

Abstract
A high-throughput assay for real-time measurement of translation rates in cell-free protein synthesis (SNAP assay) is described. The SNAP assay enables quantitative, real-time measurement of overall translation rates in vitro via the synthesis of O6-alkylguanine DNA O6-alkyltransferase (SNAP). SNAP production is continuously detected by fluorescence produced by the reaction of SNAP with a range of quenched fluorogenic substrates. The capabilities of the assay are exemplified by measurements of the activities of Escherichia coli MRE600 ribosomes and fluorescently labeled E. coli mutant ribosomes in the PURExpress translation system and by determination of the 50% inhibitory concentrations (IC50) of three common macrolide antibiotics.
Keywords: SNAP, in vitro translation, assay, single-molecule fluorescence, IC50, ribosome activity, translation rate
INTRODUCTION
The development of reliable and inexpensive cell-free protein synthesis has enabled not only the production of hard-to-express recombinant proteins (Jackson et al. 2004) but also the investigation of translation dynamics in vitro, allowing discovery of novel translation-inhibiting antibiotics (Brandi et al. 2007) and measurements of kinetic parameters of key steps in translation (Underwood et al. 2005). Recent studies have used fluorescence resonance energy transfer (FRET) experiments at the single-molecule scale to measure the dynamics of translation initiation and elongation directly in real time (Tsai et al. 2012; Chen et al. 2013a). Time-resolved bulk detection of protein synthesis is required to validate these results.
Current methods to measure translation rates detect properties of a translation product reporter, such as luciferase luminescence (Hale 2000), green fluorescent protein (GFP) fluorescence (Nomura et al. 2001), chloramphenicol acetyltransferase activity (Shaw 1975; Kim and Swartz 2001; Harvey et al. 2012), and 14C radioactivity (Bayley and Griffiths 1968). The throughput and generality of these methods are limited by the requirement for multiple steps of processing or specialized equipment to obtain a translation signal. An elegant high-throughput method has been reported in which synthesis of fast-maturing Emerald GFP (EmGFP) produces a detectable fluorescent signal upon translation (Rosenblum et al. 2012). However, this method is limited by the requirement of oxygen for maturation of GFP, which interferes with fluorescence detection at the single-molecule level. As a result, translation of GFP in corresponding single-molecule FRET experiments must be interrupted with oxygenation prior to visualization (Uemura et al. 2008), which complicates the comparison between the bulk assay and single-molecule FRET data.
Here we circumvent these limitations by developing an assay in which cell-free protein synthesis of a fast-reacting variant of the human suicide enzyme O6-alkylguanine DNA O6-alkyltransferase (SNAPf) is monitored in real time by its subsequent reaction with a quenched fluorogenic guanine substrate, producing covalently modified fluorescent SNAPf molecules that signal translation of a folded, active protein (Supplemental Fig. S1). Fusion of a C-terminal SecM stall sequence to SNAPf limits ribosome turnover, greatly simplifying the downstream analysis of translation kinetics. A wide variety of fluorogenic substrates are available (Sun et al. 2011), thereby enabling the selection of a substrate that minimizes spectral overlap with other components of the translation system. The reaction of SNAPf with a substrate can occur in anoxia, so real-time single-molecule FRET experiments of the translation of SNAPf may also proceed uninterrupted. The flexibility and high-throughput format make the SNAP assay a general real-time quantitative technique for measuring the overall translation rate of fluorescent and nonfluorescent cell-free protein synthesis and evaluating the effects of ribosomal mutations and macrolide antibiotics on ribosome activity and translation rate.
RESULTS
Designing the SNAP assay
The SNAP assay was designed to use a commercial reconstituted in vitro translation system (PURExpress ΔRibosome) as the source of cell-free protein synthesis machinery. PURExpress was chosen for its ease of use, defined formulation of purified components, and modifiability, which in the case of the ΔRibosome kit allowed us to substitute commercial ribosomes with wild-type and mutant variants prepared in-house. Wild-type ribosomes were isolated from the ribonuclease-deficient Escherichia coli strain MRE600, and the Cy3B-C30S and Cy5-Z50S mutant ribosomes are regularly used in intersubunit FRET experiments (Marshall et al. 2008; Tsai et al. 2012), thereby interfacing the SNAP assay with an established fluorescent translation system. SNAPf was used as the reporter enzyme, because of the large selection of fluorogenic substrates, suitable enzyme kinetics (see Supplemental Fig. S7), and potential ease of observation in anoxic single-molecule FRET experiments. The substrate CBG-549-QSY7 was chosen for its long half-life and low rates of nonenzymatic dequenching in a variety of reducing and nonreducing translation buffers (Supplemental Fig. S8), and the substrate CBG-488-TQ2 was selected for its rapid reactivity and limited spectral overlap with Cy3B (Sun et al. 2011). These enzyme–substrate combinations confer generality, ease of use, high throughput, and single-molecule FRET compatibility to the SNAP assay.
The SNAPf vector was constructed with a 54-nt 3′ SecM minimal arrest sequence (Nakatogawa and Ito 2002). Once translated, this 18-residue peptide interacts with the exit tunnel of the 50S subunit leading to translation stalling (Nakatogawa and Ito 2001; Yap and Bernstein 2009). The SecM arrest sequence has been shown to be an effective method of anchoring a mature protein to the ribosome immediately following translation in both single-molecule and bulk experiments (Evans et al. 2005; Uemura et al. 2008). The stalled 70S-SNAPf complex has a half-life of ∼60 min (Evans et al. 2005; Tsai et al. 2014), so turnover is still observed in longer time courses. The arrest sequence has two functions in the SNAP assay. First, the sequence limits ribosomal turnover without slowing initiation or elongation, while increasing the likelihood that the reaction between SNAPf and its substrate occurs before the ribosome can reinitiate. Second, SecM stalling is a common technique for single-molecule detection of translation products anchored to surface-immobilized ribosomes, enabling single-molecule experimentation using the same SNAPf construct as that in bulk studies.
Translation was assayed by adding ribosomes, SNAPf mRNA, and quenched fluorogenic substrate to a PURExpress ΔRibosome cell-free protein synthesis reaction and tracking the fluorescence produced by the subsequent reaction between translated SNAPf enzyme and its substrate. We first tested the response of the assay by translating SNAPf mRNA with wild-type ribosomes. The fluorescence due to protein synthesis was found to increase in intensity as a function of time, reflecting the reaction between newly synthesized SNAPf and its substrate. The rate of fluorescence production was directly proportional to ribosome concentration (Fig. 1B,C), and stable background fluorescence was observed when mRNA was withheld (Fig. 1A). The dependence of the SNAP assay on both mRNA and ribosomes in a translation reaction confirms that fluorescence intensity response is indeed correlated to SNAPf synthesis.
FIGURE 1.
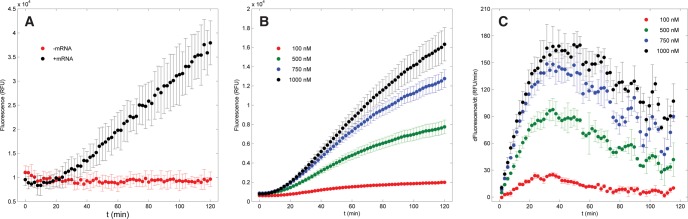
(A) Comparison of fluorescence generated in the presence or absence of SNAPf mRNA. Reaction conditions: 100 nM ribosomes, 0 or 100 nM mRNA, and 500 nM CBG-549-QSY7. (B) Fluorescence response over a range of wild-type ribosome concentrations. Reaction conditions: 100, 500, 750, or 1000 nM ribosomes, 1 μM mRNA, and 10 μM CBG-549-QSY7. (C) Differential curves for the fluorescence.
The reaction scale of the assay was configured so as to be compatible with high-throughput instrumentation. The final reaction volume of 10 μL minimizes reagent usage and is well suited for 384-well plates so that dozens of measurements could be recorded in parallel. With standard liquid-handling instruments and plate stackers, this amount can be extended to potentially hundreds of reactions that can be tested in quick succession.
Measuring translation inhibition
To show that the SNAP assay is sensitive to changes in in vitro translation, we examined the effects of three macrolide antibiotics, which inhibit translation by binding to the peptide exit tunnel, impeding nascent chain elongation (Mankin 2008). We measured SNAPf fluorescence as a function of time in in vitro translation systems in the presence of one of three common macrolides (erythromycin A, clarithromycin, or roxithromycin). A range of concentrations was tested for each macrolide, and the fluorescence curves were compared with positive (no macrolide) and negative controls (no mRNA). In Figure 2, production of SNAPf fluorescence is illustrated to be a function of both time and erythromycin A concentration. After an observation period of 2 h, SNAPf fluorescence was reduced by 24 ± 11%, 46 ± 12%, 74 ± 7%, and 80 ± 3% for treatments with 20, 40, 60, and 80 nM erythromycin A, respectively. Similar trends were observed with roxithromycin and clarithromycin (Supplemental Fig. S2). Overall, all macrolide-treated samples showed diminished fluorescence, and fluorescence was inversely proportional to macrolide concentration. This result demonstrates that the SNAP assay is sensitive to translation inhibition by small molecules.
FIGURE 2.
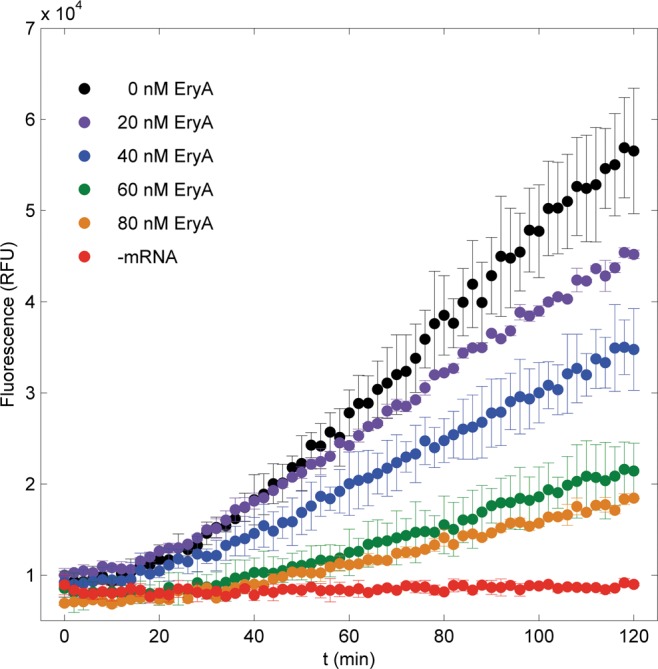
Fluorescence curves for in vitro SNAPf synthesis with and without erythromycin A treatment. Reaction conditions: 100 nM ribosomes, 100 nM mRNA, and 500 nM CBG-549-QSY7.
In addition to small-molecule inhibitors, ribosomal mutations may also cause translation dysfunction. To explore the SNAP assay's sensitivity to ribosomal mutations, cell-free protein synthesis reactions were performed with wild-type MRE600 ribosomes and the FRET-compatible mutants Cy3B-C30S and Cy5-Z50S. Fluorescence curves as functions of time were generated for four combinations of wild-type and fluorescent mutant ribosomal subunits (Fig. 3A). Intensity curves exhibited a short lag period of ∼20 min followed by exponential growth. Wild-type ribosomes produced intense fluorescence up to 13.0 ± 1.2 × 104 RFU, while SNAPf fluorescences in mutant ribosome samples maximized at 5.5 ± 0.1 × 104, 4.2 ± 0.3 × 104, and 4.6 ± 0.2 × 104 RFU for Cy5-Z50S, Cy3B-C30S, and the Cy3B-C30S/Cy5-Z50S double mutant, respectively. Numerical calculations of the differentials of these fluorescence curves followed the same trend, where the wild-type translation rate is greatest, followed by the Cy5-Z50S mutant, Cy3B-C30S/Cy5-Z50S double mutant, and Cy3B-C30S mutant, respectively (Fig. 3B). This diminution of fluorescence accumulation was similar to that of macrolide inhibition, thereby demonstrating the ability of the assay to detect changes in ribosome function.
FIGURE 3.

(A) Fluorescence curves of in vitro SNAPf translation by wild-type and mutant ribosomes. Points with error bars are fluorescence data, and solid lines are fits to the first-order kinetics model. All fits were validated by bootstrap analyses. Reaction conditions: 500 nM ribosomes, 2 μM mRNA, and 10 μM CBG-488-TQ2. (B) Differentials of fluorescence curves.
Single-molecule observation of SNAPf translation
To illustrate the compatibility of the SNAPf reporter with single-molecule fluorescence experiments, we then performed coupled in vitro transcription–translation reactions with ribosomes that had been immobilized on zero-mode waveguide (ZMW) chips (Fig. 4A). In accordance with standard protocols (Uemura et al. 2008), samples were prereacted at 37°C to allow for transcription and translation of SNAPf prior to deoxygenation and visualization. All experiments were performed with nonfluorescent C30S ribosomes that had been tethered to the chip surface via a complementary nonfluorescent biotinylated DNA oligomer, and SecM-anchored SNAPf was reacted with unquenched SNAP Surface 647 substrate. Chips were washed to remove any unreacted substrate. Full-chip average DY647 fluorescence was recorded and compared with reactions performed in the presence of an antibiotic, with the plasmid replaced with one encoding GFP, and in the absence of plasmid (Fig. 4B). A comparison of these interactions clearly shows that DY647 fluorescence is entirely dependent on the presence of the SNAPf plasmid as both conditions in which the plasmid is replaced or omitted resulted in ∼10-fold reductions in fluorescence. Furthermore, translation inhibition with chloramphenicol also abrogated SNAPf production for about a fivefold loss in fluorescence.
FIGURE 4.

(A) Experimental setup for ZMW-based single-molecule SNAPf experiments. (B) Full-chip average fluorescence values for SNAPf translation detected by ZMW. When the reaction is treated with a translation inhibitor, GFP is substituted for SNAPf, or plasmid is omitted, fluorescence decreases significantly. (C) The fluorescent signal from an individual ZMW well. Colocalization of red Cy5 signal (Cy5-Z50S) to green DY549 signal (SNAPf) is observed, and the dyes photobleach after 200 and 100 sec, respectively.
Using the SNAP Surface 549 substrate, experiments were also performed in which the fluorescence of each well of the chip was recorded and compared with plasmid-omitted controls. When translated with wild-type MRE600 50S ribosomes, 26.8% of ZMW wells showed DY549 fluorescence, as opposed to 9.2% when plasmid is omitted (signal is 2.9× background). The 50S subunit was then replaced with the Cy5-Z50S mutant in order to cross-reference DY549 and Cy5 fluorescences and to select against wells with background fluorescence due to nonspecific fluorophore–chip interactions. Figure 4C depicts one representative fluorescence pattern under these conditions; the presence of both green (DY549) and red (Cy5) signals in the well indicates an immobilized C30S-Z50S-SNAPf ternary complex, and both dyes photobleach after relatively long lifetimes of 100 and 200 sec, respectively. Translation reactions with Cy5-Z50S mutants produced colocalized DY549 and Cy5 fluorescence in 8.0% of ZMW wells, whereas only 2.7% showed colocalized signal in the absence of plasmid (signal is 3.0× background). As anticipated, two-dye colocalization reduced stochastic background fluorescence by a factor of 3.5, although the reduced Cy5-Z50S activity also reduced the amount of SNAPf fluorescence by the same amount. Thus, the signal-to-noise ratio remained ∼3 in both experiments. The statistically significant detection of SNAPf fluorescence via ZMW validates the compatibility of the SNAPf reporter with single-molecule fluorescence experiments.
DISCUSSION
Quantification of wild-type ribosome activity
The SNAP assay measures the composite of all translation-related processes, starting with mRNA recruitment during initiation and ending with the post-translational production of covalently modified fluorescent SNAPf. The SNAP reaction has been measured at rates that compare favorably with the maturation rate of EmGFP (see Supplemental Information; Iizuka et al. 2011), a previously reported effective signal of productive translation (Rosenblum et al. 2012). Also, the SecM minimal arrest sequence almost ensures that the SNAPf reaction occurs prior to the completion of termination. It is then reasonable to assume that the process of translation is the limiting step in the SNAP assay, and the composite of all translation-related processes and the SNAPf reaction simplifies to the sum of translation initiation, elongation, and termination through a pseudo-first-order kinetic model with respect to ribosome concentration.
Correspondingly, in vitro translation fluorescence curves (Fig. 3A) exhibit a short lag time, followed by an inflection point after 20 min that marks the beginning of an exponential first-order reaction curve. On this basis, starting at the inflection point, the curves were fitted to the following simple kinetic model:
In this function, a is a normalization parameter, b is the first-order rate constant, c represents the composite of all slow, nonenzymatic dequenching reactions, and d is an x-axis offset due to the aforementioned lag time. The translation activity is calculated from the fit as the product of the normalization parameter and rate constant, resulting in 2530 ± 480 RFU/min for wild-type ribosomes (Supplemental Table S1).
A standard curve of prereacted, commercial SNAPf protein of varying concentrations (Supplemental Fig. S6) allowed the conversion of fluorescence data to that of translated protein concentrations. As with the fluorescence curves, the converted data were fitted to the first-order kinetics model starting at f(20) (Fig. 5A). The activity for wild-type ribosomes was calculated as 60.8 ± 5.6 nM SNAPf/min. Accounting for the reaction mixture volume (10 µL) and ribosome concentration (500 nM), the specific activity may be expressed as 0.122 ± 0.011 pmol SNAPf/pmol ribosome/min (Supplemental Table S2). With the approximation that translation is rate-limiting in cell-free protein synthesis, this activity means that the process for one wild-type 70S ribosome to synthesize one molecule of SNAPf requires ∼8.2 ± 0.8 min.
FIGURE 5.
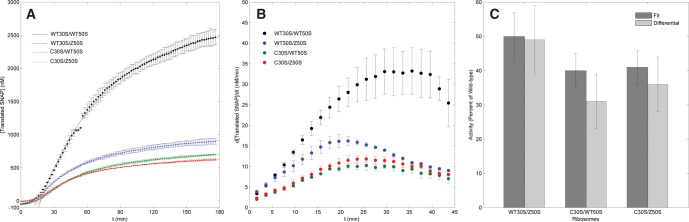
(A) SNAPf synthesis curve for wild-type and mutant ribosomes. SNAPf concentrations are data points with error bars, and the first-order kinetic fits are solid lines. Reaction conditions same as Figure 3A. (B) Differential analysis for SNAPf synthesis data. (C) Comparison of wild-type and mutant activities calculated from the first-order kinetics model and differential analysis.
It is also a useful exercise to compare this measured translation rate with previously reported in vivo and in vitro E. coli ribosome elongation rates. Since our calculated value represents the entire process of translation, a comparison requires the bold assumption that elongation is the rate-limiting step in this process. Correspondingly, under this model, the 233-residue SNAPf construct would have an elongation rate of almost 0.5 amino acids (aa)/sec. This rate is an order of magnitude slower than measurements of elongation in vivo (Kennel and Riezman 1977), but 0.5 aa/sec is comparable with other in vitro elongation measurements (Wagner et al. 1982). Given that this highly simplistic model provides a strict lower limit for elongation in the SNAP assay, actual elongation rates are likely on-par with values that have been previously reported.
The concentration curves were further analyzed by numerical calculations of the differentials of the data (Fig. 5B). Since derivatives represent the rates of SNAPf synthesis at each point in time, translation activities may be calculated independently from the fit models. For wild-type ribosomes, the maximum translation rate was calculated to be 33.2 ± 5.5 nM SNAPf/min, yielding a specific activity of 0.066 ± 0.011 pmol SNAPf/pmol ribosome/min, total translation time of 15.2 ± 2.6 min, and approximate elongation rate of 0.25 aa/sec. Compared with the activity calculated from the fit model, this activity differs only by a factor of 2 and remains within the reasonable approximations for in vitro translation rates.
Calculation of macrolide IC50 values
By benchmarking the kinetics of uninhibited wild-type translation, inhibition of translation due to macrolides can next be quantified. To calculate 50% inhibitory concentrations (IC50) for each of the antibiotics, end-point fluorescence values for each concentration of macrolide were normalized to the positive and negative controls to generate values that are percentages of the untreated reactions. These percentages were plotted as functions of macrolide concentration, allowing a linear fit to determine the concentration at which SNAPf fluorescence would be half of the untreated control. The IC50 values were calculated as 42 ± 10, 27 ± 7, and 14 ± 14 nM, for erythromycin A, roxithromycin, and clarithromycin, respectively (Fig. 6). Dissociation constants for these antibiotics have been previously reported as 36 ± 1.8, 20 ± 1, and 8 ± 0.5 nM, respectively (Dinos et al. 2003). The marked similarities between the IC50 values and dissociation constants serve to validate further the ability of the assay to quantify inhibition of translation by antibiotics.
FIGURE 6.

IC50 values calculated for the macrolide antibiotics erythromycin A, roxithromycin, and clarithromycin.
It is interesting to note that all three IC50 values fall below the theoretical lower limit of half of the ribosome concentration. This result supports a model of macrolide activity in which the macrolide induces a translation arrest state with a lifetime exceeding the binding time of the drug. Macrolides with 14-membered rings, such as the three tested here, have been found to have relatively rapid dissociation rates that are comparable with the rate of peptidyl-tRNA drop-off (Lovmar et al. 2004). Thus, the macrolide can dissociate immediately following peptidyl-tRNA drop-off, leaving the mRNA-70S complex stalled in an inactive state while the macrolide is free to bind to other ribosomes. Furthermore, elongating ribosomes with filled exit tunnels and terminating ribosomes stalled by the SecM arrest sequence are likely to be immune to macrolide action (Mankin 2008), leaving the remaining population susceptible to a comparably high concentration of free macrolide. Combined, these properties lend credence to the IC50 values measured using the SNAP assay.
Since the SNAP assay is insensitive to the mechanism of inhibition, a wide variety of known and novel antibiotics, regardless of class, can be evaluated in the future. The procedure for measuring translation inhibition with the SNAP assay is simple and does not require the interruption of translation for visualization, which is often the case for assays that detect luminescence, enzyme redox activity, and radioactivity. Thus, the SNAP assay is an uncomplicated, broad method for evaluating translation inhibition due to small-molecule inhibitors.
Quantification of mutant ribosome activities
Application of the first-order kinetics model to the mutant ribosome fluorescence curves allowed quantification of the impact of mutations and fluorescent labeling on in vitro translation activity. The translation activities calculated from these fits are 1400 ± 10 RFU/min for Cy5-Z50S mutants, 1090 ± 90 RFU/min for Cy3B-C30S mutants, and 1100 ± 180 RFU/min for Cy3B-C30S/Cy5-Z50S double mutants (Supplemental Table S1). Converting the fluorescence curves to SNAPf concentrations yields activities of 30.3 ± 1.5, 24.2 ± 0.9, and 24.7 ± 1.0 nM SNAPf/min, respectively, and normalization to sample volume and ribosome concentration results in specific activities of 0.061 ± 0.003, 0.048 ± 0.002, and 0.049 ± 0.002 pmol SNAPf/pmol ribosome/min, respectively (Supplemental Table S2). These rates correspond to 15.6 ± 0.8, 20.8 ± 0.8, and 20.4 ± 0.8 min, respectively, required for one 70S ribosome to translate one molecule of SNAPf protein. Using the simplified model of elongation-limited translation, the mutant activities would correspond to minimum elongation rates of ∼0.25, 0.20, and 0.20 aa/sec, respectively, which are also comparable with previously reported in vitro elongation rates (Wagner et al. 1982).
Differential analyses of the mutant ribosome data resulted in activities of 16.2 ± 1.0, 10.3 ± 1.2, and 11.9 ± 0.9 nM SNAPf/min; specific activities of 0.032 ± 0.002, 0.021 ± 0.002, and 0.024 ± 0.002 pmol SNAPf/pmol ribosome/min; translation times of 31.3 ± 2.0, 47.6 ± 4.5, and 41.7 ± 3.5 min; and elongation rates of 0.12, 0.08, and 0.09 aa/sec for Cy3B-C30S, Cy5-Z50S, and Cy3B-C30S/Cy5-Z50S, respectively. As noted with the wild-type data, differential rate analysis yielded activities that are half of those calculated from the first-order kinetics model. Despite this discrepancy, normalization of mutant translation activities to wild-type activities for each method reveals very similar relative values (Fig. 5C). Thus, the kinetics model and differential analysis independently support the quantification of ribosome activity.
These measurements mark the first evaluation of the impact of rRNA mutations and fluorescent labeling on ribosome activity. Calculations of specific activities are only possible with continuous, long-term detection using a noninterfering reporter signal that is not rate-limiting. The SNAP assay achieves this by using a fluorescent reporter with interchangeable fluorophore, rapid reaction kinetics, and SecM-limited ribosome turnover. Unlike most nonfluorescent translation assays, the reaction is not interrupted by measurement, and the flexibility of SNAPf substrate selection enables the user to avoid interference with fluorescently labeled translation components. However, the inability to deconvolute initiation and elongation dysfunction with mutant ribosomes illustrates the main disadvantage of the SNAP assay, which only detects active SNAPf product and is therefore agnostic to the mechanism of translation inhibition or dysfunction.
Single-molecule observation of SNAPf translation
The current method for visualizing the completion of protein synthesis in single-molecule fluorescence experiments uses GFP, which is separately translated and oxygenated prior to detection (Uemura et al. 2008). As a result, the progress of translation and the completion of protein synthesis are not simultaneously detected in real time. By pretranslating and reacting SNAPf on ZMW chips, followed by subsequent observation of SNAPf and ribosome fluorescence, we have demonstrated that SNAPf meets this standard of protein synthesis completion signals.
Furthermore, Uemura et al. (2008) have reported a time course of colocalization between Cy3-C30S ribosomes and SecM-anchored GFPuv fluorescent product using a PURExpress translation system observed by total internal reflection fluorescence (TIRF) microscopy, which provides a point of comparison between our bulk and their single-molecule measurements of C30S translation activity. The reported time constant for the single-exponential fit of the translation of GFP by C30S and wild-type 50S ribosomes was 19.2 min, or a rate of 5.21 × 10−3/min (Uemura et al. 2008). Our calculated rate of bulk SNAPf fluorescence from a similar single-exponential fit for the translation of SNAPf by C30S and wild-type 50S ribosomes is 3.33 ± 0.03 × 10−3/min (Supplemental Table S1). These values lie within an order of magnitude and indicate that SNAPf translation occurs at a time-scale relevant for single-molecule observation.
In contrast to GFP, which is limited by the requirement for molecular oxygen to induce fluorescence, SNAPf reacts in anoxia and is only limited by nonspecific substrate–surface interactions that raise background fluorescence. Improved additives that hinder these nonspecific adsorption events (Luk et al. 2008) will allow the use of the SNAPf protein reactivity in anoxia to perform long-term, real-time single-molecule FRET experiments that couple dynamic FRET detection during translation elongation with the SNAPf completion signal.
CONCLUSION
Here we have reported the use of SNAPf as a reporter for quantitative measurements of translation activity in cell-free protein synthesis. The SNAP assay has been applied to benchmark the specific activity of wild-type E. coli MRE600 ribosomes and evaluate changes in translation rates due to antibiotic macrolide treatment and ribosomal mutations. Wild-type activity was comparable with other in vitro extract translation rates, and antibiotic IC50 values followed the trend of their dissociation constants. Unlike other common translation assays, such as the luciferase, chloramphenicol acetyltransferase, and 14C radioactivity assays, translation is not interrupted by measurement, and fluorescent translation components do not interfere with fluorescence detection. Since the SNAPf reaction can occur with a variety of fluorescent substrates and in anoxia, the assay has also been demonstrated to couple effectively to single-molecule FRET experiments featuring in vitro translation of SNAPf as a reporter for the completion of translation.
Applied to single-molecule fluorescence studies of translation dynamics (Chen et al. 2013b; Rosenblum et al. 2013), the SNAP assay will be particularly effective for benchmarking the effects of fluorescently labeled translation components on cell-free protein synthesis. The flexible choice of fluorophores for the SNAP assay confers an advantage in this respect in that fluorescent translation machinery does not interfere with the SNAPf signal. Furthermore, SNAPf is superior to conventional fluorescent proteins as a single-molecule reporter of translation completion due to the preferable photophysical properties of rhodamine and cyanine dye substrates and the ability to generate fluorescence in anoxia. Overall, the SNAP assay is a general, flexible, and sensitive technique for measuring translation and translation inhibition.
MATERIALS AND METHODS
Construction and preparation of SNAPf mRNA for in vitro translation
A pUC57 plasmid was custom designed with T7 promoter, SNAPf sequence, GlySer linker, SecM stall, and BlpI restriction site and synthesized by GenScript (Supplemental Fig. S3). The flanking T7 promoter and BlpI restriction site are required for run-off transcription of the plasmid, and the SecM stall sequence was introduced to limit ribosome turnover and for downstream single-molecule studies. The plasmid was transformed into DH5α E. coli cells and harvested by Gigaprep (Qiagen). Harvested plasmid was transcribed in vitro with His-tagged T7 RNA polymerase purified from recombinant expression in E. coli (Grodberg and Dunn 1998), and the mRNA was extracted from the solution by phenol/chloroform partitioning and purified by size-exclusion chromatography according to established protocols (Lukavsky and Puglisi 2004).
The cell-free protein synthesis system
The PURExpress ΔRibosome kit (E3313S, New England Biolabs, Inc.) provided the basic materials for cell-free protein synthesis. In all translation experiments, the kit components (excluding ribosomes) were assembled according to the manufacturer's recommended concentrations. Purified SNAPf enzyme was purchased as SNAP-tag purified protein (P9312S, New England Biolabs, Inc.), and all fluorogenic SNAP substrates were provided by New England Biolabs as lyophilized powders, which were reconstituted to 1 mM in DMSO. The CBG-549-QSY7 substrate consists of guanine conjugated to fluorophore DY549P1 at the C6 position and to quencher QSY7 at C8, while CBG-488-TQ2 contains fluorophore ATTO488 and quencher TQ2.
Construction of ribosome mutants and preparation of ribosomes for in vitro translation
C30S mutants with extended 16S rRNA Domain 3′m helix 44 and Z50S mutants with extended 23S rRNA Domain VI helix 101 (Supplemental Fig. S4) were generated as reported previously (Dorywalska et al. 2005; Marshall et al. 2008). 30S, 50S, and 70S ribosome complexes of mutant and wild-type strains were isolated according to standard procedures (Robertson and Wintermeyer 1981; Powers and Noller 1991).
Parameters for fluorescent measurements and analysis
All measurements were performed on an Infinite M1000 or M1000 Pro (Tecan) microplate reader with Magellan control software. Sample volumes of 10 μL were overlaid with 15 μL of mineral oil and read in triplicate on Corning 384-well black flat clear bottom or flat black low volume plates. Fluorescence was read in 1- or 2-min intervals for up to 3 h with 400 Hz flash frequency and 20 μsec integration time. The reading chamber temperature was maintained at 37°C. Experiments with CBG-549-QSY7 used excitation and emission wavelengths/filters of 555/5 and 575/5 nm, respectively, while experiments with CBG-488-TQ2 used 509/5 and 526/5 nm, respectively.
Baseline assay response measurements
Translation reactions with 100 nM MRE600 70S ribosomes and 500 nM CBG-549-QSY7 in the PURExpress ΔRibosome reaction mixture were incubated at 37°C for 2 h in the presence or absence of 100 nM SNAPf mRNA to validate mRNA dependence for fluorescence. In addition, translation reactions with 100, 500, 750, or 1000 nM MRE600 70S ribosomes; 1 μM SNAPf mRNA; and 10 μM CBG-549-QSY7 in the PURExpress ΔRibosome reaction mixture were incubated at 37°C for 2 h to verify the assay's sensitivity to ribosome concentration. Derivatives of fluorescence curves were numerically calculated as the quotients of the difference in y-values (fluorescence) and the difference in x-values (time) of consecutive datapoints.
Measurements of translation activity
Translation reactions with 500 nM MRE600 30S/MRE600 50S, C30S/MRE600 50S, MRE600 30S/Z50S, or C30S/Z50S ribosomes; 2 μM SNAPf mRNA; and 10 μM CBG-488-TQ2 in the PURExpress ΔRibosome reaction mixture were incubated at 37°C for ∼3 h. Spec68-Cy3B and Spec22-Cy5 fluorescently labeled DNA oligomers, which are complementary to the extended helices 44 and 101, were also added to samples containing the mutant C30S and Z50S subunits, respectively (Marshall et al. 2008). Control experiments with CBG-488-TQ2 and Cy3B did not indicate any change in ATTO488 fluorescence due to the presence of Cy3B, as would be expected from the limited spectral overlap between ATTO488 and Cy3B (Supplemental Fig. S5). Fluorescence data as a function of time were fitted to the first-order kinetics model starting immediately after the lag period. Activities (in RFU/min) were calculated from the product of normalization factor a and first-order rate b terms and compared with the wild-type MRE600 30S/MRE600 50S control.
Linear fits from a standard curve of 0, 100, 250, 500, and 750 nM of SNAP purified protein prereacted for 2 h with 10 μM of CBG-488-TQ2 at 37°C in the PURExpress ΔRibosome reaction mixture (Supplemental Fig. S6) were used to quantify product formation as a function of time. The curve of product as a function of time was fitted to the first-order kinetics model starting immediately after the lag period. The activity (in pmol SNAPf/pmol ribosome/min) was calculated from the product of normalization a and first-order rate b terms.
Derivatives of fluorescence and SNAPf concentration curves were numerically calculated as the quotients of the difference in y-values (fluorescence or concentration) and the difference in x-values (time) of consecutive data points. Owing to noise within the data, these quotients can be locally erratic, so the data were smoothed by calculating the derivatives of data points separated by three points (steps); i.e., the differential across the ith and i + 4th data points. Tests with a range of step values indicated that the calculated translation rates were not affected by the step size.
Quantification of macrolide activity
Erythromycin A was purchased from Fisher Scientific (BP920-25), and roxithromycin (30425) and clarithromycin (10404) were purchased from Crescent Chemical Co. Translation reactions with 100 nM MRE600 70S ribosomes, 100 nM SNAPf mRNA, and 500 nM CBG-549-QSY7 in the PURExpress ΔRibosome reaction mixture were incubated at 37°C for ∼2 h in the presence of a defined concentration of erythromycin A, clarithromycin, or roxithromycin. Fluorescence data were normalized to untreated and nontranslating samples. Error bars were calculated using standard methodologies in error propagation. Error propagation due to normalization to the negative control was calculated as
where Δa is the propagated error due to subtraction, σs is the standard deviation of the sample, and σn is the standard deviation of the negative control. Error propagation due to normalization to the positive control was calculated as
where Δb is the propagated error due to division, s is the average of the sample, n is the average of the negative control, p is the average of the positive control, and σp is the standard deviation of the positive control. The normalized average values ((s−n)/(p−n)) their positive-error values ((s−n)/(p−n)+Δb) and their negative-error values ((s−n)/(p−n)−Δb) were each linearly fitted to determine the average IC50, positive-error IC50, and negative-error IC50 in Figure 6.
Observing translation of SNAP by single molecule
Ribosomes were immobilized via a 3′-biotinylated DNA oligonucleotide complementary to helix 44 of the C30S 16S rRNA that bound neutravidin on the chip surface, as described previously (Uemura et al. 2008). After immobilization of the C30S subunit, a translation mixture containing the PURExpress components, as well as 50S subunits and a plasmid coding for SNAPf, was added to the surface and incubated for 45 min, to allow both transcription and translation. Then the reaction mixture was removed, and a buffered solution containing SNAP Surface 549 or 647 (New England Biolabs, Inc.) substrate was added. After incubation for 30 min, the labeling solution was removed and the chip washed to prevent background fluorescence due to noncovalently bound SNAP fluorophore. For full-chip fluorescence intensity measurements, the SNAPf plasmid was replaced with a pUC57 vector encoding T7 promoter, GFPuv protein, SecM minimal arrest sequence, and T7 terminator. Chloramphenicol was purchased with Fisher Scientific (BP904-100).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. David Solow-Cordero and Mr. Jason Wu at the Stanford School of Medicine High-Throughput Bioscience Center (HTBC) for training and assistance with the platereaders and Ms. Marie Maher, Mr. Armen Mekhdijian, and Mr. Jin Chen for helpful discussions. This work was supported by National Institutes of Health (NIH) grant GM51266 and GM099687 to J.D.P., and a Stanford University Center for Molecular Analysis and Design Fellowship to M.C.C.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.047159.114.
Freely available online through the RNA Open Access option.
REFERENCES
- Bayley ST, Griffiths E 1968. Codon assignments and fidelity of translation in a cell-free protein-synthesizing system from an extremely halophilic bacterium. Can J Biochem 46:937–944. [DOI] [PubMed] [Google Scholar]
- Brandi L, Fabbretti A, Milon P, Carotti M, Pon CL, Gualerzi CO 2007. Methods for identifying compounds that specifically target translation. Methods Enzymol 431:229–267. [DOI] [PubMed] [Google Scholar]
- Chen C, Zhang H, Broitman SL, Reiche M, Farrell I, Cooperman BS, Goldman YE 2013a. Dynamics of translation by single ribosomes through mRNA secondary structures. Nat Struct Mol Biol 20:582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Petrov A, Tsai A, O'Leary SE, Puglisi JD 2013b. Coordinated conformational and compositional dynamics drive ribosome translocation. Nat Struct Mol Biol 20:718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinos GP, Connel SR, Nierhaus KH, Kalpaxis DL 2003. Erythromycin, roxithromycin, and clarithromycin: use of slow-binding kinetics to compare their in vitro interaction with a bacterial ribosomal complex active in peptide bond formation. Mol Pharmacol 63:617–623. [DOI] [PubMed] [Google Scholar]
- Dorywalska M, Blanchard SC, Gonzalez RL Jr, Kim HD, Chu S, Puglisi JD 2005. Site-specific labeling of the ribosome for single-molecule spectroscopy. Nucleic Acids Res 33:182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MS, Ugrinov KG, Frese MA, Clark PL 2005. Homogeneous stalled ribosome nascent chain complexes produced in vivo or in vitro. Nat Methods 2:757–762. [DOI] [PubMed] [Google Scholar]
- Grodberg J, Dunn JJ 1998. ompT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification. J Bacteriol 170:1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale ML 2000. Microtiter-based assay for evaluating the biological activity of ribosome-inactivating proteins. Pharmacol Toxicol 88:255–260. [PubMed] [Google Scholar]
- Harvey CJB, Puglisi JD, Pande VS, Cane DE, Khosla C 2012. Precursor directed biosynthesis of an orthogonally functional erythromycin analogue: selectivity in the ribosome macrolide binding pocket. J Am Chem Soc 134:12259–12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka R, Yamagishi-Shirasaki M, Funatsu T 2011. Kinetic study of de novo chromophore maturation of fluorescent proteins. Anal Biochem 414:173–178. [DOI] [PubMed] [Google Scholar]
- Jackson AM, Boutell J, Cooley N, He M 2004. Cell-free protein synthesis for proteomics. Brief Funct Genomic Proteomic 2:308–319. [DOI] [PubMed] [Google Scholar]
- Kennel D, Riezman H 1977. Transcription and translation initiation frequencies of the Escherichia coli operon. J Mol Biol 114:1–21. [DOI] [PubMed] [Google Scholar]
- Kim D-M, Swartz JR 2001. Regeneration of adenosine triphosphate from glycolytic intermediates for cell-free protein synthesis. Biotechnol Bioeng 74:309–316. [PubMed] [Google Scholar]
- Lovmar M, Tenson T, Ehrenberg M 2004. Kinetics of macrolide action: the josamycin and erythromycin cases. J Biol Chem 279:53506–53515. [DOI] [PubMed] [Google Scholar]
- Luk VN, Mo GCH, Wheeler AR 2008. Pluronic additives: a solution to sticky problems in digital microfluidics. Langmuir 24:6382–6389. [DOI] [PubMed] [Google Scholar]
- Lukavsky PJ, Puglisi JD 2004. Large-scale preparation and purification of polyacrylamide-free RNA oligonucleotides. RNA 10:889–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankin AS 2008. Macrolide myths. Curr Opin Microbiol 11:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall RA, Dorywalska M, Puglisi JD 2008. Irreversible chemical steps control intersubunit dynamics during translation. Proc Natl Acad Sci 105:15364–15369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Ito K 2001. Secretion monitor, SecM, undergoes self-translation arrest in the cytosol. Mol Cell 7:185–192. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Ito K 2002. The ribosomal exit tunnel functions as a discriminating gate. Cell 108:629–636. [DOI] [PubMed] [Google Scholar]
- Nomura Y, Tanaka H, Poellinger L, Higashino F, Kinjo M 2001. Monitoring of in vitro and in vivo translation of green fluorescent protein and its fusion proteins by fluorescence correlation spectroscopy. Cytometry 44:1–6. [PubMed] [Google Scholar]
- Powers T, Noller HF 1991. A functional pseudoknot in 16S ribosomal RNA. EMBO J 10:2203–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JM, Wintermeyer W 1981. Effect of translocation on topology and conformation of anticodon and D loops of tRNAPhe. J Mol Biol 151:57–79. [DOI] [PubMed] [Google Scholar]
- Rosenblum G, Chen C, Kaur J, Cui X, Goldman YE, Cooperman BS 2012. Real-time assay for testing components of protein synthesis. Nucleic Acids Res 40:e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum G, Chen C, Kaur J, Cui X, Zhang H, Asahara H, Chong S, Smilansky Z, Goldman YE, Cooperman BS 2013. Quantifying elongation rhythm during full-length protein synthesis. J Am Chem Soc 135:11322–11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw WV 1975. Chloramphenicol acetyltransferase from chloramphenicol-resistant bacteria. Methods Enzymol 43:737–755. [DOI] [PubMed] [Google Scholar]
- Sun S, Zhang A, Baker B, Sun L, Howard A, Buswell J, Maurel D, Masharina A, Johnsson K, Noren CJ, et al. 2011. Development of SNAP-tag fluorogenic probes for wash-free fluorescence imagining. Chembiochem 12:2217–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai A, Petrov A, Marshall RA, Korlach J, Uemura S, Puglisi JD 2012. Heterogeneous pathways and timing of factor departure during translation initiation. Nature 487:390–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai A, Kornberg G, Johansson M, Chen J, Puglisi JD 2014. The dynamics of SecM-induced translational stalling. Cell Rep 7:1521–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura S, Iizuka R, Ueno T, Shimizu Y, Taguchi H, Ueda T, Puglisi JD, Funatsu T 2008. Single-molecule imaging of full protein synthesis by immobilized ribosomes. Nucleic Acids Res 36:e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood KA, Swartz JR, Puglisi JD 2005. Quantitative polysome analysis identifies limitations in bacterial cell-free protein synthesis. Biotechnol Bioeng 91:425–35. [DOI] [PubMed] [Google Scholar]
- Wagner EG, Jelenc PC, Ehrenberg M, Kurland CG 1982. Rate of elongation of polyphenylalanine in vitro. Eur J Biochem 122:193–197. [DOI] [PubMed] [Google Scholar]
- Yap MN, Bernstein HD 2009. The translational regulatory function of SecM requires the precise timing of membrane targeting. Mol Microbiol 81:540–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.