

Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a rare and potential life-threatening clinical syndrome that results from uncontrolled activation of the immune system. Secondary HLH, more commonly observed in adult patients, is seen in the context of underlying triggering conditions. Epstein-Barr virus (EBV) has been recognized as the leading infectious cause and is associated with a poor outcome. As clinical and laboratory features of HLH could overlap with septic shock syndrome in most patients, the diagnosis of HLH, especially in adults, is the most challenging aspect of the disease that results in delayed recognition and treatment of rapidly progressive multiorgan system failure. We report a case of Hemophagocytic lymphohistiocytosis in a patient who presented with signs of septic shock syndrome and we review the literature on the topic.
1. Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a rare life-threatening syndrome of immune dysregulation typified by extreme immunologic response and tissue infiltration of cytokine-activated tissue macrophages (histiocytes) as well as cytotoxic T-lymphocytes that can lead to multiorgan system failure. It can sometimes occur as a primary disorder traditionally described in pediatric population where it is usually driven by autosomal recessive gene defects of granulocyte-associated cytotoxic pathways, ultimately resulting in exuberant T-cell and macrophage response that is deregulated, causing indiscriminate phagocytosis of peripheral blood elements of resident tissues [1, 2].
Secondary HLH, more commonly observed in adult patients, is seen in the context of underlying triggering conditions ranging from inflammatory processes such as autoimmune and rheumatologic disorders and neoplastic disorders particularly hematologic malignancies such as lymphoid cancers and leukemias to various forms of commonly observed, but not limited to, viral infections. Triggering infections are usually viral in etiology, especially the herpesviridae family, human immunodeficiency virus (HIV), adenovirus, and parvovirus. Less commonly, bacteria, fungi, and parasites have been reported as well. Epstein-Barr virus (EBV) has been recognized as the leading infectious cause [3, 4].
The diagnosis of HLH is based on fulfilling the diagnostic criteria described in the HLH-2004 protocol [5]; however, not all patients with HLH fulfill the criteria. In order to avoid delayed diagnosis which often leads to delays in initiation of therapy, hence higher mortality, modified diagnostic criteria have been proposed [6]. Although hemophagocytosis represents an integral part of the diagnostic criteria, it is not required to make the diagnosis of HLH based on either of the two criteria.
A standard established treatment for HLH is described in the HLH-94 protocol and later modified in HLH-2004. Principles of treatment include elimination of the trigger and suppression of the inflammation and activated immune cells, as well as supportive measures [5, 7]. Nonetheless, treatment of HLH is challenging and mortality from HLH remains high. Patients are at increased risk of infections and should be monitored very closely during treatment and a subsequent follow-up [8].
2. Case Report
A 40-year-old Hispanic man with no significant medical history presented with sharp right upper quadrant and epigastric pain, dizziness, and fever as high as 102 Fahrenheit for 4 days. He also reported vomiting over 10 times a day and severe green watery diarrhea multiple times a day. Three weeks earlier he was seen by a primary care physician for cellulitis of the lower extremities and was prescribed empiric antibiotic course. Given worsening symptoms, he presented for admission, and he was found to be febrile (102 F), hypotensive (79/54 mmHg), and tachycardic (132 bpm). On physical examination he had icteric skin, tenderness on right upper quadrant, and right lower extremity swelling with mid-tibial erythema. The rest of the physical exam was unremarkable.
Laboratory workup showed white blood cell (WBC) 2500/μL, hemoglobin (Hgb) 10.9 g/dL, platelet count 25,000/μL, prothrombin time 14.9 seconds; partial thromboplastin time 60 seconds, d-dimer 6.19 ug/mL, and fibrinogen 180 mg/dL, serum creatinine 2.9 mg/dL, blood urea nitrogen (BUN) 75 mg/dL, alanine aminotransferase (ALT) 373 U/L, aspartate aminotransferase (AST) 669 U/L, alkaline phosphatase 119 U/L, total bilirubin 4.3 mg/dL (direct bilirubin 3.0 mg/dL), normal lipase, triglyceride 378 mg/dL, and lactate dehydrogenase (LDH) 2,829 U/L (Table 1).
Table 1.
Trend of liver function test, ferritin levels, blood counts, and creatinine during the initial hospital stay.
| Days from admission | 1 | 2 | 4 | 6 | 8 | 12 | 17 |
|---|---|---|---|---|---|---|---|
| Temperature (F) | 102.7 | 102.7 | 100.3 | 98.9 | 97.8 | 98.1 | 98.5 |
| ALT (U/L) | 373 | 225 | 214 | 188 | 160 | 270 | 126 |
| AST (U/L) | 669 | 444 | 568 | 640 | 314 | 198 | 45 |
| ALP (U/L) | 119 | 59 | 79 | 169 | 236 | 340 | 318 |
| Total bilirubin (mg/dL) | 4.3 | 3.6 | 7.6 | 9.2 | 7.7 | 3.7 | 1.6 |
| Ferritin (ng/dL) | — | 75285 | 119440 | 216048 | 128888 | 15550 | — |
| Creatinine (mg/dL) | 2.9 | 2.4 | 2.3 | 5.6 | 5.1 | 3.9 | 4.5 |
| WBC (/μL) | 2500 | 1400 | 2200 | 1500 | 900 | 1400 | 900 |
| ANC (/μL) | 2240 | 1300 | 2090 | 1370 | 830 | 1210 | 730 |
| Hgb (g/dL) | 10.6 | 8.9 | 9.4 | 7.3 | 6.7 | 6.8 | 7.9 |
| Platelet (/μL) | 25000 | 20000 | 18000 | 30000 | 33000 | 47000 | 77000 |
ALT: alanine aminotransferase; ALP: alkaline phosphatase; ANC: absolute neutrophil count; AST: aspartate aminotransferase; Hgb: hemoglobin; WBC: white blood cells.
On abdominal ultrasound, his liver measured 16.3 cm with no focal lesions; spleen was mildly enlarged at 13.7 cm. He was admitted directly to the intensive care unit for presumed septic shock and early goal-directed therapy. Intravascular fluid was started, blood cultures were sent, and broad-spectrum antibiotics (vancomycin, cefepime, azithromycin, and metronidazole) were initiated. Abdominal/pelvic computed tomography (CT) without contrast revealed an enlarged spleen and no further abnormalities.
Hematology service was consulted for evaluation of pancytopenia on day 2 given the decrease in WBC and Hgb to 1,400/μL and 8.9 g/dL, respectively. Peripheral blood smear showed numerous echinocytes, few ovalocytes, and no schistocytes. Routine serum iron was 139 ug/dL with iron saturation 55%, while serum ferritin measured 72,285 ng/dL. Given that initial blood and urine cultures were negative at that point, our clinical suspicion for HLH was very high. After careful assessment of the diagnostic criteria, 5 out of 8 were met by our patient (fever, pancytopenia, high triglycerides, elevated ferritin, and splenomegaly). Bone marrow biopsy was performed on day 3 and it revealed 3 hemophagocytes (Figure 1(a)), further confirming the diagnosis. Therapy for HLH was started on the same day by the model of HLH-94 protocol with dexamethasone 20 mg IV daily and 75% dose-reduced etoposide twice a week due to liver and kidney dysfunction. Doxycycline 100 mg PO every 12 h was added for coverage of atypical microorganisms. Patient's daily liver transaminases and temperature are represented on Figure 2(a) and in Table 1. Variation of the serum ferritin level and total bilirubin during the hospitalization is shown on Figure 2(b) and in Table 1.
Figure 1.
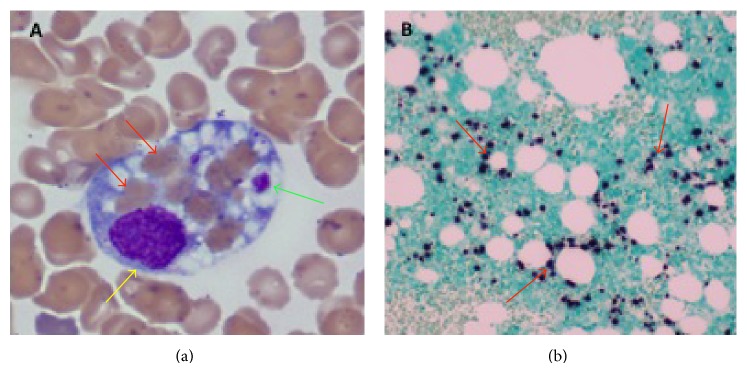
Images of the bone marrow. (a) Bone marrow aspirate smear, 1000x magnification, shows a hemophagocyte which engulfed 7 separate red blood cells (red arrows) and 2 platelets (green arrow). Yellow arrow points to a nuclei of histiocyte; (b) bone marrow clot section, EBV-encoded RNA (EBER) staining of the bone marrow revealing extensive EBV infected lymphocytes in the background (dark nuclear stain).
Figure 2.

Trend of liver enzymes, temperature (a), total bilirubin, and ferritin (b) during the initial hospital stay. Dotted line indicates the start of therapy on day 3 (ALT: alanine aminotransferase; AST: aspartate aminotransferase; T bili: total bilirubin).
A comprehensive rheumatologic and infectious workup was sent to identify any potential underlying triggers of HLH. Rheumatologic workup included anti-Sm, anti-Scl-70, ANA, ANCA, anti-dsDNA, anti- RNP, anti-cardiolipin, anti-SSA, and anti-SSB antibodies, all of which were negative. Infectious workup was negative for cytomegalovirus, herpes simplex viruses 1 and 2, Brucella, parvovirus B19, cryptococcus, Histoplasma, HIV, and hepatitis B and hepatitis C. On day 5, Serum EBV IgM was found positive and the patient was started on acyclovir 5 mg/kg IV every 24 h. Cefepime, vancomycin, and metronidazole were discontinued. On day 8, serum EBV PCR (qualitative test) was reported positive also. Review and EBER staining of the bone marrow revealed extensive infiltration of the marrow by the EBV (Figure 1(b)). Following the administration of HLH-94 therapy as well as IV Acyclovir, the patient's condition started improving and his fever subsided. Rituximab was not given for EBV infection due to the dramatic nature of his improvement on the initial therapy (dexamethasone, Etoposide, and antimicrobials).
On day 9, Q fever phase II serology was found to be positive at 1 : 64 titers. On further questioning, patient recalled having had a contact with his dog that had delivered puppies 3 months prior to development of his symptoms. Ciprofloxacin 500 mg PO daily was added. Patient continued with progressive clinical improvement and, on day 11, he was transferred out of the ICU to the medical floor for further management.
He was found to have acute tubular necrosis from systemic involvement of HLH and required intermittent hemodialysis for metabolic optimization. By day 17, patient had completed 14 days of IV acyclovir, 4 doses of etoposide, 14 days of doxycycline, and 9 days of ciprofloxacin. Repeat Q fever phase II serology was negative. On day 17, patient was discharged home on ciprofloxacin, further acyclovir course, fluconazole, trimetoprim/sulfamethoxazole, and steroid taper.
He continued to show improvement on outpatient laboratory follow-up visits. On hematology follow-up visit on day 28, renal function and liver function showed improvement and his blood cell counts showed full recovery (Table 2). On the same visit, acyclovir, fluconazole, and ciprofloxacin were discontinued given resolution of neutropenia. Prophylactic trimetoprim/sulfamethoxazole was continued along with dexamethasone.
Table 2.
Trend of laboratory work after discharge and during the second hospital stay.
| Days from initial admission | 18 | 21 | 28 | 35 | 36 |
|---|---|---|---|---|---|
| Second admission | |||||
| ALT (U/L) | 128 | 110 | 108 | 383 | 3657 |
| AST (U/L) | 44 | 35 | 38 | 296 | 6197 |
| ALP (U/L) | 287 | 279 | 232 | 341 | 418 |
| Total bilirubin (mg/dL) | 1.7 | 1.5 | 1.1 | 2.3 | 10.5 |
| Ferritin (ng/dL) | 4963 | ||||
| Creatinine (mg/dL) | 4.5 | 3.6 | 1.7 | 0.8 | 1.2 |
| WBC (/μL) | 900 | 1100 | 3800 | 7500 | 10000 |
| ANC (/μL) | 650 | 690 | 2550 | 62600 | 7900 |
| Hgb (g/dL) | 8.3 | 7.8 | 8.1 | 8.0 | 9.1 |
| Platelet (/μL) | 113000 | 158000 | 197000 | 46000 | 24000 |
ALT: alanine aminotransferase; ALP: alkaline phosphatase; ANC: absolute neutrophil count; AST: aspartate aminotransferase; Hgb: hemoglobin; WBC: white blood cells.
On day 35, patient presented with shortness of breath and acute lower extremity pain. His vital signs were stable. He had scleral icterus and his strength was decreased in all extremities. Laboratory investigations showed thrombocytopenia (48,000/μL), high ferritin (4,963 ng/dL), and elevated transaminases (Table 2). CT angiogram of the chest did not show pulmonary embolism. Patient was admitted and supportive measures with oxygen, electrolyte replacement, and pain management were initiated. By the next day his respiratory distress worsened, he became hypotensive, liver function deteriorated progressively, and platelet count dropped to 24,000/μL (Table 2). He developed severe metabolic acidosis with serum pH 7.2, bicarbonate 13 mmol/L, and lactic acid 11 mmol/L. Blood cultures were drawn and patient was started on a broad-spectrum empiric antibiotic coverage with vancomycin, cefepime, and metronidazole. He was transferred to medical ICU and was intubated for airway protection. Gancyclovir was added for potential EBV-associated hepatitis given prior documentation of EBV-related HLH. Despite these measures, he progressed to fulminant hepatic failure with significant distributive shock. Given the patient's high ferritin, though significantly better than previous ferritin of 15,550 ng/dL on day 12, thrombocytopenia, and negative urine and blood cultures, he was treated with high dose methylprednisolone (1 g/d) and intravenous immunoglobulin (1 g/kg) for possible relapsed or refractory HLH, while etoposide was omitted due to fulminant liver failure (bilirubin 10.5 mg/dL).
On the evening of day 2 of readmission, he developed cardiac arrest due to ventricular tachycardia and cardiopulmonary resuscitation was unsuccessful. His autopsy revealed multiorgan failure consistent with clinical history of severe lactic acidosis, shock, and occasional cells suspicious of hemophagocytosis in the bone marrow.
3. Discussion
HLH is a rare and potential life-threatening clinical syndrome that results from uncontrolled activation of the immune system. Historically, HLH is more common in children or adolescences; however, adult onset of HLH is becoming increasingly more recognized [9]. Currently, the precise pathophysiology of HLH remains unclear. Dysregulation of cytotoxic T cells and natural killer cells that lead to activation and proliferation of histiocytes with excessive cytokine release has been proposed as the underlying pathophysiologic mechanisms [10]. Primary HLH is a familial disorder that commonly occurs in children with predisposed genetic defects involving lymphocytic granule-mediated cytotoxic pathway [11]. Secondary HLH is an acquired syndrome that most commonly occurs in adulthood and is usually triggered by an underlying malignancy, autoimmune disease, or an infection [3]. Etiologies associated with secondary HLH are described in Table 3.
Table 3.
Etiologies associated with secondary HLH.
| Malignancy | |
| Hematologic | Lymphoma, multiple myeloma, ALL |
| Nonhematologic | Prostate cancer, lung cancer, hepatocellular carcinoma |
| Autoimmune disease | Systemic juvenile, SLE, Kawasaki disease, seronegative spondyloarthropathies |
| Infection | |
| Bacterial | Coxiella burnetii, Staphylococcus aureus, Campylobacter spp., Chlamydia spp., Legionella spp., Mycobacterium spp., Mycoplasma spp., Salmonella spp., Brucella spp., Ehrlichia spp., Borrelia spp., Clostridium spp., Listeria spp. |
| Fungal | Candida spp., Cryptococcus spp., Histoplasma spp., Pneumocystis spp., Aspergillus spp., Fusarium spp. |
| Parasitic | Plasmodium spp., Toxoplasma spp., Babesia spp., Strongyloides spp., Leishmania spp. |
| Viral | Herpes virus (EBV, CMV, HHV-8, HSV), HIV, HTLV, HAV, HBV, HCV, HEV, Influenza, mumps, measles, rubella, dengue, hantavirus, parvovirus B19, enterovirus |
ALL: acute lymphocytic leukemia; SLE: systemic lupus erythematosus; EBV: Epstein-Barr virus; CMV: cytomegalovirus; HHV-8: human herpesvirus 8; HSV: herpes simplex virus; HIV: human immunodeficiency virus; HTLV: human T-lymphotropic virus; HAV: hepatitis A virus; HBV: hepatitis B virus; HCV: hepatitis C virus; HEV: hepatitis E virus.
Recent studies suggest that late-onset familial HLH occurs more commonly than was suspected previously. In a retrospective review of 175 adults (age range 18 to 75 years) with clinical diagnosis of HLH, 14% were found to have gene mutations, most commonly defects in Perforin gene [12]. EBV, the most common infectious cause of secondary HLH, may also trigger HLH in patients with any form of familial disease. Despite this, HLH in adults is commonly attributed to an infectious etiology and a full genetic workup may not be performed [13]. Given the adult age of our patient, lack of corroborating or suspicious family history for HLH, and extensive infiltration of the bone marrow by EBV as the likely culprit as well as conscious choice to adopt a judicious use of health care resources, genetic testing was not undertaken for our patient.
As clinical and laboratory features of HLH could overlap with septic shock syndrome in most patients, the diagnosis of HLH, especially in adults, is the most challenging aspect of the disease that results in delayed recognition and treatment of rapidly progressive multiorgan system dysfunction/failure [14]. Early recognition and prompt treatment is critical and could be a key to prevention of fatal outcomes in HLH. A retrospective study in the critical care setting suggested that the HLH 2004 criteria might not be a reliable tool to differentiate between HLH and sepsis syndrome [15]. Some case reports suggest that ferritin level could be a useful test in this clinical scenario; when higher than 10,000 ng/mL, it is 90% sensitive and 96% specific for HLH [16]. Ferritin, an acute phase reactant, is a nonspecific biomarker that could be elevated in many circumstances. However, the ferritin level higher than 10,000 ng/mL is known to be associated with only a few other disorders such as systemic juvenile rheumatoid arthritis, adult still's disease, and histiocytic malignancies [17, 18]. In the appropriate clinical setting, extremely high ferritin level should alert physicians to search for other clinical and laboratory parameters to determine if the diagnostic criteria for HLH are met. The concentration of circulating soluble interleukin- (IL-) 2 receptor, also known as soluble CD25, is a very helpful diagnostic tool in HLH [19]. Since soluble IL-2 receptor reflects degree of T-cell activation, it can also be used to evaluate severity of the disease during follow-up period. Unfortunately, at present, this test is not readily available, is very expensive, and usually takes up to 2 weeks to process [20].
Two HLH treatment protocols, HLH-94 and HLH-2004, were developed based on the experience in children with primary HLH [5, 7]. These treatments include dexamethasone, etoposide, cyclosporine, and intravenous immunoglobulin. Although it remains unclear whether these protocols are the appropriate treatments for adults with secondary EBV-related HLH, most physicians refer back to them for initial therapeutic choice. Some studies suggested that early initiation of etoposide provided survival benefit in adults with secondary HLH [21, 22]. Risk and benefit of etoposide in this group of patients should be carefully evaluated given that severe liver and renal dysfunctions are commonly encountered in HLH patients. In patients with HLH and multiorgan failure, successful treatments with reduced-dose etoposide with close monitor have been reported [23]. In contrast, a few reports advocate more conservative and supportive management with corticosteroid/cyclosporine and suggested that it might be adequate in some patients [24, 25]. In our patient, dose-reduced etoposide was given along with steroid therapy during the initial hospitalization, which led to a striking improvement in the liver function and clinical status of the patient. On the second admission, due to his rapid and fulminant liver failure, etoposide was omitted and patient was given high dose steroids with IVIG, however unfortunately without benefit. Given the improvement in ferritin compared to his previous admission, he likely had septic shock, which might have resulted in exacerbation or relapse of his HLH. Patient's clinical and laboratory data together with autopsy report were also consistent with septic shock with multiorgan failure.
In a 16-year literature review of HLH cases in children up to 18 years of age, of the 198 cases diagnosed with infection-associated HLH, 52% died, mostly from severe infections, organ failure, or disseminated intravascular coagulation. EBV was found to be associated with the worst outcome (73% mortality) [3]. In retrospective studies, high EBV-DNA viral load (>1,000 copies/mL), hyperbilirubinemia (>1.8 mg/dL), and highly elevated ferritin level (>20,300 ng/mL) were proposed as factors associated with poor outcome in patients with EBV-related HLH [26, 27]. Our patient had at least two of the above factors (bilirubin and ferritin levels) making his prognosis unfavorable. Rituximab can be effective for EBV infection [28], but, due to our patient's rapid improvement with the initial therapy, we did not feel the need to treat him with this agent at that time. However, if he did not improve with our initial regimen or if he had evidence of worsening EBV infection, we would have offered this therapy to him.
Although mortality rate of HLH has been reported as high as 52%, rapid initiation of treatment can possibly reverse the devastating course of the disease [3]. Given the extremely high ferritin level found in our patient, diagnosis was made relatively promptly and appropriate therapy was initiated on the 3rd day of hospitalization. We believe checking ferritin level in all patients with clinical picture of sepsis could help raise initial suspicion index, which would trigger urgent involvement of hematology subspecialty team that can attempt to make a timely diagnosis. We therefore suggest that a screening ferritin level be included in early sepsis protocols; if ferritin level is found to be above 10,000, hematology consultation should be sought urgently in order to identify the cases of HLH in a timely manner.
Unfortunately the reported patient returned with severe sepsis and possible relapse of HLH, succumbing to the disease in short period of time. We therefore believe that patients should be followed up very frequently and with earnest vigilance with continuation of broad-spectrum prophylactic antibiotics even in the absence of neutropenia until the full spectrum of organ function is restored.
4. Conclusion
Despite being primarily a pediatric disorder, HLH does also occur in adults, particularly secondary to an underlying triggering condition. HLH is a true hematologic emergency; prompt diagnosis and treatment are crucial to avoid imminent multiorgan dysfunction resulting in a fatal outcome. Checking ferritin level as part of a standard sepsis protocols can help in raising the initial index of suspicion as it did in our case, guiding the medical care providers to seek an emergent hematology consultation for early initiation of treatment.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Janka G. E. Familial and acquired hemophagocytic lymphohistiocytosis. Annual Review of Medicine. 2012;63:233–246. doi: 10.1146/annurev-med-041610-134208. [DOI] [PubMed] [Google Scholar]
- 2.Stepp S. E., Dufourcq-Lagelouse R., Le Deist F., et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286(5446):1957–1959. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- 3.Janka G., Imashuku S., Elinder G., Schneider M., Henter J.-I. Infection- and malignancy-associated hemophagocytic syndromes: secondary hemophagocytic lymphohistiocytosis. Hematology/Oncology Clinics of North America. 1998;12(2):435–444. doi: 10.1016/s0889-8588(05)70521-9. [DOI] [PubMed] [Google Scholar]
- 4.Rouphael N. G., Talati N. J., Vaughan C., Cunningham K., Moreira R., Gould C. Infections associated with haemophagocytic syndrome. The Lancet Infectious Diseases. 2007;7(12):814–822. doi: 10.1016/s1473-3099(07)70290-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henter J.-I., Horne A., Aricó M., et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatric Blood and Cancer. 2007;48(2):124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 6.Filipovich A. H. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology/American Society of Hematology. Education Program. 2009;2009(1):127–131. doi: 10.1182/asheducation-2009.1.127. [DOI] [PubMed] [Google Scholar]
- 7.Henter J. I., Arico M., Egeler R. M., et al. HLH-94: a treatment protocol for hemophagocytic lymphohistiocytosis. HLH study Group of the Histiocyte Society. Medical and pediatric oncology. 1997;28(5):342–347. doi: 10.1002/(SICI)1096-911X(199705)28:5x003C;342::AID-MPO3x0003e;3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 8.Trottestam H., Horne A., Aricò M., et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118(17):4577–4584. doi: 10.1182/blood-2011-06-356261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishii E., Ohga S., Imashuku S., et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. International Journal of Hematology. 2007;86(1):58–65. doi: 10.1532/IJH97.07012. [DOI] [PubMed] [Google Scholar]
- 10.Risma K., Jordan M. B. Hemophagocytic lymphohistiocytosis: updates and evolving concepts. Current Opinion in Pediatrics. 2012;24(1):9–15. doi: 10.1097/mop.0b013e32834ec9c1. [DOI] [PubMed] [Google Scholar]
- 11.Egeler R. M., Shapiro R., Loechelt B., Filipovich A. Characteristic immune abnormalities in hemophagocytic lymphohistiocytosis. Journal of Pediatric Hematology/Oncology. 1996;18(4):340–345. doi: 10.1097/00043426-199611000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Zhang K., Jordan M. B., Marsh R. A., et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood. 2011;118(22):5794–5798. doi: 10.1182/blood-2011-07-370148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang K., Filipovich A. F., Johnson J., et al. Familial Hemophagocytic Lymphohistiocytosis. Seattle, Wash, USA: University of Washington, Seattle; 2013. [PubMed] [Google Scholar]
- 14.Stéphan F., Thiolière B., Verdy E., Tulliez M. Role of hemophagocytic histiocytosis in the etiology of thrombocytopenia in patients with sepsis syndrome or septic shock. Clinical Infectious Diseases. 1997;25(5):1159–1164. doi: 10.1086/516086. [DOI] [PubMed] [Google Scholar]
- 15.Raschke R. A., Garcia-Orr R. Hemophagocytic lymphohistiocytosis: a potentially underrecognized association with systemic inflammatory response syndrome, severe sepsis, and septic shock in adults. Chest. 2011;140(4):933–938. doi: 10.1378/chest.11-0619. [DOI] [PubMed] [Google Scholar]
- 16.Allen C. E., Yu X., Kozinetz C. A., McClain K. L. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatric Blood & Cancer. 2008;50(6):1227–1235. doi: 10.1002/pbc.21423. [DOI] [PubMed] [Google Scholar]
- 17.Pelkonen P., Swanljung K., Siimes M. A. Ferritinemia as an indicator of systemic disease activity in children with systemic juvenile rheumatoid arthritis. Acta Paediatrica Scandinavica. 1986;75(1):64–68. doi: 10.1111/j.1651-2227.1986.tb10158.x. [DOI] [PubMed] [Google Scholar]
- 18.Esumi N., Ikushima S., Hibi S., Todo S., Imashuku S. High serum ferritin level as a marker of malignant histiocytosis and virus-associated hemophagocytic syndrome. Cancer. 1988;61(10):2071–2076. doi: 10.1002/1097-0142(19880515)61:10<2071::AID-CNCR2820611023>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 19.Imashuku S., Hibi S., Sako M., et al. Soluble interleukin-2 receptor: a useful prognostic factor for patients with hemophagocytic lymphohistiocytosis. Blood. 1995;86(12):4706–4707. [PubMed] [Google Scholar]
- 20.Chandrakasan S., Filipovich A. H. Hemophagocytic lymphohistiocytosis: advances in pathophysiology, diagnosis, and treatment. The Journal of Pediatrics. 2013;163(5):1253–1259. doi: 10.1016/j.jpeds.2013.06.053. [DOI] [PubMed] [Google Scholar]
- 21.Imashuku S., Hibi S., Ohara T., et al. Effective control of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis with immunochemotherapy. Histiocyte Society. Blood. 1999;93(6):1869–1874. [PubMed] [Google Scholar]
- 22.Imashuku S., Kuriyama K., Teramura T., et al. Requirement for etoposide in the treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Journal of Clinical Oncology. 2001;19(10):2665–2673. doi: 10.1200/JCO.2001.19.10.2665. [DOI] [PubMed] [Google Scholar]
- 23.Buyse S., Teixeira L., Galicier L., et al. Critical care management of patients with hemophagocytic lymphohistiocytosis. Intensive Care Medicine. 2010;36(10):1695–1702. doi: 10.1007/s00134-010-1936-z. [DOI] [PubMed] [Google Scholar]
- 24.Chen R.-L., Lin K.-H., Lin D.-T., et al. Immunomodulation treatment for childhood virus-associated haemophagocytic lymphohistiocytosis. British Journal of Haematology. 1995;89(2):282–290. doi: 10.1111/j.1365-2141.1995.tb03302.x. [DOI] [PubMed] [Google Scholar]
- 25.Belyea B., Hinson A., Moran C., Hwang E., Heath J., Barfield R. Spontaneous resolution of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Pediatric Blood and Cancer. 2010;55(4):754–756. doi: 10.1002/pbc.22618. [DOI] [PubMed] [Google Scholar]
- 26.Ahn J.-S., Rew S.-Y., Shin M.-G., et al. Clinical significance of clonality and Epstein-Barr virus infection in adult patients with hemophagocytic lymphohistiocytosis. American Journal of Hematology. 2010;85(9):719–722. doi: 10.1002/ajh.21795. [DOI] [PubMed] [Google Scholar]
- 27.Kogawa K., Sato H., Asano T., et al. Prognostic factors of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in children: report of the Japan Histiocytosis Study Group. Pediatric Blood and Cancer. 2014;61(7):1257–1262. doi: 10.1002/pbc.24980. [DOI] [PubMed] [Google Scholar]
- 28.Chellapandian D., Das R., Zelley K., et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. British Journal of Haematology. 2013;162(3):376–382. doi: 10.1111/bjh.12386. [DOI] [PMC free article] [PubMed] [Google Scholar]