We then discarded the small RNA reads that did not match the T. cancriformis genomic contigs. In this step, the lengths of these small RNA had a trimodal distribution, with distinct peaks at 22 nt, at ∼27 nt, and at 32 nt (Fig. 1B). Based on previous reports (Kim et al. 2009; Wei et al. 2012), we presumed that the peak at 22 nt corresponded predominantly to miRNAs, whereas the peak at ∼27 nt primarily corresponded to piRNAs in T. cancriformis. The third peak at 32 nt consisted of many transfer RNA (tRNA) fragments (Y Hirose, KT Ikeda, E Noro, K Hiraoka, M Tomita, and A Kanai, in prep.). In the egg stage, the number of reads corresponding to small RNAs of 18–20 nt was larger (∼3% of small RNAs matched the T. cancriformis genomic contigs) than the numbers in the other stages (<1%) (Fig. 1B). More than 10% of these reads in the egg were identical to the 3′ end of T. cancriformis 28S ribosomal RNA (rRNA), suggesting that 28S rRNA was specifically degraded to small fractions in the egg stage. Small RNA reads from the 18–24-nt miRNA fraction were then extracted, and those that matched known T. cancriformis cDNA sequences were discarded. To extract candidate miRNAs, we predicted the T. cancriformis miRNA candidates would share high sequence similarity with those of other species, and we selected major sequences as conserved candidate miRNAs from several miRNA isoforms. After extracting the genomic DNA sequences corresponding to conserved candidate miRNAs, we examined whether these genomic DNA sequences folded into secondary structures commensurate with precursor miRNAs. In this way, we identified 87 conserved mature miRNAs and 71 putative miRNA precursors (Supplemental Tables S2,S3). Among the conserved miRNAs, two different putative precursor sequences of the mature tcf-miR-2a miRNA were found, and we designated them tcf-miR-2a and tcf-miR-2a-2. To clarify the miRNA sequence quality, we assigned a reliability level to individual T. cancriformis miRNAs based on a previous study (Kozomara and Griffiths-Jones 2013) by applying the following criteria: (1) The mature miRNA is detected in ≥10 reads, with no mismatches to the T. cancriformis genome contigs; (2) each strand of the miRNA precursor must pair with the 0–4 nt overhang at its 3′ end; (3) at least 50% of reads mapping to each strand of the precursor must have the same 5′ end; (4) the free energy of the predicted RNA secondary structure must be <0.2 kcal/mol/nt; and (5) at least 60% of the bases in the mature sequence must be paired in the predicted secondary structure. We categorized the reliability levels in T. cancriformis from level 1 to level 4 (i.e., level 4 represents the highest reliability), using the four criteria (2–5) described above, and all miRNAs must meet criterion (1). The miRNAs were then classified according to the number of these criteria they met (e.g., if miR-X met three criteria (3–5), it was categorized at level 3, and if miR-Y met two criteria (4–5), it was categorized at level 2). Of the 88 conserved miRNAs (including miR-2a-2), 76 were categorized into reliability level 4. Furthermore, 84 of 87 were found in the experimental data from the deep-sequencing analysis of the small RNA libraries from the six developmental stages of T. cancriformis (data not shown), suggesting strong reproducibility of the conserved miRNA expression. These results show that our method efficiently screened the conserved miRNAs in T. cancriformis.
We then predicted the novel candidate miRNAs in T. cancriformis using miRDeep2 (Friedlander et al. 2012), inputting the 87 conserved mature T. cancriformis miRNAs, 71 putative T. cancriformis miRNA precursors, and 30,424 known mature miRNAs registered in miRBase release 20.0 (Kozomara and Griffiths-Jones 2013). After evaluating the scores obtained for the output of miRDeep2 (see Materials and Methods), we identified 93 novel candidate miRNAs and 98 putative precursor sequences for the novel candidate miRNAs (Supplemental Tables S4,S5). Sixty-six of the 98 putative precursor sequences of the novel candidate miRNAs were categorized into reliability level 4, suggesting that many reliable novel miRNAs were successfully predicted by miRDeep2.
When we compared the annotated miRNA sequences of 18–24 nt in the six developmental stages (Fig. 1C), we found that the proportion of conserved miRNAs was higher than the proportion of novel candidate miRNAs in all stages (30.5%–58.2% conserved miRNAs and 1.0%–1.8% novel candidate miRNAs). The average read number for the conserved miRNAs was 30,823.6, whereas that of the novel miRNAs was only 756.0, supporting the previous finding that the number of miRNAs conserved among a wide range of species tends to be high (Watanabe et al. 2008). Because both the variety and proportion of conserved miRNAs increased from the egg to the first instar (Fig. 1C; Supplemental Table S1), we presumed that these conserved miRNAs are related to the morphogenesis of the first instar larva. In contrast, the proportion of novel candidate miRNAs decreased from the egg to the first instar, although their variety increased. Because four novel candidate miRNAs (tcf-miR-n501, tcf-miR-n502, tcf-miR-n503, and tcf-miR-n504) were expressed in the egg stage with high read counts (≥10,000), these novel candidate miRNAs may be related to the release from dormancy.
Changes in the expression of miRNAs during T. cancriformis development
It has been reported that miRNAs are intimately related to development (Wienholds and Plasterk 2005; Kloosterman and Plasterk 2006), so we hypothesized that miRNA expression will change markedly with the dramatic morphological changes that occur of the early larvae of T. cancriformis. First, the expression of 87 conserved T. cancriformis miRNAs was analyzed during the six developmental stages based on the read counts for each miRNA. miRNA expression was normalized using spike reads (Supplemental Tables S6,S7), and these conserved miRNAs were then roughly clustered into seven groups based on their expression patterns (Groups 1-I to 1-VII in Fig. 2A), and showed that the expression patterns of the conserved miRNAs varied throughout the six developmental stages of T. cancriformis. Some miRNAs were stage-specifically expressed in the egg, fourth instar larval, and adult stage (Groups 1-I to 1-III in Fig. 2A). To validate the expression of the conserved miRNAs, Northern blotting analyses were performed, and 13 miRNAs (tcf-let-7-5p, tcf-miR-1, tcf-miR-2b, tcf-miR-12, tcf-miR-34, tcf-miR-87, tcf-miR-125, tcf-miR-133, tcf-miR-184-3p, tcf-miR-276-3p, tcf-miR-279a, tcf-miR-375, and tcf-miR-750) were detected, at ∼20–25 nt in length, in the adult stage (Fig. 2B; Supplemental Fig. S1), suggesting that these 13 miRNAs are actually expressed in the adult stage of T. cancriformis. The expression patterns of six (tcf-let-7-5p, tcf-miR-2b, tcf-miR-12, tcf-miR-34, tcf-miR-87, and tcf-miR-125) of these 13 miRNAs were then investigated in the six developmental stages (Fig. 2B). A strong correlation was observed between the Northern blotting data and the read counts from the deep-sequencing analysis (tcf-let-7-5p: r = 0.95, P = 0.00273; tcf-miR-2b: r = 0.96, P = 0.00192; tcf-miR-12: r = 0.82, P = 0.04336; tcf-miR-34: r = 0.98, P = 0.00016; tcf-miR-87: r = 0.81, P = 0.04738; and tcf-miR-125: r = 0.98, P = 0.00027) (see Materials and Methods). These results show that our method efficiently extracted the conserved miRNAs and that their expression profiles based on read numbers are reliable.
FIGURE 2.
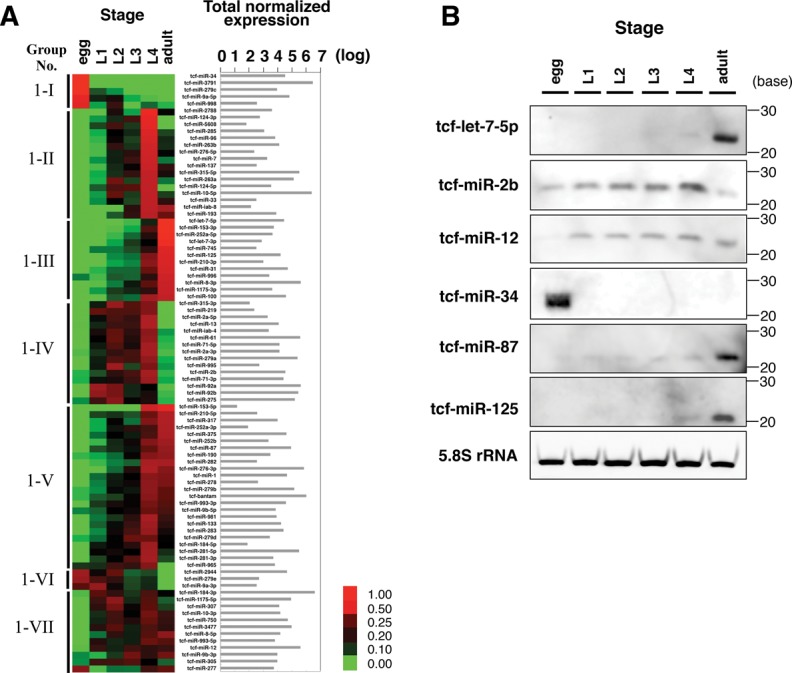
Expression profiles of conserved T. cancriformis miRNAs through the six developmental stages. (A) Expression profiles of conserved miRNAs based on read numbers in the deep-sequencing analysis. miRNA reads were normalized to the spike reads in each stage. A more intense red color indicates a more strongly expressed miRNA, whereas a more intense green color indicates a more weakly expressed miRNA. The conserved miRNAs were categorized into seven groups based on their expression patterns (Groups 1-I to 1-VII). The total normalized expression for each of the six stages is listed. (B) Northern blotting analysis of six conserved miRNAs during T. cancriformis development. A typical pattern of 5.8S rRNA expression is also shown as the loading control.
We then investigated whether the expression patterns of the six conserved miRNAs were common to T. cancriformis and D. melanogaster, a representative model arthropod species. We used Northern blotting data from a previous study of D. melanogaster (Sempere et al. 2003). The expression patterns of two (tcf-let-7-5p and tcf-miR-125) of the six conserved miRNAs were similar, and increased toward the adult stages of T. cancriformis and D. melanogaster (Group 1-III in Fig. 2A,B). In contrast, the expression patterns of four conserved miRNAs (tcf-miR-2b, tcf-miR-12, tcf-miR-87, and tcf-miR-34) were quite different in these species. Triops cancriformis tcf-miR-2b was predominantly expressed in the larval stages, especially increasing toward the fourth instar (Group 1-IV in Fig. 2A,B), whereas in D. melanogaster, dme-miR-2 was expressed throughout the developmental stages, and especially in the egg stage. tcf-miR-12 was expressed from the first instar to the adult stage (Group 1-VII in Fig. 2A,B), whereas the expression of dme-miR-12 decreased toward the adult stage. tcf-miR-34 was only expressed in the egg stage (Group 1-I in Fig. 2A,B), whereas dme-miR-34 was strongly expressed in the adult stage. tcf-miR-87 was expressed throughout all six stages, but was especially strongly expressed in the adult stage (Group 1-V in Fig. 2A,B), whereas dme-miR-87 was detected in the egg, first and second instar larval, pupal, and adult stages. This inconsistency in the expression patterns of the conserved miRNAs suggests that although these miRNAs have very similar sequences (≥80%), they may play different roles in different species.
To characterize these novel candidate miRNAs, an expression analysis was conducted with the same method used for the conserved miRNAs (Supplemental Tables S6,S8). Based on their expression patterns during development, the candidate miRNAs were roughly clustered into nine groups (Groups 2-I to 2-IX in Fig. 3A), showing that expression of these novel candidate miRNAs also varied throughout the six developmental stages. In particular, we found that some miRNAs were stage-specifically expressed in the egg, first instar, second instar, fourth instar larval, or adult stage (Groups 2-I to 2-V in Fig. 3A), although each developmental stage of T. cancriformis is only short (0–0.5 h for first instar, 3–8 h for second instar, 13–22 h for third instar, and 26–37 h for fourth instar larvae), in accordance with the dramatic morphological changes in T. cancriformis larvae. To confirm the expression of these novel candidate miRNAs, a Northern blotting analysis of eight of them (from tcf-miR-n501 to tcf-miR-n508) was performed. As a consequence, the expression of five of the eight miRNAs was detected in one stage that showed higher read counts for each candidate miRNA (egg stage for tcf-miR-n502, tcf-miR-n503, and tcf-miR-n504; fourth instar larval stage for tcf-miR-n505; and adult stage for tcf-miR-n501) (Fig. 3B). These results show that at least these five novel miRNAs are actually expressed in the cells. Because the expression of these five miRNAs changed dynamically throughout the six developmental stages, they may be involved in stage-specific gene regulation. For instance, tcf-miR-n505 was increasingly expressed toward the fourth instar (Group 2-VI in Fig. 3A), with high read counts (4599 read counts) in fourth instar larvae (Supplemental Table S4). Of the 93 novel candidate miRNAs, 10 were strongly expressed in fourth instar larvae (three in Group 2-IV, seven in Group 2-VI in Fig. 3A). Several morphological characteristics of T. cancriformis change in the fourth instar larvae, when the compound eyes expand and the number of body segments increases. We speculate that some miRNAs that are strongly expressed in the fourth instar larvae, including tcf-miR-n505, are involved in these differentiation processes. To understand the exact functions of these miRNAs, an expression analysis at the tissue level must be performed in future work.
FIGURE 3.
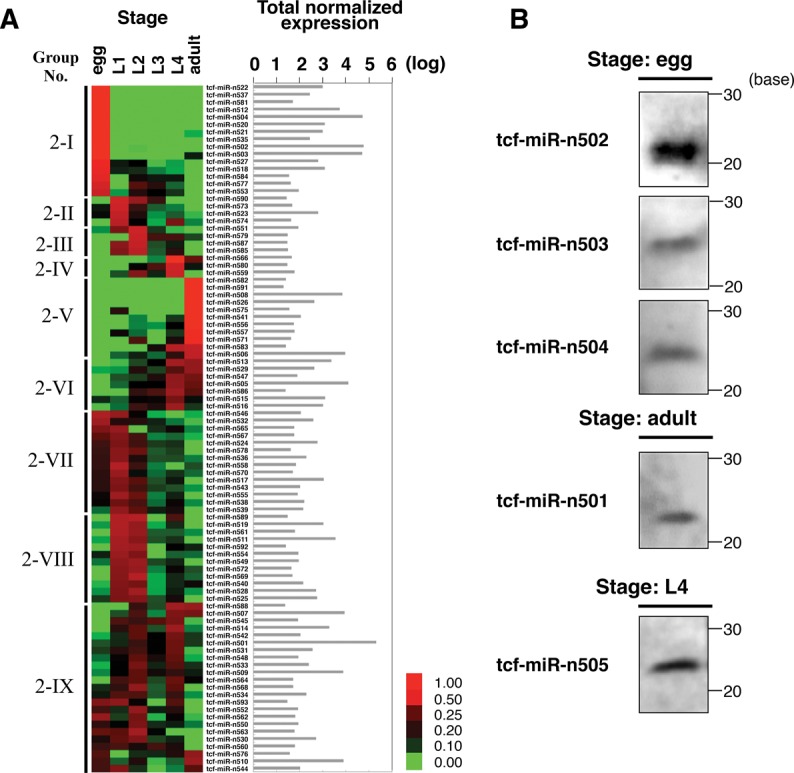
Expression profiles of novel candidate T. cancriformis miRNAs through the six developmental stages. (A) Expression profiles of novel candidate miRNAs in the deep-sequencing analysis. miRNA reads were normalized to the spike reads in each stage. A more intense red color indicates a strongly expressed miRNA, whereas a more intense green color indicates a more weakly expressed miRNA. The novel candidate miRNAs were categorized into nine groups based on their expression patterns (Groups 2-I to 2-IX). The total normalized expression for each of the six stages is listed. (B) Northern blotting analysis of the expression of five novel miRNAs.
Evolutionary conservation analysis of T. cancriformis miRNA sequences and miRNA clusters
To clarify the evolutionary position of T. cancriformis, a phylogenetic tree was constructed based on the 18S rRNA sequences of T. cancriformis and 12 model species from a wide range of Metazoa. The phylogenetic analysis showed that T. cancriformis is closely related to Daphnia pulex, in the crustaceans (Fig. 4A). The branching order of T. cancriformis was very similar to that on a phylogenetic tree constructed in a previous study (Bourlat et al. 2008). To investigate the evolution of the T. cancriformis miRNA sequences, we compared the 87 conserved T. cancriformis miRNAs found in this study with 7634 miRNAs previously reported in 12 model species and registered in miRBase release 20.0. These conserved miRNAs were roughly classified into six groups based on their sequence conservation (Groups 3-I to 3-VI in Fig. 4B), and 81 of the 87 T. cancriformis miRNAs shared sequence similarity (≥80%), based on seed matching, with those of arthropod species (Groups 3-I to 3-III and 3-V in Fig. 4B, and pink circle in Fig. 4C), although the organism is called a “living fossil.” Among these 81 miRNAs, 26 were also conserved in vertebrates (Groups 3-I to 3-III in Fig. 4B, and the overlapping region between the pink and light blue circles in Fig. 4C). Five of these 26 miRNAs (tcf-let-7-5p, tcf-miR-9a-5p, tcf-miR-125, tcf-miR-100, and tcf-miR-133) shared identical sequences with those of a wide range of Bilateria (indicated with arrows in Fig. 4B). In particular, tcf-miR-9a-5p had an identical sequence from organisms ranging from vertebrates to arthropods. Five miRNAs (tcf-miR-8-3p, tcf-miR-12, tcf-miR-276-3p, tcf-miR-993-3p, and tcf-miR-iab-4) also shared identical sequences with various arthropods (indicated with arrowheads in Fig. 4B).
FIGURE 4.

Evolution of the conserved miRNAs found in T. cancriformis. (A) Phylogenetic tree of T. cancriformis and 12 metazoan animals constructed from 18S rRNA sequences. The evolutionary position of T. cancriformis is shown in red. Bootstrap support values are indicated near the branches. (B) Distributions of conserved miRNAs across metazoan animals. The metazoan miRNAs that share sequence similarity (≥80%) and complete seed matches with T. cancriformis miRNAs are shown with colors. Based on miRNA conservation, 87 conserved miRNAs were classified into six groups (Groups 3-I to 3-VI). Arrows and arrowheads indicate the miRNAs highly conserved among bilaterians and arthropods, respectively. (hsa) H. sapiens, (mmu) M. musculus, (gga) G. gallus, (xtr) X. tropicalis, (dre) D. rerio, (ame) A. mellifera, (bmo) B. mori, (dme) D. melanogaster, (tcf) T. cancriformis, (dpu) Daphnia pulex, (cel) C. elegans, (nve) N. vectensis, (aqu) Amphimedon queenslandica. (C) Conserved miRNAs occurring in T. cancriformis and other metazoan animals. The numbers indicate the conserved T. cancriformis miRNAs that share sequence similarity with the miRNAs of vertebrates, arthropods, nematodes, cnidaria, or other species. (D) Nucleotide sequence comparison of let-7 in bilaterian animals. A sequence alignment of bilaterian let-7 is shown. The colored nineteenth nucleotide, “G” or “A,” shows the difference in the let-7 sequences of vertebrates and ecdysozoans. Underlining indicates the let-7 sequence of T. cancriformis.
According to the literature, 95 miRNAs of Marsupenaeus japonicus, which belongs to the Crustacea, have been reported (Ruan et al. 2011; Huang et al. 2012). Therefore, we comparatively analyzed the 180 T. cancriformis miRNAs (87 conserved miRNAs and 93 novel candidate miRNAs) and the 95 M. japonicus miRNAs. Based on seed matching, 55 of the 95 M. japonicus miRNAs shared sequence similarities (≥80%) with the T. cancriformis conserved miRNAs, whereas no M. japonicus miRNAs shared sequence similarity with the 93 novel T. cancriformis candidate miRNAs. Of these 55 miRNAs, 13 shared identical sequences and 44 shared ≥90% sequence similarity. The number of M. japonicus miRNAs that shared ≥90% sequence similarity with those of T. cancriformis was almost equivalent to the number it shares with other arthropods (from 36 to 42) (Fig. 4B), suggesting that M. japonicus and T. cancriformis are phylogenetically related. Sequence alignments of these well-conserved miRNAs in M. japonicus and other model species identified interesting characteristics of tcf-let-7. Generally, vertebrates have multiple copies of let-7, whereas ecdysozoans have only a single copy, and ecdysozoans let-7 is identical in vertebrates let-7a, except at a single nucleotide, one base from the 3′ end. In T. cancriformis, a single copy of let-7 was detected that is identical to vertebrate let-7c, which differs from arthropod let-7 at the nineteenth nucleotide, which is altered from “A” to “G” (e.g., dme-let-7-5p: UGAGGUAGUAGGUUGUAUAGU; hsa-let-7c-5p and tcf-let-7: UGAGGUAGUAGGUUGUAUGGUU) (Fig. 4D), suggesting that T. cancriformis let-7 evolved in a unique way. Further analysis is required to clarify why the T. cancriformis let-7 sequence is more similar to those of the vertebrates than to those of the arthropods.
Next, we investigated whether the 93 novel T. cancriformis candidate miRNAs share sequence similarity with the genomic DNA sequences of related model species, such as C. elegans, D. melanogaster, and Daphnia pulex. Forty-eight of the 93 novel T. cancriformis candidate miRNAs share ≥80% sequence similarity, based on complete seed matching, with the genomic DNA sequences of these organisms (Supplemental Fig. S2A). Of these 48 candidates, two in C. elegans, one in D. melanogaster, and six in Daphnia pulex potentially form secondary structures typical of precursor miRNAs (Supplemental Fig. S2B,C,S3A) and meet reliability criteria (2), (4), and (5). In the C. elegans genomic DNA sequence, two predicted miRNAs (cel-miR-n534 and cel-miR-n581), which are located in the exon region of clec-41 and a tRNA pseudogene, respectively (data not shown), share sequence similarity with tcf-miR-n534 and tcf-miR-n581, respectively. It has been reported that most miRNAs are located in intergenic and intronic regions. However, exonic miRNAs located within the exons of host protein-coding genes are rarely reported (Wang 2010), suggesting that cel-miR-n534 and cel-miR-n581 are unlikely to be expressed as miRNAs in C. elegans. In contrast, dme-miR-n539, which shares sequence similarity with tcf-miR-n539, is located in the intronic region of Toll-9 in the D. melanogaster genomic sequence (data not shown), suggesting that dme-miR-n539 is expressed as an miRNA in the fly. Daphnia pulex, which belongs to the crustaceans like T. cancriformis, has six candidate miRNAs that share sequence similarity with novel T. cancriformis miRNAs (Supplemental Fig. S3A). From a sequence alignment of these six miRNA sequences, three miRNAs (miR-n503, miR-n504, and miR-n512) are seen to have the same seed sequences (5′-UUGCACU-3′) in both T. cancriformis and Daphnia pulex (Supplemental Fig. S3B). Furthermore, these three miRNAs are closely located on genomic contigs (i.e., form an miRNA cluster) and are possibly encoded by the same pri-miRNA at both species (Supplemental Fig. S3C,D). tcf-miR-n503 and tcf-miR-n504 were also detected in our Northern blotting analysis (Fig. 3B), and most importantly, the expression of the miRNAs forming this miRNA cluster correlated significantly (r > 0.98, P ≤ 0.001; Supplemental Table 9). These data support the existence of an miRNA cluster in T. cancriformis.
We next checked whether another miRNA cluster exists in the T. cancriformis draft genome, which was determined in this study. In D. melanogaster, several miRNAs encoded within 10 kb on the same genomic strand are defined as an miRNA cluster (Marco et al. 2013). Because there is currently no information about the transcriptional unit in T. cancriformis, we set a more stringent criterion to define an miRNA cluster in this organism: several miRNAs encoded within 2 kb on the same genome strand. Our analysis showed that T. cancriformis had 16 miRNA clusters, consisting of 2–8 miRNAs (Supplemental Table S9). It has been reported that 74 (31%) of all annotated miRNAs in the genome of D. melanogaster are clustered (Marco et al. 2013), whereas 48 (28%) of the annotated miRNAs in the draft genome of T. cancriformis are clustered, suggesting that the proportions of miRNAs forming clusters are almost the same in the two species. We also noted that the expression of miRNAs forming 11 of these clusters was significantly correlated (P ≤ 0.05; Supplemental Table S9). These results suggest that the miRNAs in each cluster are transcribed from the same transcriptional unit. Next, we compared the compositions of the T. cancriformis miRNA clusters with those of the arthropod D. melanogaster and Tribolium castaneum clusters (Marco et al. 2010, 2013). Of the 11 miRNAs clusters of T. cancriformis, the compositions of four (tcf-miR-2a-2/2a/13/2b/71, tcf-miR-305/275, tcf-miR-92a/92b, tcf-let-7/miR-100) were (partly) conserved relative to those of D. melanogaster, whereas the compositions of five clusters (tcf-miR-2a-2/2a/13/2b/71, tcf-miR-2788/193, tcf-miR-305/275, tcf-miR-92a/92b, and tcf-let-7/miR-100) were (partly) conserved relative to those of Tribolium castaneum.
Phylogenetic evolutionary analysis of T. cancriformis DICER and AGO family proteins
A bioinformatics analysis showed that T. cancriformis has only one DICER (dicer1), three AGO (ago1–3), one PIWI (piwi), and one AUB (aubergine) (Figs. 5,6) (see Materials and Methods). These were predicted using DICER and AGO family proteins from Homo sapiens, D. melanogaster, M. japonicus, and Daphnia pulex as the query sequences (see Materials and Methods). We refer to each protein as “tcf XXX” (e.g., tcf DICER1) hereafter. Triops cancriformis dicer1 encodes the tcf DICER1 protein of 2048 amino acids (aa), and we predicted five domains: DEAD (E-value = 1.51 × 10−21), helicase C-terminal (E-value = 3.00 × 10−16), PAZ (E-value = 1.53 × 10−24), RNase III 1 (E-value = 2.47 × 10−23), and RNase III 2 (E-value = 1.45 × 10−47) in tcf DICER1. The dsrm domain, which is the RNA-binding domain in the C-terminal region, was not predicted in our first prediction because the E-value was high. However, by taking into consideration the conservation of amino acid sequences, dsrm can be regarded a domain of tcf DICER1 (Fig. 5A; Supplemental Fig. S4). Tcf DICER1 shows 44.9% amino acid identity and 76.4% similarity with Daphnia pulex DICER1, and 20.5% identity and 56.8% similarity with D. melanogaster DCR1. The six functional domains are particularly well conserved among these related species (Supplemental Fig. S4).
FIGURE 5.
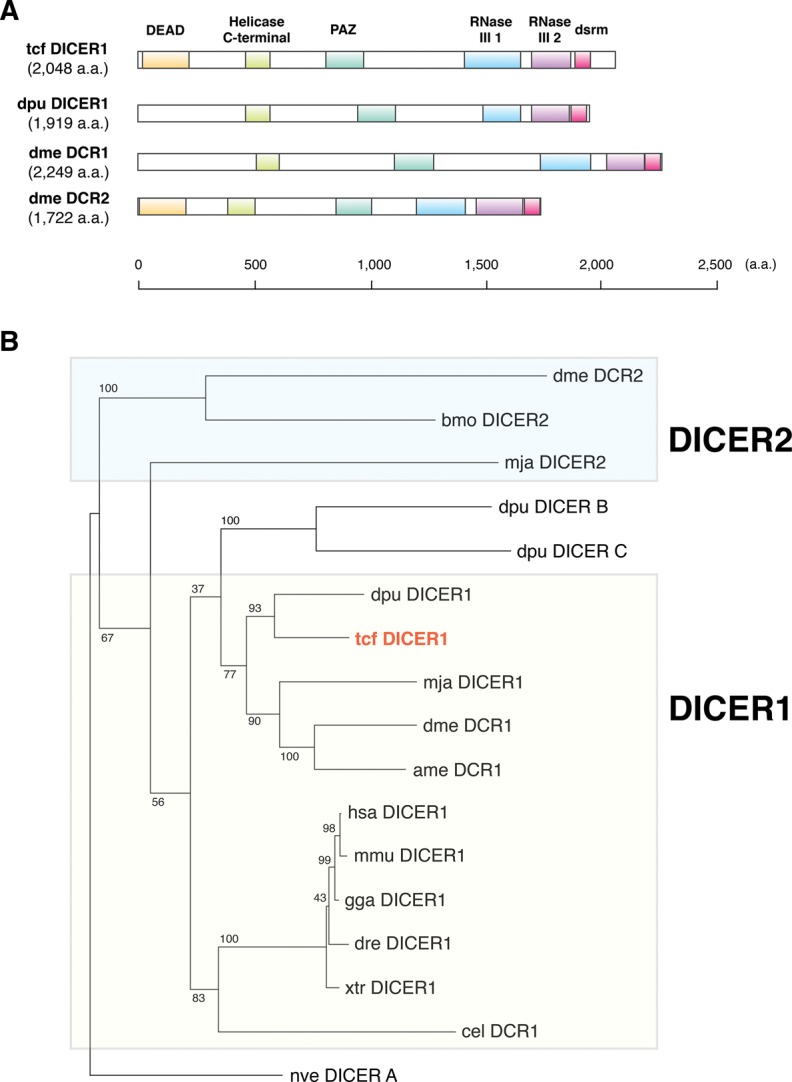
Evolution of T. cancriformis DICER proteins. (A) Comparison of the domain structures of the DICER proteins of T. cancriformis, Daphnia pulex, and D. melanogaster. Each domain structure is based on UniProt and SMART predictions. (B) Phylogenetic tree of DICER proteins from several metazoan species. The evolutionary position of tcf DICER1 is shown in red. Bootstrap support values are indicated near the branches.
FIGURE 6.

Evolution of T. cancriformis AGO family proteins. (A) Comparison of the domain structures of the AGO family proteins of T. cancriformis, Daphnia pulex, and D. melanogaster. Each domain structure is based on UniProt and SMART predictions. (B) Phylogenetic tree of the AGO family proteins of several metazoan species. The evolutionary positions of the tcf AGO family proteins are shown in red. Bootstrap support values are indicated near the branches.
To investigate the evolution of the tcf DICER1 protein, a phylogenetic tree was constructed using the amino acid sequences of DICER proteins from T. cancriformis and several model species. On the phylogenetic tree, tcf DICER1 was positioned close to those of the crustacean Daphnia pulex (Fig. 5B). It has been reported that most arthropods, including D. melanogaster, express both DICER1 and DICER2 proteins, and that most arthropod DICER1 protein have lost the DEAD domain (Mukherjee et al. 2013). In contrast, vertebrates and C. elegans have single DICER proteins (DICER1), containing the DEAD domain. Our analysis showed that T. cancriformis has a single DICER1 protein containing the DEAD domain with the DECH motif (Supplemental Fig. S4), suggesting that this domain is functional. DEAD is the RNA helicase domain in the N-terminal region of DICER1, and may be involved in the autoinhibition of the DICER activity (Ma et al. 2008). We found that tcf DICER1 is also more similar to the DICER1 proteins in the vertebrates than to those in the arthropods, like the let-7 miRNA, suggesting that miRNA systems of T. cancriformis evolved in a unique fashion. Because only the DICER1 protein was found in T. cancriformis, we looked for the DICER2 protein among the DICER proteins of other species, such as Bombyx mori and Litopenaeus vannamei (see Materials and Methods). However, we found no DICER2 protein containing the PAZ, RNase III 1, and RNase III 2 domains encoded in the T. cancriformis genomic DNA contigs. However, there is still a slight possibility that T. cancriformis has a DICER2 protein in which the domains differ considerably from those of previously identified DICER proteins, or that T. cancriformis DICER2 will be found when more precise genomic sequence information becomes available, because the current data are draft genome sequences that still include many gaps.
The AGO family proteins play important roles in small-RNA-guided gene-silencing processes (Meister 2013), and are basically subdivided into the AGO subfamily and PIWI subfamily. In this study, we identified tcf AGO1 (884 aa) and tcf AGO2 (786 aa) of the AGO subfamily, and tcf AGO3 (744 aa), tcf PIWI (782 aa), and tcf AUB (1291 aa) of the PIWI subfamily in T. cancriformis. We predicted that all these tcf AGO family proteins contain PAZ and Piwi domains, known from the AGO family proteins of other species (Fig. 6A). Most of the PAZ and Piwi domains were predicted with E-values of <1.00 × 10−19, with the exception of the PAZ domains in AGO1 (E-value = 5.41 × 10−3) and AGO2 (E-value = 0.46), indicating that the prediction of these AGO family proteins is reliable. It has been reported that the AGO3, PIWI, and AUB proteins are involved in piRNA biogenesis (Brennecke et al. 2007; Gunawardane et al. 2007). Although the presence of piRNAs in T. cancriformis has yet to be demonstrated, it is highly likely that T. cancriformis has a piRNA regulatory system. When we examined the sequence similarities in the AGO family proteins, the tcf AGO1 protein shared a high degree of similarity with those of Daphnia pulex and D. melanogaster (96.6% and 89.9% amino acid identity, respectively), and most of the amino acid sequences of the PAZ and Piwi domains are identical to the corresponding domains in these species (Supplemental Fig. S5A). However, the tcf AGO2 protein showed 35.8% and 19.2% amino acid identity to the Daphnia pulex and D. melanogaster protein, respectively, and other tcf AGO family proteins showed 15.6%–46.3% identity with the proteins of these species. The D. melanogaster AGO proteins reportedly play distinct roles. For instance, AGO1 is involved in miRNA-directed RNA cleavage in RNAi, and AGO2 is involved in siRNA-directed RNAi (Okamura et al. 2004). In contrast, H. sapiens AGO1–4 are involved in miRNA biogenesis (Peters and Meister 2007). Our phylogenetic analysis positioned T. cancriformis AGO1 close to AGO1 from Daphnia pulex, M. japonicus, D. melanogaster, and AGO1–4 from H. sapiens, whereas the other AGO family proteins were diverged from the corresponding proteins in other species (Fig. 6B). Because the miRNA regulatory system is largely conserved and plays important roles in eukaryotes, it is conceivable that the AGO1 proteins are under selective pressure to maintain their functional domains and their functions. In conclusion, throughout this paper, we have shown the importance of nonmodel species, as well as model species, in the evolutionary analysis of miRNAs. We have also provided detailed sequence information previously unknown to the scientific community involved in RNA research. This study provides a foundation for further discussion of the evolution of miRNAs and the components of the RNAi machinery.
MATERIALS AND METHODS
Triops cancriformis culture
Triops cancriformis (adults and eggs) were obtained from two rice fields (Sakata, Yamagata, Japan and Higashitagawa-gun, Yamagata, Japan). Hundreds of eggs were placed in water and exposed to light for 24 h to enhance the hatching efficiency (Takahashi 1975). After hatching, the larvae were incubated at 20°C and kept under a 14.5 h light:9.5 h dark cycle. According to a previous study (Igarashi 1971), the developmental stages of T. cancriformis are defined based on average body length (first instar: 530 μm; second instar: 590 μm; third instar: 800 μm; and fourth instar: 1180 μm). More concretely, the T. cancriformis larvae at each developmental stage were harvested after the appropriate incubation period after hatching (0–0.5 h for first instar, 3–8 h for second instar, 13–22 h for third instar, and 26–37 h for fourth instar larvae). Cultured T. cancriformis specimens (1 mo after hatching; body length: 1–3 cm) were used as the adult samples.
Deep sequencing of T. cancriformis small RNA and genomic DNA
To construct the small RNA libraries, 500 eggs, 500 first instar larvae, 500 second instar larvae, 500 third instar larvae, 500 fourth instar larvae, and eight adults were used. Each sample was ground with a mortar and pestle, and the total RNAs were extracted with TRIzol Reagent (Invitrogen), according to manufacturer's instructions. To normalize the reads among the developmental stages, two small RNA spikes (spike1: p-AACUGUGUCUUUUCUGAAUAGA; and spike2: p-UAUUUAGAAUGGCGCUGAUCUG) corresponding to mammal-specific small RNAs (Wang et al. 2011) were added at different concentrations (spike 1, 20 fmol; spike 2, 0.5 fmol) to 20 μg of total RNA (or 30 μg for egg RNA only). The small RNA fraction (∼12–45 nt), including miRNAs, was isolated and purified from these total RNA samples by gel electrophoresis. Takara Bio Incorporated constructed the small RNA libraries and performed the deep-sequencing analysis of the libraries. A cDNA library was constructed as described in a previous study (Pfeffer et al. 2005). Briefly, each small RNA was directly joined to the 3′ adaptor and 5′ adaptor with T4 RNA ligase. The ligation product was reverse transcribed and amplified with polymerase chain reaction (PCR). The nucleotide sequences were determined with the Illumina HiSeq 2000 (Illumina).
To construct the genomic DNA library, two adult specimens were used. After the specimens were ground, the genomic DNA was extracted with the GNOME DNA Isolation Kit (BIO101), according to the manufacturer's protocol, and treated with ribonuclease mix solution (Wako Pure Chemical Industries) for 30 min at 37°C. Takara Bio Incorporated constructed the genomic DNA library and performed the deep-sequencing analysis with Illumina Hiseq 2000.
Computational extraction of conserved T. cancriformis miRNAs
A six-step filtering approach was used to extract the reliable T. cancriformis candidate miRNAs from the small RNA deep-sequencing data. In step 1, unique sequences with their associated count numbers were obtained and low-quality reads, sequence errors containing the character “N,” low-quality base calls, and low count reads (<5) were discarded. In step 2, the small RNA reads that completely matched the genomic DNA contigs obtained from our deep sequences were retained. In step 3, sequence reads whose lengths corresponded to the miRNA fraction (18–24 nt) were extracted. In step 4, the sequence reads that mapped to the T. cancriformis cDNAs (3981 expressed sequence tags and 579 genes) from the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov, October 2013), or to the internal transcribed spacer (ITS) region (AB930494) between 18S rRNA and 28S rRNA were removed. In step 5, the BLASTN program (Camacho et al. 2009) was used to search the T. cancriformis miRNAs, and sequence reads were compared based on sequence similarities to already reported miRNA sequences from other species registered in miRBase Release 20.0 (June, 2013) (Kozomara and Griffiths-Jones 2013). Specifically, sequence reads with ≥80% sequence similarity to other reported miRNAs and with a complete seed match with at least two species (one of which was T. cancriformis) were extracted. In this study, we defined the seed sequence as occurring at either nucleotide positions 1–7, 2–8, or 3–9 from the 5′ side of the miRNA. In step 6, each genomic contig corresponding to the sequence read was analyzed with an RNA-folding program (Hofacker 2003) to determine whether the nucleotide sequence could form the potential secondary structure typical of precursor miRNAs. In this process, small RNA reads were mapped to the genomic DNA sequences corresponding to the miRNA precursor sequences. We confirmed that most abundant small RNA reads were identical to the candidate miRNAs, and that the small RNA mapping patterns against the genomic DNA sequences did not resemble the degradation products of mRNAs. Finally, the conserved T. cancriformis miRNAs were designated “tcf-miR-XX,” with identifying numbers (XX; e.g., tcf-miR-1) according to Ambros et al. (2003).
Prediction of novel T. cancriformis candidate miRNAs
Novel T. cancriformis candidate miRNAs were predicted with miRDeep2 (Friedlander et al. 2012) using the sequence reads that remained after the removal of reads that did not match the T. cancriformis cDNAs. To improve the performance of miRDeep2, mature and precursor T. cancriformis candidate miRNAs and previously reported miRNAs were used as the positive input data set. After prediction, we used three thresholds to detect the reliable novel candidate miRNAs: (1) the lowest miRDeep2 score cutoff (5.0) that yielded the highest signal-to-noise ratio (5.3); (2) a significant Randfold P value (equal to or lower than 0.05) for the potential miRNA precursor; and (3) novel nonredundant miRNA precursor candidates were permitted no mismatches between the small RNA reads and genomic DNA contigs. Novel T. cancriformis candidate miRNAs were designated “tcf-miR-n5XX” with identifying numbers (XX; e.g., tcf-miR-n501).
Expression profiles of T. cancriformis candidate miRNAs
To compare the relative levels of small RNA expression in the six different developmental stages, read normalization was performed using two spike RNAs, as described previously (Wang et al. 2011). The read number obtained from the second instar larvae was defined as the standard by setting it to 20 million, and the read numbers for the other stages were normalized to it. Read normalization was as described previously (Wang et al. 2011). The normalized expression profiles of T. cancriformis miRNAs are listed with their total normalized expression. The miRNA profiles were clustered using Cluster 3.0 (Eisen et al. 1998), followed by manual adjustment, and visualized with Java Treeview using a pixel setting value of 2.0 (Saldanha 2004).
Northern blot analysis
Total RNA was extracted from each developmental stage of T. cancriformis with TRIzol Reagent (Invitrogen), according to the manufacturer's instructions. The total RNA (0.05–20 μg) was separated on denaturing 12.5% polyacrylamide gel containing 8 M urea, and transferred to Hybond-N+ membrane (GE Healthcare) by electroblotting. After UV crosslinking, the membrane was prehybridized in DIG Easy Hyb buffer (Roche Diagnostics) for 30 min at 30°C or 37°C, depending on the oligodeoxynucleotide used. In some cases, hybridization buffer made in-house containing 0.6 M sodium citrate, 0.06 M NaCl, Denhardt's solution (1% Ficoll, 1% polyvinylpyrrolidone, and 1% bovine serum albumin), 0.1 mg/mL UltraPure Salmon Sperm DNA Solution (Invitrogen), and 0.5% SDS, was used. A biotin-labeled antisense oligodeoxynucleotide was prepared using the Biotin 3′ End DNA Labeling Kit (Pierce Biotechnology), and hybridization was performed in the same buffer with the labeled antisense oligodeoxynucleotide overnight at 30°C or 37°C. The membrane was then washed with buffer containing 0.6 M sodium citrate, 0.06 M NaCl, and 0.5% SDS at either 30°C or 37°C. The nonisotopic blots were visualized with ECF Substrate (GE Healthcare) and the images were captured with a Molecular Imager FX Pro (Bio-Rad Laboratories). The band intensities were also analyzed with Molecular Imager FX Pro. Using the quantified band intensities and read counts from the deep-sequencing data, we calculated Pearson's correlation with the StatPlus:mac LE.2009 software (AnalystSoft, Inc.). The probes used to detect T. cancriformis miRNAs were as follows:
tcf-let-7-5p_comp: 5′-AACCATACAACCTACTACCTCA-3′
tcf-miR-87_comp: 5′-ACGCACCTGAAGCTTTGCTCAA-3′
tcf-miR-125_comp: 5′-TCACAAGTTAGGGTCTCAGGGA-3′
tcf-miR-1_comp: 5′-CTCCATACTTCTTTACATTCCA-3′
tcf-miR-2b_comp: 5′-CTCGTCAAAGCTGGCTGTGATA-3′
tcf-miR-12_comp: 5′-CCAGTACCTGATGTAATACTCA-3′
tcf-miR-34_comp: 5′-CAACCAGCTAACCACACTGCCA-3′
tcf-miR-133_comp: 5′-ACAGCTGGTTGAAGGGGACCAA-3′
tcf-miR-184-3p_comp: 5′-GCCCTTATCAGTTCTCCGTCCA-3′
tcf-miR-276-3p_comp: 5′-AGAGCACGGTATGAAGTTCCTA-3′
tcf-miR-279a_comp: 5′-TGGATGAGTGTGGATCTAGTCA-3′
tcf-miR-375_comp: 5′-TAACTCGAGCCGAACGAACAAA-3′
tcf-miR-750_comp: 5′-TGAGCTGGAAGAGATAGATCTGG-3′
tcf-miR-n501_comp: 5′-AGCTGTCAATCATATAACCAAGT-3′
tcf-miR-n502_comp: 5′-CAGGATGAACCGCACCCAGTGA-3′
tcf-miR-n503_comp: 5′-TCGCCTCGAACCATACAGTGCAA-3′
tcf-miR-n504_comp 5′-AAGCCCACTACCGGTTAGTGCAA-3′
tcf-miR-n505_comp: 5′-ACGCACCTGATGATTTGCTCAC-3′
tcf_5.8S_comp: 5′-CAGCGTTCTTCATCGATCCACGAGCCGAGTGATCC-3′
Evolutionary conservation of T. cancriformis miRNAs
To identify the conserved T. cancriformis miRNAs, we first collected the miRNA sequences from miRBase release 20.0 for 12 model species: H. sapiens, Mus musculus, Gallus gallus, Xenopus tropicalis, Danio rerio, Apis mellifera, B. mori, D. melanogaster, Daphnia pulex, C. elegans, N. vectensis, and Amphimedon queenslandica. The BLASTN program was used to compare the distributions of the miRNAs across the species. The conservation criteria were defined as ≥80% sequence identity shared across the miRNAs of all 12 species, with complete seed matching.
We examined the conservation of novel T. cancriformis candidate miRNAs in model species. The T. cancriformis miRNA sequences of novel candidates were compared with the genomic DNA sequences of model species (D. melanogaster, Daphnia pulex, and C. elegans) using BLASTN. The D. melanogaster and C. elegans genomes were downloaded from the University of California at Santa Cruz (UCSC) genome browser (http://genome.ucsc.edu/) and the Daphnia pulex genome was obtained from Joint Genome Institute (JGI) (http://www.jgi.doe.gov/). If those small RNAs in related species shared ≥80% sequence similarity with the novel miRNA sequences of T. cancriformis with complete seed matching, were predicted to form the appropriate secondary structure, and met the reliability criteria (2), (4), and (5), these small RNA sequences were considered potential candidate miRNAs in the corresponding species.
Construction of a phylogenetic tree
A maximum likelihood tree of 13 animal species, including T. cancriformis, was constructed based on 18S rRNA sequences (including partial 18S rRNA sequences) obtained from NCBI. Seaview version 4.4.0 (Gouy et al. 2010) was used to align the sequences and construct the phylogenetic tree. The multiple alignment of these 18S rRNAs was generated with MUSCLE version 3.8.31 (Edgar 2004) and the phylogenetic analysis was performed with phyML 3.0 (Guindon et al. 2010) with the GTR model. Support values were calculated with 1000 bootstrap replications. The amino acid sequences of the DICER proteins and AGO family proteins (including the predicted DICER and AGO family proteins) were obtained from either previous studies (Grimson et al. 2008; Schurko et al. 2009; Mukherjee et al. 2013), JGI, or UniProt (http://www.uniprot.org, October 2013), and multiple alignments of these amino acid sequences were generated with MUSCLE. The aligned sequences were edited manually and the gaps were trimmed. The phylogenetic trees were constructed using the maximum likelihood method with phyML 3.0 with the LG model for the DICER proteins, and with the distance neighbor-joining method for the AGO family proteins. Support values were calculated with 1000 bootstrap replications.
Prediction of the components of the RNAi machinery
The T. cancriformis genomic DNA contigs were first searched for sequences encoding the RNAi machinery components (DICER and AGO family proteins) with TBLASTN using the default parameters. As the query sequences, we used the amino acid sequences of the DICER and AGO family proteins and their functional domains from H. sapiens, D. melanogaster, M. japonicus, and Daphnia pulex. For H. sapiens, D. melanogaster and M. japonicus, these query sequences were obtained from UniProt (H. sapiens DICER1, Q9UPY3; AGO1, Q9UL18; AGO2, Q9UKV8; AGO3, Q9H9G7; AGO4, Q9HCK5; PIWIL1, Q96J94; PIWIL2, Q8TC59; PIWIL3, Q7Z3Z3; PIWIL4, Q7Z3Z4; for D. melanogaster DCR1, Q9VCU9; DCR2, A1ZAW0; AGO1, Q27IR0; AGO2, Q9VUQ5; AGO3, Q7PLK0; PIWI, Q9VKM1; AUB, O76922; and for M. japonicus DICER1, D2XYX5; DCR2, H6WZT1; AGO1, J7I7H1; AGO2, J7MCI3). For Daphnia pulex, the amino acid sequences of these proteins were obtained from previous studies (Schurko et al. 2009; Mukherjee et al. 2013) and JGI (DICER1, EFX72380; DICER B, EFX69538; DICER C, EFX86072; AGO1, 305022; AGO2, 311791; AGO3, 442510; AUB A, 239845; AUB B, 220987; AUB C, 308681; AUB F, 195225). The domain sequences of these proteins were predicted with SMART version 7.0 (Letunic et al. 2012). When DNA contigs that were partly similar to the deduced DICER or AGO proteins were identified, the exonic regions were predicted with AUGUSTUS ver. 2.7 (Stanke et al. 2006) and GENSCAN 1.0 (Burge and Karlin 1997). The domain information for each protein was checked with SMART version 7.0. We chose the candidates that contained PAZ, RNase III 1, and RNase III 2 domains for the DICER proteins, and the PAZ and Piwi domains for the AGO family proteins. In the DICER2 search, we looked for further homologous sequences. A TBLASTN search was performed against the T. cancriformis genomic DNA contigs with the default parameters. As query sequences, the amino acid sequences of the proteins and the functional domains of DICER2 were used in four species: B. mori (D7UT11), Schmidtea mediterranea (XP_002574802), L. vannamei (F5AW47), and Tribolium castaneum (NP_001107840). The amino acid sequences of these proteins were obtained from a previous study (Mukherjee et al. 2013) or UniProt, and their domain sequences were predicted with SMART version 7.0. The amino acid sequences of the proteins and the functional domains of Daphnia pulex DICER B and DICER C, and tcf DICER1 were also used to extract any other DICER proteins containing their functional domains.
DATA DEPOSITION
The nucleotide sequences of the T. cancriformis small RNAs and genomic DNA have been deposited in the DNA Data Bank of Japan (DDBJ) (http://www.ddbj.nig.ac.jp/index-e.html) under accession numbers PRJDB1672 and PRJDB1662, respectively.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Keiji Igarashi (Tohoku University of Community Service and Science, Japan) for sharing his extensive knowledge of the tadpole shrimp. We also thank Dr. Yoshiki Ikeda (Institute for Advanced Biosciences, Keio University, Japan) for valuable discussions and technical assistance, and all the members of the RNA group at the Institute for Advanced Biosciences, Keio University, Japan, for their insightful discussions. This research was supported, in part, by research funds from the Yamagata Prefectural Government and Tsuruoka City, Japan, and a research grant from the Japan Society for the Promotion of Science (JSPS).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.045799.114.
NOTE ADDED IN PROOF
After the acceptance of this manuscript, it was found that triops novel candidate miRNA tcf-miR-n-505 is very similar to the miR-87 in Culex quinquefasciatus (Skalsky et al. 2010) and Aedes aegypti (Li et al. 2009).
REFERENCES
- Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, et al. 2003. A uniform system for microRNA annotation. RNA 9:277–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297. [DOI] [PubMed] [Google Scholar]
- Bartel DP 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourlat SJ, Nielsen C, Economou AD, Telford MJ 2008. Testing the new animal phylogeny: a phylum level molecular analysis of the animal kingdom. Mol Phylogenet Evol 49:23–31. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128:1089–1103. [DOI] [PubMed] [Google Scholar]
- Burge C, Karlin S 1997. Prediction of complete gene structures in human genomic DNA. J Mol Biol 268:78–94. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li Q, Wang J, Guo X, Jiang X, Ren Z, Weng C, Sun G, Wang X, Liu Y, et al. 2009. Identification and characterization of novel amphioxus microRNAs by Solexa sequencing. Genome Biol 10:R78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D 1998. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci 95:14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N 2012. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27:221–224. [DOI] [PubMed] [Google Scholar]
- Grimson A, Srivastava M, Fahey B, Woodcroft BJ, Chiang HR, King N, Degnan BM, Rokhsar DS, Bartel DP 2008. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature 455:1193–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. [DOI] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, Siomi MC 2007. A slicer-mediated mechanism for repeat-associated siRNA 5′ end formation in Drosophila. Science 315:1587–1590. [DOI] [PubMed] [Google Scholar]
- Hofacker IL 2003. Vienna RNA secondary structure server. Nucleic Acids Res 31:3429–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, Xu D, Zhang X 2012. Characterization of host microRNAs that respond to DNA virus infection in a crustacean. BMC Genomics 13:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi K 1971. Ecological studies on Triops longicaudatus (LE CONTE) inhabiting Shonai District, Japan. (III) Some features of the young individuals. J Yamagata Agric For Soc 28:4–5. [Google Scholar]
- Kim VN, Han J, Siomi MC 2009. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10:126–139. [DOI] [PubMed] [Google Scholar]
- Kloosterman WP, Plasterk RH 2006. The diverse functions of microRNAs in animal development and disease. Dev Cell 11:441–450. [DOI] [PubMed] [Google Scholar]
- Korn M, Rabet N, Ghate HV, Marrone F, Hundsdoerfer AK 2013. Molecular phylogeny of the Notostraca. Mol Phylogenet Evol 69:1159–1171. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S 2013. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaman D, Chen PY, Fak J, Yalcin A, Pearce M, Unnerstall U, Marks DS, Sander C, Tuschl T, Gaul U 2005. Antisense-mediated depletion reveals essential and specific functions of microRNAs in Drosophila development. Cell 121:1097–1108. [DOI] [PubMed] [Google Scholar]
- Letunic I, Doerks T, Bork P 2012. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res 40:D302–D305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB 2003. Prediction of mammalian microRNA targets. Cell 115:787–798. [DOI] [PubMed] [Google Scholar]
- Li S, Mead EA, Liang S, Tu Z 2009. Direct sequencing and expression analysis of a large number of miRNAs in Aedes aegypti and a multi-species survey of novel mosquito miRNAs. BMC Genomics 10:581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma E, MacRae IJ, Kirsch JF, Doudna JA 2008. Autoinhibition of human dicer by its internal helicase domain. J Mol Biol 380:237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A, Hui JH, Ronshaugen M, Griffiths-Jones S 2010. Functional shifts in insect microRNA evolution. Genome Biol Evol 2:686–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A, Ninova M, Ronshaugen M, Griffiths-Jones S 2013. Clusters of microRNAs emerge by new hairpins in existing transcripts. Nucleic Acids Res 41:7745–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathers TC, Hammond RL, Jenner RA, Hänfling B, Gómez A 2013. Multiple global radiations in tadpole shrimps challenge the concept of ‘living fossils’. Peer J 1:e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G 2013. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 14:447–459. [DOI] [PubMed] [Google Scholar]
- Mukherjee K, Campos H, Kolaczkowski B 2013. Evolution of animal and plant dicers: early parallel duplications and recurrent adaptation of antiviral RNA binding in plants. Mol Biol Evol 30:627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahvi A, Shoemaker CJ, Green R 2009. An expanded seed sequence definition accounts for full regulation of the hid 3′ UTR by bantam miRNA. RNA 15:814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Ishizuka A, Siomi H, Siomi MC 2004. Distinct roles for Argonaute proteins in small RNA-directed RNA cleavage pathways. Genes Dev 18:1655–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters L, Meister G 2007. Argonaute proteins: mediators of RNA silencing. Mol Cell 26:611–623. [DOI] [PubMed] [Google Scholar]
- Pfeffer S, Lagos-Quintana M, Tuschl T 2005. Cloning of small RNA molecules. Curr Protoc Mol Biol Chapter 26: Unit 26 24. [DOI] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G 2000. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403:901–906. [DOI] [PubMed] [Google Scholar]
- Ruan L, Bian X, Ji Y, Li M, Li F, Yan X 2011. Isolation and identification of novel microRNAs from Marsupenaeus japonicus. Fish Shellfish Immunol 31:334–340. [DOI] [PubMed] [Google Scholar]
- Saldanha AJ 2004. Java Treeview—extensible visualization of microarray data. Bioinformatics 20:3246–3248. [DOI] [PubMed] [Google Scholar]
- Schurko AM, Logsdon JM Jr, Eads BD 2009. Meiosis genes in Daphnia pulex and the role of parthenogenesis in genome evolution. BMC Evol Biol 9:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sempere LF, Sokol NS, Dubrovsky EB, Berger EM, Ambros V 2003. Temporal regulation of microRNA expression in Drosophila melanogaster mediated by hormonal signals and broad-Complex gene activity. Dev Biol 259:9–18. [DOI] [PubMed] [Google Scholar]
- Siomi MC, Sato K, Pezic D, Aravin AA 2011. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol 12:246–258. [DOI] [PubMed] [Google Scholar]
- Skalsky RL, Vanlandingham DL, Scholle F, Higgs S, Cullen BR 2010. Identification of microRNAs expressed in two mosquito vectors, Aedes albopictus and Culex quinquefasciatus. BMC Genomics 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Tzvetkova A, Morgenstern B 2006. AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol 7Suppl 1:S11.1–S11.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi F 1975. Effect of light on the hatching of eggs in Triops granarius (Notostraca: Triopsidae). Environ Control Biol 13:29–33. [Google Scholar]
- Takane K, Fujishima K, Watanabe Y, Sato A, Saito N, Tomita M, Kanai A 2010. Computational prediction and experimental validation of evolutionarily conserved microRNA target genes in bilaterian animals. BMC Genomics 11:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z 2010. MicroRNA: a matter of life or death. World J Biol Chem 1:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Czech B, Crunk A, Wallace A, Mitreva M, Hannon GJ, Davis RE 2011. Deep small RNA sequencing from the nematode Ascaris reveals conservation, functional diversification, and novel developmental profiles. Genome Res 21:1462–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Tomita M, Kanai A 2008. Perspective in the evolution of human microRNAs: copy number expansion and acquisition of target gene specialization. Prog Theor Phys Suppl 173:219–228. [Google Scholar]
- Wei C, Salichos L, Wittgrove CM, Rokas A, Patton JG 2012. Transcriptome-wide analysis of small RNA expression in early zebrafish development. RNA 18:915–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienholds E, Plasterk RH 2005. MicroRNA function in animal development. FEBS Lett 579:5911–5922. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.