

Abstract
Cigarette smoking contributes to the development of pulmonary hypertension (PH) complicated with chronic obstructive pulmonary disease (COPD), and the pulmonary vascular remodeling, the structural basis of PH, could be attributed to abnormal proliferation of pulmonary artery smooth muscle cells (PASMCs).In this study, morphometrical analysis showed that the pulmonary vessel wall thickness in smoker group and COPD group was significantly greater than in nonsmokers. In addition, we determined the expression patterns of connective tissue growth factor (CTGF) and cyclin D1 in PASMCs harvested from smokers with normal lung function or mild to moderate COPD, finding that the expression levels of CTGF and cyclin D1 were significantly increased in smoker group and COPD group. In vitro experiment showed that the expression of CTGF, cyclin D1 and E2F were significantly increased in human PASMCs (HPASMCs) treated with 2% cigarette smoke extract (CSE), and two CTGF siRNAs with different mRNA hits successfully attenuated the upregulated cyclin D1 and E2F, and significantly restored the CSE-induced proliferation of HPASMCs by causing cell cycle arrest in G0. These findings suggest that CTGF may contribute to the pathogenesis of abnormal proliferation of HPASMCs by promoting the expression of its downstream effectors in smokers with or without COPD.
It is generally agreed that cigarette smoking is one of the most important risk factors for COPD and pulmonary hypertension1. Pulmonary hypertension is a complex medical condition of pulmonary arteries, characterized by sustained vasoconstriction, thickening of pulmonary artery walls, vascular remodeling, and progressive increase in pulmonary vascular resistance, leading to right ventricular failure and finally death2. Abnormal proliferation of PASMCs is believed to be responsible for medial hypertrophy, artery remodeling and vascular lumen narrowing.
As an important complication of COPD, pulmonary hypertension is an independent risk factor that significantly affects the course of the disease. A growing body of evidence indicates that cigarette smoking induces pulmonary vascular remodeling in patients with mild-to-moderate COPD as well as smokers with normal lung function3,4, both of which share documented similar gene expression profiles5. These findings suggested that cigarette smoking might be a direct cause of pulmonary vascular remodeling at the initial stage of COPD6. However, the precise mechanisms underlying this process remain unclear.
Our previous studies have shown that smoking exposure evidently induced pulmonary artery remodeling in rats by accelerating the proliferation of PASMCs via up-regulating CTGF, and shRNA-based down-regulation of CTGF significantly attenuated the induced pulmonary artery remodeling7. Firstly identified in conditioned medium of human umbilical vein endothelia cells, connective tissue growth factor (CTGF) is a 38 kDa, cysteine rich protein8, and it has been found to function as a major molecular mediator of cell adhesion, migration, proliferation, extracellular matrix (ECM) synthesis in a variety of cell types, including vascular endothelial cells, fibroblasts, osteoblastic cells, and smooth muscle cells9,10,11,12,13,14. The following study by our group further confirmed the proliferation-promoting effect of CTGF and found that ectopic introduction of CTGF significantly induced expression of cyclin D1 in rat PASMCs15.
It has been widely accepted that cell cycle progression is precisely regulated at various biological checkpoints by cyclins, cyclin-dependent kinases (CDKs) and CDK inhibitors16. Among the cyclin family members, cyclin D1 is a critical regulator in the control of cell cycle progression, and functions as a ‘mitogenic sensor’ as well as a cell proliferation enhancer by driving target cells through the restriction point in the G1 phase of the cell cycle17,18,19. In recent years, accumulating evidence points to a vital role of CTGF in the control of cell cycle progression20,21, and Cyclins, including D type cyclins, may act together with their CDK partners to control the mammalian cell proliferation22. Moreover, CTGF has been reported to activate the cyclin D1 in various cell types, such as human lung fibroblasts, human glomerular mesangial cells, and esophageal squamous cell carcinoma cells. Nevertheless, the roles of CTGF and cyclin D1 in regulating the cell cycle progression and cell proliferation, and the interaction of the two genes in human hPASMCs remain largely elusive.
Based on the above evidence, we hypothesized that upregulation of CTGF contributes to smoking-induced pulmonary artery remodeling by promoting G1/S transition in a cyclin D1-dependent manner. To test it, we determined and compared the expression patterns of CTGF and cyclin D1 as well as structural alternation in pulmonary vessels harvested from smokers with normal lung function or mild to moderate COPD patients. Additionally, we examined the influence of CSE on the expression of CTGF in the HPASMCs and investigated whether CTGF contributes to the CSE-induced proliferation of HPASMCs by upregulating cyclin D1 and E2F in vitro.
Results
Clinical data analysis
All patients in the three groups were male. No differences were identified between the three groups with regard to age and oxygen pressure in arterial blood (PaO2) (P > 0.05). Smoking history was similar in smoker group and COPD group (P > 0.05). As expected from the selection criteria, the values of forced expiratory volume in 1 second (FEV1) (% predicted) and FEV1/forced vital capacity (FVC) (%) in COPD group were significantly lower than in nonsmoker group or in smoker group (P < 0.01) (Table 1).
Table 1. Characteristics of the Recruited Subjects.
| Groups | Nonsmoker (n = 24) | Smoker (n = 30) | COPD (n = 27) |
|---|---|---|---|
| Age (years) | 62.14 ± 4.26 | 59.23 ± 5.81 | 62.19 ± 5.31 |
| Smoking (packets-years) | 0 | 38.28 ± 10.97* | 45.88 ± 11.23* |
| BMI (kg/m2) | 21.94 ± 3.57 | 20.86 ± 3.23 | 20.11 ± 3.62 |
| FEV1% predicted | 98.21 ± 7.59 | 94.39 ± 9.51 | 74.59 ± 10.83* |
| FEV1/FVC% | 79.65 ± 6.34 | 82.81 ± 5.95 | 61.65 ± 6.39* |
| PaO2 (mmHg) | 92.11 ± 7.29 | 87.61 ± 8.35 | 82.93 ± 8.02 |
Abbreviations: BMI = body mass index; FEV1 = forced expiratory volume in 1 second; PaO2 = arterial partial pressure of oxygen. *P < 0.05 versus nonsmoker group.
Morphometric measurements
To evaluate the artery remodeling status in each group, lung tissue of similar site and depth in each subject was biopsied with a focus on “intermediate vessels” (50–500 μm) as described in Materials and Methods section. As expected, the pulmonary arteries walls in nonsmoker group were physiologically thin with relative large lumens. Conversely, lumens of pulmonary arteries in smoker group and COPD group were much narrower due to substantially thickened vessel walls compared with the nonsmokers'. As shown in Figure 1, pulmonary vessel wall thickness were comparable between smoker group and COPD group (P > 0.05), and both of them were significantly higher than that of nonsmokers (P < 0.01).
Figure 1. Morphological features of pulmonary artery of nonsmoker group, smoker group, and COPD group.

(A). The lung sections were stained with H&E to evaluate the vessel wall thickness (Scale bars: 20 μm). (B). Pulmonary vessel wall thickness (WT) was expressed as % total diameter in pulmonary arteries. *P < 0.05 versus nonsmoker group.
Exposure to CSE substantially promoted proliferation of HPASMCs
To investigate the effect of CSE on the proliferation of HPASMCs in vitro, HPASMCs were challenged with 0%, 1%, 2%, 5%, or 10% CSE and cell proliferation was evaluated. CSE at concentrations of 1% and 2% caused a significant increase in cell survival compared with the control, and the peak survival was observed in the cells treated with 2% CSE. In contrast, CSE at higher concentrations, such as 5% and 10%, did not promote, but unexpectedly suppress cell proliferation by introducing apoptosis to more portions of the cells (Figure 2). Thus, the following experiments were performed with 2% CSE.
Figure 2. Effects of CSE on the HPASMCs proliferation were evaluated by cell counting.

The cells were seeded in 24-well plates and incubated with CSE at different concentrations (1–10%) for 24 h. 0% group was considered as control group. *P < 0.05 versus control group.
Determination of expression of CTGF and cyclin D1 in explants of intrapulmonary arteries
Expression patterns of CTGF and cyclin D1 were determined in explants of intrapulmonary arteries by using real-time quantitative PCR (Figure 3A and 3B) and western blot (Figure 3C–F), respectively. The mRNA and protein expression levels of CTGF and cyclin D1 in explants of intrapulmonary arteries were similarly elevated in smoker group and COPD group in comparison to nonsmoker group. In line with this, CTGF and cyclin D1 mRNA and protein expression levels were both enhanced in HPASMCs exposed to 2% CSE, as compared with the control, as shown in figure 4 and 5.
Figure 3.

The expression patterns of CTGF were determined in pulmonary vessels collected from smokers, patients with mild to moderate COPD as well as normal controls by real-time quantitative PCR (A) and western blot (C). The results of western blots were densitometrically analyzed (E). The expression patterns of cyclin D1 were determined in pulmonary vessels collected from smokers, patients with mild to moderate COPD as well as normal controls by real-time quantitative PCR (B) and western blot (D). The results of western blots were densitometrically analyzed (F). *P < 0.05 versus control group; #P < 0.05 versus 2% CSE group.
Figure 4. HPASMCs were treated with CSE and/or anti-CTGF siRNA#1 (#1, differently designed mRNA hit from #2), and the mRNA and protein expression levels were examined by real-time PCR and western blot.

(A). Upper panel: mRNA expression level of CTGF in the cells treated with control, CSE only, CSE plus anti-CTGF siRNA#1, and control plus anti-CTGF siRNA#1; Middle panel: Protein expression level of CTGF in the cells treated with control, CSE only, CSE plus anti-CTGF siRNA#1, and control plus anti-CTGF siRNA#1; Lower panel: Densitometry analysis of the western blots. (B). Upper panel: mRNA expression level of cyclin D1 in the cells treated with control, CSE only, CSE plus anti-CTGF siRNA#1, and control plus anti-CTGF siRNA#1; Middle panel: Protein expression level of cyclin D1 in the cells treated with control, CSE only, CSE plus anti-CTGF siRNA#1, and control plus anti-CTGF siRNA#1; Lower panel: Densitometry analysis of the western blots. (C). Upper panel: mRNA expression level of E2F in the cells treated with control, CSE only, CSE plus anti-CTGF siRNA#1, and control plus anti-CTGF siRNA#1; Middle panel: Protein expression level of E2F in the cells treated with control, CSE only, CSE plus anti-CTGF siRNA#1, and control plus anti-CTGF siRNA#1; Lower panel: Densitometry analysis of the western blots. *P < 0.05 versus control group; #P < 0.05 versus 2% CSE group.
Figure 5. HPASMCs were treated with CSE and/or anti-CTGF siRNA#2 (#2, differently designed mRNA hit from #1), and the mRNA and protein expression levels were examined by real-time PCR and western blot.

(A). Upper panel: mRNA expression level of CTGF in the cells treated with control, CSE only, CSE plus anti-CTGF siRNA#2, and control plus anti-CTGF siRNA#2; Middle panel: Protein expression level of CTGF in the cells treated with control, CSE only, CSE plus anti-CTGF siRNA#2, and control plus anti-CTGF siRNA#2; Lower panel: Densitometry analysis of the western blots. (B). Upper panel: mRNA expression level of cyclin D1 in the cells treated with control, CSE only, CSE plus anti-CTGF siRNA#2, and control plus anti-CTGF siRNA#2; Middle panel: Protein expression level of cyclin D1 in the cells treated with control, CSE only, CSE plus anti-CTGF siRNA#2, and control plus anti-CTGF siRNA#2; Lower panel: Densitometry analysis of the western blots. (C). Upper panel: mRNA expression level of E2F in the cells treated with control, CSE only, CSE plus anti-CTGF siRNA#2, and control plus anti-CTGF siRNA#2; Middle panel: Protein expression level of E2F in the cells treated with control, CSE only, CSE plus anti-CTGF siRNA#2, and control plus anti-CTGF siRNA#2; Lower panel: Densitometry analysis of the western blots. *P < 0.05 versus control group; #P < 0.05 versus 2% CSE group.
Effect of downregulation (by siRNAs) and upregulation (by ectopic expression) of CTGF on the expression of cyclin D1 and E2F
To mutually confirm the silencing effect of the siRNAs on CTGF as well as consequential influence on cyclin D1 and E2F, two anti-CTGF siRNAs with different hits in the mRNA of CTGF were used for the following “loss-of-function” analysis. We found that the two CTGF siRNAs similarly and effectively restored the enhanced mRNA and protein expression of CTGF, cyclin D1 and E2F in HPASMCs treated with 2% CSE, as shown in Figure 4 and 5. Consistently, the protein expression levels of cyclin D1 and E2F were substantially promoted following the upregulation of CTGF by ectopic overexpression (Figure 6).
Figure 6. “Gain-of-function” analysis was performed to evaluate the effect of overexpressed CTGF on the downstream effectors such as cyclin D1 and E2F.
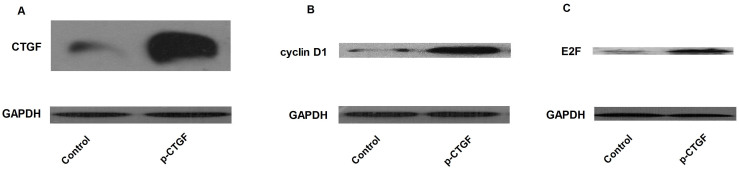
Ectopic overexpression of CTGF in HPASMCs (A) substantially induced the expression of cyclin D1(B), as well as its downstream effector, E2F (C). The activation of CTGF-cyclin D1-E2F axis might be responsible for the enhanced proliferation of HPASMCs.
Effect of downregulation (by siRNAs) and upregulation (by ectopic expression) of CTGF on the proliferation of HPASMCs exposed to CSE
To assess the effect of downregulation (by siRNAs) and upregulation (by ectopic expression) of CTGF on the proliferation of HPASMCs exposed to CSE, we carried out cell counting and BrdU incorporation assay to evaluate the survivals of CSE-treated PASMCs transfected with CTGF siRNAs or pcDNA3.1-CTGF, finding that, as shown in Figure 7, both CTGF siRNAs markedly decreased the survivals of HPASMCs, while ectopic overexpression of CTGF significantly promoted the survival of the cells. The results of BrdU assay exhibited the similar tendency. No significant difference was observed between control group and CTGF siRNA-treated control group.
Figure 7. The effect of CTGF siRNA on HPASMCs proliferation induced by 2% CSE.

(A). Quantification of cell number after treated with control, CSE, CSE + CTGF siRNA#1, control ++ CTGF siRNA#1, CSE + CTGF siRNA#2, control ++ CTGF siRNA#2, and overexpressed CTGF. Results are presented as percentages of the control group. (B). Percentages of BrdU-positive cells after treated with control, CSE, CSE + CTGF siRNA#1, control ++ CTGF siRNA#1, CSE + CTGF siRNA#2, control ++ CTGF siRNA#2, and overexpressed CTGF. Results are presented as percentages of the control group. The cells were equally aliquoted and treated with control (C), CSE (D), CSE + CTGF siRNA#1 (E), control ++ CTGF siRNA#1 (F), CSE + CTGF siRNA#2 (G), control ++ CTGF siRNA#2 (H), and overexpressed CTGF(I), and the cell proliferation status of each group was presented. *P < 0.05 versus control group; #P < 0.05 versus 2% CSE group.
Effect of downregulation (by siRNAs) and upregulation (by ectopic expression) of CTGF on the cell cycle progression in HPASMCs exposed to CSE
To further explore the molecular mechanism underlying the effect of up- or down-regulation of CTGF on the survival of PASMCs, we further investigated the influence of CTGF siRNAs or ectopic overexpression of CTGF on cell cycle status. According to flowcytometry analysis, we found that 2%CSE reduced the proportion of cells in G0/G1 phase (Figure 8), and ectopic overexpression of CTGF similarly reduced the cells in G0/G1 phase. However, both CTGF siRNAs similarly caused G0/G1 cell cycle arrest in HPASMCs.
Figure 8. CTGF siRNA caused the G0/G1 cell cycle arrest.

The percentages of differently treated cells in G0/G1 (A) and S phase (B). The flowcytometry analysis results of the cells treated with control (C), CSE (D), CTGF siRNA#1 (E), CTGF siRNA#2 (F), and overexpressed CTGF (G). *P < 0.05 versus control group; #P < 0.05 versus 2% CSE group.
Identification of interaction between the PASMCs and Fibroblast cells
To study the possible interaction between the PASMCs and pulmonary fibroblast cells, we evaluated the expression level of CTGF and cyclin D1 in CSE-treated HPASMCs cultured in conditioned medium harvested from fibroblast cells transfected CTGF siRNA, and found that the expected silencing effect was not observed in HPASMCs cultured in conditioned medium harvested from fibroblast cell transfected with CTGF siRNA, compared with the control (HPASMCs cultured in conditioned medium harvested from fibroblast cells transfected with negative control siRNA) (Data not shown).
Discussion
In this study, we demonstrated that cigarette smoking significantly induced pulmonary vascular remodeling, as well as upregulation of CTGF and cyclin D1 in human pulmonary vessels. In addition, we found that exposure to CSE induced a profound increase in DNA synthesis and cell survival of cultured HPASMCs and CSE-induced proliferation was found to be dependent on CTGF upregulation and downstream mitogenic signaling pathway activation. These data confirmed the critical role of CTGF in the control of HPASMCs proliferation and pulmonary vascular remodeling induced by cigarette smoking.
Cigarette smoking is a major preventable public health problem and known to be associated with a wide spectrum of diseases, including some vascular diseases, such as pulmonary hypertension. The aberrantly enhanced proliferation of PASMCs is one of the key factors leading to medial hypertrophy, artery remodeling and vascular lumen narrowing, all of which constitute the pathological changes in pulmonary hypertension23. In the present study, we found that low concentrations CSE (such as 1% and 2%) significantly enhanced the proliferation of HPASMCs. In contrast, higher concentration CSE suppressed the cells growth by introducing more apoptosis. These results were in line with some previous studies7,15,24, but appeared to be contradictory to others25,26,27, and such discrepancy could be attributed to either different cell types or different process of CSE preparation.
CTGF has been reported to play a regulatory role in a variety of biological activities such as cell proliferation, adhesion, and migration28. Recent studies showed that CTGF is one of the key components mediating the biological effects of cigarette smoking29,30. In this study, we further extended these findings by showing that the expression of CTGF was substantially upregulated in smokers with or without deteriorated lung function. Simultaneously, we have demonstrated that upregulation of CTGF significantly contributed to the enhanced cigarette smoke exerted its biological effects via CTGF. We extended this finding and revealed that CTGF expression was increased in smoker with or without normal lung function. Furthermore, we demonstrated in the in vitro study that CTGF was involved in the proliferation of primary HPASMCs induced by CSE in vitro. To identify the mechanism underlying the regulatory role of CTGF in the control of cell proliferation, we further evaluated the effect of CTGF on cell cycle distribution by using flowcytometry. Our study showed that upregulation of CTGF induced by exposure to CSE or ectopic introduction promoted cell cycle transition progression from G0/G1 to S phase, and accordingly, downregulation of CTGF by transfection of two different siRNAs significantly attenuated CSE-induced proliferation of HPASMCs by causing cell cycle arrest at G0/G1 phase.
Furthermore, we explored the molecular mechanism underlying the abnormally enhanced proliferation of HPASMCs and thickened pulmonary vessel wall by comparing the gene expression pattern in HPASMCs collected from smokers with normal lung function or mild to moderate COPD patients and normal control, and identified a substantial increase in cyclin D1 expression in the former group. To confirm such regulatory relationship, two CTGF siRNAs with different mRNA hits were used to downregulate CTGF, and explored its effect on the expression of cyclin D1 and E2F, and we showed that CTGF siRNAs introduction led to a significant decrease in cyclin D1 and E2F levels, and inhibited the cell proliferation in HPASMCs exposed to CSE by causing G0 phase arrest. Consistently, ectopic overexpression of CTGF markedly caused an increase in both cyclin D1 and E2F expression, and promoted the proliferation of the cells by promoting G0/S transition. Therefore, it was evident that aberrantly enhanced proliferation of HPASMCs directly resulted from upregulation of cyclin D1 as well as its downstream effector, E2F, which was mediated by altered expression of CTGF. Our results are consistent with previous studies from other cell types which showed that CTGF activated cyclin D131,32. Cyclin D1 is a critical regulator in the control of cell cycle progression, and plays a key role in regulating G1/S transition17. Our previous study reported that cyclin D1 specific plasmid-based shRNA successfully inhibited the PASMCs proliferation and attenuated CSE-induced pulmonary vascular remodeling in rats33. Meanwhile, E2F is a downstream effector of cyclin D1, and its activity is necessary for S phase progression34. Taken together, the above data support the hypothesis that CTGF is likely involved in the control of HPASMCs proliferation by regulating cell cycle progression, and cyclin D1 and E2F may be the downstream effectors of CTGF in the pathogenesis of the disease.
The pulmonary artery is made up of several resident cell types including the endothelial cells fibroblast cells, and smooth muscle. Dysregulated cellular activities of those cells are central to the pathology of PH in animal models as well as in humans35, and the communication and interaction between the vascular cell types have been repeatedly reported and well documented36. In this study, we examined the expression level of CTGF in the CSE-treated HPASMCs cultured with conditioned medium harvested from fibroblasts transfected with CTGF siRNA, whereas we did not identify any detectable change in the expression of CTGF in the cells. We reasoned that such observation could be attributed to rapid degradation of siRNA in vitro, or distinct work pattern in different cell types.
In the present study, several issues deserve comment. Firstly, although the lung resection and pulmonary artery dissection were performed at the most distant point and were completely free of tumor tissue, we cannot completely rule out a possible influence of the inflammatory paraneoplastic microenvironment; Secondly, neither of the two CTGF siRNAs completely abolished the proliferation of HPASMCs induced by CSE, even though CTGF siRNA markedly suppressed the CTGF expression, indicating that other mediators or effectors might be involved in the mechanism underlying CSE-induced HPASMCs proliferation, such as endothelin and protein kinase C37,38. Moreover, it is noteworthy that CTGF siRNA indeed reduced, but could not completely abolish the upregulated cyclin D1 and E2F expression induced by CSE, suggesting that although CTGF is clearly involved in cyclin D1-mediated HPASMCs proliferation, other intracellular signaling pathways, such as MAPK/β-catenin and PI3K/Akt, may be also involved in regulating the process39,40. These molecules are also implicated in regulating the expression of cyclin D1, which could be an explanation to the observation that knockdown of CTGF in HPASMCs resulted in a reduction, but not a complete inhibition of cyclin D1 expression and proliferation of HPASMCs.
In summary, we concluded that cigarette smoke exposure significantly promoted proliferation of HPASMCs and induced pulmonary artery remodeling, and such changes could be attributed, at least in part, to the activation of CTGF-cyclin D1-E2F signaling axis. Therefore, CTGF inhibition may prove to be a novel target for the treatment of pulmonary artery hypertension, especially in smokers.
Methods
Case selection
Resected lung samples were obtained from 81 patients who underwent pneumonectomy in our hospitals from January to December, 2012. Pulmonary function tests were performed within days prior to surgical treatment in all patients. According to the guideline of COPD41 and smoking history of patients, patients were divided into three groups: 24 nonsmokers with normal lung function (nonsmoker group), 30 smokers with normal lung function (smoker group), and 27 smokers with mild to moderate COPD (COPD group). The clinical and lung functional characteristics in the three groups of patients are shown in Table 1. None of the patients had history of bronchial asthma, respiratory tract infectious diseases one month prior to the study, no cardiac, renal, hepatic, gastrointestinal, and endocrine diseases. Control subjects were taking no medications. COPD patients were receiving bronchodilators only, and no corticosteroid of any form was being taken. Their medical therapy consisted of a combination of long-acting (salmeterol) and short-acting (salbutamol) β2-agonists as needed, an anticholinergic (Tiotropium Bromide), and a xanthine preparation (theophylline). All patients were considered to be clinically stable because none of them had required medical attention and/or any change in their regular therapy within 3 months prior to the enrollment. This study was approved by the research ethics committee of Anhui medical university, and informed consent was obtained from all participants. The study was performed in accordance with the guidelines established by the Science Council of China.
Tissue preparation and morphometric analysis of pulmonary vessels
Tissue samples were taken at a site more than 5 cm away from carcinoma, and the biopsies were performed at similar sites or similar depth. Each tissues specimen was fixed in 4% paraformaldehyde. After dehydration, it was embedded in paraffin and selected for 5μm-thick serial sectioning. intrapulmonary artery of intermediate size (50–500μm) branches were isolated under optical microscope, and then the endothelial surface and the surrounding adventitia were carefully dissected, the medial layer was immediately stored at −80°C. This study was approved by the research ethics committee of Anhui medical university, and informed consent was obtained from all patients. This study was performed in accordance with the guidelines established by the Science Council of China.
Sections were deparaffinized and stained with hematoxylin and eosin (H&E). Then the H&E-stained tissue sections were observed under a light microscope. For each patient, five arteries with external diameter between 50–500μm and structurally integral laminas were randomly selected and measured. The pulmonary vessel wall thickness was expressed as the percentage of the external diameter [(2 × measured wall thickness/external diameter) × 100].
Cell culture
Segments of pulmonary artery (50–500μm external diameter) from nonsmoker group were incubated in Hanks' solution containing collagenase (1.5 mg/mL) for 20 minutes. After incubation, a thin layer of adventitia was carefully stripped off with a fineforcep, and the endothelium was removed by gentle scratching of the intimal surface with a surgical blade. The remaining smooth muscle was then digested with collagenase (2.0 mg/mL) and elastase (0.5 mg/mL) for 35–45 minutes at 37°C. Cells were cultured in Dulbecco's modified Eagle's Medium (DMEM) (Life Technologies Inc., USA) containing 10% fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 mg/mL) and cultured in a humidified incubator at 37°C. The cells were passaged by trypsinization with 0.05% trypsin–EDTA and used for experiments at passages 3–8. Cells were identified by immunochemistry staining of α-SM-actin antibody. For all experiments, cells were made quiescent by serum-free media for 24 h before exposure to CSE or DMEM.
Preparation of CSE solution
Cigarette smoke extract (CSE) was prepared freshly for each experiment, using Marlboro cigarettes. CSE was prepared as previously described15. Briefly, cigarette smoke derived from one cigarette was drawn slowly into a 50 ml syringe and bubbled through 30 ml of DMEM. One cigarette yielded five draws of the syringe, with each individual draw taking approximately 10 s to complete. The resulting solution, which was considered ‘‘100%’’ strength, was then filtered before being diluted in DMEM to the required strength for application to HPASMCs. Cells were divided into severn groups randomly: control group, 2%CSE group, CTGF siRNA#1-treated 2%CSE group, CTGF siRNA#1-treated control group, CTGF siRNA#2-treated 2%CSE group, CTGF siRNA#2-treated control group, and plasmid-based extopic overexpression group. All groups, except control group and plasmid-based ectopic overexpression group, were challenged with CSE for 24 hours.
siRNA transfection
Two anti-CTGF siRNAs (siRNA#1: 5′-GCACCAGCAUGAAGACAUACCdTdT-3′ and 5′-GGUAUGUCUUCAUGCUGGUGCdTdT-3′; siRNA#2: 5′-CCAAGCCUAUCAAGUUUGAGCdTdT-3′ and 5′-GCUCAAACUUGAUAGGCUUGGdTdT-3′, and control siRNA: 5′-UUCUCCGAACGUGUCACGUdTdT-3′ and 5′-ACUCCUCGCAGCAUUUCCCGGdTdT-3′), as described previously21, were used to knockdown the expression of CTGF in HPASMCs. For transfection, cells were seeded into plates, incubated overnight, and then transfected using Lipofectamine 2000 transfection reagent according to the manufacturer's instructions at 40–60% cell confluence.
Cell counting
Cells were seeded in 24-well plates (5,000 cells/well) and cultured overnight. Cells were then maintained in serum-free media for 24 h, followed by different treatments. At the end-point, cells were harvested and counted using a hemocytometer.
BrdU incorporation assays
BrdU was added 2 h before culture termination. At the end of culture, cells were immunoblocked with goat serum, and incubated overnight at 4°C with the anti-BrdU antibody. Then, they were incubated for 60 min with biotinylated goat anti-mouse IgG (1:200), and stained with DAB. The percentage of stained cells was determined by counting the number of positively stained cells within a microscopic vision field, divided by the total number of cells in that same field.
RNA extraction and quantitative real-time PCR
Total RNA was extracted by using Trizol (Invitrogen) according to the manufacturer's protocol. Then, first-strand cDNA was synthesized by using the First-Strand cDNA synthesis kit (ReverTra Ace-α-,FSK-100;Toyobo) according to the manufacturer's instructions. Quantitative real-time PCR was performed using the SYBR Green Realtime PCR Master Mix (QPK-201, Toyobo) with the Light Cycler Instrument (Roche Diagnostics Corp., Basel, Suisse). The cDNA fragments were denatured at 95°C for 15 s, annealed at 58°C for 15 s, and extended at 72°C for 45 s for 40 cycles. Each sample was examined in triplicate and the amounts of the PCR products produced were normalized to that of GAPDH which served as internal control. The primers used in the study were as follows: for CTGF, forward (5′-ACTATGATTAGAGCCAACTG-3′) and reverse (5′-TGTTCTCTTCCAGGTCAG-3′); for cyclin D1, forward (CGTGGGCTCTAAGATGAAGG-3′) and reverse (5′-TGCGGATGATCTGTTTGTTC-3′); for E2F, forward (5′-GCGCAGGACGCCAGCGCCC-3′) and reverse (5′-GCCTGGGCGCCTCAGTCAA-3′); for GAPDH, forward (5′-GAAGGTGAAGGTCGGAGTC-3′) and reverse (5′-GAAGATGGTGATGGGATTTC-3′). Relative transcript levels of each gene were calculated using the ΔΔCt method, using GAPDH as the housekeeping gene42.
Western blot analysis
Total protein was extracted from cultured HPASMCs or intrapulmonary arteries. Total protein concentration was determined by the Bradford method using Bradford reagent. Equivalent amounts of protein (50μg) were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). The proteins were transferred to polyvinylidene difluoride membranes and blocked for 2 hours in PBS with 0.1% Tween (PBS-T) containing 5% nonfat dried milk. The membranes were then incubated with primary antibody, CTGF (1:200)(Santa cruz), E2F (1:500) (Cell signaling) or cyclin D1 (1:500) (Cell signaling), overnight and then washed three times with PBS-T. The membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (1:10,000) for 1 hour, then washed three times with PBS-T. Immunoreactivity was detected using an enhanced chemiluminescence Western blotting detection kit according to the manufacturer's instructions and exposed to X-ray film. Immunoblots were scanned using a GS-800 densitometer and protein bands were quantified with Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA). To control for loading, blots were stripped and reanalyzed for GAPDH.
Cell cycle analysis
To estimate the proportions of cells in different phases of the cell cycle, cellular DNA contents were measured by flow cytometry. After treatments, cells were harvested, washed twice with cold PBS, and then fixed overnight at −20°C in 70% ethanol. Immediately before flow cytometry, the cells were resuspended in PBS containing PI (50 m g/ml) and DNase-free RNase (10 m g/ml). Flow cytometry was performed using a FACScalibur (Becton Dickinson, San Diego, CA) system with CELLquest software. The percentages of cells in different phases of the cell cycle were determined using the ModFit software.
Construction of the vector containing CTGF
The coding sequence of CTGF was PCR-amplified using the following primer set: 5′-CGGGATCCATGACCGCCGCCAGTATGGGC-3′ and 5′-ATAAGAATGCGGCCGCTCATGCCATGTCTCCGTACATC-3′. The PCR product was subsequently purified, double digested, and insert into pcDNA3.1 between two restrictive sites, BamHI and NotI. The sequence of the insert was confirmed by direct Sanger sequencing prior to the following functional analysis. For transfection, cells were seeded into plates, incubated overnight, and then transfected using Lipofectamine 2000 transfection reagent according to the manufacturer's instructions at 70–80% cell confluence.
Generation of conditioned medium
Human pulmonary fibroblast cells were purchased from Shanghai Cell Bank of Chinese Academy of Sciences (Shanghai, China). After 24-hour incubation in serum-free DMEM, the medium in 60-mm dishes was collected from the fibroblast cells transfected with CTGF siRNA. The medium from fibroblast cells transfected with control siRNA for 24 hours was used as the control. Conditioned media were centrifuged at 15,000 g for 5 minutes at 4°C. The supernatants were then aliquoted and used in subsequent experiments.
Proliferation of HPASMCs in response to conditioned medium
CSE-treated HPASMCs were plated at 1.5 × 104 cells/well in 24- well plates, overnight. The following day, the cultured medium was replaced with conditioned medium collected from pulmonary fibroblast cells transfected with CTGF siRNA or the control. The expression of CTGF and cyclin D1 was evaluated as described above, and proliferation of HPASMCs was evaluated by cell counting or BrdU incorporation assays. Each experiment was repeated at least thrice.
Statistical analysis
All data were expressed as means ± SEM. Independent t-test (for two-group comparison), and one-way ANOVA (for multiple-group comparison) followed by the least significant difference (LSD) test were used to compare between groups using software SPSS 12.0 (Chicago, IL). All experiments were performed in triplicate, and data were considered statistically significant when P < 0.05.
Author Contributions
All authors reviewed the manuscript. G.Y.S. and R.W. designed research; S.J.Z., M.L., D.X.Z., Z.M.Z., X.W.H., Y.H.L., R.W. and G.Y.S. performed the experiments, R.W. and G.Y.S. analyzedthe data; S.J.Z., M.L., R.W. and S.G.Y. wrote manuscript; G.Y.S. and R.W. reviewed the manuscript.
Acknowledgments
This research was supported by the fund from the Natural Science Foundation of China (No.81300041, No.81100038), the fund from the Natural Science Foundation of the Anhui Higher Education Institutions of China (No.KJ2012Z184), the fund for the academic backbone of the excellent young and middle-age people of Anhui medical university (2013), the fund from the first affiliated hospital of Anhui medical university for reserve talents (2014).
References
- Wright J. L. et al. Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 295, L1–15 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humber M. t. et al. Treatment of pulmonary arterial hypertension. N Engl J Med. 351, 1425–1436 (2004). [DOI] [PubMed] [Google Scholar]
- Peinado V. I. et al. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am J Physiol. 274, L908–913 (1998). [DOI] [PubMed] [Google Scholar]
- Santos S. et al. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J. 19, 632–638 (2002). [DOI] [PubMed] [Google Scholar]
- Llinàs L. et al. Similar gene expression profiles in smokers and patients with moderate COPD. Pulm Pharmacol Ther. 24, 32–41 (2011). [DOI] [PubMed] [Google Scholar]
- Weitzenblum E. et al. Pulmonary hypertension in chronic obstructive pulmonary disease and interstitial lung diseases. Semin Respir Crit Care Med. 30, 458–470 (2009). [DOI] [PubMed] [Google Scholar]
- Wang R. et al. Knockdown of connective tissue growth factor by plasmid-based short hairpin RNA prevented pulmonary vascular remodeling in cigarette smoke-exposed rats. Arch Biochem Biophys. 508, 93–100 (2011). [DOI] [PubMed] [Google Scholar]
- Bradham D. M. et al. Connective tissue growth factor: a cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. J Cell Biol. 114, 1285–1294 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babic A. M. et al. Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin alphavbeta3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol Cell Biol. 19, 2958–2966 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishikawa K. et al. Overexpression of connective tissue growth factor gene induces apoptosis in human aortic smooth muscle cells. Circulation. 100, 2108–2112 (1999). [DOI] [PubMed] [Google Scholar]
- Shimo T. et al. Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J Biochem. 126, 137–145 (1999). [DOI] [PubMed] [Google Scholar]
- Takigawa M. CTGF/Hcs24 as a multifunctional growth factor for fibroblasts, chondrocytes and vascular endothelial cells. Drug News Perspect. 16, 11–21 (2003). [DOI] [PubMed] [Google Scholar]
- Nishida T. et al. Effects of CTGF/Hcs24, a hypertrophic chondrocyte-specific gene product, on the proliferation and differentiation of osteoblastic cells in vitro. J Cell Physiol. 184, 197–206 (2000). [DOI] [PubMed] [Google Scholar]
- Bonniaud P. et al. Connective tissue growth factor is crucial to inducing a profibrotic environment in "fibrosis-resistant" BALB/c mouse lungs. Am J Respir Cell Mol Biol. 31, 510–516 (2004). [DOI] [PubMed] [Google Scholar]
- Wang R. et al. CCN2 promotes cigarette smoke-induced proliferation of rat pulmonary artery smooth muscle cells through upregulating cyclin D1 expression. J Cell Biochem. 113, 349–359 (2012). [DOI] [PubMed] [Google Scholar]
- Morgan D. O. Principles of CDK regulation. Nature 374, 131–134 (1995). [DOI] [PubMed] [Google Scholar]
- Sherr C. J. G1 phase progression: cycling on cue. Cell 79, 551–555 (1994). [DOI] [PubMed] [Google Scholar]
- Fu M. et al. Minireview: Cyclin D1: normal and abnormal functions. Endocrinology. 145, 5439–5447 (2004). [DOI] [PubMed] [Google Scholar]
- Qin X. Q. et al. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci U S A. 91, 10918–10922 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahab N. et al. Connective tissue growth factor and regulation of the mesangial cell cycle: Role in cellular hypertrophy. J Am Soc Nephrol. 13, 2437–2445 (2002). [DOI] [PubMed] [Google Scholar]
- Jing J. et al. RNA Interference Targeting Connective Tissue Growth Factor Inhibits the Transforming Growth Factor- Induced Proliferation in Human Tenon Capsule Fibroblasts. J ophthalmol. 2013, 354798 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffler L. et al. Growth inhibition in G(1) and altered expression of cyclin D1 and p27(kip-1)after forced connexin expression in lung and liver carcinoma cells. J Cell Biochem. 79, 347–354 (2000). [DOI] [PubMed] [Google Scholar]
- Morrell N. W. et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 54, S20–31 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luppi F. et al. Effects of cigarette smoke condensate on proliferation and wound closure of bronchial epithelial cells in vitro: role of glutathione. Respir Res. 6, 140 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nana-Sinkam S. P. et al. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. Am J Respir Crit Care Med. 175, 676–685 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miglino N. et al. Cigarette smoke inhibits lung fibroblast proliferation by translational mechanisms. Eur Respir J. 39, 705–711 (2012). [DOI] [PubMed] [Google Scholar]
- Chang Y. et al. Genetic deletion of IL-17A reduces cigarette smoke-induced inflammation and alveolar type II cell apoptosis. Am J Physiol Lung Cell Mol Physiol. 306, L132–143 (2014). [DOI] [PubMed] [Google Scholar]
- Cicha L. et al. Connective tissue growth factor: context-dependent functions and mechanisms of regulation. Biofactors. 35, 200–208 (2009). [DOI] [PubMed] [Google Scholar]
- Churg A. et al. Cigarette smoke drives small airway remodeling by induction of growth factors in the airway wall. Am J Respir Crit Care Med. 174, 1327–1334 (2006). [DOI] [PubMed] [Google Scholar]
- Wang R. D. et al. Transforming growth factor-beta(1) drives airway remodeling in cigarette smoke-exposed tracheal explants. Am J Respir Cell Mol Biol. 33, 387–393 (2005). [DOI] [PubMed] [Google Scholar]
- Jiang C. G. et al. Downregulation of connective tissue growth factor inhibits the growth and invasion of gastric cancer cells and attenuates peritoneal dissemination. Mol Cancer. 10, 122 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J. E. et al. Proliferation is not increased in airway myofibroblasts isolated from asthmatics. Eur Respir J. 32, 362–371 (2008). [DOI] [PubMed] [Google Scholar]
- Zeng D. X. et al. Suppression of cyclin D1 by plasmid-based short hairpin RNA ameliorated experimental pulmonary vascular remodeling. Microvasc Res. 90, 144–149 (2013). [DOI] [PubMed] [Google Scholar]
- Li Qin L. et al. Inhibition of Smooth Muscle Cell Proliferation by Ezetimibe via the Cyclin D1-MAPK Pathway. J Pharmacol Sci. 125, 283–91 (2014). [DOI] [PubMed] [Google Scholar]
- Nogueira-Ferreira R. et al. Cellular interplay in pulmonary arterial hypertension: implications for new therapies. Biochim Biophys Acta. 1843, 885–893 (2014). [DOI] [PubMed] [Google Scholar]
- Qin Yao. et al. Micro-RNA-21 regulates TGF-b-induced myofibroblast differentiation by targeting PDCD4 in tumor-stroma interaction. Int J Cancer. 128, 1783–1792 (2011). [DOI] [PubMed] [Google Scholar]
- Milara J. et al. Bosentan inhibits cigarette smoke-induced endothelin receptor expression in pulmonary arteries. Eur Respir J. 39, 927–938 (2012). [DOI] [PubMed] [Google Scholar]
- Xiang M. et al. Cigarette smoke extract promotes human pulmonary artery smooth muscle cells proliferation through protein kinase C alpha-dependent induction of cyclin D1. Chin Med J (Engl). 123, 3663–3670 (2010). [PubMed] [Google Scholar]
- Wu S. H. et al. Lipoxin A(4) inhibits proliferation of human lung fibroblasts induced by connective tissue growth factor. Am J Respir Cell Mol Biol. 34, 65–72 (2006). [DOI] [PubMed] [Google Scholar]
- Deng Y. Z. et al. Connective tissue growth factor is overexpressed in esophageal squamous cell carcinoma and promotes tumorigenicity through beta-catenin-T-cell factor/Lef signaling. J Biol Chem. 282, 36571–36581 (2007). [DOI] [PubMed] [Google Scholar]
- Vestbo J. et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 187, 347–365 (2013). [DOI] [PubMed] [Google Scholar]
- Winer J. et al. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem. 270, 41–49 (1999). [DOI] [PubMed] [Google Scholar]